
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlling thermal expansion within mixed cocrystals by tuning molecular motion capability†
Xiaodan
Ding
 a,
Daniel K.
Unruh
a,
Ryan H.
Groeneman
b and
Kristin M.
Hutchins
*a
a,
Daniel K.
Unruh
a,
Ryan H.
Groeneman
b and
Kristin M.
Hutchins
*a
aDepartment of Chemistry and Biochemistry, Texas Tech University, Lubbock, Texas 79409, USA. E-mail: kristin.hutchins@ttu.edu
bDepartment of Biological Sciences, Webster University, St. Louis, Missouri 63119, USA
First published on 5th June 2020
Abstract
Controlling thermal expansion (TE) behaviors of organic materials is challenging because several mechanisms can govern TE, such as noncovalent interaction strength and structural motions. Here, we report the first demonstration of tuning TE within organic solids by using a mixed cocrystal approach. The mixed cocrystals contain three unique molecules, two of which are present in variable ratios. These two molecules either lack or exhibit the ability to undergo molecular motion in the solid state. Incorporation of higher ratios of motion-capable molecules results in larger, positive TE along the motion direction. Addition of a motion-incapable molecule affords solids that undergo less TE. Fine-tuned TE behavior was attained by systematically controlling the ratio of motion-capable and -incapable molecules in each solid.
Introduction
Thermal expansion (TE) is the response of a material to changes in temperature. TE is classified as either positive, negative, or zero, and can occur along an axis or volumetrically.1 Positive TE (PTE) is the most common behavior in materials2,3 and involves an increase in size upon heating. Alternatively, negative TE (NTE) is rarer4–6 and corresponds to a decrease in size upon heating. Lastly, near-zero or zero TE materials are quite rare7–9 and are characterized by minimal to no change in response to temperature.TE behaviours for inorganic materials like concrete, glass, and metal-based solids are relatively well understood.10 In general, inorganic solids are typically held together in all three dimensions through covalent or well-defined metal–organic coordination bonds. The response of these bonds to temperature is often predictable due to the defined nature of the network and strength of bonds holding the solid together, although anomalous TE behaviours have been reported.11–13 On the other hand, designing and controlling TE behaviours for purely organic molecular materials remains challenging.14–17 Organic solids are sustained by noncovalent interactions and controlling these forces in each crystallographic dimension is not trivial. For molecular solids, the strength of the interactions holding the solid together is one key factor that affects TE behavior.18,19
Many recent studies have demonstrated that organic molecules are capable of motion in the crystalline state including rotation,20–22 flipping,23 and isomerization.24 These studies motivated us to investigate organic molecules functionalized with groups capable of undergoing molecular motion in the solid state and determine if such motion impacts TE. We have demonstrated molecular pedal motion, which resembles the motion of pedaling a bicycle,25 gives rise to large PTE that occurs along the direction where motion happens26,27 (Scheme 1). Importantly, motion occurrence is quantifiable through variable-temperature X-ray diffraction experiments.
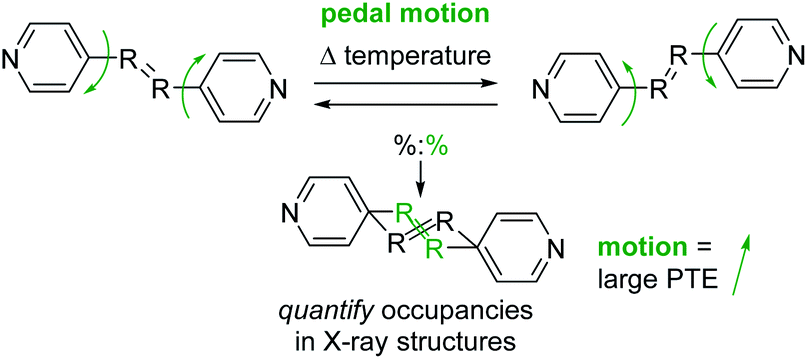 | ||
| Scheme 1 Molecular pedal motion affords large PTE. Bipyridine derivative shown above. The motion group and rings are both disordered in the X-ray structures. | ||
Cocrystallization, incorporation of at least two unique compounds into a single-phase solid, offers the opportunity to modify solid-state structures and achieve desirable properties.28,29 The properties of the cocrystal typically differ from the individual components. Most cocrystals contain two components (Scheme 2), although higher order cocrystals have recently been synthesized.30 The bulk properties of cocrystals such as solubility,31 reactivity,32 mechanical,33,34 thermal,35 or optical36 are often modified by systematically changing one of the components.
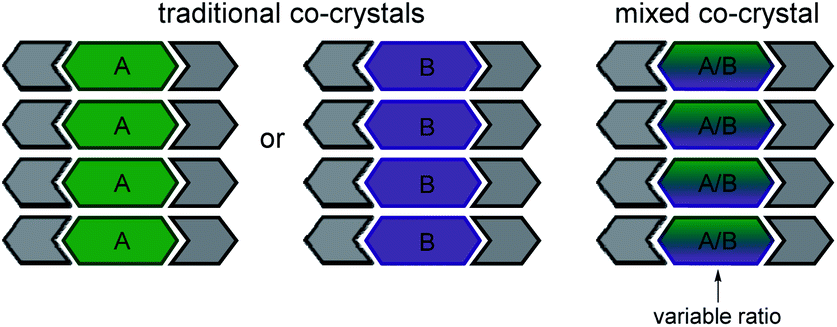 | ||
| Scheme 2 Comparison of traditional and mixed cocrystals. | ||
Mixed cocrystals (i.e. cocrystal solid solutions) are crystalline solids wherein the structural function of one conformational isomer is taken up by two or more molecules present in a variable ratio37,38 (Scheme 2). The ability to synthesize mixed cocrystals is achieved by using similarly shaped molecules that can readily replace each other at equivalent crystallographic positions. The field of traditional cocrystals has seen significant growth in the past two decades; however, mixed cocrystals remain underdeveloped. Mixed cocrystals could provide a novel strategy for systematically tuning TE behavior of organic solids in a modular fashion. Rather than changing one component of the material entirely, multiple components are incorporated and each component lends a unique property to the material. The approach is similar to using multiple ligands in a metal–organic framework4 that impart different functions.
Specifically, we sought to synthesize mixed cocrystals featuring components that can or cannot undergo pedal motion, which would afford larger or smaller TE, respectively, as a function of pedal motion ability. Moreover, controlling the ratio of each component allows the TE of the material to be fine-tuned. Azo (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) and ethylene (CC) groups are capable of pedal motion,39 while acetylene (C
N) and ethylene (CC) groups are capable of pedal motion,39 while acetylene (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C) groups are not. Recently, we demonstrated that bipyridines 4,4′-azopyridine (AP) and trans-1,2-bis(4-pyridyl)ethylene (BPE) undergo molecular pedal motion in hydrogen-bonded cocrystals with resorcinol (res), while 1,2-bis(4-pyridyl)acetylene (BPA) is motion incapable.26 Importantly, AP and BPE do not undergo pedal motion as single-component solids.40,41 In the cocrystals with res, AP underwent pedal motion over the entire temperature range, BPE experienced motion, but became ordered at 210 K, and BPA acted as a control. Cocrystals containing AP experienced the largest PTE, and cocrystals containing BPA experienced the least TE along the motion direction.26
C) groups are not. Recently, we demonstrated that bipyridines 4,4′-azopyridine (AP) and trans-1,2-bis(4-pyridyl)ethylene (BPE) undergo molecular pedal motion in hydrogen-bonded cocrystals with resorcinol (res), while 1,2-bis(4-pyridyl)acetylene (BPA) is motion incapable.26 Importantly, AP and BPE do not undergo pedal motion as single-component solids.40,41 In the cocrystals with res, AP underwent pedal motion over the entire temperature range, BPE experienced motion, but became ordered at 210 K, and BPA acted as a control. Cocrystals containing AP experienced the largest PTE, and cocrystals containing BPA experienced the least TE along the motion direction.26
Here, we describe the first use of mixed cocrystals as a platform for controlling and tuning TE behavior in organic materials. We synthesized a series of mixed cocrystals based on res and a mixture of hydrogen-bond acceptors, AP, BPE, or BPA (Scheme 3). A total of nine mixed cocrystals were synthesized, namely, res·(AP)x·(BPA)y, res·(BPE)x·(BPA)y, and res·(AP)x·(BPE)y, using three different ratios for each system (x + y = 1) to investigate and control the impact each component has on the overall TE behavior of the material. We demonstrate TE along the direction of pedal motion can be fine-tuned by controlling the amount of motion-capable (AP or BPE) or motion-incapable (BPA) molecules in the mixed cocrystal because motion affords larger PTE.
 | ||
| Scheme 3 Components and synthesis of mixed cocrystals. | ||
Experimental section
The nine mixed cocrystals were synthesized by incorporating two bipyridines and res. The molar ratio between res and the total of the two bipyridines was kept at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The bipyridine in each mixed cocrystal was incorporated at three different percentages, 25%, 50%, or 75% and the total amount of the two bipyridine components sums to 100%. The nine cocrystals are, res·(AP)0.75·(BPA)0.25, res·(AP)0.50·(BPA)0.50, res·(AP)0.25·(BPA)0.75, res·(BPE)0.75·(BPA)0.25, res·(BPE)0.50·(BPA)0.50, res·(BPE)0.25·(BPA)0.75, res·(AP)0.75·(BPE)0.25, res·(AP)0.50·(BPE)0.50, and res·(AP)0.25·(BPE)0.75. Each mixed cocrystal was synthesized by dissolving all three components individually; the resulting solutions were combined into one vial and allowed to evaporate slowly. For each mixed cocrystal, ethanol was used as the solvent for res, BPE, and AP and toluene was required for BPA. Crystals suitable for single-crystal X-ray diffraction formed within three days. For each mixed cocrystal, six full crystallographic data sets were collected on the same crystal at 290, 270, 250, 230, 210 and 190 K (Tables S1–S18†) to quantify the molecular pedal motion and calculate the TE parameters. There are 54 unique X-ray data sets in total. 1H NMR spectroscopy was used to quantify the percentages of each component in the crystalline solids. Single crystals were removed from each vial for analysis before all the solvent evaporated (Fig. S10–S18†).
:1 ratio. The bipyridine in each mixed cocrystal was incorporated at three different percentages, 25%, 50%, or 75% and the total amount of the two bipyridine components sums to 100%. The nine cocrystals are, res·(AP)0.75·(BPA)0.25, res·(AP)0.50·(BPA)0.50, res·(AP)0.25·(BPA)0.75, res·(BPE)0.75·(BPA)0.25, res·(BPE)0.50·(BPA)0.50, res·(BPE)0.25·(BPA)0.75, res·(AP)0.75·(BPE)0.25, res·(AP)0.50·(BPE)0.50, and res·(AP)0.25·(BPE)0.75. Each mixed cocrystal was synthesized by dissolving all three components individually; the resulting solutions were combined into one vial and allowed to evaporate slowly. For each mixed cocrystal, ethanol was used as the solvent for res, BPE, and AP and toluene was required for BPA. Crystals suitable for single-crystal X-ray diffraction formed within three days. For each mixed cocrystal, six full crystallographic data sets were collected on the same crystal at 290, 270, 250, 230, 210 and 190 K (Tables S1–S18†) to quantify the molecular pedal motion and calculate the TE parameters. There are 54 unique X-ray data sets in total. 1H NMR spectroscopy was used to quantify the percentages of each component in the crystalline solids. Single crystals were removed from each vial for analysis before all the solvent evaporated (Fig. S10–S18†).
Results and discussion
Single-crystal X-ray analyses revealed all nine mixed cocrystals crystallized in the centrosymmetric, triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and are isostructural. The asymmetric unit of each mixed cocrystal contains one res and one bipyridine-based molecule. The bipyridine site is occupied by two different acceptors (with AP and BPE being disordered at various temperatures). The two bipyridines in each mixed cocrystal were successfully modelled, and the abundance of each bridge group was determined by a free variable refinement. All site occupancies of the bipyridine-based molecules were refined to a total sum of one with SIMU, RIGU, and DFIX restraints and constraints applied to maintain reasonable ADP values and bond lengths. Akin to the traditional two-component cocrystals,26,42 each mixed cocrystal is dominated by discrete four-component assemblies held together by four O–H⋯N hydrogen bonds (Fig. 1 and Table S23†).
and are isostructural. The asymmetric unit of each mixed cocrystal contains one res and one bipyridine-based molecule. The bipyridine site is occupied by two different acceptors (with AP and BPE being disordered at various temperatures). The two bipyridines in each mixed cocrystal were successfully modelled, and the abundance of each bridge group was determined by a free variable refinement. All site occupancies of the bipyridine-based molecules were refined to a total sum of one with SIMU, RIGU, and DFIX restraints and constraints applied to maintain reasonable ADP values and bond lengths. Akin to the traditional two-component cocrystals,26,42 each mixed cocrystal is dominated by discrete four-component assemblies held together by four O–H⋯N hydrogen bonds (Fig. 1 and Table S23†).
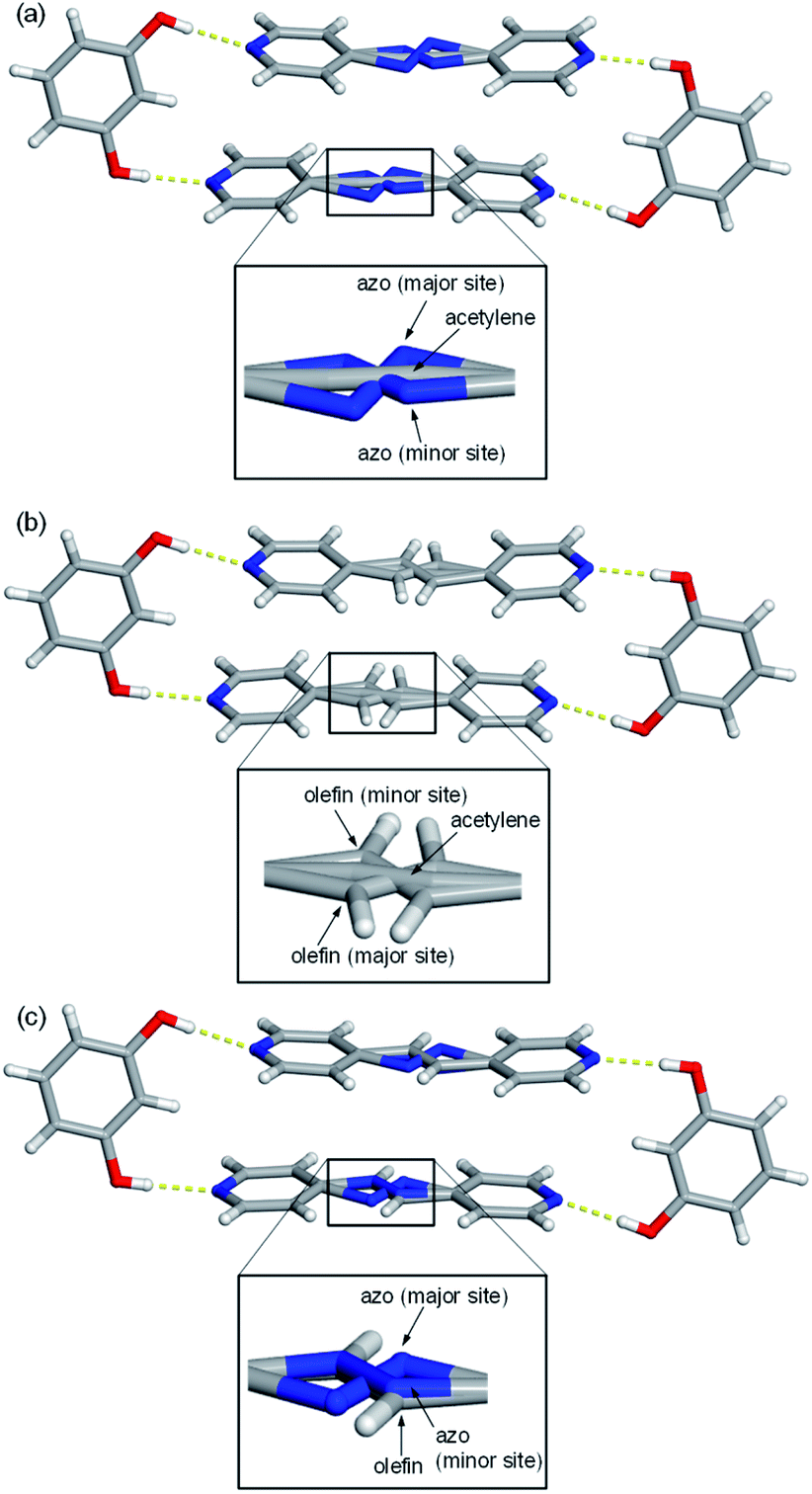 | ||
| Fig. 1 Representative hydrogen-bonded assemblies highlighting the three unique components of the bipyridine portion within: (a) res·(AP)x·(BPA)y, (b) res·(BPE)x·(BPA)y, (c) res·(AP)x·(BPE)y. Hydrogen bonds shown with yellow dashed lines. Ring disorder was omitted for clarity. | ||
In addition to the two main sites for the two unique bipyridines in each mixed cocrystal, AP and BPE are also capable of molecular pedal motion. Thus, in many of the mixed cocrystals, there are three sites for the bipyridine portion: the major site of the motion-capable molecule, the minor site of the motion-capable molecule, and the site of the second component (Fig. 1). For motion-capable molecules, the summation of the major and minor sites is equal to the total percentage of that molecule within the mixed cocrystal. The composition of each mixed cocrystal at 290 K based on synthetic feed, X-ray diffraction, and 1H NMR spectroscopy is outlined in Table 1. All results demonstrate good agreement with the theoretical ratios.
| Mixed cocrystal | Theoretical ratio from synthetic feed | Ratio determined by single-crystal X-ray diffraction at 290 K | Ratio determined by 1H NMR | ||
|---|---|---|---|---|---|
| First component (major/minor site) | First component (total occupancy) | Second component | |||
| res·(AP)0.75·(BPA)0.25 | 75:25 |
61/15 | 76 | 24 | 73:27 |
| res·(AP)0.50·(BPA)0.50 | 50:50 |
45/10 | 55 | 45 | 47:53 |
| res·(AP)0.25·(BPA)0.75 | 25:75 |
29/5 | 34 | 66 | 21:79 |
| res·(BPE)0.75·(BPA)0.25 | 75:25 |
66/9 | 75 | 25 | 80:20 |
| res·(BPE)0.50·(BPA)0.50 | 50:50 |
43/8 | 51 | 49 | 50:50 |
| res·(BPE)0.25·(BPA)0.75 | 25:75 |
30/0 | 30 | 70 | 28:72 |
| res·(AP)0.75·(BPE)0.25 | 75:25 |
64/13 | 77 | 23 | 74:26 |
| res·(AP)0.50·(BPE)0.50 | 50:50 |
37/11 | 48 | 52 | 47:53 |
| res·(AP)0.25·(BPE)0.75 | 25:75 |
14/9 | 23 | 77 | 22:78 |
To determine if the motion-capable molecule in each mixed cocrystal is undergoing pedal motion, the site occupancies at each temperature are quantified and compared (Tables S19–S21†). If the site occupancies change as a function of temperature, the molecule is undergoing dynamic pedal motion. If the numbers remain nearly constant or there is no disorder, then pedal motion is not occurring. Furthermore, if there is only one site, i.e. the minor site is equal to 0%, pedal motion is either not occurring or has ceased (Fig. 2).
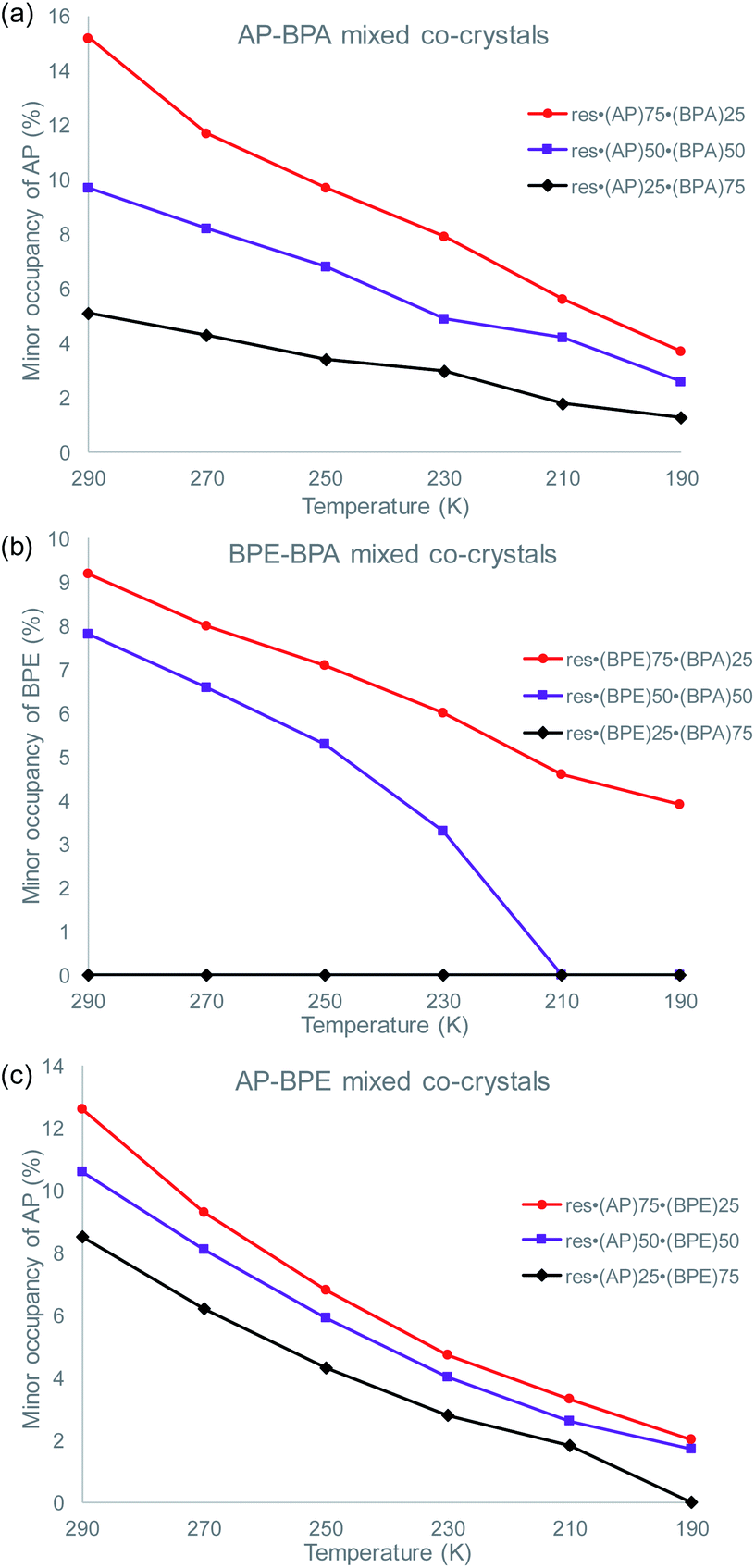 | ||
| Fig. 2 Change in the minor site occupancies of the motion-capable group as a function of temperature for: (a) res·(AP)x·(BPA)y, (b) res·(BPE)x·(BPA)y, and (c) res·(AP)x·(BPE)y. | ||
In the original two-component cocrystals over the temperature range of 290–190 K, AP underwent pedal motion over the whole range and BPE became ordered at 210 K.26 In the mixed cocrystals res·(AP)x·(BPA)y, AP undergoes dynamic pedal motion in all three systems (Fig. 2a). When AP is present in the lowest percentage (25%) the amount of pedal motion is lower as the minor site occupancies range from 1–5%. On the other hand, for the res·(BPE)x·(BPA)y mixed cocrystals, BPE only undergoes pedal motion when it is present at high percentages (75 and 50%, Fig. 2b). When BPE is present at 50%, the olefin becomes fully ordered at 210 K, and motion ceases. When BPE is present at only 25%, pedal motion was not detectable in the solid. Lastly, in the res·(AP)x·(BPE)y mixed cocrystals, only the AP component was modelled as undergoing pedal motion. The electron density for the minor component of AP was clearly apparent at 290 K. It is possible that the BPE component is undergoing pedal motion as well, and the minor site of BPE would overlap with the major site of AP. The AP component undergoes pedal motion over the entire range, similar to when it is in mixed cocrystals with BPA (Fig. 2c).
The extended crystal packing of all nine mixed cocrystals are also isostructural and correlate with the pure two-component cocrystals. The discrete four-component hydrogen-bonded assemblies stack slightly offset along the a-axis, and the pyridine rings of one assembly lie over a neighbouring assembly's bridge group (Fig. 3a). The stacked assemblies pack into offset layers extending along the b-axis (Fig. 3b). The layers interact through C–H(pyridine)⋯O and C–H(pyridine)⋯π(res) forces.
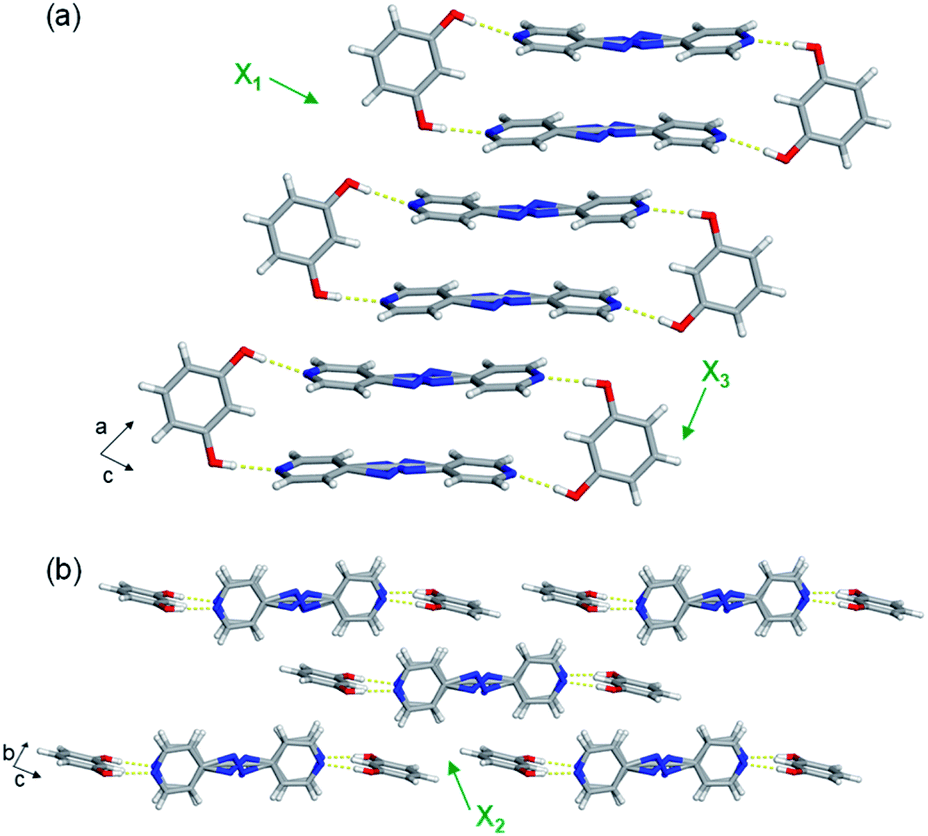 | ||
| Fig. 3 Representative (a) stacking and (b) layering of discrete hydrogen-bonded assemblies. Mixed cocrystal res·(AP)x·(BPA)y is shown as an example. Hydrogen bonds shown with yellow dashed lines. Ring disorder omitted for clarity. Directions of the principal axes shown with green arrows. | ||
To investigate the impact of multiple components and molecular pedal motion on the TE behaviours, PASCal43 was used to calculate the directions of the principal axes and linear TE coefficients for res·(AP)x·(BPA)y, res·(BPE)x·(BPA)y, and res·(AP)x·(BPE)y over the temperature range of 290 to 190 K using the unit cell parameters (Fig. S1–S9†). The values ranged from slight NTE to ‘colossal’44 PTE (Table 2).
| Mixed cocrystal | α X 1 (MK−1) [axis] | α X 2 (MK−1) [axis] | α X 3 (MK−1) [axis] | α V (MK−1) |
|---|---|---|---|---|
| res·(AP)0.75·(BPA)0.25 | −20 (1) [−1 −5 −4] | 84 (2) [1 −2 2] | 140 (2) [3 1 0] | 205 (3) |
| res·(AP)0.50·(BPA)0.50 | −13 (1) [1 4 4] | 82 (1) [1 6 −4] | 112 (2) [−5 −1 −1] | 183 (4) |
| res·(AP)0.25·(BPA)0.75 | −9 (1) [0 1 1] | 77 (1) [0 3 −2] | 110 (1) [−4 −1 −1] | 179 (1) |
| res·(BPE)0.75·(BPA)0.25 | −14 (1) [1 3 3] | 70 (1) [0 3 −2] | 131 (3) [−6 −2 −1] | 188 (3) |
| res·(BPE)0.50·(BPA)0.50 | −11 (1) [1 3 3] | 71 (1) [−1 8 −5] | 123 (2) [−6 −2 −1] | 183 (2) |
| res·(BPE)0.25·(BPA)0.75 | −9 (1) [1 4 5] | 70 (1) [0 4 −3] | 114 (1) [−6 −2 −1] | 177 (2) |
| res·(AP)0.75·(BPE)0.25 | −10 (1) [1 3 3] | 82 (1) [−1 4 −3] | 136 (5) [−3 −1 0] | 210 (4) |
| res·(AP)0.50·(BPE)0.50 | −12 (1) [1 3 4] | 79 (1) [0 2 −1] | 132 (2) [−6 −2 −1] | 200 (3) |
| res·(AP)0.25·(BPE)0.75 | −13 (1) [1 2 2] | 77 (1) [0 2 −1] | 127 (3) [−4 −1 0] | 192 (3) |
The least expansion occurs along the X1 axis, which coincides with the strongest noncovalent interactions, the O–H⋯N hydrogen bonds (Fig. 3a and Table S23†). In organic solids sustained by noncovalent bonds, the strongest forces are typically least affected by temperature.18,19 In each case, X1 was determined to be slightly negative, ranging from −9 to −20 MK−1. All nine mixed cocrystals exhibit similar TE coefficients along X1, which was expected because changing the bridge group does not drastically affect the hydrogen bonds lying along X1. In the mixed cocrystals, the O–H⋯N separations (between res and the major sites) increase slightly upon heating or decrease slightly upon heating by ca. 0.01 Å from 190–290 K (Table S23†). Thus, the net change in each case is minimal. The C–H(β)⋯O interactions between layers also contribute slightly to the expansion along X1, and the distances increase by 0.04 Å upon heating each mixed cocrystal. The slightly negative TE for all the mixed cocrystals is due to a small decrease in the distance between res molecules within an assembly, within a layer, and between layers upon heating.
All nine mixed cocrystals undergo moderate PTE along the X2 axis, and the direction encompasses the interactions between layers of the hydrogen-bonded assemblies (Fig. 3b). The interactions include C–H(pyridine)⋯O and C–H(pyridine)⋯π(res). The TE coefficients along X2 range from 70 to 84 MK−1, and are similar within a series of mixed cocrystals and between all nine mixed cocrystals. Thus, the bridge group does not significantly influence the expansion along the X2 direction. The C–H(pyridine)⋯O hydrogen bond distances increase upon heating for all mixed cocrystals by ca. 0.04–0.05 Å on average (Table S24†). The C–H(pyridine)⋯π(res) interactions increase by ca. 0.04 Å on average for res·(AP)x·(BPA)y and res·(AP)x·(BPE)y and by 0.03 Å for res·(BPE)x·(BPA)y upon heating (Table S24†). The TE coefficients along X2 are slightly smaller for res·(BPE)x·(BPA)y, which correlates to the smaller changes in interaction distances.
The largest and tunable difference in TE occurred along X3, which lies along the direction of molecular pedal motion (Table 2 and Fig. 3a). The TE coefficients along X3 are all ‘colossal’, ranging from 112 to 140 MK−1, but there are key differences between samples (Fig. 4a). The difference in these TE values correlates with the tendency of the bridge group to undergo molecular pedal motion, and the amount of motion-capable molecule within each mixed cocrystal.
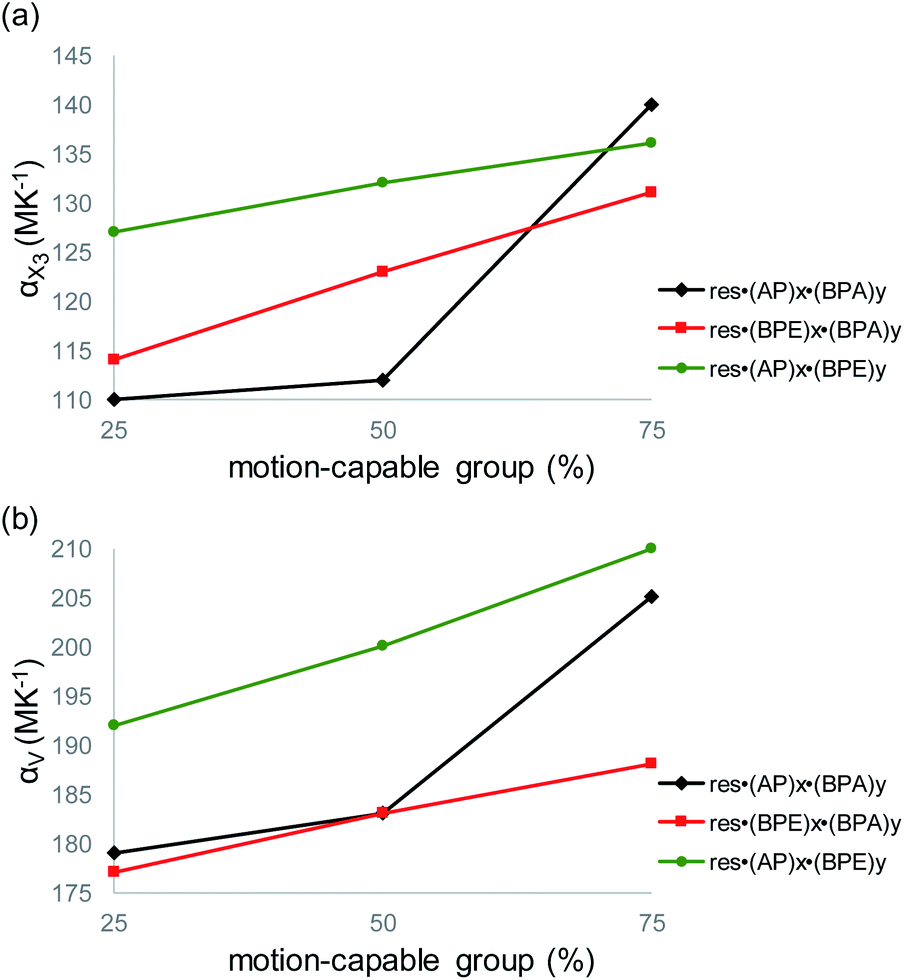 | ||
| Fig. 4 Variation in (a) TE coefficient along X3 and (b) volumetric TE coefficient as a function of the amount of motion-capable molecule within the mixed cocrystals. For res·(AP)x·(BPE)y, the motion-capable group (plotted along the x-axis) refers to AP. | ||
Mixed cocrystals yielding the largest expansion along X3 contain large ratios of motion-capable molecules and smaller ratios of BPA. The TE coefficients are 140 MK−1 for res·(AP)0.75·(BPA)0.25 and 131 MK−1 for res·(BPE)0.75·(BPA)0.25. Less expansion occurs along X3 in mixed cocrystals containing 75% or 50% BPA, which is unable to undergo pedal motion. Specifically, the TE coefficients are 110 and 114 MK−1 for res·(AP)0.25·(BPA)0.75 and res·(BPE)0.25·(BPA)0.75, respectively. The formation of mixed cocrystals with both motion-capable molecules, AP and BPE, removes BPA and should yield larger TE along X3. Indeed, the TE coefficients increase gradually from 127 to 132 to 136 MK−1 as the amount of AP is increased from 25% to 50% to 75%.
Taking standard deviations into account, there is a small overlap in the expansion along X3 when smaller amounts of motion-capable molecules are used for res·(AP)x·(BPA)y (Table 2). The most significant differences appear in mixed cocrystals when high percentages of motion-capable molecules are incorporated. Although BPA cannot undergo pedal motion, it can still contribute to TE in the solids along X3, primarily through changes in π–π stacking distances as a function of temperature. In the case of mixed cocrystals containing two motion-capable groups, there is also overlap in the expansion values along X3 when standard deviations are considered. This is likely due to the fact that both molecules can undergo motion in the solid and similar degrees of motion were observed in all three mixed cocrystals (Fig. 2c). Thus, the TE behavior along X3 can be tuned most readily by controlling the amount of motion-capable or -incapable molecules in the mixed cocrystals.
The supramolecular interactions contributing to the colossal expansion along X3 include primarily weak π–π stacking forces within and between assemblies (Fig. 3a). The π–π stacking distances within the four-component assemblies increase by ca. 0.04–0.05 Å in each mixed cocrystal upon heating, with res·(AP)0.75·(BPA)0.25 undergoing the most significant change (Table S25†). The π–π stacking distances between hydrogen-bonded assemblies also increase by ca. 0.04–0.05 Å upon heating. The occurrence of motion could also weaken intermolecular interaction strengths, allowing more expansion to occur along that direction.
The volumetric TE coefficient for each mixed cocrystal is ‘colossal’ and ranges from 177 to 210 MK−1 (Table 2 and Fig. 4b). The volume of each mixed cocrystal increases linearly upon warming (Fig. S19–S27†), and the volumetric expansion coefficients are largest for mixed cocrystals containing higher ratios of motion-capable molecules. The volumetric TE coefficients are consistently larger when the solid contains two motion-capable molecules, i.e.res·(AP)x·(BPE)y.
We also investigated the changes in the unit cell parameters at higher temperature data sets (290–250 K) versus lower temperature data sets (230–190 K, Tables S1–S18†) to determine if the TE differs due to different percentages of the motion-capable groups. For three of the mixed cocrystals, res·(AP)0.50·(BPA)0.50, res·(AP)0.25·(BPA)0.75, and res·(BPE)0.25·(BPA)0.75, there is less TE at lower temperatures (230–190 K). These systems feature lower percentages of motion-capable groups and higher percentages of motion-incapable BPA. Less TE at lower temperatures in these systems is in good agreement with our overall observations that higher percentages of motion-capable molecules afford larger PTE.
Saha and co-workers recently reported the influence of molecular width on TE.14 In particular, they observed that the smallest TE occurred along the direction of greatest molecular width. Treating the four-component hydrogen-bonded assembly in each mixed cocrystal as a rigid unit, the longest molecular width is the assembly including the O–H⋯N hydrogen bonds (Fig. 3a), which undergoes the smallest TE. In contrast, the greatest TE occurs along the π-stacked bipyridine layers, which correlates to pedal motion and represents shortest molecular width. These widths also correlate with the interaction strengths. Along the assembly width, stronger O–H⋯N hydrogen bonds dominate, while along the π-stacked direction, weaker π–π interactions dominate. Thus, the strength of interactions within a given width correlates to the degree of expansion.
Conclusion
In summary, we have demonstrated the ability to tune TE behaviours in organic materials by using a mixed cocrystal approach and integrating motion-capable or -incapable molecules. Incorporation of larger ratios of motion-capable molecules in the solids affords more significant PTE along the motion direction. Addition of a motion-incapable molecule, BPA, yields less TE in the solids. Mixed cocrystals offer a novel strategy for controlling TE behavior in organic solids, and this approach is applicable to both organic and metal–organic materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. M. H. gratefully acknowledges financial support from Texas Tech University.Notes and references
- D. Das, T. Jacobs and L. J. Barbour, Nat. Mater., 2010, 9, 36–39 CrossRef CAS PubMed
.
-
R. S. Krishnan, R. Srinivasan and S. Devanarayanan, Thermal Expansion of Crystals, Pergamon, Oxford, U.K., 1979 Search PubMed
- J. L. Korčok, M. J. Katz and D. B. Leznoff, J. Am. Chem. Soc., 2009, 131, 4866–4871 CrossRef PubMed
- S. J. Baxter, A. Schneemann, A. D. Ready, P. Wijeratne, A. P. Wilkinson and N. C. Burtch, J. Am. Chem. Soc., 2019, 141, 12849–12854 CrossRef CAS PubMed
- L. Wu, B. Li and J. Zhou, ACS Appl. Mater. Interfaces, 2016, 8, 17721–17727 CrossRef CAS PubMed
- Q. Gao, J. Wang, A. Sanson, Q. Sun, E. Liang, X. Xing and J. Chen, J. Am. Chem. Soc., 2020, 142, 6935–6939 CrossRef CAS PubMed
- Z. Ren, R. Zhao, X. Chen, M. Li, X. Li, H. Tian, Z. Zhang and G. Han, Nat. Commun., 2018, 9, 1–6 CrossRef CAS PubMed
- F. Qin, J. Chen, U. Aydemir, A. Sanson, L. Wang, Z. Pan, J. Xu, C. Sun, Y. Ren, J. Deng, R. Yu, L. Hu, G. J. Snyder and X. Xing, Inorg. Chem., 2017, 56, 10840–10843 CrossRef CAS PubMed
- E. Phillips, A. G. J. Halder, K. W. Chapman, A. L. Goodwin and C. J. Kepert, J. Am. Chem. Soc., 2010, 132, 10–11 CrossRef PubMed
- B. K. Saha, A. Rather and S. A. Saha, Eur. J. Inorg. Chem., 2017, 28, 3390–3394 CrossRef
- P. Lama, A. Hazra and L. J. Barbour, Chem. Commun., 2019, 55, 12048–12051 RSC
- M. K. Gupta, B. Singh, R. Mittal, M. Zbiri, A. B. Cairns, A. L. Goodwin, H. Schober and S. L. Chaplot, Phys. Rev. B, 2017, 96, 214303 CrossRef
- R. K. Das, H. Aggarwal and L. J. Barbour, Inorg. Chem., 2015, 54, 8171–8173 CrossRef CAS PubMed
- S. A. Rather, V. G. Saraswatula, D. Sharada and B. K. Saha, New J. Chem., 2019, 43, 17146–17150 RSC
- A. Janiak, C. Esterhuysen and L. J. Barbour, Chem. Commun., 2018, 54, 3727–3730 RSC
- V. G. Saraswatula, S. Bhattacharya and B. K. Saha, New J. Chem., 2015, 39, 3345–3348 RSC
- M. K. Panda, R. Centore, M. Causà, A. Tuzi, F. Borbone and P. Naumov, Sci. Rep., 2016, 6, 29610 CrossRef CAS PubMed
-
A. I. Kitaigorodsky, Molecular Crystals and Molecules, Academic Press, New York, 1973, Physical Chemistry Series, vol. 29 Search PubMed
- V. G. Saraswatula and B. K. Saha, New J. Chem., 2014, 38, 897–901 RSC
- L. Catalano, S. Perez-Estrada, H. Wang, A. J.-L. Ayitou, S. I. Khan, G. Terraneo, P. Metrangolo, S. Brown and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2017, 139, 843–848 CrossRef CAS PubMed
- M. E. Howe and M. A. Garcia-Garibay, J. Org. Chem., 2019, 84, 9835–9849 CrossRef CAS PubMed
- W. Danowski, T. Van Leeuwen, S. Abdolahzadeh, D. Roke, W. R. Browne, S. J. Wezenberg and B. L. Feringa, Nat. Nanotechnol., 2019, 14, 488–494 CrossRef CAS PubMed
- T. Takeda, M. Ozawa and T. Akutagawa, Angew. Chem., Int. Ed., 2019, 58, 10345–10352 CrossRef CAS PubMed
- B. Shao, H. Qian, Q. Li and I. Aprahamian, J. Am. Chem. Soc., 2019, 141, 8364–8371 CAS
- J. Harada and K. Ogawa, Chem. Soc. Rev., 2009, 38, 2244–2252 RSC
- K. M. Hutchins, D. K. Unruh, F. A. Verdu and R. H. Groeneman, Cryst. Growth Des., 2018, 18, 566–570 CrossRef CAS
- N. Juneja, D. K. Unruh, E. Bosch, R. H. Groeneman and K. M. Hutchins, New J. Chem., 2019, 43, 18433–18436 RSC
- C. A. Gunawardana and C. B. Aakeröy, Chem. Commun., 2018, 54, 14047–14060 RSC
- N. K. Duggirala, M. L. Perry, Ö. Almarsson and M. J. Zaworotko, Chem. Commun., 2016, 52, 640–655 RSC
- N. A. Mir, R. Dubey and G. R. Desiraju, Acc. Chem. Res., 2019, 52, 2210–2220 CrossRef CAS PubMed
- O. E. Y. Kilinkissa, K. K. Govender and N. B. Báthori, CrystEngComm, 2020, 22, 2766–2771 RSC
- T. Friščić, E. Elacqua, S. Dutta, S. M. Oburn and L. R. MacGillivray, Cryst. Growth Des., 2020, 20, 2584–2589 CrossRef
- S. Saha and G. R. Desiraju, J. Am. Chem. Soc., 2018, 140, 6361–6373 CrossRef CAS PubMed
- S. Dey, S. Das, S. Bhunia, R. Chowdhury, A. Mondal, B. Bhattacharya, R. Devarapalli, N. Yasuda, T. Moriwaki, K. Mandal, G. D. Mukherjee and C. M. Reddy, Nat. Commun., 2019, 10, 3711 CrossRef PubMed
- J. C. Gamekkanda, A. S. Sinha and C. B. Aakeröy, Cryst. Growth Des., 2020, 20, 2432–2439 CrossRef CAS
- K. Zheng, S. Gao, M. Chen, A. Li, W. Wu, S. Qian and Q. Pang, CrystEngComm, 2020, 22, 1404–1413 RSC
- M. Lusi, CrystEngComm, 2018, 20, 7042–7052 RSC
- M. Lusi, Cryst. Growth Des., 2018, 18, 3704–3712 CrossRef CAS
- J. Harada and K. Ogawa, Cryst. Growth Des., 2014, 14, 5182–5188 CrossRef CAS
- J. Vansant, G. Smets, J. P. Declercq, G. Germain and M. Van Meerssche, J. Org. Chem., 1980, 45, 1557–1565 CrossRef CAS
- K. M. Hutchins, K. A. Kummer, R. H. Groeneman, E. W. Reinheimer, M. A. Sinnwell, D. C. Swenson and L. R. MacGillivray, CrystEngComm, 2016, 18, 8354–8357 RSC
- L. R. MacGillivray, J. L. Reid and J. A. Ripmeester, J. Am. Chem. Soc., 2000, 122, 7817–7818 CrossRef CAS
- M. J. Cliffe and A. L. Goodwin, J. Appl. Crystallogr., 2012, 45, 1321–1329 CrossRef CAS
- A. L. Goodwin, M. Calleja, M. J. Conterio, M. T. Dove, J. S. O. Evans, D. A. Keen, L. Peters and M. G. Tucker, Science, 2008, 319, 794–797 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, X-ray crystallographic data, thermal expansion results, selected intermolecular interaction distances, expansivity indicatrix images, and 1H NMR spectra for all mixed cocrystals. CCDC 1995475–1995480, 1995482–1995493, 1995550–1995555, 1995687–1995704, and 1995706–1995717. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02795b |
| This journal is © The Royal Society of Chemistry 2020 |