
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Integrating single Ni sites into biomimetic networks of covalent organic frameworks for selective photoreduction of CO2†
Xin
Chen‡
a,
Qiang
Dang‡
a,
Rongjian
Sa
 b,
Liuyi
Li
*a,
Lingyun
Li
a,
Jinhong
Bi
ac,
Zizhong
Zhang
c,
Jinlin
Long
c,
Yan
Yu
*a and
Zhigang
Zou
*ad
b,
Liuyi
Li
*a,
Lingyun
Li
a,
Jinhong
Bi
ac,
Zizhong
Zhang
c,
Jinlin
Long
c,
Yan
Yu
*a and
Zhigang
Zou
*ad
aKey Laboratory of Eco-materials Advanced Technology, College of Materials Science and Engineering, Fuzhou University, Fuzhou 350108, China. E-mail: lyli@fzu.edu.cn; yuyan@fzu.edu.cn; zgzou@nju.edu.cn
bInstitute of Oceanography, Ocean College, Minjiang University, Fuzhou, Fujian 350108, China
cState Key Laboratory of Photocatalysis on Energy and Environment, College of Chemistry, Fuzhou University, Fuzhou 350108, China
dEco-materials and Renewable Energy Research Center, College of Engineering and Applied Sciences, Nanjing University, Nanjing 210093, China
First published on 9th June 2020
Abstract
Selective photoreduction of CO2 into a given product is a great challenge but desirable. Inspired by natural photosynthesis occurring in hierarchical networks over non-precious molecular metal catalysts, we demonstrate an integration of single Ni sites into the hexagonal pores of polyimide covalent organic frameworks (PI-COFs) for selective photoreduction of CO2 to CO. The single Ni sites in the hexagonal pores of the COFs serve as active sites for CO2 activation and conversion, while the PI-COFs not only act as a photosensitizer to generate charge carriers but also exert a promoting effect on the selectivity. The optimized PI-COF with a triazine ring exhibits excellent activity and selectivity. A possible intra- and inter-molecular charge-transfer mechanism was proposed, in which the photogenerated electrons in PI-COFs are efficiently separated from the central ring to the diimide linkage, and then transferred to the single Ni active sites, as evidenced by theoretical calculations.
Introduction
Selective photoconversion of CO2 to a target industrial product is an intriguing approach to simultaneously enrich solar energy and utilize CO2.1,2 However, efficient photoreduction of CO2 with high selectivity, particularly in aqueous solution, is a considerable challenge because of the multi-electron reaction process and the competing H2 evolution in the CO2 reduction reaction.3,4 So far, many molecular metal complex based photocatalytic systems have been developed to selectively reduce CO2 into solar fuels with high efficiency. Molecular complexes as precursors of single active sites with tailorable and versatile coordination possess maximum efficiency of catalytic sites in the reaction.5–8 However, most of the systems suffer from insufficiently stable and expensive photosensitizers to achieve high performance.9 The utilization of semiconductors would be a promising alternative approach for photocatalysis,10,11 as they always possess higher photostability.12–14 In practical applications, the photocatalytic performance of semiconductor-based catalytic systems is still limited because of their exterior surface catalytic mechanism, always leading to a limited utilization of photogenerated charges. Taking inspiration from nature, where photocatalysis for converting solar energy into chemical energy occurs in the hierarchical networks in plants' leaves with non-precious metal catalysts,15,16 the combination of single metal sites and a hierarchical porous semiconductor may offer an applicable approach towards the development of photocatalytic systems for selective conversion of CO2.17 In this context, it is desirable to explore novel semiconductors with intrinsic hierarchical porosity to accommodate single active sites and maximize the transfer of photogenerated charges to the active sites.The development of covalent organic frameworks (COFs)18 provides a promising platform for photocatalysis.19–24 The periodic and permanent porosity endow COFs with a nature-mimicking architecture, while the diverse compositions and synthetic approaches allow COFs with tunable microstructures and optical and electronic structures.25–29 The highly conjugated structure in-plane in COFs can ensure the mobility of photoinduced charges.30–33 Moreover, COFs with tunable porosity can accommodate guest molecules for target applications.34–39 In terms of catalysis, the microenvironment in the cavity of a heteroatom-rich COF may impose complicated effects on the active sites and its catalytic performance.40 Compared to metal–organic frameworks, organic polymers and inorganic networks, COFs possess a metal-free skeleton and periodic porosity to form biomimetic microenvironments as in plants' leaves for the accommodation of metal molecular catalysts. Profiting from the unique characteristics of COFs, it is reasonable to expect the integration of single metal sites in photoactive COFs for photoreduction of CO2.
Herein, we report an integration of single Ni sites in the biomimetic channels in polyimide covalent organic frameworks (PI-COFs) for selective photoreduction of CO2 to CO (Fig. 1). The excellent catalytic performance mainly arises from the synergistic effects of single Ni sites and the PI-COFs, in which the engineered PI-COFs via adjusting the building units not only act as hosts for accommodating single Ni sites but also are responsible for the generation and separation of charge carriers.
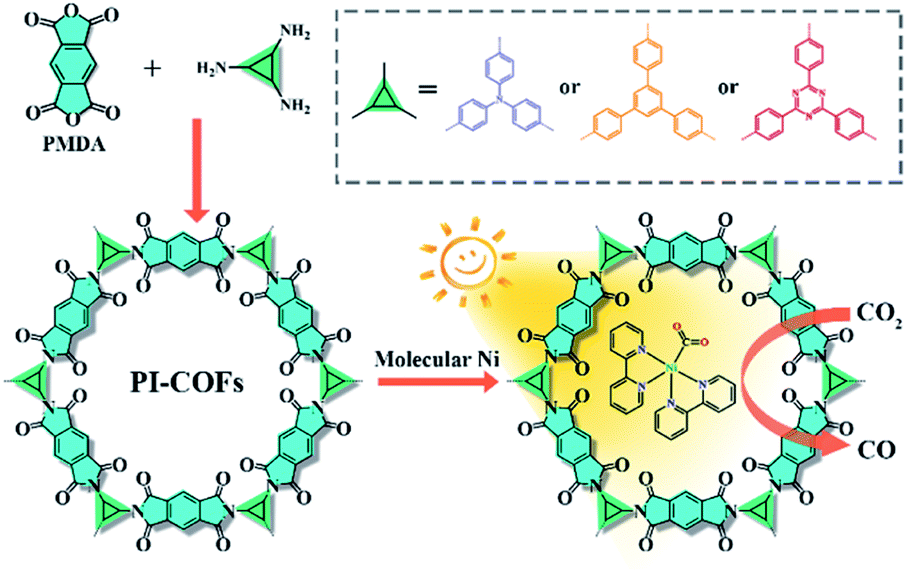 | ||
| Fig. 1 Synthesis of PI-COFs and the schematic illustration of photocatalytic selective reduction of CO2 by PI-COFs with a molecular Ni catalyst. | ||
Results and discussion
Three polyimide covalent organic frameworks (PI-COFs) were synthesized by coupling pyromellitic dianhydride (PMDA) with tris(4-aminophenyl)amine (TAPA), 1,3,5-tris(4-aminophenyl)benzene (TAPB) and 1,3,5-tris(4-aminophenyl)triazine (TAPT), respectively, and are denoted as PI-COF-1, PI-COF-2 (ref. 41) and PI-COF-TT,42 respectively. The formation of the COFs was assessed by Fourier transform infrared (FT-IR), solid-state 13C NMR spectroscopy and powder X-ray diffraction (PXRD). FT-IR spectra of all three COFs showed strong peaks at 1720–1725 cm−1 (Fig. S1–S3†), confirming the formation of five-membered imide rings. In the solid-state 13C NMR spectroscopy, PI-COFs showed the characteristic signal for the carbonyl carbon of the imide ring at 165 ppm (Fig. S4†).43 The overlapping peaks from 116.5 to 144.7 ppm were attributed to phenyl carbons. The PXRD patterns of PI-COFs show prominent peaks, indicating their crystalline nature. The experimental profiles of PI-COFs match well with their simulated PXRD patterns indicating a serrated stacking bnn net with adjacent sheets slipping by 1/4 of the unit cell distances (Fig. 2a–c).41 All PI-COFs exhibit similar geometries, whereas the linkages in PI-COF-2 and PI-COF-1 twist rather than remaining planar as in PI-COF-TT, and the phenyl group is tilted by 48.44 and 41.37° with respect to the diimide plane, respectively. The N2 adsorption/desorption measurements show that all as-synthesized PI-COFs have a pore size in the range of 1.5 to 3.5 nm calculated using the nonlocal density functional theory (NLDFT), which is in agreement with the pore sizes predicted from the theoretical crystal structures. The Brunauer–Emmett–Teller (BET) surface areas of PI-COF-1, PI-COF-2 and PI-COF-TT are calculated to be 475, 1175 and 825 m2 g−1, respectively (Fig. S5–S7†). All three PI-COFs exhibit good CO2 uptake behavior due to the polarity of the polymer surface44 (Fig. S8–S10†), and PI-COF-TT shows the highest isosteric heat (Qst) (29.76 kJ mol−1) for CO2 adsorption at low coverage (Fig. S11†). This may be attributed to the dipole–quadrupole interactions between CO2 molecules and the imide groups and triazine rings in the framework.44,45 Scanning electron microscopy (SEM) images show that PI-COF-1 and PI-COF-2 exhibit a “sphere-like” morphology (Fig. S12†), while PI-COF-TT is a cross-linked network (Fig. S13†). Crystalline domains were identified in PI-COF-TT in the transmission electron microscopy (TEM) images (Fig. 2d). The presence of regular lines with a spacing of ∼3.0 nm in TEM images was consistent with the interatomic distances of the [100] plane inferred from PXRD and computational models. TEM images of PI-COF-1 and PI-COF-2 showed that the dense nanospheres were comprised of small nanoparticles (Fig. S14†). PI-COFs are stable in common organic solvents, water and acidic aqueous solutions (Fig. S15 and S16†). Thermogravimetric analysis (TGA) showed that PI-COFs can be stable up to 450 °C in argon (Fig. S17†), and no obvious changes of crystal intensity were observed after heating to 300 °C (Fig. S18†).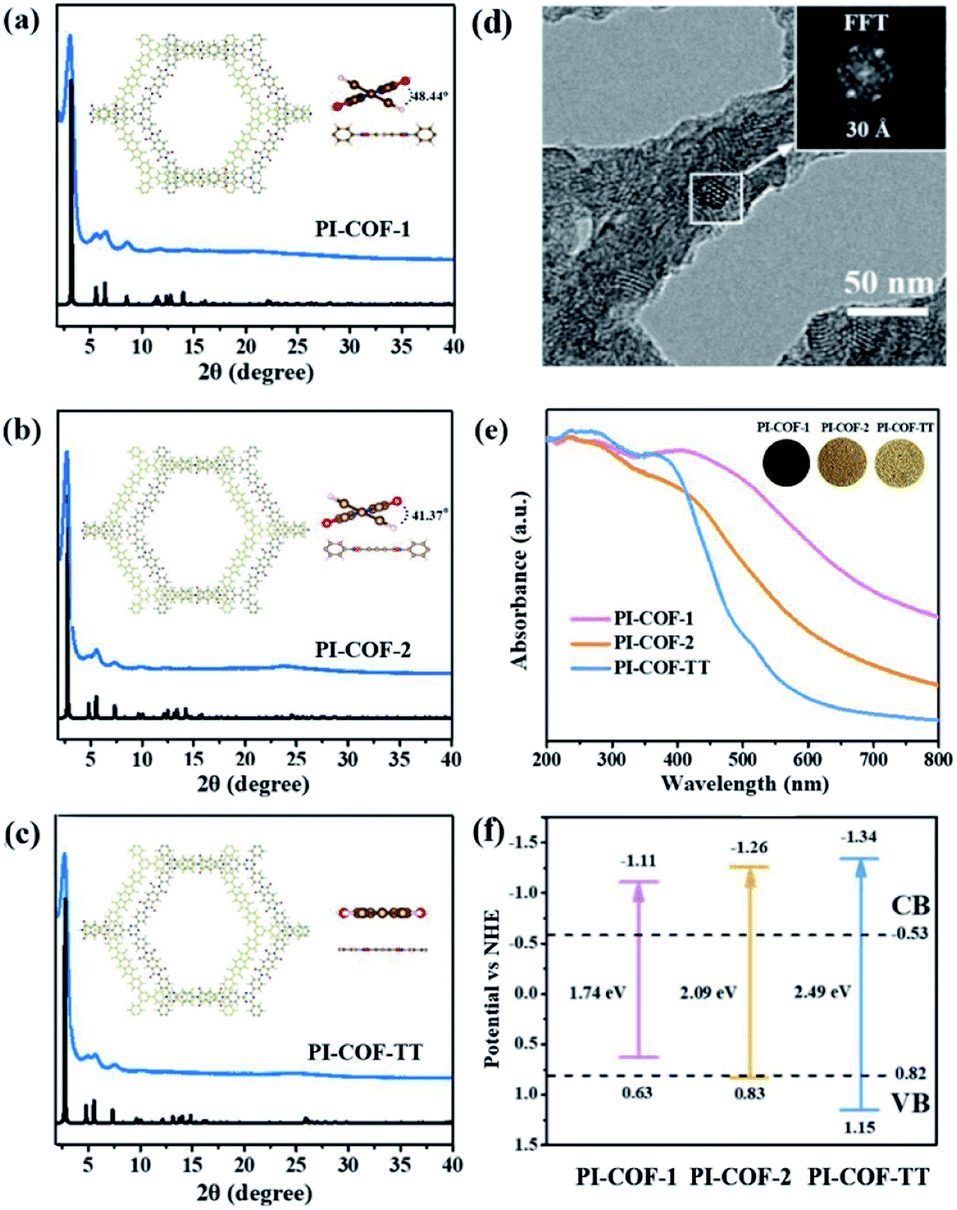 | ||
| Fig. 2 (a–c) PXRD patterns that were experimentally observed (blue) and the simulated pattern (black) of PI-COF-1, PI-COF-2 and PI-COF-TT, respectively. Top inset: the structures of PI-COFs; (d) TEM images of PI-COF-TT; (e) UV-vis diffuse reflectance spectra and (f) band gap structures of PI-COFs. | ||
UV-vis diffuse reflectance spectra (DRS) show that all three PI-COFs absorb light in the ultraviolet and parts of the visible region (Fig. 2e), suggesting the optical band gaps of 1.74, 2.09 and 2.49 eV for PI-COF-1, PI-COF-2 and PI-COF-TT (Fig. S19†), respectively. The intrinsic absorption band edge varies with the central ring of the COFs. From adjusting triazine, phenyl and tertiary amine units as the central ring, the electron-rich property of the central ring increased, so a remarkable red-shifted absorption of PI-COFs was observed. The band structures of the as-synthesized PI-COFs were obtained from the combination of optical absorption spectra and Mott–Schottky plots (Fig. S20–S22†), and are shown in Fig. 2f. Obviously, the changes of the electronic and steric properties of the central ring result in a progressively enlarged band gap and the corresponding conduction band (CB) and valence band (VB) positions. It is noteworthy that the reduction potentials of the CB electrons in PI-COFs are much more negative than the reduction potentials of CO2 to various hydrocarbon fuels, making them suitable for reduction of CO2.
Although Re-modified COFs have been previously employed in photocatalytic reduction of CO2,46,47 it is desirable to develop non-precious metal-based catalytic systems.48 The [Ni(bpy)3]2+ complex has been shown to reduce CO2 actively in electrocatalysis49 or in homogeneous photocatalysis.50 The combination of photoactive COFs and [Ni(bpy)3]2+ as photocatalytic systems may offer a viable approach for conversion of CO2. The photocatalytic activity of PI-COFs toward reduction of CO2 in aqueous solution was studied by using self-assembled [Ni(bpy)3]2+ (bpy is 2,2′-bipyridyl) as a precursor of active sites. The [Ni(bpy)3]2+ complex can be easily formed upon addition of a bpy ligand and Ni(II) salt as evidenced by UV-vis absorption spectra. A remarkable red-shift of bpy in UV-vis absorption spectra indicates the coordination of bpy with Ni ions (Fig. 3a). The encapsulation of molecular Ni complexes into the COFs (denoted as Ni@PI-COF-TT) was confirmed by aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM), energy dispersive X-ray (EDX) mapping, X-ray photoelectron spectroscopy (XPS), PXRD and N2 sorption measurements. HAADF-STEM images clearly show the crystalline domain in Ni@PI-COF-TT (Fig. 3b and c). The fast-Fourier transform (FFT) image confirms the crystalline nature of Ni@PI-COF-TT. Evidently, the bright spots corresponding to the Ni single sites are distributed in the hexagonal pores of Ni@PI-COF-TT (Fig. 3d), and not on the external surface of the stacking layers. EDX elemental mapping and TEM images reveal that Ni ions are uniformly dispersed in the framework (Fig. 3e and S23†). XPS investigations with Ar-ion etching were conducted on Ni@PI-COF-TT. The peak at 856.0 eV was assigned to Ni2+ (Fig. 3f). After Ar+ etching, the intensity of the characteristic peaks of Ni2+ relatively increased, further confirming the encapsulation of the molecular Ni complex in the channels of Ni@PI-COF-TT. A new peak at 854.1 eV is attributed to the reduction of Ni2+ to Ni0, which was caused by Ar ion bombardment. Compared to PI-COF-TT, the PXRD peak of Ni@PI-COF-TT at 2.7° corresponding to the (110) facet weakened, due to the disorder induced by the Ni complexes inside the channels of Ni@PI-COF-TT (Fig. S24†). The PXRD peak intensity can be recovered after the removal of [Ni(bpy)3]2+ from the COF by washing with water. The BET surface area of Ni@PI-COF-TT is obviously reduced yet can be recovered after [Ni(bpy)3]2+ removal (Fig. S25†). The reduction of porosity in Ni@PI-COF-TT is mainly attributed to the occupancy of molecular Ni complexes in the channels.
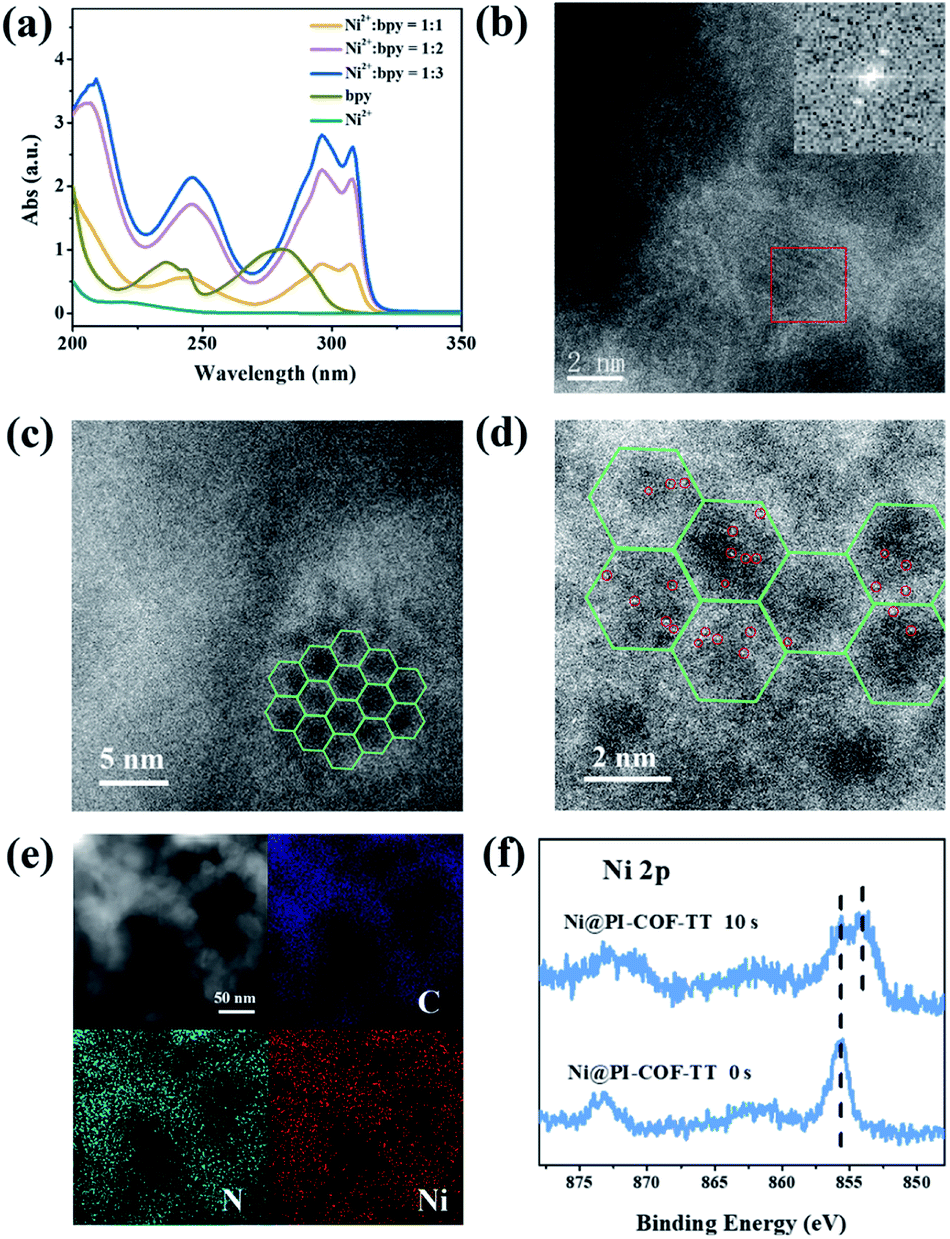 | ||
| Fig. 3 (a) The UV-vis absorption spectra of the in situ formed [Ni(bpy)3]2+ by the consecutive addition of bpy to a solution of Ni[(ClO)4]2 in acetonitrile. (b–d) Aberration-corrected HAADF-STEM images of Ni@PI-COF-TT, and the FFT image is shown in the inset in (b). (e) EDX elemental mapping image of Ni@PI-COF-TT; (f) Ni 2p XPS upon Ar+ sputtering of Ni@PI-COF-TT. | ||
In the photocatalytic CO2 reduction reaction, under the optimized reaction conditions, PI-COF-TT can generate 1933 μmol g−1 CO with a 93% selectivity over H2 production in a 4 h reaction (Fig. 4a), while PI-COF-1 and PI-COF-2 showed relatively low catalytic activities (Fig. 4b). It is noteworthy that the activity of PI-COF-TT is lower than that of the previously reported Ni-TpBpy-based catalytic system, mainly as a result of the absence of precious metal sensitizers. Nevertheless, the catalytic performance of PI-COF-TT is among the best compared to other noble-metal-free catalytic systems that have been reported so far (Table S1†). Control experiments show that the photocatalytic reduction of CO2 results from the coexistence of PI-COF-TT and [Ni(bpy)3]2+ (Fig. 4c). Cycling experiments indicate the catalytic and structural stability of PI-COF-TT in the photocatalysis (Fig. 4d and S26†). To confirm the origin of the as-formed CO, 13CO2 labelling experiments were performed. A major signal at a mass/charge ratio of 29 on the spectrum corresponding to 13CO appears, confirming that the generated CO comes from the reduction of CO2 (Fig. 4e). There were no detectable hydrocarbon products such as HCOOH and CH3OH when analyzing the reaction solvent through 1H NMR and high-performance liquid chromatography (HPLC) (Fig. S27†). The trend of CO production matches well with the optical absorption spectrum of PI-COF-TT (Fig. 4f), suggesting that the CO2 reduction is indeed induced by PI-COF-TT. The quantum efficiency (AQE) of PI-COF-TT was estimated to be 0.55% at 380 nm. Acetonitrile was found to be a favourable reaction solvent for CO2 reduction with a relatively high activity and selectivity (Fig. 5a), mainly because of its appropriate coordination ability to Ni, which can not only contribute to the stabilization of the Ni active sites, but also retain the accessibility to the Ni center. Other metal complexes were tested under similar reaction conditions (Fig. 5b). The results show that the Co complex could also reduce CO2 to produce CO efficiently but with a low selectivity for CO compared to H2. H2O in the reaction could significantly influence the catalytic activity and product selectivity. Upon increasing the amount of H2O in the reaction, the production of both CO and H2 decreases (Fig. 5c), probably due to the intrinsic hydrophobicity of PI-COFs. With the increment of the amount of Ni2+ in the reaction the reduction production gradually increased, further confirming that the Ni single sites act as catalytically active sites (Fig. 5d).
 | ||
| Fig. 4 (a) Kinetic profile of CO production by PI-COF-TT; (b) catalytic performance of PI-COFs; (c) control experiments using PI-COF-TT in 2 h CO2 photoreduction; (d) recycling test for CO2 reduction by PI-COF-TT in 2 h; (e) GCMS measurements of the gas production from 13CO2 photoreduction; (f) wavelength dependence of CO evolution in a 1 h reaction. | ||
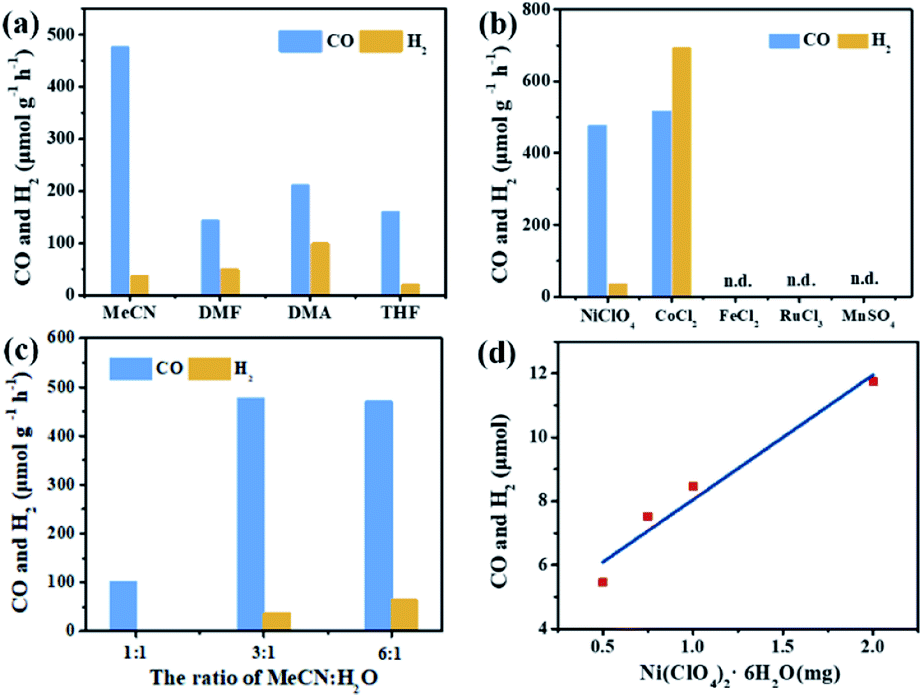 | ||
| Fig. 5 Effect of solvents (a), metal salts (b), H2O concentration (c) and Ni concentration (d) on CO2 reduction over PI-COF-TT in a 2 h reaction. | ||
The photoelectrochemical properties of the PI-COFs were investigated by electrochemical impedance spectroscopy (EIS) and transient photocurrent measurements. Nafion solution was used as an additive to PI-COF powder to form the active layer,51 which can form a homogeneous ink with the COF and further help to attach onto the surface of electrodes. Control experiments showed negligible effects of Nafion on the current density of electrodes (Fig. S28†). As shown in Fig. 6a, Nyquist curves show that all three PI-COFs exhibit two semicircles. The small semicircles in the high-frequency region correspond to the charge transfer resistance, while the large ones in the low-frequency region are related to diffusion resistance. The radius of the semicircles in the high frequency region of PI-COF-TT is smaller than that of other two PI-COFs, suggesting that the triazine ring in the PI-COFs obviously improves the rate of charge transfer.52 Linear potential sweep measurements of PI-COF-TT showed a higher current density of 10.35 μA cm−2 at 0.1 V vs. RHE compared to PI-COF-1 and PI-COF-2 (Fig. S29†). The current density is directly correlated with the increase in the catalytic performance. In addition, PI-COF-TT exhibits higher photocurrent intensity than PI-COF-1 and PI-COF-2 (Fig. 6b), which probably contributes to the improved photocatalytic activity.53 To show the morphology of the active photosensitizer layer, SEM of the PI-COF electrodes was performed. Top view SEM images revealed that the FTO glass electrodes can be well covered by PI-COFs (Fig. S30†). All PI-COF deposits show interstitial voids and textural porosity which may also contribute to their catalytic performance.54,55
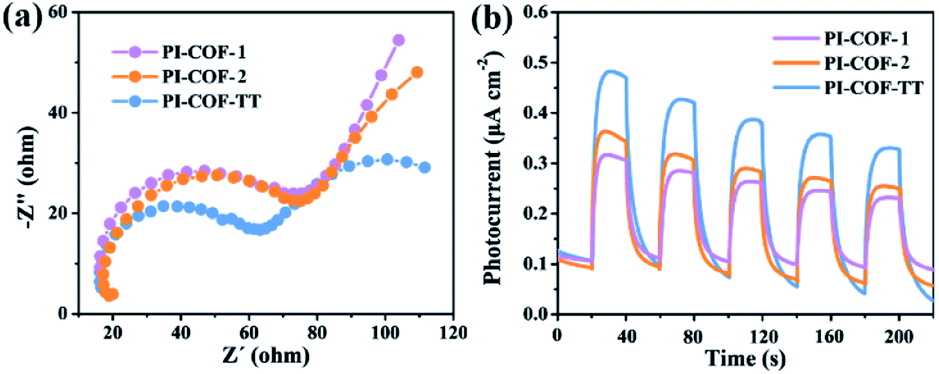 | ||
| Fig. 6 (a) EIS Nyquist plots of bulk PI-COFs; (b) photocurrent responses of PI-COFs. | ||
An interesting photochromic phenomenon was observed in the photoreduction of CO2 over PI-COFs (Fig. S31†). When the catalytic system was irradiated under a CO2 atmosphere, as shown in Fig. 7a and b, the color of PI-COF-TT changed from the original yellow to green to yellow to orange. After the introduction of air into the reaction, the color of PI-COF-TT changed from orange back to the original yellow. The color change of PI-COF-TT takes place in the presence of triethanolamine (TEOA) under light irradiation (Table S2†). The electron paramagnetic resonance (EPR) spectrum of the suspension of PI-COF-TT and TEOA in acetonitrile shows a prominent increment of EPR signals after light illumination (Fig. 7c and d), indicating charge generation upon photoexcitation,56 and leading to the photochromic phenomenon. In comparison with PI-COF-TT, the EPR signal intensity of Ni@PI-COF-TT decreased predominantly under identical conditions, revealing the electron transfer from PI-COF-TT to [Ni(bpy)3]2+. The solid-state photoluminescence (PL) emission spectra of PI-COFs show negligible peaks around 400–700 nm compared to the strong peak of PMDA at about 430 nm (Fig. S32–S34†). The weak emission of PI-COFs mainly results from the intramolecular charge transfer from the central rings acting as the electron donor to pyromellitic diimide units acting as the electron acceptor in PI-COFs.31,57 Cyclic voltammetry (CV) and optical tests of pyromellitic diimide, N,N′-bis(phenyl)pyromellitimide, triphenylamine, triphenylbenzene and triphenyltriazine as model compounds of building blocks were performed to reveal their relative energy levels (Fig. S35†). The LUMO levels of N,N′-bis(phenyl)pyromellitimide are very close to those of triphenylamine, triphenylbenzene and triphenyltriazine, which means that the connection of pyromellitic diimide and triphenylamine, triphenylbenzene or triphenyltriazine affording an extended conjugation framework could presumably contribute to the electron transfer. When a physical mixture of pyromellitic diimide and triphenyltriazine was employed in the catalytic reaction, no reduction products such as CO, CH4 and H2 were observed (Fig. S36†), which further reveals the vital role of the formation of the conjugated structure in PI-COFs for charge transfer. Density functional theory (DFT) calculations show that the CB and VB wave functions of PI-COFs are separately localized on the diimide units and the central rings, respectively (Fig. 7e). Due to their narrow band gaps, the PI-COFs can be excited to form electron–hole pairs by light irradiation. The photogenerated electrons transfer from the central rings to the diimide units. The planar π-conjugated structure in PI-COF-TT brings a superior separation of photogenerated electrons and holes compared to PI-COF-1 and PI-COF-2,58 thus contributing to the enhanced catalytic efficiency.
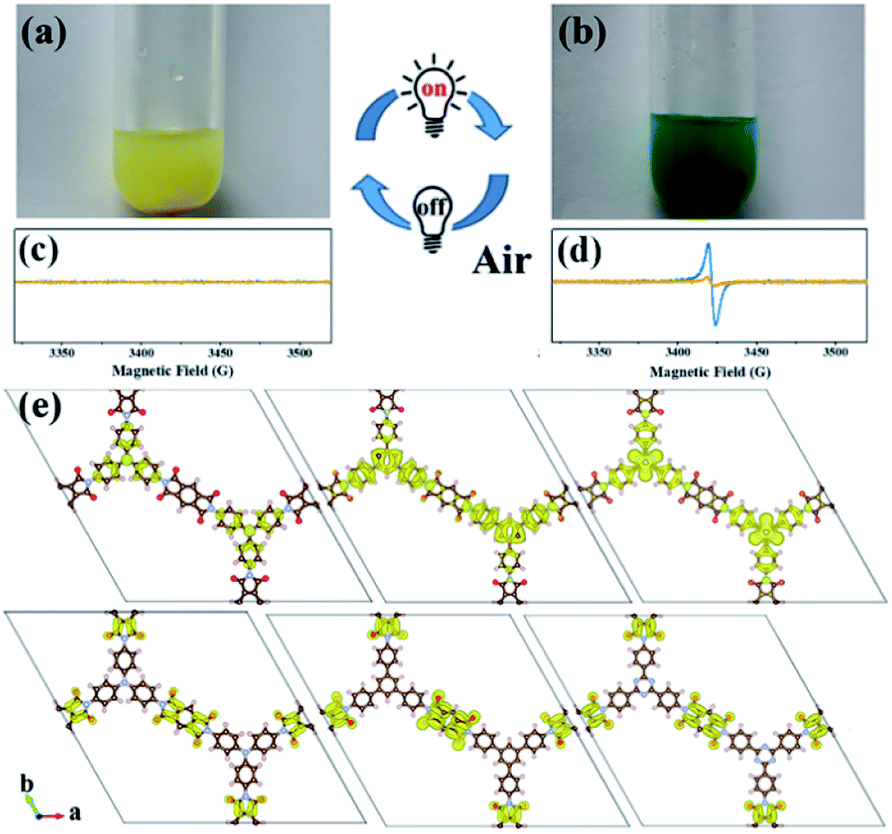 | ||
| Fig. 7 Photos and corresponding EPR spectra of PI-COF-TT (blue) and Ni@PI-COF-TT (yellow) with TEOA in acetonitrile solution before (a and c), and after (b and d) 10 min light illumination; charge density distribution for PI-COF-1, PI-COF-2 and PI-COF-TT at the CB minimum and the VB maximum (e) (drawn at the isosurface level of 0.0001 e Å−3). | ||
It is noteworthy that an obvious induction period accompanies the photochromic phenomenon. Thus, DFT calculations and control experiments were carried out to further explore the reaction mechanism. The in situ formed molecular Ni complexes preferred to be adsorbed to the diimide unit and not to the triazine ring, as evidenced by DFT calculations (Fig. 8a). However, the distance between the Ni complexes and framework (3.196 Å) indicates only weak interactions between PI-COF-TT and Ni complexes. The relatively weak signals of Ni 2p XPS (Fig. 8b) and EDX elemental mapping (Fig. 8c and S37†) of the recovered PI-COF-TT with bpy from the catalytic reaction than those of the recovered PI-COF-TT from the reaction without bpy confirmed the weak interactions between the molecular Ni active sites and the framework. PI-COFs with direct impregnation of [Ni(bpy)3]2+ showed significantly lower activity than PI-COFs with in situ formed [Ni(bpy)3]2+ (Fig. S38†). In the in situ formed [Ni(bpy)3]2+ case, the assembly of Ni ions and the bpy ligand occurred in the pores of PI-COFs, resulting in the uniform distribution of [Ni(bpy)3]2+ in PI-COFs. However, in the direct impregnation of [Ni(bpy)3]2+ case, the [Ni(bpy)3]2+ complexes with a relatively large molecular volume cannot diffuse effectively into the pores of PI-COFs, leading to the inefficient catalytic activity. The recovered PI-COF-TT does not generate the CO product in the additional reaction run in the absence of Ni complexes, further confirming that Ni active sites are free in the channel and only physically adsorbed on the pore surfaces.59 Additionally, when a bpy ligand with large substituents was used, the production of CO and H2 was not detected under similar reaction conditions (Fig. 8d). This could be due to the steric hindrance of the substituent on the bpy ligand, leading to inefficient contact between the Ni center and COF wall. These noncovalent interactions are always considered to be conducive for retaining their intrinsic properties as well as the sufficient interfacial mass transport.48,60 Thus, the induction period could be mainly ascribed to the collisional electron transfer from the COF to molecular Ni complexes to form molecular Ni active sites,48 which is verified by our previous report showing that [Ni(bpy)2]0 formed from the reduction of [Ni(bpy)3]2+ in the photocatalytic reaction serves as the active site for the activation and conversion of CO2.40 In this case, the molecular [Ni(bpy)2]0 active sites are produced from the reduction of [Ni(bpy)3]2+ by photogenerated electrons of PI-COF-TT under UV-vis light irradiation (Fig. S39 and S40†). Since single Ni0 atoms are active, excess bpy is necessary to stabilize single Ni sites through the formation of [Ni(bpy)2]0via coordination of bpy to Ni.
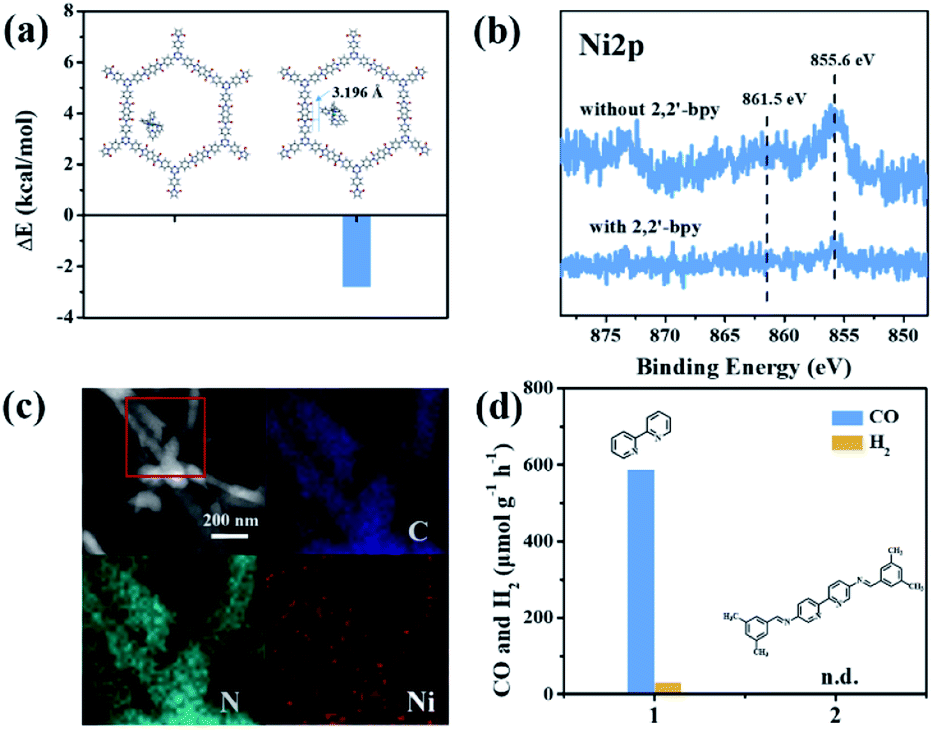 | ||
| Fig. 8 (a) The adsorption energy (ΔE) of Ni(bpy)32+ in PI-COF-TT. The distance between the Ni complex and the framework (inset); (b) Ni 2p XPS of the recovered PI-COF-TT with and without 2,2′-bipyridyl from the catalytic reaction; (c) EDX elemental mapping of the recovered PI-COF-TT from the photoreduction of CO2 with 2,2′-bipyridyl; (d) 2,2′-bipyridyl (1) and modified 2,2′-bipyridyl (2) performance in the photoreduction of CO2 over PI-COF-TT in 2 h. | ||
In order to understand the selectivity trends between the CO and H2 products, the adsorption energies of CO2 and H2O onto [Ni(bpy)2]0 with and without PI-COF-TT were calculated (Fig. S41 and Table S3†). As shown in Fig. 9a, the adsorption energy of CO2 on [Ni(bpy)2]0 in the presence of PI-COF-TT was significantly lower than that of H2O, implying the stronger affinity of molecular [Ni(bpy)2]0 toward CO2, which would facilitate the formation of the key intermediate Ni–CO2 adducts and afford selectivity for the CO2 reduction product rather than the H2 product. It is noteworthy that the adsorption energy of CO2 on Ni active sites was obviously reduced by PI-COF-TT through the hydrogen bonding interactions between the hydrogen atom in the PI unit and the activated CO2 molecule (Fig. 9a, inset). Accordingly, the selective photoreduction of CO2 was greatly dependent on the selective adsorption and activation of CO2 on the metal active sites and the special reaction microenvironment. The synergistic photocatalytic system containing PI-COFs and single Ni sites facilitates the selective activation of CO2 and inhibits the competitive H2 evolution, leading to the enhanced catalytic activity and selectivity.
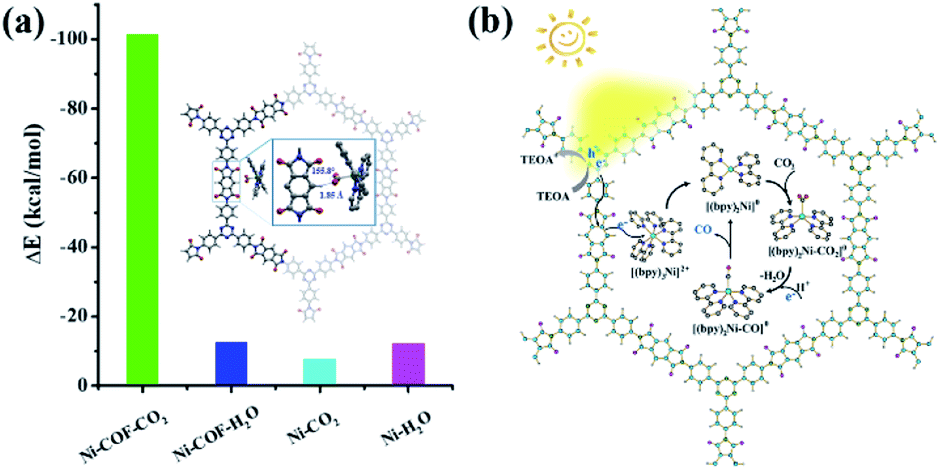 | ||
| Fig. 9 (a) DFT-calculated adsorption energy (ΔE, kcal mol−1) of CO2 and H2O on [Ni(bpy)2]0 with and without PI-COF-TT (inset: the activation of CO2 by [Ni(bpy)2]0 in PI-COF-TT). Ni–COF–CO2, Ni–COF–H2O, Ni–CO2 and Ni–H2O are the complexes of [Ni(bpy)2]0 with CO2 and/or H2O without and/or with PI-COF-TT, respectively. (b) Proposed reaction mechanism for photoreduction of CO2 over PI-COF-TT. | ||
Based on the above results, a possible mechanism for the selective photoreduction of CO2 over PI-COF-TT with molecular Ni complexes was proposed (Fig. 9b). Light illumination on PI-COF-TT generates electron–hole pairs. The holes are reductively quenched by TEOA. The electrons move from the triazine ring to the PI unit and subsequently transfer to the accommodated molecular [Ni(bpy)3]2+ complexes to form [Ni(bpy)2]0 active sites, which can couple with CO2 to produce CO. The open channel of PI-COF-TT facilitates the processes of photogeneration of molecular Ni active sites and the subsequent activation and conversion of CO2. Besides, the strong adsorption affinity of PI-COF-TT for CO2 may increase the local concentration of CO2 in the channels, thereby promoting the formation of Ni–CO2 adducts and facilitating the selective reduction of CO2.
Conclusions
In summary, we demonstrated a design of an integration of PI-COFs with Ni single sites for selective photoreduction of CO2. The electronic properties of PI-COFs can be facilely tuned for photocatalysis. The photogenerated electron–hole pairs in PI-COFs under irradiation can be efficiently separated through an intra- and inter-molecular charge-transfer mechanism to drive the reduction of CO2. The excellent catalytic performance of the COF-based catalytic system mainly arises from the synergistic effects of the photoactive PI-COF and single Ni sites, in which the PI-COF with a nature-mimicking architecture serves as the functionalized host for single Ni sites to promote the reaction. This work presents an integration of Ni single sites in PI-COFs for CO2 reduction and a deep understanding of the electron-transfer mechanism in COFs. Based on the diversity of COFs and molecular metal complexes, we believe, inspired by our work, there would be more functionalized COFs developed for robust and efficient catalytic systems for sustainable energy conversion.Experimental
Synthesis of PI-COF-TT
TAPT (35.1 mg, 0.10 mmol) and PMDA (32.7 mg, 0.15 mmol) were placed in a mixed solution of mesitylene/NMP/isoquinoline (0.5 mL/0.5 mL/0.05 mL). The tube was flash frozen at 77 K (liquid N2 bath) and degassed by pump–thaw three times. The tube was sealed and heated at 200 °C for 5 days, giving a yellow precipitate. The precipitate was purified by Soxhlet extraction using tetrahydrofuran overnight, and finally dried under vacuum at 80 °C to give PI-COF-TT (yield 66%).Photocatalytic reduction of CO2
A mixed solution of acetonitrile, H2O and triethanolamine (TEOA) (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1, 5 mL) containing PI-COFs (10 mg), Ni(ClO4)2·6H2O (2 mg, 5.5 μmol) and 2,2′-bipyridyl (15 mg, 0.1 mmol) was purged with CO2 for 15 min. The solution was then irradiated under UV-Vis light (300 W Xe lamp, PLS-SEX 300/300UV, 780 mW cm−2) at 313 K. After each reaction time, the generated gas in the headspace of the reaction vessel was sampled with a gas-tight syringe and determined by gas chromatography (Agilent 7890B) with both TCD and FID detectors. H2 was detected using a TCD detector. CO was converted to CH4 in a methanation reactor and then analyzed using an FID detector. The apparent quantum efficiency (AQE) at 380 nm was calculated by the following equation: AQE = (2 × amount of CO molecules evolved in 1 h/number of incident photons in 1 h) × 100%.
:1:1, 5 mL) containing PI-COFs (10 mg), Ni(ClO4)2·6H2O (2 mg, 5.5 μmol) and 2,2′-bipyridyl (15 mg, 0.1 mmol) was purged with CO2 for 15 min. The solution was then irradiated under UV-Vis light (300 W Xe lamp, PLS-SEX 300/300UV, 780 mW cm−2) at 313 K. After each reaction time, the generated gas in the headspace of the reaction vessel was sampled with a gas-tight syringe and determined by gas chromatography (Agilent 7890B) with both TCD and FID detectors. H2 was detected using a TCD detector. CO was converted to CH4 in a methanation reactor and then analyzed using an FID detector. The apparent quantum efficiency (AQE) at 380 nm was calculated by the following equation: AQE = (2 × amount of CO molecules evolved in 1 h/number of incident photons in 1 h) × 100%.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (No. U1905215, 51672046, 51672047), Natural Science Foundation of Fujian Province of China (No. 2019J01226, 2019J01648), and Open Project Program of the State Key Laboratory of Photocatalysis on Energy and Environment (No. SKLPEE-KF201815, SKLPEE-KF201813).Notes and references
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119(6), 3962–4179 CrossRef CAS PubMed.
- W. Tu, Y. Zhou and Z. Zou, Adv. Mater., 2014, 26, 4607–4626 CrossRef CAS PubMed.
- J. Ran, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef PubMed.
- K. Niu, Y. Xu, H. Wang, R. Ye, H. L. Xin, F. Lin, C. Tian, Y. Lum, K. C. Bustillo, M. M. Doeff, M. T. M. Koper, J. Ager, R. Xu and H. Zheng, Sci. Adv., 2017, 3, e1700921 CrossRef PubMed.
- J. W. Wang, W. J. Liu, D. C. Zhong and T. B. Lu, Coord. Chem. Rev., 2019, 378, 237–261 CrossRef CAS.
- D. Hong, Y. Tsukakoshi, H. Kotani, T. Ishizuka and T. Kojima, J. Am. Chem. Soc., 2017, 139, 6538–6541 CrossRef CAS PubMed.
- T. Ouyang, H. H. Huang, J. W. Wang, D. C. Zhong and T. B. Lu, Angew. Chem., Int. Ed., 2017, 56, 738–743 CrossRef CAS PubMed.
- X. Zhang, M. Cibian, A. Call, K. Yamauchi and K. Sakai, ACS Catal., 2019, 9(12), 11263–11273 CrossRef CAS.
- N. Elgrishi, M. B. Chambers, X. Wang and M. Fontecave, Chem. Soc. Rev., 2017, 46, 761–796 RSC.
- M. F. Kuehnel, K. L. Orchard, K. E. Dalle and E. Reisner, J. Am. Chem. Soc., 2017, 139, 7217–7223 CrossRef CAS PubMed.
- P. Huang, J. Huang, S. A. Pantovich, A. D. Carl, T. G. Fenton, C. A. Caputo, R. L. Grimm, A. I. Frenkel and G. Li, J. Am. Chem. Soc., 2018, 140, 16042–16047 CrossRef CAS PubMed.
- T. Kajiwara, M. Fujii, M. Tsujimoto, K. Kobayashi, M. Higuchi, K. Tanaka and S. Kitagawa, Angew. Chem., Int. Ed., 2016, 55, 2697–2700 CrossRef CAS PubMed.
- Q. Han, X. Bai, Z. Man, H. He, L. Li, J. Hu, A. Alsaedi, T. Hayat, Z. Yu, W. Zhang, J. Wang, Y. Zhou and Z. Zou, J. Am. Chem. Soc., 2019, 141, 4209–4213 CrossRef CAS PubMed.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS PubMed.
- X. Zheng, G. Shen, C. Wang, Y. Li, D. Dunphy, T. Hasan, C. J. Brinker and B. L. Su, Nat. Commun., 2017, 8, 14921 CrossRef CAS PubMed.
- Y. Li, Z.-Y. Fu and B.-L. Su, Adv. Funct. Mater., 2012, 22, 4634–4667 CrossRef CAS.
- H. Zhou, P. Li, J. Liu, Z. Chen, L. Liu, D. Dontsova, R. Yan, T. Fan, D. Zhang and J. Ye, Nano Energy, 2016, 25, 128–135 CrossRef CAS.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed.
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef.
- Y. Fu, X. Zhu, L. Huang, X. Zhang, F. Zhang and W. Zhu, Appl. Catal., B, 2018, 239, 46–51 CrossRef CAS.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W.-H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180–1189 CrossRef CAS PubMed.
- J. Thote, H. B. Aiyappa, A. Deshpande, D. Diaz Diaz, S. Kurungot and R. Banerjee, Chem.–Eur. J., 2014, 20, 15961–15965 CrossRef CAS PubMed.
- J. Ming, A. Liu, J. Zhao, P. Zhang, H. Huang, H. Lin, Z. Xu, X. Zhang, X. Wang, J. Hofkens, M. B. J. Roeffaers and J. Long, Angew. Chem., 2019, 131, 18458–18462 CrossRef.
- X. Kang, X. Wu, X. Han, C. Yuan, Y. Liu and Y. Cui, Chem. Sci., 2020, 11, 1494–1502 RSC.
- N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 1, 16068 CrossRef CAS.
- L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789–2793 RSC.
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Nat. Commun., 2015, 6, 8508 CrossRef CAS PubMed.
- Z. Zhou, W. Zhong, K. Cui, Z. Zhuang, L. Li, L. Li, J. Bi and Y. Yu, Chem. Commun., 2018, 54, 9977–9980 RSC.
- L. Y. Li, Z. M. Zhou, L. Y. Li, Z. Y. Zhuang, J. H. Bi, J. H. Chen, Y. Yu and J. G. Yu, ACS Sustainable Chem. Eng., 2019, 7, 18574–18581 CrossRef CAS.
- G. Lin, H. Ding, R. Chen, Z. Peng, B. Wang and C. Wang, J. Am. Chem. Soc., 2017, 139, 8705–8709 CrossRef CAS PubMed.
- J. Lv, Y. X. Tan, J. Xie, R. Yang, M. Yu, S. Sun, M. D. Li, D. Yuan and Y. Wang, Angew. Chem., Int. Ed., 2018, 57, 12716–12720 CrossRef CAS PubMed.
- R. Chen, J. L. Shi, Y. Ma, G. Lin, X. Lang and C. Wang, Angew. Chem., Int. Ed., 2019, 58, 6430–6434 CrossRef CAS.
- M. Lu, J. Liu, Q. Li, M. Zhang, M. Liu, J.-L. Wang, D.-Q. Yuan and Y.-Q. Lan, Angew. Chem., 2019, 131, 12522–12527 CrossRef.
- S. Nandi, S. K. Singh, D. Mullangi, R. Illathvalappil, L. George, C. P. Vinod, S. Kurungot and R. Vaidhyanathan, Adv. Energy Mater., 2016, 6, 1601189 CrossRef.
- D. Mullangi, V. Dhavale, S. Shalini, S. Nandi, S. Collins, T. Woo, S. Kurungot and R. Vaidhyanathan, Adv. Energy Mater., 2016, 6, 1600110 CrossRef.
- K. Cui, W. Zhong, L. Li, Z. Zhuang, L. Li, J. Bi and Y. Yu, Small, 2019, 15, 1804419 Search PubMed.
- Y. He, X. Chen, C. Huang, L. Li, C. Yang and Y. Yu, Chin. J. Catal., 2021, 42, 123–130 CrossRef CAS.
- W. Liu, X. Li, C. Wang, H. Pan, W. Liu, K. Wang, Q. Zeng, R. Wang and J. Jiang, J. Am. Chem. Soc., 2019, 141, 17431–17440 CrossRef CAS PubMed.
- X. Yang, Y. He, L. Li, J. Shen, J. Huang, L. Li, Z. Zhuang, J. Bi and Y. Yu, Chem.–Eur. J., 2020, 26, 1864–1870 CrossRef CAS.
- W. Zhong, R. Sa, L. Li, Y. He, L. Li, J. Bi, Z. Zhuang, Y. Yu and Z. Zou, J. Am. Chem. Soc., 2019, 141, 7615–7621 CrossRef CAS PubMed.
- Q. Fang, Z. Zhuang, S. Gu, R. B. Kaspar, J. Zheng, J. Wang, S. Qiu and Y. Yan, Nat. Commun., 2014, 5, 4503 CrossRef PubMed.
- X. Zhu, S. An, Y. Liu, J. Hu, H. Liu, C. Tian, S. Dai, X. Yang, H. Wang, C. W. Abney and S. Dai, AIChE J., 2017, 63, 3470–3478 CrossRef CAS.
- C. Zhang, S. Zhang, Y. Yan, F. Xia, A. Huang and Y. Xian, ACS Appl. Mater. Interfaces, 2017, 9, 13415–13421 CrossRef CAS PubMed.
- M. R. Liebl and J. Senker, Chem. Mater., 2013, 25, 970–980 CrossRef CAS.
- P. Arab, E. Parrish, T. İslamoğlu and H. M. El-Kaderi, J. Mater. Chem. A, 2015, 3, 20586–20594 RSC.
- S. Yang, W. Hu, X. Zhang, P. He, B. Pattengale, C. Liu, M. Cendejas, I. Hermans, X. Zhang, J. Zhang and J. Huang, J. Am. Chem. Soc., 2018, 140, 14614–14618 CrossRef CAS PubMed.
- Z. Fu, X. Wang, A. M. Gardner, X. Wang, S. Y. Chong, G. Neri, A. J. Cowan, L. Liu, X. Li, A. Vogel, R. Clowes, M. Bilton, L. Chen, R. S. Sprick and A. I. Cooper, Chem. Sci., 2020, 11, 543–550 RSC.
- T. Banerjee, F. Haase, G. Savasci, K. Gottschling, C. Ochsenfeld and B. V. Lotsch, J. Am. Chem. Soc., 2017, 139, 16228–16234 CrossRef CAS PubMed.
- S. Daniele, P. Ugo, G. Bontempelli and M. Fiorani, J. Electroanal. Chem., 1987, 219, 259–271 CrossRef CAS.
- Y. J. Yuan, H. W. Lu, J. R. Tu, Y. Fang, Z. T. Yu, X. X. Fan and Z. G. Zou, ChemPhysChem, 2015, 16, 2925–2930 CrossRef CAS PubMed.
- F. R. Pomilla, A. Brunetti, G. Marcì, E. I. García-López, E. Fontananova, L. Palmisano and G. Barbieri, ACS Sustainable Chem. Eng., 2018, 6, 8743–8753 CrossRef CAS.
- E. Liu, C. Jin, C. Xu, J. Fan and X. Hu, Int. J. Hydrogen Energy, 2018, 43, 21355–21364 CrossRef CAS.
- E. Liu, J. Chen, Y. Ma, J. Feng, J. Jia, J. Fan and X. Hu, J. Colloid Interface Sci., 2018, 524, 313–324 CrossRef CAS.
- J. M. Rotter, S. Weinberger, J. Kampmann, T. Sick, M. Shalom, T. Bein and D. D. Medina, Chem. Mater., 2019, 31, 10008–10016 CrossRef CAS.
- X. Zhao, P. Pachfule, S. Li, T. Langenhahn, M. Ye, C. Schlesiger, S. Praetz, J. Schmidt and A. Thomas, J. Am. Chem. Soc., 2019, 141, 6623–6630 CrossRef CAS.
- E. Jin, Z. Lan, Q. Jiang, K. Geng, G. Li, X. Wang and D. Jiang, Chem, 2019, 5, 1632–1647 CAS.
- A. C. Jakowetz, T. F. Hinrichsen, L. Ascherl, T. Sick, M. Calik, F. Auras, D. D. Medina, R. H. Friend, A. Rao and T. Bein, J. Am. Chem. Soc., 2019, 141, 11565–11571 CrossRef CAS.
- S. Jin, M. Supur, M. Addicoat, K. Furukawa, L. Chen, T. Nakamura, S. Fukuzumi, S. Irle and D. Jiang, J. Am. Chem. Soc., 2015, 137, 7817–7827 CrossRef CAS.
- B. P. Biswal, H. A. Vignolo-González, T. Banerjee, L. Grunenberg, G. Savasci, K. Gottschling, J. Nuss, C. Ochsenfeld and B. V. Lotsch, J. Am. Chem. Soc., 2019, 141, 11082–11092 CrossRef CAS PubMed.
- Q. Sun, Y. Tang, B. Aguila, S. Wang, F.-S. Xiao, P. K. Thallapally, A. M. Al-Enizi, A. Nafady and S. Ma, Angew. Chem., 2019, 131, 8762–8767 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and characterization. See DOI: 10.1039/d0sc01747g |
| ‡ X. Chen and Q. Dang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |