
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of heterocycle content on metal binding isostere coordination†
Benjamin L.
Dick
 ,
Ashay
Patel
and
Seth M.
Cohen
*
,
Ashay
Patel
and
Seth M.
Cohen
*
Department of Chemistry and Biochemistry, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0358, USA. E-mail: scohen@ucsd.edu
First published on 17th June 2020
Abstract
Bioisostere replacement is a core concept in modern medicinal chemistry and has proven an invaluable strategy to address pharmacodynamic and pharmacokinetic limitations of therapeutics. The success of bioisostere replacement is often dependent on the scaffold that is being modified (i.e., “context dependence”). The application of bioisostere replacement to a picolinic acid fragment was recently demonstrated as a means to expand a library of metal-binding pharmacophores (MBPs) to modulate their physicochemical properties, while retaining their metal binding and metalloenzyme inhibitory activity. Here, metal binding isosteres (MBIs) with different nitrogen-containing heteroarenes is explored. This resulted in a number of new MBIs that were evaluated for their physicochemical properties and metal binding features. It was observed that the coordination behavior of an MBI is dependent on the identity and arrangement of the heteroatoms within each heteroarene. To further understand the observed coordination chemistry trends, density functional theory (DFT) calculations were performed. Theory indicates that preferences in coordination geometry are largely determined by the electronic character of the heteroarene scaffold. These results provide important insights into the development of novel MBI scaffolds that can serve to broaden the scope of scaffolds for metalloenzyme inhibitor development.
Introduction
Metalloenzymes are an undervalued class of therapeutic targets relative to the entirety of clinical targets.1 Clinically approved metalloenzyme inhibitors are limited in scope, specifically with respect to the metal binding motif, which are often dominated by a select few metal binding functional groups.1,2 This overreliance on a limited set of metal binding functional groups (a.k.a., metal-binding pharmacophores, MBPs) capable of binding active site metal ions is a barrier to developing next-generation metalloenzyme inhibitors. Efforts to apply fragment-based drug discovery (FBDD) to metalloenzymes has produced new libraries of potential MBPs.3–5 However, some new MBPs may possess physiochemical properties that limit their viability for use in drug candidates.6,7In medicinal chemistry, functional groups that adversely modulate the pharmacological properties of a drug candidate or introduce clear pharmacological liabilities are often replaced with alternate functional groups with broadly similar properties referred to as bioisosteres.8,9 For example, carboxylic acids are often replaced with other acidic functional groups, such as tetrazoles that possess some similarities (i.e., pKa), but differ in other aspects (i.e., lipophilicity).10,11 In an effort to increase the chemical diversity and to improve the drug-likeness of MBPs, carboxylic acid bioisostere replacement was applied to the pyridine-2-carboxylic acid (picolinic acid) MBP, generating a novel set of metal binding isosteres (MBIs).12 This exercise in bioisostere replacement was successful in generating a set of viable metal coordinating motifs varying in their physicochemical properties (i.e., pKa, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P, logD7.4) without compromising their metalloenzyme inhibitory activity.
P, logD7.4) without compromising their metalloenzyme inhibitory activity.
Herein, we report a diverse series of nitrogen heteroarene scaffolds containing a carboxylic acid that were explored for bioisostere replacement. MBIs were synthesized and their metal binding ability assessed via examining bioinorganic model complexes. Certain MBIs had their physicochemical properties determined and coordination chemistry behaviour analysed via computational methods. This expanded study examines a wide variety of heteroarene scaffolds allowing for examination of the effects of bioisostere replacement on the coordination abilities of the resulting MBIs, and shows that the effect of bioisostere replacement is context dependent.13 The findings presented here demonstrate that while the application of bioisostere replacement to MBPs may be a straightforward concept, the coordination behaviour and physiochemical properties are dependent on the parent scaffold and require consideration when employed in metalloenzyme drug discovery.
Results and discussion
Scaffold selection and MBI synthesis
Carboxy-substituted heteroarene MBPs were examined as possible candidates for MBI development. The candidates for study included indazole-3-carboxylic acid (1), benzimidazole-2-carboxylic acid (2), 1,2-benzisothiazole-3-carboxylic acid (3), benzothiazole-2-carboxylic acid (4), and 1,2-benzisoxazole-3-carboxylic acid (5) (Fig. 1). Benzoxazole-2-carboxylic acid would be a logical addition to this series, but was not included as the scaffold is known to readily decarboxylate.14 Compounds 1, 2, 4, and 5 were commercially available, while 3 was synthesized according to a published procedure.15 A series of both acyclic (A and B series) and cyclic (C and D series) bioisosteres were selected for synthesis. For the acyclic MBIs the starting material was generally the corresponding carboxylic acid (1, 2, 3, 4, and 5). The corresponding O-methyl indazole-3-hydroxamic acid (1a) and O-methyl benzimidazole-2-hydroxamic acid (2a) were generated from the carboxylic acid using peptide coupling conditions. O-Methyl 1,2-benzisothiazole-3-hydroxamic acid (3a) and O-methyl benzothiazole-2-hydroxamic acid (4a) were synthesized from the carboxylic acid using acid chloride coupling conditions. The indazole-3-hydroxamic acid (1b) and benzimidazole-2-hydroxamic acid (2b) were generated from the carboxylic acid using peptide coupling conditions. 1,2-Benzisothiazole-3-hydroxamic acid (3b) was synthesized from the carboxylic acid using acid chloride coupling. Benzothiazole-2-hydroxamic acid (4b) was synthesized from ethyl benzothiazole-2-carboxylate and hydroxylamine under basic conditions. The O-methyl 1,2-benzisoxazole-3-hydroxamic acid (5a) and 1,2-benzisoxazole-3-hydroxamic acid (5b) were both synthesized from 5 by generating the acid chloride and combining it with the corresponding hydroxyl amine.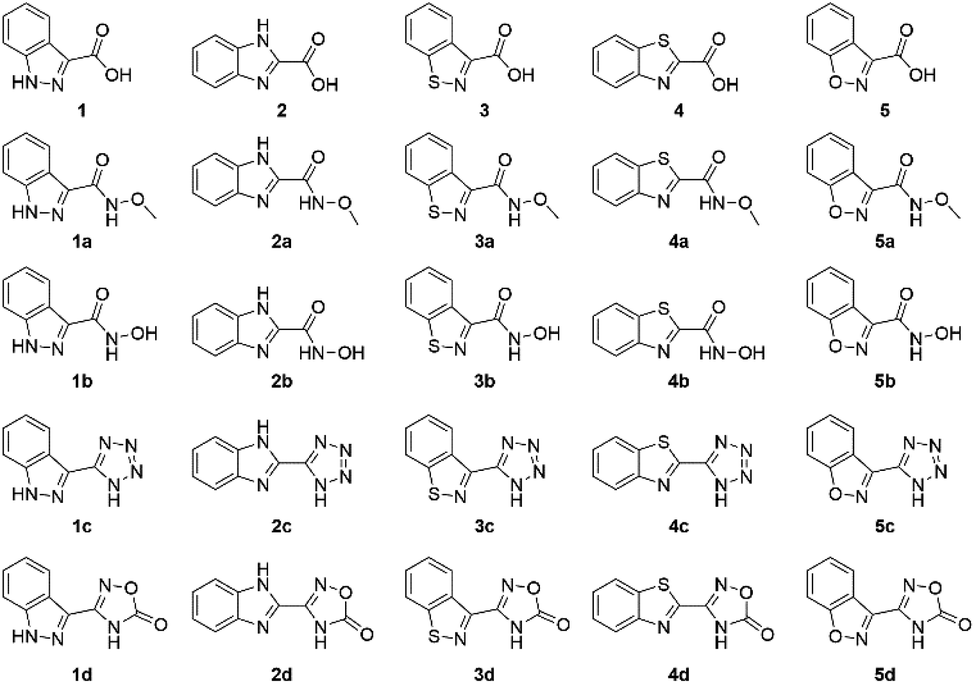 | ||
| Fig. 1 Heterocyclic MBPs (top row, compounds 1–5) that were chosen for MBI development (all other rows). | ||
The cyclic MBIs (C and D series, Fig. 1) of 1, 2,163,17 and 418 were all synthesized starting from the commercially available or published synthesis of the corresponding nitrile derivatives. The nitrile derivative of 5 was prepared in a similar fashion starting from 5 (synthetic details can be found in the ESI†). The tetrazoles were synthesized from the corresponding nitriles via a 3 + 2 cycloaddition with sodium azide yielding the indazole-3-tetrazole (1c), benzimidazole-2-tetrazole (2c), 1,2-benzisothiazole-3-tetrazole (3c), benzothiazole-2-tetrazole (4c), and 1,2-benzisoxazole-3-tetrazole (5c). To obtain the oxadiazolone derivatives, the N-hydroxy amidine was first synthesized for each scaffold from the respective nitrile using hydroxylamine, which was isolated and then cyclized with ethyl chloroformate yielding the indazole-3-oxadiazolone (1d), benzimidazole-2-oxadiazolone (2d), 1,2-benzisothiazole-3-oxadiazolone (3d), benzothiazole-2-oxadiazolone (4d), and 1,2-benzisoxazole-3-oxadiazolone (5d).
Synthesis and characterization of model complexes
To determine the metal binding ability of each MBI in a bioinorganic context, TpPh,MeZn(MBI) (TpPh,Me = hydrotris(5,3-methylphenylpyrazolyl)borate) complexes were prepared. These model complexes are mimetic of a tris(histidine) Zn2+ metalloenzyme active site and are useful for characterizing MBI coordination chemistry.12,19 TpPh,MeZn(MBP) complexes of 1, 2, 3, 4, and 5 were prepared and crystallized. It was found that all of these MBPs coordinate in a bidentate fashion via the nitrogen atom of the heteroarene and the carboxylate oxygen (Fig. 2). The only exception was for TpPh,MeZn(5), which coordinated to the Zn2+ ion in a monodentate fashion only through the carboxylate donor (Fig. 2), indicating the 1,2-benzisoxazole scaffold has significantly decreased coordination ability. Interestingly, the Zn–heteroarene nitrogen bond distances for the N,S heteroarenes (3 and 4) were slightly longer than the corresponding N,N heteroarenes (1 and 2), by about 0.10 to 0.15 Å, while the Zn–carboxylate bond distances exhibited a smaller deviation (∼0.05 Å) (Table S6†). This difference in bond lengths suggest an overall decrease in the donor ability of the N,S heterocycle scaffolds relative to the N,N scaffolds.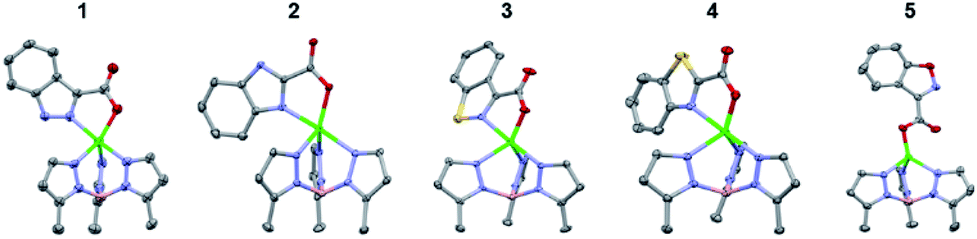 | ||
| Fig. 2 Crystal structure of TpPh,MeZn(MBP) complexes of 1, 2, 3, 4, and 5. Phenyl groups of TpPh,Me have been removed for clarity. | ||
Next, the coordination ability of the O-methyl hydroxamate MBIs (A series) was analysed (Fig. 3). The X-ray structures of TpPh,MeZn(MBI) with 2a, 3a, and 4a exhibit the same bidentate coordination (through the heterocycle and the carbonyl oxygen of the O-methyl hydroxamate) that was observed for the pyridine scaffold.12 Interestingly, the structures of the TpPh,MeZn(MBI) complexes of 1a and 5a show bidentate coordination only through the oxygen donor atoms of the O-methyl hydroxamate. The bidentate coordination through the O-methyl hydroxamate has only rarely been observed crystallographically and not in the presence of alternative coordination motifs.20,21 This bidentate coordination through an ether-like coordination motif (1a and 5a), when there is a suitable nitrogen heterocycle donor available, suggests that the coordination ability of the nitrogen atoms are compromised due to electronic effects. Additionally, it is unlikely that sterics play a significant role in the observed coordination chemistry, as in the case of comparing the difference in coordination between 1a to 3a, the only difference in both structures is a single atom substitution (N versus S). To further probe the effect of bioisostere replacement on each scaffold, the hydroxamate series of bioisosteres was analysed crystallographically, as the hydroxamate is a stronger ligand than the respective O-methyl hydroxamate.
 | ||
| Fig. 3 Crystal structure of TpPh,MeZn(MBI) complexes of 1a, 2a, 3a, 4a, and 5a. Phenyl groups of TpPh,Me have been removed for clarity. | ||
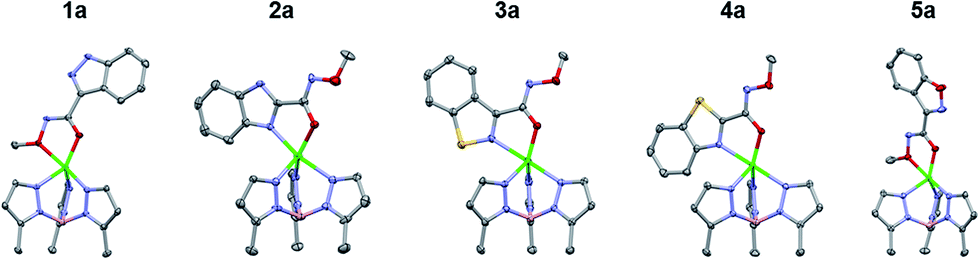
The TpPh,MeZn(MBI) complexes with the hydroxamate series (1b, 2b, 3b, 4b, and 5b) exhibited similar trends as was observed with the O-methyl hydroxamate series, with one exception. Compounds 1b and 5b were observed to coordinate through the hydroxamate only and 2b and 4b were bound through the heterocycle and the hydroxamate (Fig. 4), which are all consistent with the O-methyl hydroxamate series. In contrast, 3b did not coordinate in the same manner as 3a, but rather was coordinated to the Zn2+ center through the hydroxamic acid in a bidentate fashion (like 1b and 5b). The combined results of the O-methyl hydroxamate and hydroxamate series (A and B series) suggests significant electronic differences not only depend on heteroarene heteroatom content, but also the arrangement of the heteroatoms in the ring system.
 | ||
| Fig. 4 Crystal structure of TpPh,MeZn(MBI) complexes of 1b, 2b, 3b, 4b, and 5b. Phenyl groups of TpPh,Me have been removed for clarity. | ||
To gain further insight into the coordinative ability of these MBIs, heterocyclic MBIs (series C and D) were analysed crystallographically (Fig. 5 and 6). Interestingly, the coordination mode of 1c was almost monodentate in character with a Zn–heterocycle bond distance of 2.61 Å. The coordination mode of 2c was clearly bidentate in character, with a Zn–heterocycle bond distance of 2.28 Å, as had been observed before with the pyridine-2-tetrazole complex.12 The complex with 3c again showed monodentate binding with the Zn–heterocycle distance ∼3.05 Å, while the complex with 4c exhibited bidentate binding with an elongated Zn–heterocycle distance of 2.48 Å. The TpPh,MeZn(MBI) complex with 5c again presented monodentate coordination with a Zn–heterocycle distance of 3.17 Å. To evaluate whether these coordination trends were due to a steric effect, a more sterically bulky heterocycle bioisostere, oxadiazolone, was investigated. The oxadiazolone TpPh,MeZn(MBI) complexes (Fig. 6) exhibited the same coordination motifs and bond distances as the tetrazole complexes, suggesting an important electronic contribution to the observed coordination trends and no significant steric impediments.
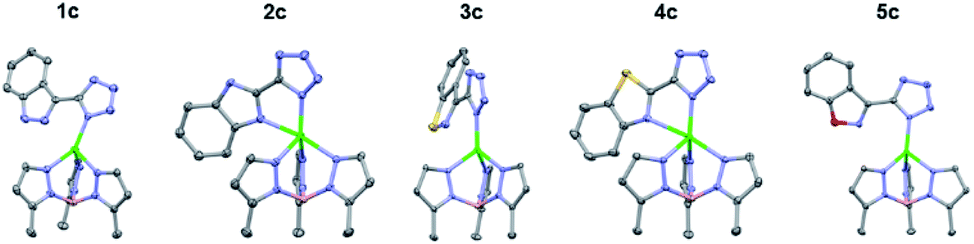 | ||
| Fig. 5 Crystal structure of TpPh,MeZn(MBI) complexes of 1c, 2c, 3c, 4c, and 5c. Phenyl groups of TpPh,Me have been removed for clarity. | ||
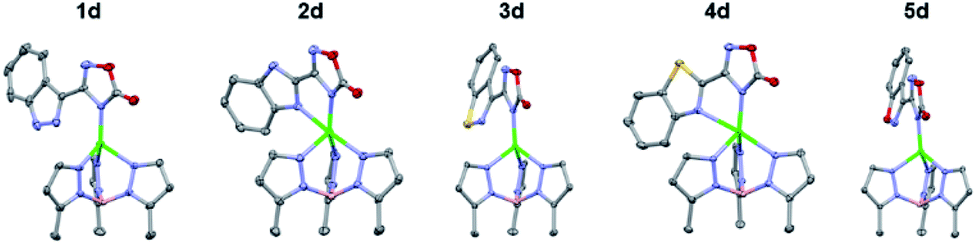 | ||
| Fig. 6 Crystal structure of TpPh,MeZn(MBI) complexes of 1d, 2d, 3d, 4d, and 5d. Phenyl groups of TpPh,Me have been removed for clarity. | ||
Based on the TpPh,MeZn(MBI) structures of both the acyclic and cyclic MBI complexes, it is evident that the heterocyclic scaffold greatly influences MBI binding. The heteroarenes presenting a 1,3-heteroatom arrangement (series 2 and 4) appear to retain bidentate coordination and short metal–ligand bond distances. In contrast, the heteroarenes bearing a 1,2 arrangement of heteroatoms vary significantly in their preferred coordination mode and bond distances. For example, the series based on the 1,2-benzisoxazole scaffold (5a–d) exhibited either monodentate coordination or coordination through the bioisostere replacement alone (5a and 5b). These results suggest that retention of the coordination mode upon carboxylate replacement is dependent on the coordination character/ability of the parent scaffold, which appears to be affected by heteroatom identity and pattern within the heteroarene scaffold. Taken together, the crystallographic data suggest the TpPh,MeZn(MBI) model complex is a useful evaluative tool for metal binding, and may be useful to recapitulate coordination behaviour of similar substitutions in metalloenzyme inhibitors.22 To further understand the electronic effect of each bioisostere replacement, the physicochemical properties of the MBIs were measured.
Physicochemical analysis
Upon determining that the MBIs of the different series have different coordination abilities to the TpPh,MeZn(MBI) bioinorganic model system, an evaluation of physicochemical properties was performed. The acidity (pKa) of the bioisostere groups of the O-methyl hydroxamic ester (1b–5b) and oxadiazolone MBIs (1d–5d) were determined as representative sets. Acidity was measured using a potentiometric method with or without methanol as a cosolvent (Table 1).23 The compounds were also evaluated for their lipophilicity (logP) via a potentiometric method, which was combined with the measured pKa value to determine logD7.4 (Table 1).
The physicochemical values between MBIs vary significantly, even in cases where the only difference between compounds is a single atom replacement or arrangement in the heteroarene. Comparing isomeric MBIs such as, 1a and 2a, yields an interesting trend with the 1,3-heteroatom arrangement (2a) MBIs possessing a substantially lower pKa value than the corresponding 1,2-isomer (1a). Interestingly, single atom substitutions had a strong effect on the logP values, causing a significant shift in some cases (2dversus4d). These observed differences were also manifest in the logD7.4 values, as they are derived from both measurements. Overall, the pKa values are significantly affected by the heteroatom positioning and identity, yielding a set of substituted heteroarene MBIs that span a range of two pKa units, suggesting the heteroarene scaffold strongly affects the electronics of the bioisostere replacement and vice versa. To better understand how electronic effects are influenced by the positioning and identity of the heteroatoms within these heteroarene rings, computational analysis of the coordination complexes was performed.
Computational analysis
To understand the electronics of the MBIs and energetics of the resulting TpPh,MeZn(MBI) model complexes, an evaluation of each complex via density functional theory (DFT) was performed. All computations were performed using ωB97x-D/def2-TZVPP as it generally recapitulates the geometries observed experimentally.24 An added benefit of performing these calculations is to exclude the effect of noncovalent interactions present in the crystal structures that may influence the observed modes of coordination. A phenyl truncation of the TpPh,MeZn(MBI) models was used to reduce computational cost, with the phenyl substituents present on the TpPh,Me ligand being replaced with a hydrogen atom (i.e., TpMe). Additionally, the benzoxazole scaffold (Fig. S26†) was investigated even though it could not be accessed synthetically.14 Further details regarding the computational methodology can be found in the Experimental section below.In agreement with the bond lengths determined from X-ray crystallography (Table S6†), computations show that the carboxylate/isostere–metal bonds of TpMeZn(MBP) complexes are generally shorter than the heteroarene–metal (i.e., N–Zn) bonds (Table 2). The greatest deviation between the experimentally and computationally determined bond lengths is observed when the ligating heteroarene engages in long range interactions with the Zn (3c, 3d, 5c, and 5d). However, the computational results recapitulated the experimental results with respect to the observed trends in Zn–MBI bond distances, with 1,3-heteroatom arrangement MBIs (e.g.2, 2c, 4, 4c) producing shorter Zn–heteroarene bond distances than their 1,2-isomers (e.g.1, 1c, 3, 3c).
| MBP | Zn–carboxylate (Å) | Zn–N (heteroarene, Å) |
|---|---|---|
| 1 | 1.953 | 2.255 |
| 2 | 2.021 | 2.161 |
| 3 | 1.959 | 2.261 |
| 4 | 1.994 | 2.193 |
| 5 | 1.955 | 2.319 |
| Benzoxazole-2-carboxylic acid | 1.996 | 2.199 |
| MBI | Zn–tetrazole (Å) | Zn–N (heteroarene, Å) |
|---|---|---|
| 1c | 1.948 | 2.784 |
| 2c | 2.033 | 2.283 |
| 3c | 1.951 | 2.706 |
| 4c | 2.004 | 2.348 |
| 5c | 1.953 | 2.959 |
| Benzoxazole-2-tetrazole | 1.985 | 2.496 |
| MBI | Zn–oxadiazolone (Å) | Zn–N (heteroarene, Å) |
|---|---|---|
| 1d | 1.942 | 2.776 |
| 2d | 2.004 | 2.391 |
| 3d | 1.949 | 2.740 |
| 4d | 1.976 | 2.491 |
| 5d | 1.940 | 2.910 |
| Benzoxazole-2-oxadiazolone | 1.957 | 2.738 |
Computed free energies (1 atm at 298 K) determined at the ωB97x-D/def2-TZVPP level of theory indicate that bidentate coordination through both the heteroarene and carboxylate (N,O) in the TpMeZn(1) complex is 4.7 kcal mol−1 lower in electronic energy than isomeric bidentate coordination through only the carboxylate (O,O), whereas the analogous coordination modes of the isomeric TpMeZn(5) complexes are closer to being isoenergetic (ΔG = 1.2 kcal mol−1) (Fig. 7). These differences in relative stabilities can be attributed to differences in electronic structure of the heterocyclic ring systems. The 1,2-benzisoxazole (5) contains a more electronegative oxygen atom adjacent to the ligating nitrogen atom than the indazole (1), making it a poorer electron donor than 1. Furthermore, the negative charge of the carboxylate is more effectively withdrawn in 5 due to the greater electron-withdrawing character of the 1,2-benzisoxazole.25 Differences in the electronic character of heteroarene ligands 1–5, 1a–5a, 1b–5b, 1c–5c, and 1d–5d, not only modulate ligand–metal bond lengths, but ultimately influence the actual modes by which these ligands coordinate metal ions.
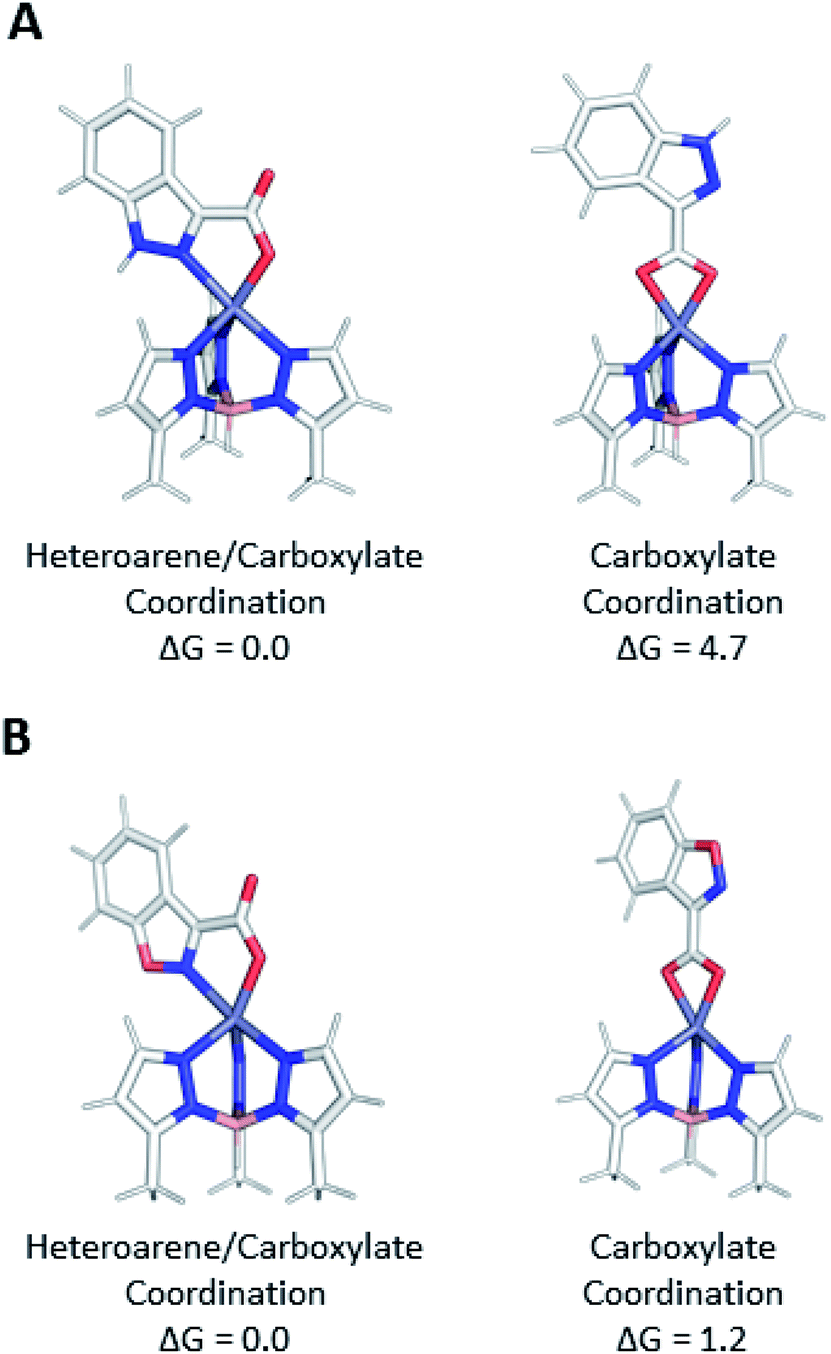 | ||
| Fig. 7 A comparison of the structures and relative Gibbs free energies (kcal mol−1) for carboxylate only (O,O) and isomeric heteroarene/carboxylate (N,O) coordination of (A) TpMeZn(1) and (B) TpMeZn(5) using ωB97x-D/def2-TZVPP level of theory. | ||
Theory shows that regardless of the nature of the heteroarene substituent, heteroarenes with a 1,3-heteroatom arrangement (2, 4, and benzoxazole-2-carboxylic acid) coordinate with shorter bond lengths to the Zn2+ center than do their 1,2-isomers (1, 3, and 5). In examining the isomeric pairs (1vs.2) the computed energy difference is largest when comparing 1,2-benzisoxazole-containing complexes, TpMeZn(5), TpMeZn(5c), and TpMeZn(5d), to their benzoxazole isomers (Fig. 8, S27 and S28†). In contrast, the corresponding energy differences between isomeric benzothiazole- and 1,2-benzisothiazole-ligated TpPh,MeZn(MBI) complexes (e.g., TpPh,MeZn(4) and TpPh,MeZn(3)) are much smaller, ranging in energy from ∼4 to ∼9 kcal mol−1. These energetic preferences can be attributed to differences in the aromaticity of the heteroarenes. For instance, benzimidazole-ligated complexes, like TpPh,MeZn(2c) are more stable than the those that featuring an indazole ligand (e.g., TpPh,MeZn(1c)) because through greater electron delocalization 2c more effectively shifts electron density toward the coordinating nitrogen atom of the heteroarene than its isomer 1c. A smaller difference in the stability of TpPh,MeZn(4c) and TpPh,MeZn(3c) pair of complexes is due to both the reduced aromaticity of the benzothiazole (4) moiety and the reduced electronegativity of sulfur.25,26 These computational data provide a rationalization for the tighter coordination, and likely better donor ability, observed for the benzimidazole and benzothiazole MBPs/MBIs (2, 4, 2c, 4c, 2d, 4d). Furthermore, ligand binding interactions were analysed by using computational data to determine the favourability of isodesmic reactions (Fig. S29, ESI†). These calculations generally show that for the 1,3-heterocyclic MBPs/MBIs coordinate more strongly than their 1,2-isomers. Interestingly, this preference does not hold when this analysis is performed on the isomeric 1,2-benzisothiazole/1,3-benzothiazole TpMeZn(MBI) complexes.
 | ||
| Fig. 8 ωB97x-D/def2-TZVPP-optimized geometries of: isomeric TpMeZn(2) and TpMeZn(1) (top and bottom left, respectively), isomeric TpMeZn(benzoxazole-2-carboxylic acid) and TpMeZn(5) (top and bottom middle, respectively), and isomeric TpMeZn(4) and TpMeZn(5) (top and bottom right, respectively). Energies (in kcal mol−1) are free energies determined at the ωB97x-D/def2-TZVPP level of theory. | ||
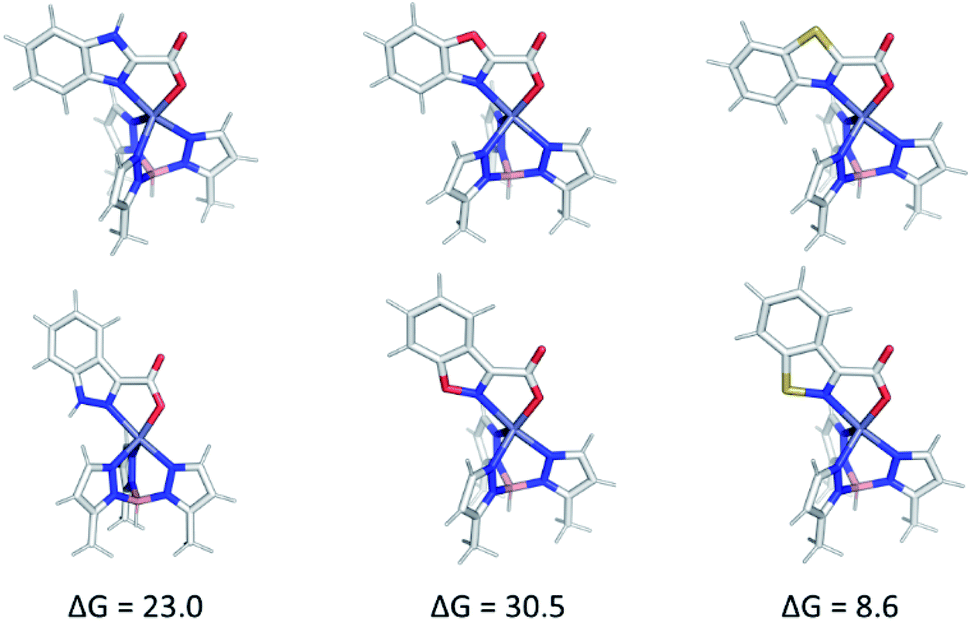
Computational analysis of the TpPh,MeZn(MBI) complexes with ligands 1a–5a and 1b–5b is complicated by the possibility of multiple coordination modes (e.g., N,O or N,N coordination), various coordination geometries, and conformational flexibility of the hydroxamate and hydroxamic methyl ester ligands. As shown in Fig. 9, computational modelling of the phenyl truncated complex TpMeZn(1a), reveals the crystallographically observed mode of coordination is the lowest energy mode of coordination predicted computationally. Because there were many potential modes of coordination with TpPh,MeZn(MBI) complexes with ligands 1a–5a and 1b–5b, geometry optimizations were performed on the complete, phenyl-substituted TpPh,MeZn(1a) complex. The inclusion of the phenyl substituents also correctly predicts that the preferred coordination geometry involving heteroarene coordination via the nitrogen atom and the oxygen atom of 1a. Interestingly, these calculations show that the phenyl substituents reduce the energetic differences between the various possible modes of coordination by ∼1 kcal mol−1, relative to the TpMeZn(1a) results. Overall, these computational analyses suggest that while the phenyl groups contribute slightly to the observed modes of coordination, the major driving force behind MBI coordination is ligand electronics.
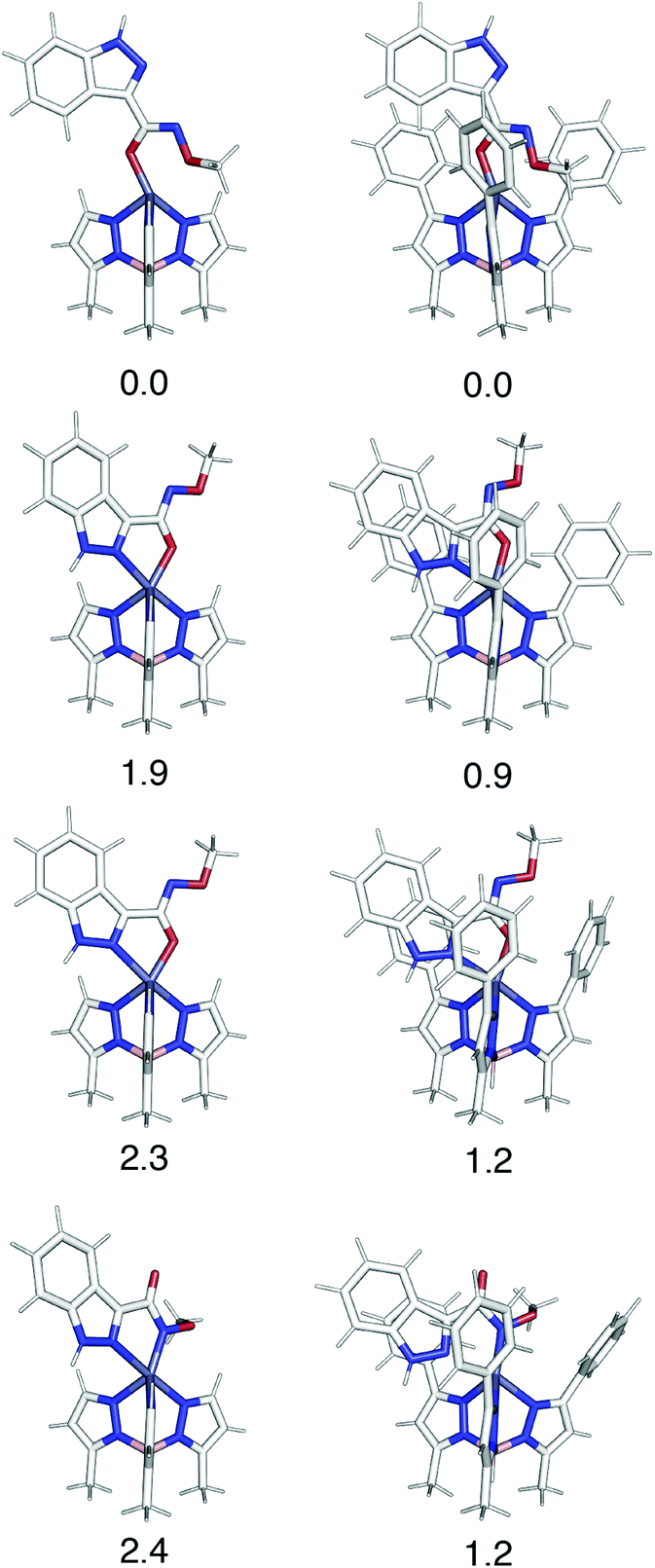 | ||
| Fig. 9 A comparison of various coordination modes of phenyl-truncated TpMeZn(1a) (left) and phenyl substituted TpPh,MeZn(1a) (right) complexes. Relative Gibbs free energies, in kcal mol−1, determined using ωB97x-D/def2-TZVPP level of theory are reported below each structure. | ||
Conclusions
The development of a series of MBIs based on a diverse range of heteroarene scaffolds was shown to produce an interesting range of expected, as well as unexpected, coordination modes. DFT calculations were able to reproduce the experimental results from X-ray crystallography. From these data it was determined that the content and arrangement of the heteroarene significantly modifies the coordination ability of the resulting MBP or MBI. Specifically, the heteroarene aromaticity and subsequent donation ability of the heteroarene donor atom(s) was determined to be essential to dictating the resulting mode of coordination to a bioinorganic model complex. This likely dependence of the success of bioisostere replacement on the electronic character of each scaffold suggests a “context dependence” in isostere replacement for MBPs. These results and analyses provided here allow for an understanding and prediction of future MBI success. In addition, the results described here can be applied to MBP development, particularly to other heteroarene MBP scaffolds. This work demonstrates that MBIs are useful scaffolds for FBDD of metalloenzymes, but the effect of bioisosteres on metal coordination must be understood to successfully implement the use of isosteres for metalloenzyme inhibition.Experimental
General information
Starting materials and solvents were purchased and used without further purification from commercial suppliers (Sigma-Aldrich, Alfa Aesar, EMD, TCI, etc.). Detailed synthetic routes for each MBI are provided in the ESI.† [(TpPh,Me)ZnOH] (TpPh,Me = hydrotris(5,3-methylphenylpyrazolyl)borate) was synthesized as reported using [KTpPh,Me], which was prepared as previously reported.27General synthesis and crystallization of TpPh,MeZn(MBI) model complexes
[(TpPh,Me)ZnOH] (60 mg, 0.11 mmol) was dissolved in 15 mL of CH2Cl2 in a 50 mL round-bottom flask. The MBP/MBI (0.11 mmol, 1 equiv.) in 10 mL of MeOH was added, and the reaction mixture was stirred overnight under a nitrogen atmosphere (note: in a select few cases, to conserve material, the amounts of [(TpPh,Me)ZnOH] and MBI were halved while keeping solvent volumes constant). The resulting mixture was evaporated to dryness via rotary evaporation and subsequently a minimal amount (∼5 mL) of benzene was added, if that was insufficient to dissolve the residue a minimal amount of MeOH was added (∼1–3 mL). The solution was filtered to remove any undissolved solids. The resulting complex in benzene or benzene/MeOH was recrystallized using vapor diffusion with pentane; crystals typically formed within one week.Physicochemical properties analysis
Physicochemical properties were determined using a Sirius T3 instrument. All titrations, both pKa and logP, were performed in 0.15 M KCl with 0.5 M HCl and KOH. The pKa of a compound was determined by analysing each MBI sample in triplicate using potentiometric titrations.23 Experiments were typically performed over a pH range of 2.0–12.0. Standard deviations were derived from fitting all three replicate experiments. For water insoluble compounds methanol was added and the obtained apparent pKa (psKa) was extrapolated to the aqueous pKa by the Yasuda–Shedlovsky procedure.28,29 logP was determined via potentiometric titrations in the presence of varying ratios of octanol and water.30 The presence of octanol shifts the pKa of ionizable species, and based on the shift, a logP can be determined. Measurements for logP determination were typically performed over a pH range of 2.0–12.0. Three experiments with varying ratios of water:octanol were performed, allowing for a standard deviation to be determined from the fitting of all measurements. MBI sample sizes were ≤0.5 mg for both pKa and logP measurements.
Computational methodology
All DFT computations were performed using Gaussian 09.31 All geometry optimizations were performed at the ωB97x-D/def2-TZVPP level of theory.24,32,33 Frequency calculations were performed to confirm that each structure was in fact a local minimum (stationary point) on the potential energy surface. All computed free energies were calculated assuming standard state conditions of 1 atm in the gas phase at 298 K. Initial coordinates for computation were prepared using Gaussview 5.0.9 and Avogadro 1.2.0.34–36 Pymol 2.3 was used to render images of the DFT-optimized structures presented herein.37Conflicts of interest
The authors declare the following competing financial interest(s): S. M. C. is a co-founder, has an equity interest, and receives income as member of the Scientific Advisory Board for Cleave Therapeutics and is a co-founder, has an equity interest, and a member of the Scientific Advisory Board for Forge Therapeutics. Both companies may potentially benefit from the research results of certain projects in the laboratory of S. M. C. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies.Acknowledgements
Support was provided by the National Institutes of Health (R01 GM098435; R01 AI149444). B. L. D. was supported by the National Institute of Health Molecular Biophysics Training Grant (T32GM008326-26). Computational studies by A. P. were supported by the National Science Foundation (MCB-1020765), National Institute of Health (R01 GM31749 and R01 GM095970), Howard Hughes Medical Institute, National Biomedical Computation Resource (NBCR), and NSF supercomputer centers through Prof. J. Andrew McCammon (U.C. San Diego); we also thank Prof. McCammon for helpful discussions. A. P. thanks M. D. Burkart for financial support through the National Institutes of Health (NIH GM095970). We thank Prof. Arnold Rheingold, Dr Milan Gembicky, and Dr Curtis Moore (U.C. San Diego) for assistance with crystallographic data collection and structure determination. We also thank Dr Yongxuan Su for mass spectrometry sample analysis at The Molecular Mass Spectrometry Facility at U.C. San Diego.References
- A. Y. Chen, R. N. Adamek, B. L. Dick, C. V. Credille, C. N. Morrison and S. M. Cohen, Chem. Rev., 2019, 119, 1323–1455 CrossRef CAS PubMed.
- Y. Yang, X. Q. Hu, Q. S. Li, X. X. Zhang, B. F. Ruan, J. Xu and C. Liao, Curr. Top. Med. Chem., 2016, 16, 384–396 CrossRef CAS PubMed.
- J. A. Jacobsen, J. L. Fullagar, M. T. Miller and S. M. Cohen, J. Med. Chem., 2011, 54, 591–602 CrossRef CAS PubMed.
- C. V. Credille, Y. Chen and S. M. Cohen, J. Med. Chem., 2017, 60, 9912 CrossRef CAS PubMed.
- C. V. Credille, C. N. Morrison, R. W. Stokes, B. L. Dick, Y. Feng, J. Sun, Y. Chen and S. M. Cohen, J. Med. Chem., 2019, 62, 9438–9449 CrossRef CAS PubMed.
- A. Y. Chen, P. W. Thomas, A. C. Stewart, A. Bergstrom, Z. Cheng, C. Miller, C. R. Bethel, S. H. Marshall, C. V. Credille, C. L. Riley, R. C. Page, R. A. Bonomo, M. W. Crowder, D. L. Tierney, W. Fast and S. M. Cohen, J. Med. Chem., 2017, 60, 7267–7283 CrossRef CAS PubMed.
- C. Perez, J. Li, F. Parlati, M. Rouffet, Y. Ma, A. L. Mackinnon, T. F. Chou, R. J. Deshaies and S. M. Cohen, J. Med. Chem., 2017, 60, 1343–1361 CrossRef CAS PubMed.
- G. A. Patani and E. J. LaVoie, Chem. Rev., 1996, 96, 3147–3176 CrossRef CAS PubMed.
- H. L. Friedman, Natl. Acad. Sci., 1951, 206, 295–362 Search PubMed.
- T. Lassila, J. Hokkanen, S. M. Aatsinki, S. Mattila, M. Turpeinen and A. Tolonen, Chem. Res. Toxicol., 2015, 28, 2292–2303 Search PubMed.
- H. Pajouhesh and G. R. Lenz, NeuroRx, 2005, 2, 541–553 CrossRef PubMed.
- B. L. Dick and S. M. Cohen, Inorg. Chem., 2018, 57, 9538–9543 CrossRef CAS PubMed.
- C. Ballatore, D. M. Huryn and A. B. Smith 3rd, ChemMedChem, 2013, 8, 385–395 CrossRef CAS PubMed.
- I. I. F. Boogaerts and S. P. Nolan, J. Am. Chem. Soc., 2010, 132, 8858–8859 CrossRef CAS PubMed.
- N. J. Hrib, J. G. Jurcak, K. L. Burgher, P. G. Conway, H. B. Hartman, L. L. Kerman, J. E. Roehr and A. T. Woods, J. Med. Chem., 1994, 37, 2308–2314 CrossRef CAS PubMed.
- D. Cundy and G. Simpson, Aust. J. Chem., 1996, 49, 199–203 CrossRef CAS.
- O. Barba, G. J. Dawson, T. M. Krulle, R. J. Rowley, D. Smyth and G. H. Thomas, Prosidion Limited, UK WO2006085118A2, 2006.
- A. L. Gryshuk and J. Perkins, Lawrence Livermore National Security, LLC, USA, US20110213124A1, 2011.
- D. T. Puerta and S. M. Cohen, Inorg. Chem., 2003, 42, 3423–3430 CrossRef CAS PubMed.
- S. M. George, J. H. Nam, G. Y. Lee, J. H. Han, B. K. Park, C. G. Kim, D. J. Jeon and T.-M. Chung, Eur. J. Inorg. Chem., 2016, 2016, 5539–5546 CrossRef CAS.
- J. H. Lee, E. A. Jung, G. Y. Lee, S. H. Han, B. K. Park, S. W. Lee, S. U. Son, C. G. Kim and T.-M. Chung, ChemistrySelect, 2018, 3, 6691–6695 CrossRef CAS.
- T. Heinrich, H. P. Buchstaller, B. Cezanne, F. Rohdich, J. Bomke, M. Friese-Hamim, M. Krier, T. Knochel, D. Musil, B. Leuthner and F. Zenke, Bioorg. Med. Chem. Lett., 2017, 27, 551–556 CrossRef CAS PubMed.
- D. Schonherr, U. Wollatz, D. Haznar-Garbacz, U. Hanke, K. J. Box, R. Taylor, R. Ruiz, S. Beato, D. Becker and W. Weitschies, Eur. J. Pharm. Biopharm., 2015, 92, 155–170 CrossRef CAS PubMed.
- J. D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- A. R. Katritzky, K. Jug and D. C. Oniciu, Chem. Rev., 2001, 101, 1421–1450 CrossRef CAS PubMed.
- A. R. Katritzky, V. Feygelman, G. Musumarra, P. Barczynski and M. Szafran, J. Prakt. Chem., 1990, 332, 870–884 CrossRef CAS.
- D. T. Puerta and S. M. Cohen, Inorg. Chim. Acta, 2002, 337, 459–462 CrossRef CAS.
- M. Yasuda, Bull. Chem. Soc. Jpn., 1959, 32, 429–432 CrossRef CAS.
- B. Pesce, Electrolytes: Proceedings of an International Symposium Held in Trieste June, 1959, Pergamon, 1962 Search PubMed.
- B. Slater, A. Mccormack, A. Avdeef and J. E. A. Comer, J. Pharm. Sci., 1994, 83, 1280–1283 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision D.01, Gaussian Inc, Pittsburg PA, 2009 Search PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- Avogadro: an open-source molecular builder and visualization tool. Version 1.2.0, http://avogadro.cc Search PubMed.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView Version 5, 2016 Search PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- PyMOL, The PyMOL Molecular Graphics System, Version 2.2 Schrödinger, LLC Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, synthetic procedures, crystal data tables, and supplementary figures and tables. CCDC 1999816–1999840. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02717k |
| This journal is © The Royal Society of Chemistry 2020 |