
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRedox-controlled chalcogen-bonding at tellurium: impact on Lewis acidity and chloride anion transport properties†
Benyu
Zhou
and
François P.
Gabbaï
 *
*
Department of Chemistry, Texas A&M University, College Station, Texas 77843-3255, USA. E-mail: francois@tamu.edu
First published on 6th July 2020
Abstract
Our interests in the chemistry of atypical main group Lewis acids have led us to devise strategies that augment the affinity of chalcogen-bond donors for anionic guests. In this study, we describe the oxidative methylation of diaryltellurides as one such strategy along with its application to the synthesis of [Mes(C6F5)TeMe]+ and [(C6F5)2TeMe]+ starting from Mes(C6F5)Te and (C6F5)2Te, respectively. These new telluronium cations have been evaluated for their ability to complex and transport chloride anions across phospholipid bilayers. These studies show that, when compared to their neutral Te(II) precursors, these Te(IV) cations display both higher Lewis acidity and transport activity. The positive attributes of these telluronium cations, which originate from a lowering of the tellurium-centered σ* orbitals and a deepening of the associated σ-holes, demonstrate that the redox state of the main group element provides a convenient handle over its chalcogen-bonding properties.
The field of chalcogen (Ch) bonding is in rapid expansion owing to applications in supramolecular chemistry, catalysis, and anion transport.1–11 The formation of chalcogen-bonds, which are defined as secondary bonding interactions between a donor (D) and a chalcogenide (ChR2, R = substituent),12 can be rationalized based on the presence of regions of positive electrostatic potential on the chalcogen atom. These regions often referred to as σ-holes, sit at the termini of Ch–R polar bonds and spatially coincide with the largest lobe of the Ch–R σ* orbitals (Fig. 1). Investigations into the origin of chalcogen-bonds point to the interplay of electrostatic, dispersion, and orbital delocalization effects.13,14 The dispersion term is accentuated in the case of softer, inherently polarizable, chalcogens, while the electrostatic term can be controlled by adjusting the polarity of Ch–R σ-bond. The orbital delocalization term corresponding to the D→ChR2 interaction responds favourably to a lowering of the Ch–R σ* orbital energy, as observed with heavy chalcogens. An analogous dependence is seen with respect to the polarity of the Ch–R bond which, when increased, leads to higher chalcogen parentage of the σ* orbital. All three bonding terms culminate at tellurium, the softest, heaviest, and most electropositive chalcogen.2,5,15,16
 | ||
| Fig. 1 (Left) Orbital and electrostatic origin of the Lewis acidity in tellurium derivatives. The intensification of the blue color illustrates an energy lowering in the case of the LUMO and a greater positive charge development at tellurium in the case of the σ-hole. | ||
(C6F5)2Te is a particularly noteworthy molecule that embodies the points discussed above. The chalcogen-bond donor properties of this derivative manifest in the short intermolecular Te–F contacts found in the crystal17 as well as in the formation of adducts with donors such as tetramethylthiourea.18 Matile brought (C6F5)2Te back into the spotlight by demonstrating its use in anion binding catalysis19 and chloride anion transport.20 The chalcogen-bond donor properties of (C6F5)2Te can be correlated to the electron-withdrawing properties of the pentafluorophenyl substituents, which deepen the σ-hole and stabilize the Te–C σ* orbital.
Motivated by these recent developments, we are charting new strategies to predictably enhance the chalcogen-bond donor properties of tellurium compounds, with the ultimate aim of reaching bona fide Lewis acidity. Based on the knowledge that telluronium cations form partially covalent halide complexes21–23 and encouraged by recent contributions on the effect of oxidation on the Lewis acidity of phosphorus,24–26 antimony,27–29 sulfur,30–32 and iodine species,33,34 we have now decided to probe the effect of oxidative alkylation of simple diaryltellurides as a way to stabilize the accepting Te–C σ* orbital while at the same time deepening the coincident σ-hole as illustrated in Fig. 1.
Considering stability, ease of synthesis, and synthetic modularity, we decided to target a series of telluronium cations obtained by direct methylation of the diaryltellurides shown in Scheme 1. While (C6F5)2Te and Mes2Te are known,17,35 we were able to obtain Mes(C6F5)Te (1) by the reaction of a C6F5MgBr solution with a solution of MesTeBr generated in situ by the combination of bromine and dimesitylditelluride. This new telluride has been characterized by traditional means, including multinuclear NMR spectroscopy and elemental analysis. These neutral tellurides were next converted into the corresponding methyltelluronium species. While the more electron-rich derivatives Mes2Te and 1 could be efficiently methylated using MeI/AgBF4 in CH2Cl2, methylation of (C6F5)2Te only proceeded with Me3O·BF4 upon heating to 90 °C in a mixture of 1,2-dichloroethane and toluene (vol. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2). The harsher conditions used for this last reaction reflects the electron-deficiency of the tellurium centre induced by the two pentafluorophenyl groups. The three new telluronium cations [Mes2TeMe]+ ([2]+), [Mes(C6F5)TeMe]+ ([3]+), and [(C6F5)2TeMe]+ ([4]+) have been isolated as their tetrafluoroborate salts and fully characterized. All three salts give well-resolved 1H and 19F-NMR spectra. Installation of the methyl group gives rise to a singlet in the 1H-NMR spectra of these derivatives. This singlet, which appears in the 3.17–3.45 ppm window for all three salts, is flanked by 125Te satellites separated by 2JTe–H = 29.1–31.2 Hz. These coupling constants are consistent with those observed for known methyltelluronium species.36 The 125Te-NMR resonance of the telluronium cations appears at 563.7 ppm for [2]+, 637.4 ppm for [3]+, and 598.1 for [4]+, a set of values that are distinctly downfield from those of the telluride precursors (258.7 ppm for Mes2Te, 280.0 ppm for 1, and 287.1 ppm for (C6F5)2Te).
:2). The harsher conditions used for this last reaction reflects the electron-deficiency of the tellurium centre induced by the two pentafluorophenyl groups. The three new telluronium cations [Mes2TeMe]+ ([2]+), [Mes(C6F5)TeMe]+ ([3]+), and [(C6F5)2TeMe]+ ([4]+) have been isolated as their tetrafluoroborate salts and fully characterized. All three salts give well-resolved 1H and 19F-NMR spectra. Installation of the methyl group gives rise to a singlet in the 1H-NMR spectra of these derivatives. This singlet, which appears in the 3.17–3.45 ppm window for all three salts, is flanked by 125Te satellites separated by 2JTe–H = 29.1–31.2 Hz. These coupling constants are consistent with those observed for known methyltelluronium species.36 The 125Te-NMR resonance of the telluronium cations appears at 563.7 ppm for [2]+, 637.4 ppm for [3]+, and 598.1 for [4]+, a set of values that are distinctly downfield from those of the telluride precursors (258.7 ppm for Mes2Te, 280.0 ppm for 1, and 287.1 ppm for (C6F5)2Te).
 | ||
| Scheme 1 Synthesis of the telluronium salts investigated in this study. | ||
The structures of these new telluronium salts have also been confirmed by X-ray crystallography (Fig. 2).37 In the solid-state, short Te–F contacts between the tellurium atom and the tetrafluoroborate counter anion are observed. The shortest contact, which is much smaller than the sum of the van der Waals radii of Te and F (Σvdw(Te–F) = 3.45 Å),38 is observed in the case of [4][BF4] which displays a Te⋯F distance of 2.6902(7) Å (Fig. 2). This distance is approximately 0.3 Å shorter than in [2][BF4] (3.066(5) Å) and 0.1 Å shorter than in [3][BF4] (2.7908(15) Å), attesting to the higher Lewis acidity of the tellurium centre in the bis(pentafluorophenyl) cation [4]+. The value of the C11–Te–F6 angle in [3][BF4] (168.56°) and C8–Te–F11 angle in [4][BF4] (167.14°) shows that the fluorine atom involved in the short contact is positioned trans from the pentafluorophenyl ring. This position corresponds to that defined by the lowest-lying σ* orbital or deepest σ-hole in these two salts, underscoring the importance of the molecular design principles presented in Fig. 1. The higher Lewis acidity of the tellurium atom in [4]+ is further reflected by the presence of a second Te⋯F short contact of 2.814(1) Å involving the tetrafluoroborate anion of a neighboring, symmetry equivalent unit. The resulting C2–Te–F12′ angle of 171.86° shows that this second contact occurs trans from the second pentafluorophenyl substituent of [4]+. It follows that [4]+ could, in principle, support interactions with two anionic guests.
 | ||
| Fig. 2 (Top) Crystal structures of [2]BF4, [3]BF4, and [4]BF4 (from left to right). Ellipsoids are drawn as 50% probability level. All the hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and bond angles (deg): [2]BF4: Te–C1 2.151(8), Te–C2 2.140(7), Te–C11 2.147(7), Te–F4 3.066(5), C1–Te–C2 109.0(3), C1–Te–C11 96.9(3), C2–Te–C11 99.2(3), C11–Te–F4, 172.8(2); [3]BF4: Te–C1 2.121(2), Te–C2 2.113(2), Te–C11 2.115(2), Te–F6 2.7908(15), C1–Te–C2 101.06(10), C1–Te–C11 95.43(9), C2–Te–C11 102.14(8), C11–Te–F6 168.56(7); [4]BF4: Te–C1 2.1212(10), Te–C2 2.1140(9), Te–C8 2.1114(9), Te–F11 2.6902(11), Te–F12′ 2.8138(8), C1–Te–C2 99.88(4), C1–Te–C8 96.00(4), C2–Te–C8 91.33(3), C8–Te–F11 167.14(3), C2–Te–F12′ 171.86(3). The optimized structures and ESP maps of [2]+, [3]+, and [4]+ are shown to the right of each corresponding crystal structures. (Bottom) Optimized structures and ESP maps of Mes2Te, Mes(C6F5)Te, and (C6F5)2Te. ESPs were drawn with an isosurface value of 0.001 au. The Vs,max are given in kcal mol−1. | ||
The telluronium cations have also been studied by Density Functional Theory (M062X functional; mixed basis set: 6-31G(d′)/6-31G+(d′)/cc-pVTZ-PP). To better compare the electrostatic potential about the tellurium centre, ESP maps were plotted on the same scale (Fig. 2). All cations feature pronounced σ-holes which can be conveniently characterized by their corresponding Vs,max values. The Vs,max values of the two σ-holes are almost identical to one another in the case of [2]+ (Vs,max = 103.1 and 104.0 kcal mol−1) and [4]+ (Vs,max = 136.0 and 138.3 kcal mol−1) which feature two identical aryl substituents. In the case of [3]+, the Vs max values of the two σ-holes (116.3 and 122.3 kcal mol−1) show a larger differentiation, with the largest value corresponding to the hole trans from the electron-withdrawing pentafluorophenyl substituent. The electron-withdrawing properties of this substituent are also responsible for the significant deepening of the σ-holes (or increase in Vs,max) upon transitioning from [2]+ to [4]+. This trend is paralleled by a decrease in the energies of the lowest unoccupied molecular orbitals observed upon sequential replacement of the mesityl substituents by pentafluorophenyl substituents. This trend is illustrated in Fig. 3. The largest energy drop is observed upon the introduction of the first pentafluorophenyl group in [3]+ whose LUMO energy is 1.00 eV lower than that of [2]+. The substitution of the second mesityl group induces a further stabilization of the LUMO by 0.75 eV, making [4]+ the most electron-deficient cation of this series. The LUMO+1 shows a similar energy ordering within this series of compounds and lay only 0.24 eV above the LUMO in the case of [2]+ and [4]+ and 0.6 eV in the case of [3]+. The LUMO and LUMO+1 of these compounds bear strong parentage from the σ*(Te–C) orbitals, thereby defining the site of expected maximum Lewis acidity in these compounds. The larger LUMO–LUMO+1 separation in [3]+ illustrates the differing electronegativity of the pentafluorophenyl and mesityl substituents in this compound. In [4]+, the presence of two closely spaced, low lying vacant orbitals indicates the presence of two accessible Lewis acidic sites, a conclusion supported by the observation of two short Te⋯F-BF3− contacts in the structure of [4][BF4].
 | ||
| Fig. 3 Energy level and pictorial representation of LUMO and LUMO+1 orbitals of the telluronium cations [2]+, [3]+, [4]+. | ||
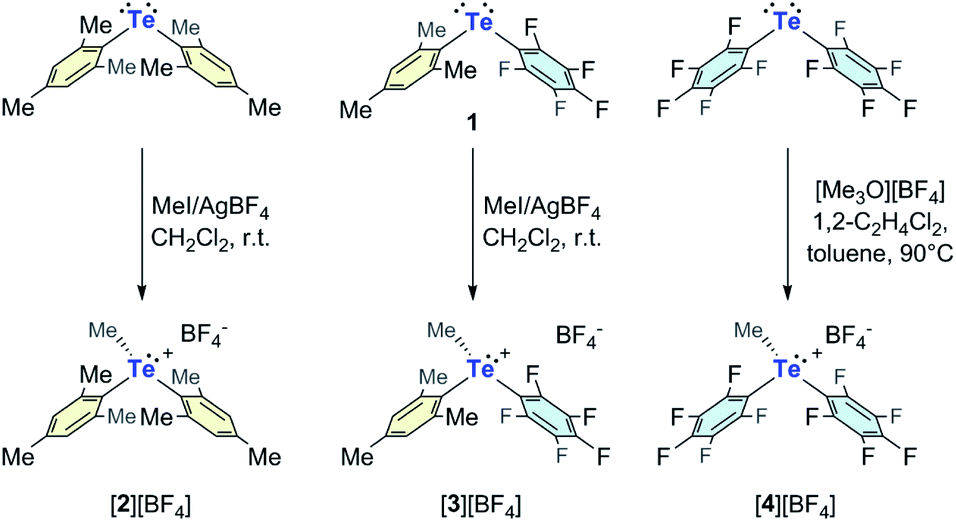
Returning to the main objective of this study which is to establish the beneficial impact of oxidation of the tellurium center on the chalcogen-bond donor properties of these derivatives, the ESP maps of [2]+, [3]+, and [4]+ can be compared to those of their neutral precursors (Fig. 2). In all cases, we observe that the Vs,max values in the cations are significantly higher than those of the neutral precursor, indicating that the cationic systems should indeed be better chalcogen-bond donors. Comparison of the LUMO energies provides a consistent picture, with those of the neutral derivatives Mes2Te, Mes(C6F5)Te, and (C6F5)2Te being >4 eV higher than those of [2]+, [3]+, and [4]+ (see ESI for details†). Because electrostatic effects are unscreened and thus magnified in the gas-phase, we have also investigated the effect of solvation on the electronic property differences observed between the telluronium cations and their neutral precursors. The results obtained with the SMD solvation model and water as a solvent are presented in Fig. 4 for 1 and [3]+. These results show that solvation dampens the extent of LUMO stabilization upon conversion of 1 into [3]+.
 | ||
| Fig. 4 (a) Changes observed in the energy of the LUMO and ESP map upon conversion of 1 into [3]+ (Vs,max unit: kcal mol−1). (b and c) 19F NMR spectra recorded upon addition of TBACl to a MeCN solution of 1 (b) and [3]BF4 (c). Cl− equiv. (eq(Cl−)) used for in the titrations: eq(Cl−) = 0, 0.2, 0.4, 0.6, 0.8, 1.0, 1.4, 1.8, 2.2, 2.6, 3.0, 3.8, 4.6, 5.4, 6.2, 7.0, 9.8 for spectra a–q recorded with 1 (panel (b)); eq(Cl−) = 0, 0.18, 0.36, 0.54, 0.72, 0.90, 1.08, 1.25, 1.44, 1.62, 1.79, 2.15, 2.51, 2.87, 3.23 for spectra a–o recorded with [3]BF4 (panel (c)). | ||
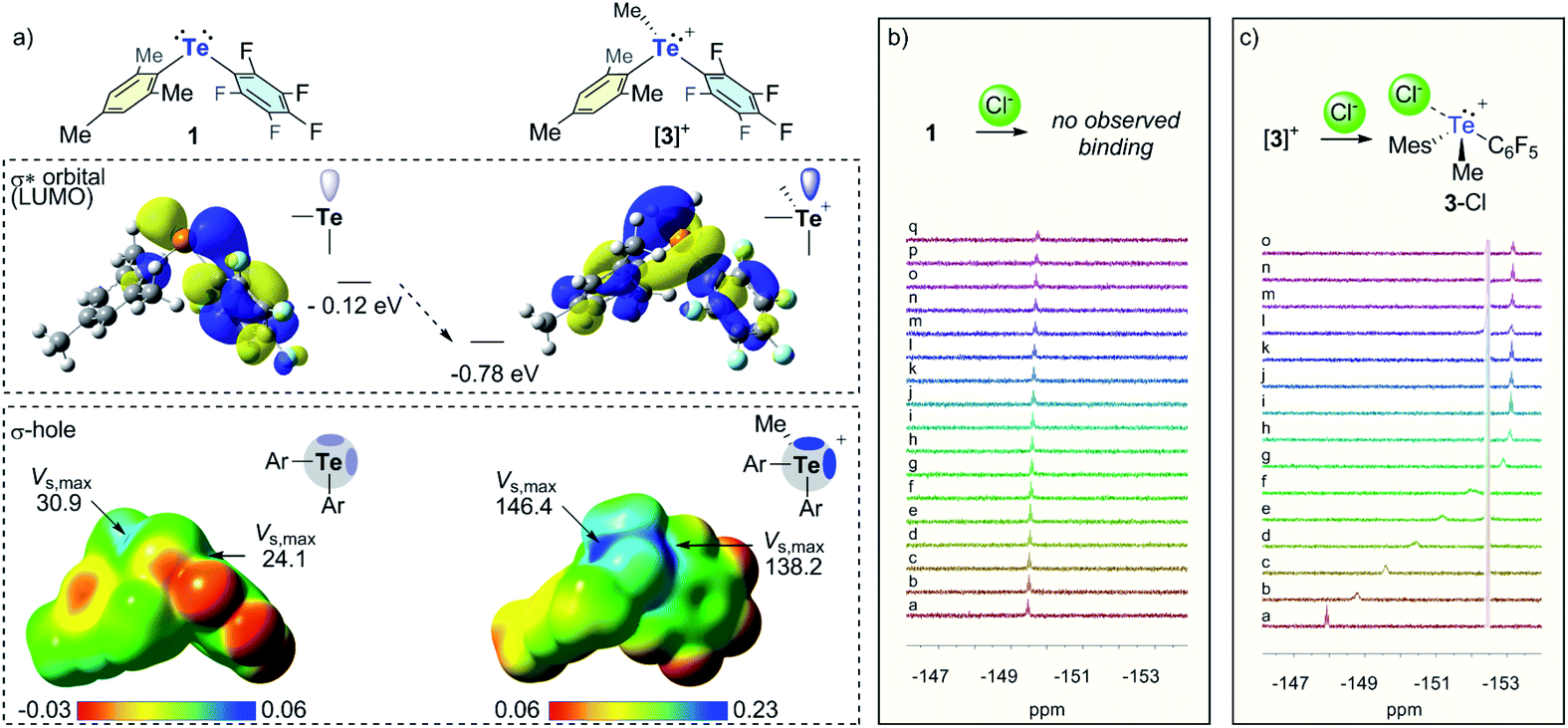
The above computational results predict that the telluronium cations should be more Lewis acidic than their neutral, divalent precursors. To verify this prediction, we became eager to compare the anion binding properties of the most electron deficient cations [3]+ and [4]+ to those of their neutral precursors Mes(C6F5)Te (1) and (C6F5)2Te, respectively. We first chose to evaluate the chloride anion affinity of [3]+ and [4]+ by carrying out 19F NMR titrations in MeCN as illustrated in Fig. 4 for [3]+. These experiments reveal that [3]+ binds one chloride anion with an association constant of 1.2 × 104 (± 6 × 103) M−1. The situation presented by [4]+ is more complicated in that it displays two binding events (Scheme 2, see ESI for additional details†). The first binding event is essentially quantitative, suggesting an association constant K1 ≥ 105. This finding is consistent with the partially covalent nature of electron-deficient triorganyltellurium(IV) chloride derivatives such as (C6F5)3TeCl.22 The first binding event is followed by a second one, which we interpret as the coordination of a second chloride anion at the remaining σ-hole with an association constant K2 = 260 (±80) M−1 (Scheme 2). The smaller value of K2 against K1 is in agreement with the expected negative cooperativity between these two Lewis acidic sites. The chloride anion affinities displayed by [3]+ and [4]+ are respectively larger than those measured for their neutral precursors 1 and (C6F5)2Te, in the same solvent. Indeed, while 1 showed no measurable chloride anion affinity in MeCN (Fig. 4), a low binding constant of 36 M−1 was measured for (C6F5)2Te. The drastically increased chloride anion affinity of the cationic derivatives highlights the benefits of the approach illustrated in Fig. 1. Last but not least, we note that (C6F5)2Te had been previously evaluated in THF, affording a binding constant of 2000 M−1. The lower binding constant measured in MeCN shows the negative influence of a polar solvent such a MeCN that can saturate the tellurium Lewis acid while also more effectively solvating the chloride anion and the receptor.
 | ||
| Scheme 2 Chloride anion binding by [4]+ as inferred from 19F NMR spectroscopy. | ||
Given our interest in the development of Lewis acidic main group cations39,40 as anion transporters,41–47 we decided to conclude this study by comparing the chloride anion transport properties of the new telluronium cations [2]+, [3]+, and [4]+ with those of their neutral precursors. The main objective of these investigations was to assess the impact of increased Lewis acidity or chalcogen-bond donor properties on the transport activity of these derivatives. As a prelude to these studies, we tested the stability of [2]+, [3]+, and [4]+ in water/DMSO (9:1) and did not observe the decomposition of the compounds (see ESI†). Next, we prepared large unilamellar vesicles (LUVs) using EYPC (egg-yolk phosphatidylcholine) as a phospholipid and a concentrated KCl solution as a cargo (Fig. 5). These freshly prepared LUVs were subjected to transporter-mediated chloride anion efflux experiments monitored with an ion-selective electrode.48 In all cases, anion transport was coupled with the addition of valinomycin as a potassium ion transporter. We first investigated the neutral derivatives with the intent of comparing the activity of Mes2Te and 1 to that of (C6F5)2Te, a derivative reported to be a potent chloride anion transporter.20 While (C6F5)2Te indeed mediated fast chloride efflux, the activity of Mes2Te and 1 could not be discerned from the background efflux induced by the addition of DMSO alone (Fig. 5). The superiority of (C6F5)2Te is assigned to the presence of two electron-withdrawing groups and their impact on the chalcogen-bond donor properties of the tellurium centre. It is also possible that the steric bulk of the mesityl group of Mes2Te and 1 has a detrimental influence, especially in the case of 1. After evaluating these neutral derivatives, we turned our attention to the telluronium cations. While [2]+ showed no activity, [3]+ promoted a very rapid release of the chloride anion cargo, reaching almost 100% after 4.5 minutes. The contrasting behaviour of [2]+ and [3]+ underscores the importance of the role played by the pentafluorophenyl group. At the same time, the greater activity of [3]+ when compared to its neutral precursor 1 illustrates the benefit that results from the oxidative methylation of the tellurium center. A comparison of the initial chloride efflux rate kini obtained for (C6F5)2Te (0.40%·s−1) and [3]+ (0.75%·s−1) indicate that the latter is distinctly more active. The superiority of [3]+ is further illustrated by a comparison of the EC50 values obtained from a Hill analysis, which shows that the value obtained for [3]+ (0.20 mol%) is ∼five times lower than that of (C6F5)2Te (1.1 mol%). Finally, we also evaluated the more Lewis acidic cation [4]+. We observed that it outperforms [3]+ as well as its neutral precursor, as indicated by the accelerated initial chloride efflux rate (kini = 1.68%·s−1) and a low EC50 value of 0.13 mol%. This last result suggests a positive correlation between the Lewis acidity and the transport activity of these tellurium derivatives. A similar correlation was established in a prior study dealing with stibonium cations as transporters.39 The higher transport activity of [3]+ and [4]+ is notably higher than that of (C6F5)2Te but not disproportionately so. It is possible that the high Lewis acidity of these two cationic compounds impedes chloride anion release, thereby dampening their transport activity.49,50 Experiments carried out with [3]+ and [4]+ with chloride-loaded DPPC (DPPC = 1,2-dipalmitoyl-sn-glycero-3-phosphtidylcholine) vesicles show a temperature dependence consistent with a carrier-type mechanism (see ESI†), as observed for other transporters based on main group cations.39,40 Finally, [3]+ and [4]+ do not induce significant leakage when administered to vesicles loaded with the self-quenching dye 5(6)-carboxyfluorescein. This last result indicates that these cationic derivatives do not act as surfactants and leave the phospholipid membrane unaffected.
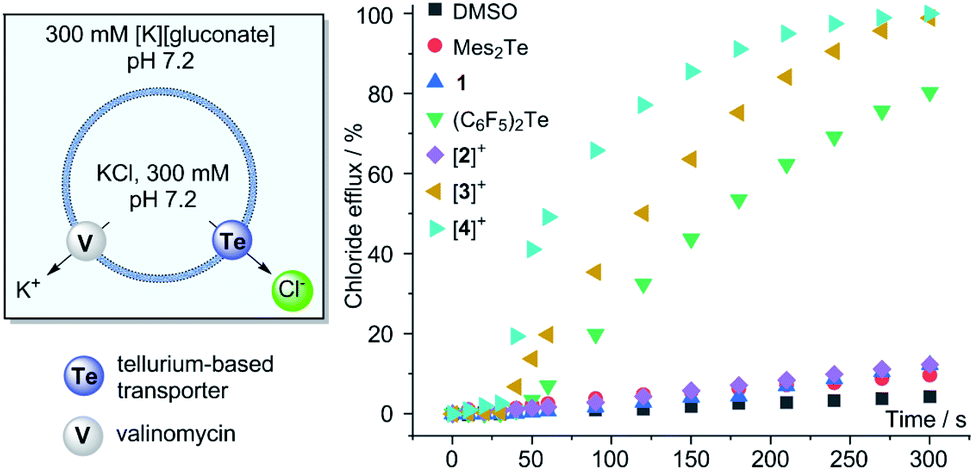 | ||
| Fig. 5 (Left) Simplified graphical summary of the chloride anion efflux experiment using EYPC LUVs in the presence of valinomycin (0.1 mol% with respect to lipid concentration). The medium was buffered using HEPES (10 mM). Transporters were added as a solution in DMSO (7 μL) to reach a final concentration of 2 mol% with respect to the lipid concentration. (Right) Chloride efflux data recorded with an ion selective electrode. The transporter is added 30 s after addition of valinomycin. | ||
In summary, this work defines the oxidative alkylation of diorganotellurides as a simple strategy for enhancing the chalcogen-bond donor properties of the tellurium center. This enhancement correlates with a lowering of the Te–C σ* orbitals and the presence of deeper σ-holes in the resulting telluronium cations. As indicated by chloride anion binding studies, the enhanced chalcogen-bond donor properties of this derivatives lead to the emergence of classical Lewis acidity. A positive outcome is also observed in the context of transmembrane chloride anion transport, with the telluronium cations displaying a higher activity than their neutral telluride precursors. We propose that this strategy may open the door to redox switchable chalcogen-bonding/group 16-centered Lewis acidity, a phenomenon that could be exploited for the design of anion transporters that respond to changes in the redox state of the medium.31 Finally, we note that the concept introduced in this paper is related to previously published strategies relying on the oxidation of a remote main group center as a means to enhance the activity of anion transporters.51,52
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Support from the Welch Foundation (Grant A-1423), the National Science Foundation (CHE-1856453), Texas A&M University (Arthur E. Martell Chair of Chemistry), and the Laboratory for Molecular Simulation at Texas A&M University (software and computational resources) is gratefully acknowledged.References
- N. Sudha and H. B. Singh, Coord. Chem. Rev., 1994, 135, 469–515 CrossRef.
- A. F. Cozzolino, P. J. W. Elder and I. Vargas-Baca, Coord. Chem. Rev., 2011, 255, 1426–1438 CrossRef CAS.
- M. Breugst, D. von der Heiden and J. Schmauck, Synthesis, 2017, 49, 3224–3236 CrossRef CAS.
- K. T. Mahmudov, M. N. Kopylovich, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Dalton Trans., 2017, 46, 10121–10138 RSC.
- R. Gleiter, G. Haberhauer, D. B. Werz, F. Rominger and C. Bleiholder, Chem. Rev., 2018, 118, 2010–2041 CrossRef CAS PubMed.
- K. Selvakumar and H. B. Singh, Chem. Sci., 2018, 9, 7027–7042 RSC.
- J. Y. C. Lim and P. D. Beer, Chem, 2018, 4, 731–783 CAS.
- J. Bamberger, F. Ostler and O. G. Mancheño, ChemCatChem, 2019, 11, 5198–5211 CrossRef CAS PubMed.
- L. Vogel, P. Wonner and S. M. Huber, Angew. Chem., Int. Ed., 2019, 58, 1880–1891 CrossRef CAS PubMed.
- K. Strakova, L. Assies, A. Goujon, F. Piazzolla, H. V. Humeniuk and S. Matile, Chem. Rev., 2019, 119, 10977–11005 CrossRef PubMed.
- N. Biot and D. Bonifazi, Coord. Chem. Rev., 2020, 413, 213243 CrossRef CAS.
- N. W. Alcock, Secondary Bonding to Nonmetallic Elements in Advances in Inorganic Chemistry and Radiochemistry, ed. H. J. Emeléus and A. G. Sharpe, Academic Press, 1972, vol. 15, pp. 1–58 Search PubMed.
- C. Bleiholder, D. B. Werz, H. Köppel and R. Gleiter, J. Am. Chem. Soc., 2006, 128, 2666–2674 CrossRef CAS.
- D. J. Pascoe, K. B. Ling and S. L. Cockroft, J. Am. Chem. Soc., 2017, 139, 15160–15167 CrossRef CAS PubMed.
- P. Wonner, T. Steinke, L. Vogel and S. M. Huber, Chem. - Eur. J., 2020, 26, 1258–1262 CrossRef CAS PubMed.
- P. Wonner, A. Dreger, L. Vogel, E. Engelage and S. M. Huber, Angew. Chem., Int. Ed., 2019, 58, 16923–16927 CrossRef CAS PubMed.
- T. M. Klapotke, B. Krumm, P. Mayer, K. Polborn and O. P. Ruscitti, Inorg. Chem., 2001, 40, 5169–5176 CrossRef CAS PubMed.
- S. Aboulkacem, D. Naumann, W. Tyrra and I. Pantenburg, Organometallics, 2012, 31, 1559–1565 CrossRef CAS.
- S. Benz, A. I. Poblador-Bahamonde, N. Low-Ders and S. Matile, Angew. Chem., Int. Ed., 2018, 57, 5408–5412 CrossRef CAS PubMed.
- L. M. Lee, M. Tsemperouli, A. I. Poblador-Bahamonde, S. Benz, N. Sakai, K. Sugihara and S. Matile, J. Am. Chem. Soc., 2019, 141, 810–814 CrossRef CAS PubMed.
- M. J. Collins, J. A. Ripmeester and J. F. Sawyer, J. Am. Chem. Soc., 1987, 109, 4113–4115 CrossRef CAS.
- T. M. Klapötke, B. Krumm, P. Mayer, K. Polborn and O. P. Ruscitti, J. Fluorine Chem., 2001, 112, 207–212 CrossRef.
- H. Y. Zhao and F. P. Gabbaï, Nat. Chem., 2010, 2, 984–990 CrossRef CAS PubMed.
- J. M. Bayne and D. W. Stephan, Chem. Soc. Rev., 2016, 45, 765–774 RSC.
- M. H. Holthausen, M. Mehta and D. W. Stephan, Angew. Chem., Int. Ed., 2014, 53, 6538–6541 CrossRef CAS PubMed.
- R. J. Andrews, S. S. Chitnis and D. W. Stephan, Chem. Commun., 2019, 55, 5599–5602 RSC.
- D. Tofan and F. P. Gabbaï, Chem. Sci., 2016, 7, 6768–6778 RSC.
- M. Yang, D. Tofan, C.-H. Chen, K. M. Jack and F. P. Gabbaï, Angew. Chem., Int. Ed., 2018, 57, 13868–13872 CrossRef CAS PubMed.
- D. You and F. P. Gabbaï, Trends Chem., 2019, 1, 485–496 CrossRef.
- Y. Kim, H. Zhao and F. P. Gabbaï, Angew. Chem., Int. Ed., 2009, 48, 4957–4960 CrossRef CAS PubMed.
- H. Zhao and F. P. Gabbaï, Org. Lett., 2011, 13, 1444–1446 CrossRef CAS PubMed.
- F. A. Tsao, A. E. Waked, L. Cao, J. Hofmann, L. Liu, S. Grimme and D. W. Stephan, Chem. Commun., 2016, 52, 12418–12421 RSC.
- F. Heinen, E. Engelage, A. Dreger, R. Weiss and S. M. Huber, Angew. Chem., Int. Ed., 2018, 57, 3830–3833 CrossRef CAS PubMed.
- T. L. Seidl and D. R. Stuart, J. Org. Chem., 2017, 82, 11765–11771 CrossRef CAS PubMed.
- M. Oba, Y. Okada, M. Endo, K. Tanaka, K. Nishiyama, S. Shimada and W. Ando, Inorg. Chem., 2010, 49, 10680–10686 CrossRef CAS PubMed.
- N. S. Dance, W. R. McWhinnie, J. Mallaki and Z. Monsef-Mirzai, J. Organomet. Chem., 1980, 198, 131–143 CrossRef CAS.
- CCDC 2004978–2004980 contain the supplementary crystallographic data for this paper.† The chiral telluronium salt [3][BF4] crystallizes in the centrosymmetrical P21/n space group, with the two enantiomers in the unit cell.
- S. Alvarez, Dalton Trans., 2013, 42, 8617–8636 RSC.
- G. Park, D. J. Brock, J.-P. Pellois and F. P. Gabbaï, Chem, 2019, 5, 2215–2227 CAS.
- G. Park and F. P. Gabbaï, Angew. Chem., Int. Ed., 2020, 59, 5298–5302 CrossRef CAS PubMed.
- A. P. Davis, D. N. Sheppard and B. D. Smith, Chem. Soc. Rev., 2007, 36, 348–357 RSC.
- J. T. Davis, O. Okunola and R. Quesada, Chem. Soc. Rev., 2010, 39, 3843–3862 RSC.
- S. Matile, A. Vargas Jentzsch, J. Montenegro and A. Fin, Chem. Soc. Rev., 2011, 40, 2453–2474 RSC.
- P. A. Gale, R. Perez-Tomas and R. Quesada, Acc. Chem. Res., 2013, 46, 2801–2813 CrossRef CAS PubMed.
- A. Vargas Jentzsch, A. Hennig, J. Mareda and S. Matile, Acc. Chem. Res., 2013, 46, 2791–2800 CrossRef CAS PubMed.
- P. A. Gale, J. T. Davis and R. Quesada, Chem. Soc. Rev., 2017, 46, 2497–2519 RSC.
- X. Wu, E. N. W. Howe and P. A. Gale, Acc. Chem. Res., 2018, 51, 1870–1879 CrossRef CAS PubMed.
- S. Matile, N. Sakai and A. Hennig, Transport Experiments in Membranes in Supramol. Chem., ed. P. A. Gale and J. W. Steed, Wiley, Chichester, U.K., 2012, vol. 2, p. 473 Search PubMed.
- J. P. Behr, M. Kirch and J. M. Lehn, J. Am. Chem. Soc., 1985, 107, 241–246 CrossRef CAS.
- A. Vargas Jentzsch, D. Emery, J. Mareda, P. Metrangolo, G. Resnati and S. Matile, Angew. Chem., Int. Ed., 2011, 50, 11675–11678 CrossRef CAS PubMed.
- S. Benz, M. Macchione, Q. Verolet, J. Mareda, N. Sakai and S. Matile, J. Am. Chem. Soc., 2016, 138, 9093–9096 CrossRef CAS PubMed.
- A. J. Plajer, J. Zhu, P. Proehm, A. D. Bond, U. F. Keyser and D. S. Wright, J. Am. Chem. Soc., 2019, 141, 8807–8815 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details. NMR spectra of novel compounds. Anion binding and transport data. Crystallographic data in CIF format. CCDC 2004978–2004980. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02872j |
| This journal is © The Royal Society of Chemistry 2020 |