
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe roles of chalcogenides in O2 protection of H2ase active sites
Xuemei
Yang
and
Marcetta Y.
Darensbourg
 *
*
Texas A&M University, Department of Chemistry, College Station, TX 77843, USA. E-mail: marcetta@chem.tamu.edu
First published on 12th August 2020
Abstract
At some point, all HER (Hydrogen Evolution Reaction) catalysts, important in sustainable H2O splitting technology, will encounter O2 and O2-damage. The [NiFeSe]-H2ases and some of the [NiFeS]–H2ases, biocatalysts for reversible H2 production from protons and electrons, are exemplars of oxygen tolerant HER catalysts in nature. In the hydrogenase active sites oxygen damage may be extensive (irreversible) as it is for the [FeFe]–H2ase or moderate (reversible) for the [NiFe]–H2ases. The affinity of oxygen for sulfur, in [NiFeS]–H2ase, and selenium, in [NiFeSe]–H2ase, yielding oxygenated chalcogens results in maintenance of the core NiFe unit, and myriad observable but inactive states, which can be reductively repaired. In contrast, the [FeFe]–H2ase active site has less possibilities for chalcogen-oxygen uptake and a greater chance for O2-attack on iron. Exposure to O2 typically leads to irreversible damage. Despite the evidence of S/Se-oxygenation in the active sites of hydrogenases, there are limited reported synthetic models. This perspective will give an overview of the studies of O2 reactions with the hydrogenases and biomimetics with focus on our recent studies that compare sulfur and selenium containing synthetic analogues of the [NiFe]–H2ase active sites.
1. Introduction to hydrogenases
Much effort in enzyme isolation and purification in the past decades has permitted protein crystallographers to open the “black boxes” of the hydrogenase enzyme active sites exposing extraordinary organoiron fragments as components of remarkable hydrogen processing catalysts.1–6 Examples of the known classes, the nickel–iron hydrogenase, or [NiFe]–H2ase, the diiron or [FeFe]–H2ase, and the mono-iron or [Fe]–H2ase, are shown in Fig. 1.1–5,7 While phylogenetically distinct, convergent evolution has found the benefits of the diatomic CO and CN ligands that are effective for π-delocalization and H-bonding, in the [FeFe]– and [NiFe]–H2ase active sites. Both are presumed to have been natural targets for their ability to maintain low spin iron, while the CN additionally offers an H-bonding anchoring effect on the iron fragment into the protein pocket. Abundant sulfur is a key feature of the structures of the bimetallic subsites.1–5 With the [NiFe]–H2ase active site this comes in the form of four cysteine connectors to the polypeptide chain. The [FeFe]–H2ase has one cysteine that links the 2Fe subsite to a 4Fe4S, redox-buffering cluster as well as two additional sulfurs within a unique bridging azadithiolate. Thiolate sulfurs bridge the two metals in both [NiFe]– and [FeFe]–H2ase and, due to orientation of intrinsic lone pairs, hold the two metals close in “butterfly” formation, of significance for M–M bonding as another tuning point for electron delocalization as needed. Note that there are two subclasses of the [NiFe]–H2ase; the major (>90%) is all sulfur-containing, while a minor contains one selenocysteine.2,3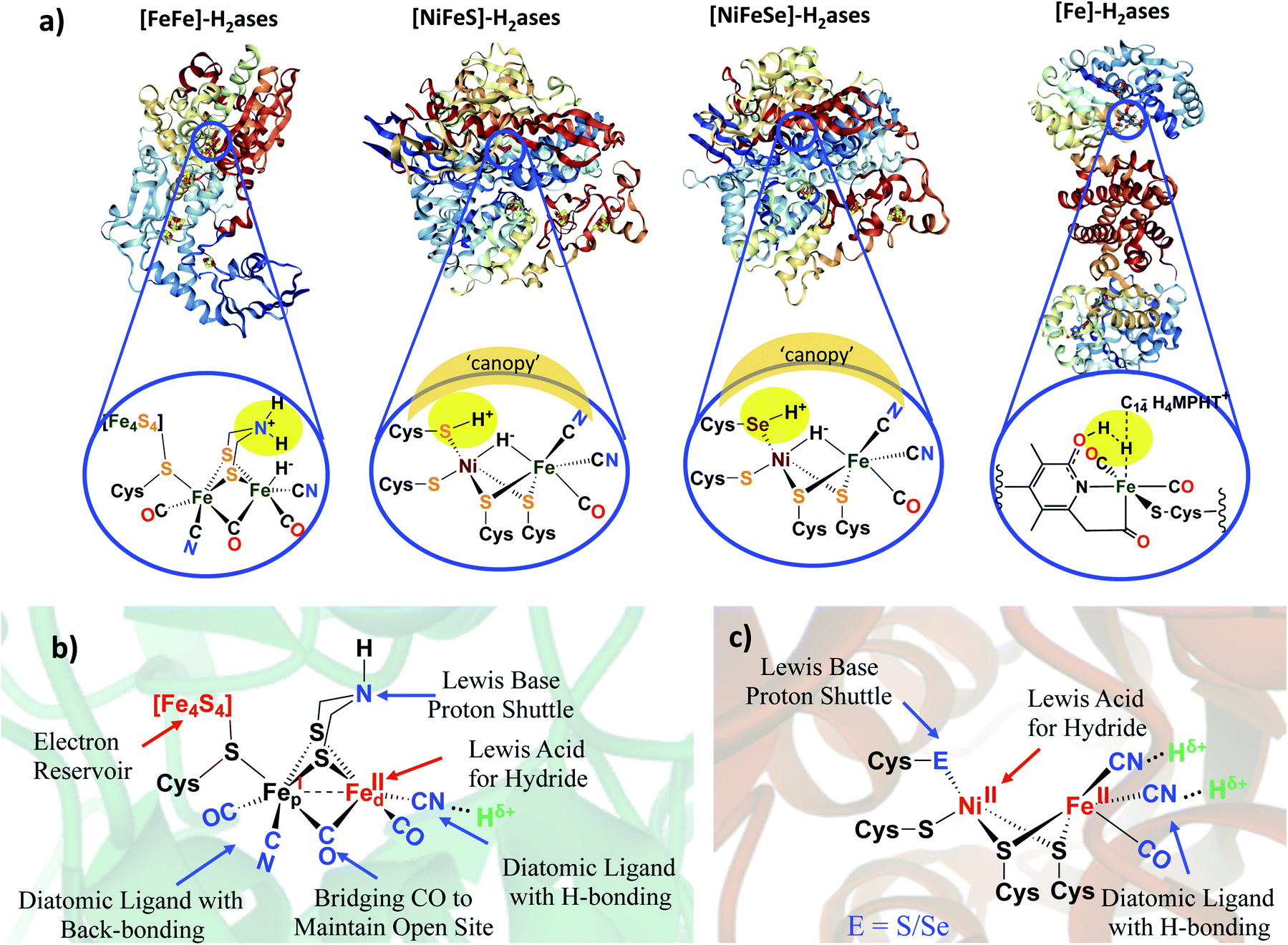 | ||
| Fig. 1 (a) Selected structures of hydrogenase proteins exemplary of the four known classes, with blow-ups of their active sites.1–5 Hydrogen-atoms in expected (or actually detected in the case of the Ni–R form of [NiFeS]–H2ase),5 idealized positions for proton–hydride coupling (or the reverse, heterolytic H2 splitting) via pendant base proton shuttles within the first coordination sphere; a “canopy” of critical immediate outer coordination sphere residues indicated by shaded area;7 (b) functional analysis of [FeFe]–H2ase active site components in second coordination spheres; (c) functional analysis of [NiFe]–H2ase active site components. | ||
In addition to creating an appropriate electronic and structural environment in the ligand fields of the metals in the hydrogenase active sites, the myriad oxidation states possible for sulfur and selenium provide repositories for adventitious O2. Reversibility in this chemistry is intertwined with abrogation of oxidative, irreversible damage at the metals. Thus, limiting the amount of reactive oxygen species is critical to the longevity of the biocatalyst.
The popularity of biomimetic research into hydrogenase active sites has been fostered by the possibility of proton reduction and hydrogen oxidation catalysis by abundant first row transition metals as molecular catalysts, as well as to further understanding of these remarkable biocatalysts.6,8 While synthetic ligand fields have yet to match the intricate and extended structures within the natural proteins, they are designed to approximate the electronic environment, and hopefully the function, of core features of those sites.9 For simplicity of interpretation, most electrocatalytic studies of model complexes for proton reduction to H2 have been carried out in the absence of O2. However, studies of the oxygen-damaged active sites, particularly the Ni-A and Ni-B states for the [NiFe]–H2ase, were seminal to the development of this field long before the precise interpretation of the various active site structures in their reduced forms was possible.6,9–11 Since that time, protein XRD has generated many structures of “oxygen-damaged” hydrogenase active sites of [NiFe]–H2ase. Our Perspective is largely limited to reports of oxygenated biomimetics of these sites. Numerous excellent reviews of the hydrogenases and models of their active sites are available such that our description of the features that control their interactions with protons or dihydrogen will be only for comparison to the more excellent electrophile, O2. The mono-iron H2ase is neglected here as its well known air sensitivity has, at this time, not been explored.
2. Strategies used by O2-tolerant [NiFeS]– and [NiFeSe]–H2ases
2.1 Deeply buried active sites and hydrophobic/hydrophilic access channels
Microorganisms that require the H2ase enzymes, likely representing Earth's earliest life forms, developed strategies to protect their exquisitely evolved, hydrogen-processing catalysts with respect to the poisonous O2 toxin as it built up in the atmosphere on the planet. An obvious strategy for the host organism is to confine itself into protective surroundings. Much like synthetic organometallic chemists that perform air-sensitive reactions in glove boxes filled with inert gas, or in special glassware on vacuum lines, many organisms that require hydrogenases operate in the low oxygen, reductive conditions at the bottom of ponds and rivers. In addition, on the molecular level within the organism, the active sites of the hydrogenase enzymes are buried deeply within the folds of their host proteins.6 Strings of iron–sulfur clusters guide electrons into and out of the active sites of the [NiFe]– and [FeFe]–H2ases. Hydrophobic and hydrophilic channels further control access or exit of H2 and H+, respectively, apparently terminating at the ideal position of reactivity.12 The hydrophobic channel(s) also provide a path for O2 access. Attesting to the validity of this conclusion are mutagenesis studies that show a dependence on oxygen damage at the active site of [NiFe]–H2ase from the Ralstonia eutropha organism with modification of specific amino acid residues that control the size of the channel.12A recent (2019) study involving the [NiFeSe]–H2ase from D. vulgaris Hildenborough challenges the conventional wisdom that hydrophobicity is considered optimal for diffusion of neutral diatomics, H2 as substrate, or O2 or CO as inhibitors.13 Pereira, Matias, Leger, et al. found that a hydrophilic channel modified by mutation of two glycine residues with alanine or serine resulted in more O2-tolerant variants, without changing the hydrogenase activity. Such mutations, expected to narrow this channel, prevented or slowed the oxidation of the active-site cysteine that lies at the end of the channel. The inevitable conclusion is that the native hydrophilic channel also allows access of O2 or reactive oxygen species (ROS).13 Nevertheless, other studies from this group notes that the difference of the oxygen sensitivity of the [NiFeSe]–H2ase, also from D. vulgaris Hildenborough, could largely be ascribed to the difference in chemical properties of the Se vs. S elements, vide infra.14 Such results indicate that the enzymes have multiple points of control available to protect the small organometallic-like catalysts encased in the polypeptide folds.
By the H+/H− placements in Fig. 1a, we noted our inclination for direct assistance by inner sphere pendant bases in the ultimate H+/H− coupling mechanism. Fig. 2 presents the currently accepted mechanism of [NiFe]–H2ase, including off-cycle Ni–Fe complexes with O-atom uptake resulting from adventitious O2.6 Further discussion of the Ni-B and Ni-A states is provided later in this Perspective. Suffice it to say that the eventual unraveling of nickel-based EPR signals from the off cycle, stable oxygenated species as related to activation was of major importance to the understanding of the competition for electron-rich sites by O2 and by H+.
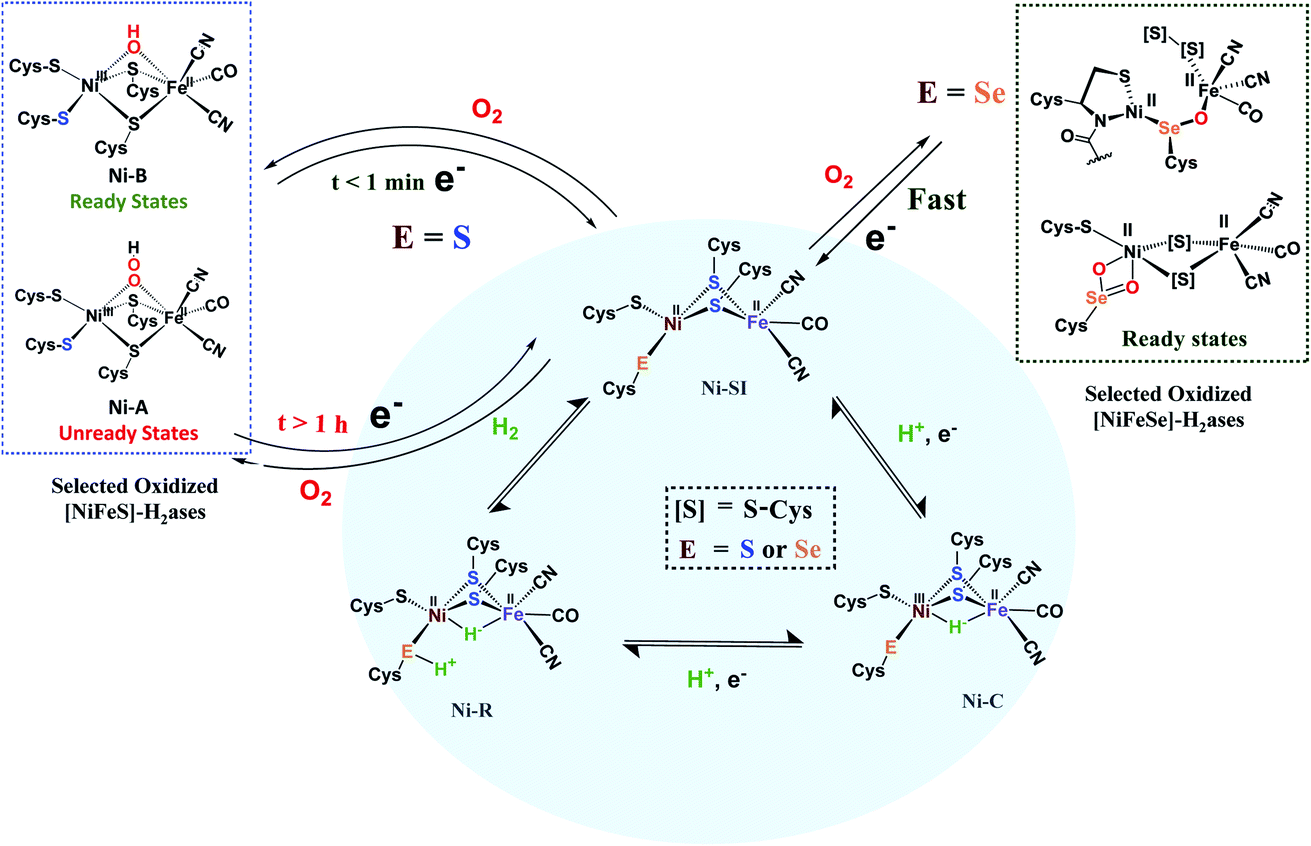 | ||
| Fig. 2 Abbreviated catalytic cycle (blue oval) of [NiFe]H2ase in all-S or Se forms, including the off-cycle oxygenates that were identified by crystallography and EPR spectroscopy.6 | ||
While the [FeFe]–H2ase is far more air sensitive than the [NiFe], a scenario has been presented wherein an exogenous sulfur ligand, derived from H2S in the Desulfovibrio desulfuricans sulfate reducing bacterium, is found to occupy the exposed open site on the “rotated”, distal iron of the H cluster, Fig. 1b. Such a position blocks both hydrogenase activity and imparts immunity from O2 attack.15
2.2 The canopy effect: residues in immediate outer coordination sphere mediate H+/H2 interconversion and O2 reactivity
As described above, the identical positions of a terminal thiolate sulfur in [NiFeS]–H2ase and a terminal selenoate selenium in [NiFeSe]–H2ase, present a strong argument that the differences in activity and responses to O2 should be ascribed to the pendant base effects of the chalcogens as crucial mediators of H+/H2 access to the metals and their interconversion. An alternate possibility that has gained considerable traction is that the ultimate mediator lies in outer coordination sphere bases, positioned in a “canopy” above the more open side of the Ni–Fe active site, as indicated in Fig. 1. Elegant experiments from Armstrong and collaborators have identified four highly conserved residues in the [NiFe]–H2ase in Escherichia coli, including an arginine that dangles a guanidine/guanidinium at a distance of ca. 4.4 Å from the critical H+ uptake center between Ni and Fe.7 In support of the hypothesis that this outer sphere base serves as the ultimate proton shuttle and directly controls reactivity are several mutagenesis studies that track with the rates of H2 oxidation. Consistently there is a decrease in O2 tolerance when the negatively charged residues in the canopy are neutralized in the variants, proposed to be due to stabilization of the oxidized resting NiIII–OH or Ni B state.7Armstrong's detailed studies are made possible by the powerful technique of protein film voltammetry on variants designed to probe the role of each residue that lines the outer shell of the active site.7 The study does not negate the possibility that the pendant chalcogens could also be intermediaries or proton depots, subsequent to residence in the canopy. The nearby chalcogens certainly demonstrate efficacy to operate as O-atom acceptors. These studies underscore the extraordinary complexity of the working parts of hydrogenase catalysts, and interesting research still engages the curiosity of scientists.
2.3 Assistance from FeS clusters: rapid conversion of O2 to hydroxide to avoid ROS
When O2 fugacity is sufficient, O2 entry to the hydrogenase active sites presents opportunity for competitive electron transfer to H+versus O2; reactive oxygen species (ROS), shown in bold in Scheme 1, derived from the O2 possibly leading to ultimate degradation of the low valent iron to inactive oxo-species. While the reductive cost of four electrons is great, rapid conversion of the bound O2 to hydroxide anion, followed by protonation yielding water, is the ultimate answer to safe O2 removal from the active sites.16–18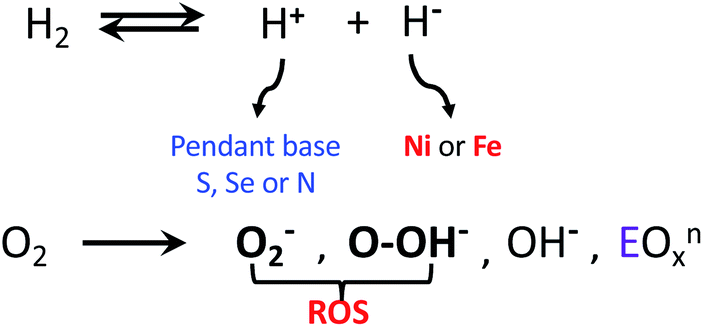 | ||
| Scheme 1 H2 and O2 reactions in hydrogenase enzymes; O2− and OOH− are designated as reactive oxygen species (ROS).6 | ||
A fascinating chemical line of defense using a rapid conversion approach has been identified for the [NiFeS]–H2ase in the membrane-bound hydrogenase (MBH) from Ralstonia eutropha.16 While the core NiFe component is identical in [NiFeS]–H2ase's from various sources, and the electron transport iron sulfur chain operates similarly, there is an impressive compositional and structural modification in the iron sulfur cluster that is proximal to the NiFe site. Identified by X-ray crystallography by Friedrich, Lenz, et al., and by Higuchi, et al., as a [4Fe3S]6Cys cluster, one structural sulfide is displaced from a normal 4Fe4S cluster, generating a [4Fe3SS′] “cubane”, where the S′ is a cysteinyl sulfur from the CysCys motif and is bridging two irons, Fig. 3.16–18 The sixth cysteine is on another iron, balancing the oxidation states within the cluster. When called upon by polarization at the NiFe site as O2 invades, the CysCys dipeptide assists the 4Fe3S cluster in providing electrons to the O2 electrophile by stabilizing the resulting superoxidized cluster, Fig. 3. This stabilization is a result of the deprotonation of the amide nitrogen and its minor shift into bonding range of the iron. The reversibility of this concomitant structural/redox change is key to the oxygen tolerance of MBH, which actually operates in the presence of O2.16–18
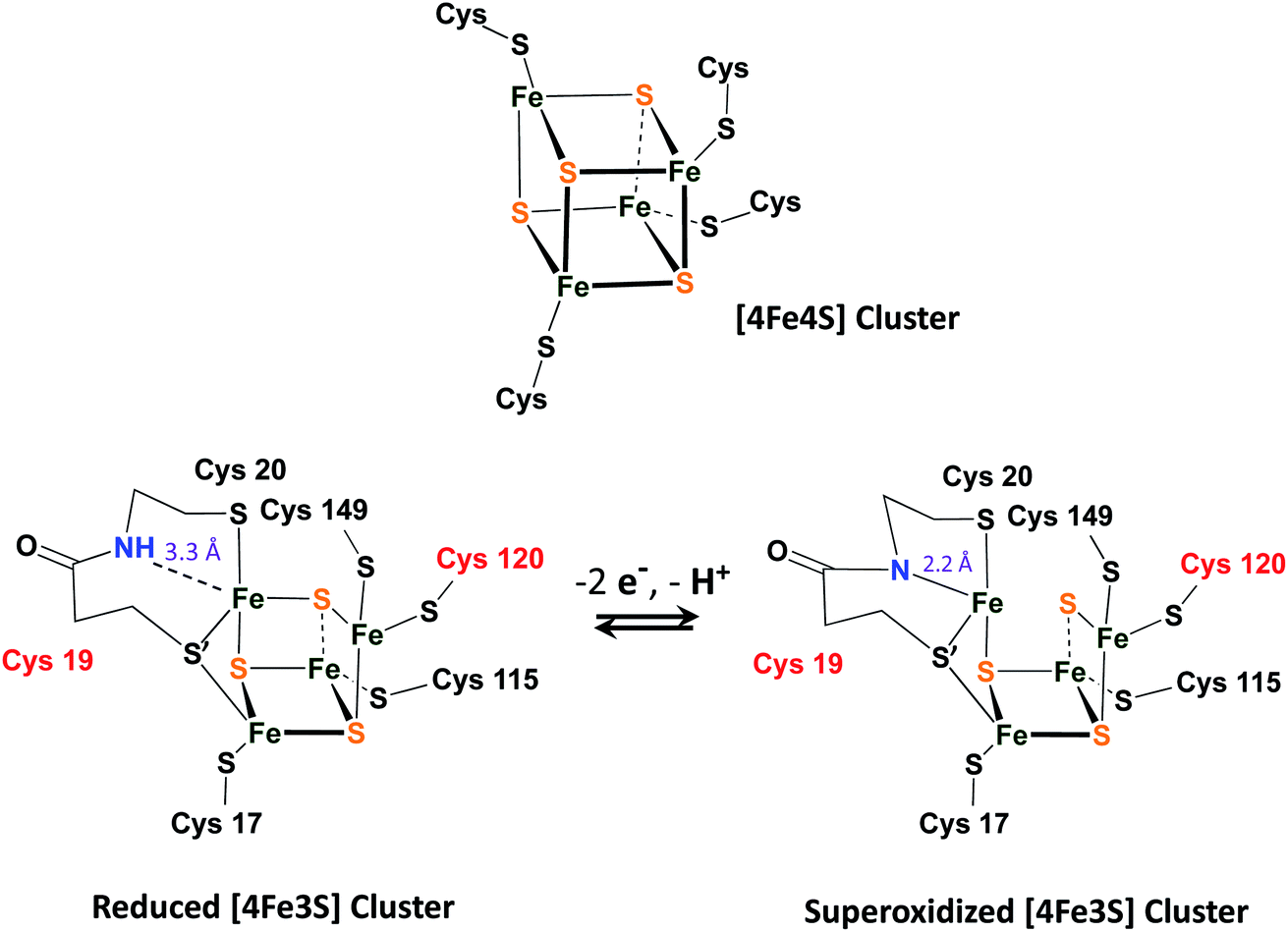 | ||
| Fig. 3 A typical [4Fe4S]4Cys cubane cluster (top) and (below) the exceptional, [4Fe3S]6Cys cluster that is proximal to the [NiFeS]–H2ase active site.16–18 | ||
2.4 Metal-protection by chalcogens
Another strategy for O2 evasion and repair of oxidation, particularly seen for the NiFe hydrogenases, is the oxygenation of the chalcogens. Found in the crystals of [NiFeS]–H2ase as early as 1995, the sulfoxygenates were eventually and convincingly associated with EPR signals attributed to Ni(III) (Ni-A and Ni-B states) that correlated with reactivity recovery.19,20 Selected samples of these crystalline-trapped Ni and S-oxygenates are shown in Scheme 2.21–24 The Ni–Fe derivatives bridged by hydroxide, representing the easiest to be reduced and returned to full activity, are still referred to as Ni-B, Fig. 2 and Scheme 2. The slow-to-recover sulfoxide species are known as Ni-A.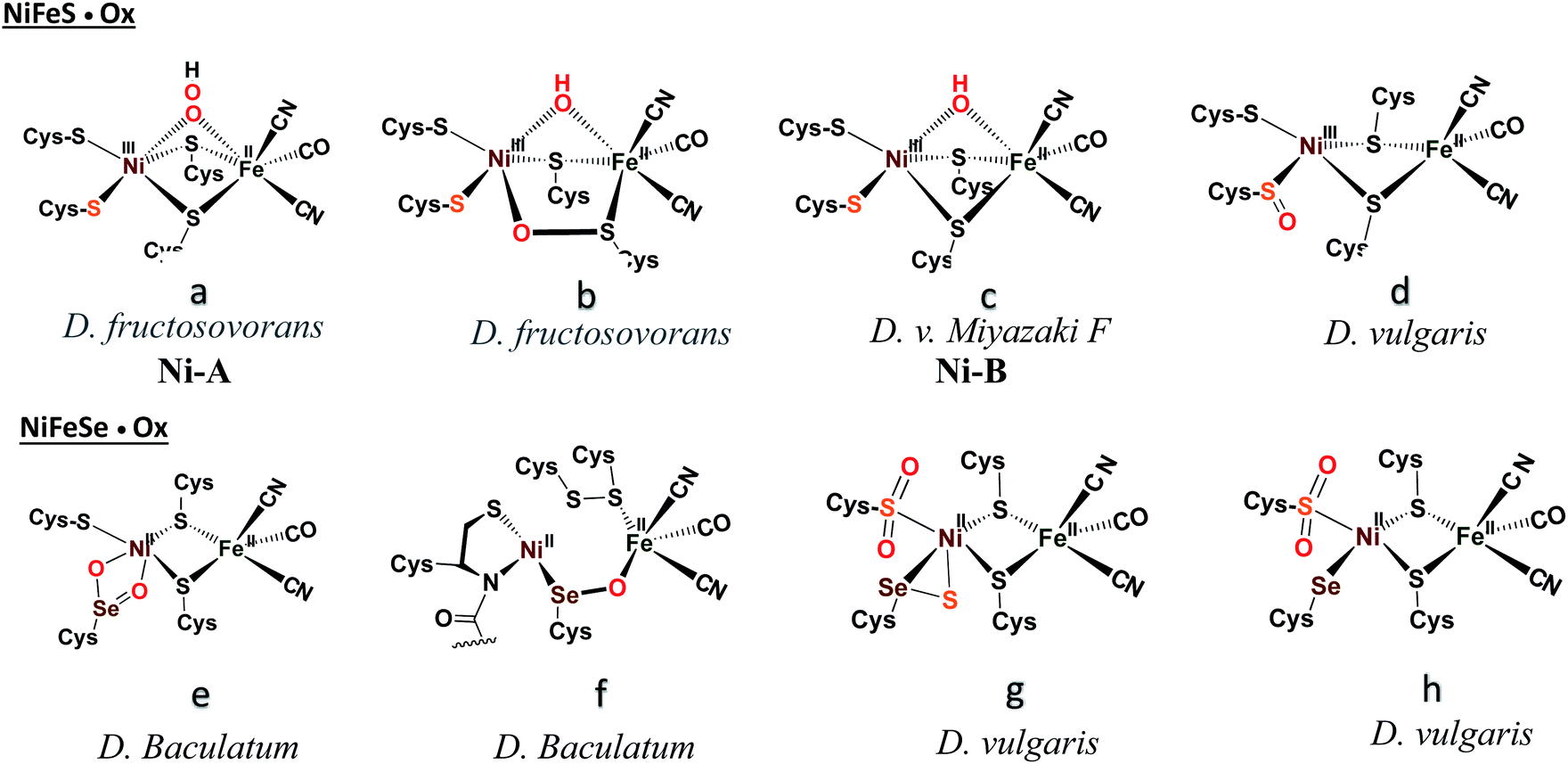 | ||
| Scheme 2 A selection of structurally characterized oxygenates of [NiFeS]– and [NiFeSe]–H2ase active sites.21–24 | ||
Note that all the oxidized/oxygenated NiFeS species contain EPR-active Ni(III); at least 20 different examples are known from gaseous O2-infused crystallization approaches. Although fewer examples have been identified, the analogous [NiFeSe]–H2ase find a richer variety of oxidation or oxygenation.23,24 None of the latter contain Ni(III) but rather chalcogenate oxidation to S–S and Se–S bonds is seen as well as the 2-oxy species in the form of sulfinates and selenoates. The latter, complex e in Scheme 2, is uncommon if not unknown in chalcogen chemistry, and likely only chemically available in the confines of the metallo-enzyme active site pocket where Se–O–Ni interactions likely stabilize the selenoate. While the Ni-A and Ni-B are proposed to be reactivated by adding electrons and protons with “O” removal as H2O, the association of the [NiFeSe]–H2ases with sulfate-reducing bacteria suggests another hypothetical repair mechanism: H2S derived from sulfate assists Se–O or S–O reduction in oxygenated [NiFeSe]–H2ases.25,26 This hypothesis is supported by the identification of H2S in the [NiFeSe] enzyme's crystal structure (∼7 Å away from active site), as well as the oxidized Se–S bound in the oxidized structure.24
3. Synthetic models for O-damaged [NiFe]–H2ases
3.1 Sulfoxygenation in nickel complexes
Sulfoxygenation of metal-bound thiolates, in synthetic compounds and in biological active sites, is actually quite common. There is a post-translational modification that generates S-oxygenates in the nitrile- and thiocyanate-hydratases containing non-heme iron or cobalt within in an N2S2 ligand field derived from Cys–Ser–Cys tripeptide motifs.27,28 The protein crystal structures of NHase's display the organization made possible through H-bonding of water to the oxygens of terminal sulfinate and sulfenate which optimally positions the substrates for hydration of the metal-bonded nitrile or thiocyanate.Fig. 4, panel (a), presents a selection of sulfinates and sulfenates from controlled oxygenation of a series of NiN2S2 explored in our laboratories in the 1990's.29–32 We stress that examples of S-oxygenation are not confined to the thiolate-modified diazacycles. A number of examples have been reported in detail from the Maroney laboratory especially.33–35 Nevertheless, as mentioned above, the Cys–X–Cys biomimetic N2S2 tetradentate ligands with contiguous S–N–N–S donor sites represent particularly a convenient platform for such reactivity studies. The rigidity of the tetradentate N2S2 ligand doubtless contributes to the thermodynamic stability that was foundational for later studies of oxygen-damaged hydrogenase enzyme active sites. The value of this platform is to tip the balance of S-based reactivity from electron transfer leading to dissociated disulfides to O-atom uptake or S-oxygenation maintaining the Ni–Fe core structure. In the confines of the protein matrix, both processes are seen in the oxygen-damaged [NiFe]H2ase active sites, with all representing unwanted states. In the nitrile hydratases, the S-oxygenates serve a purpose.
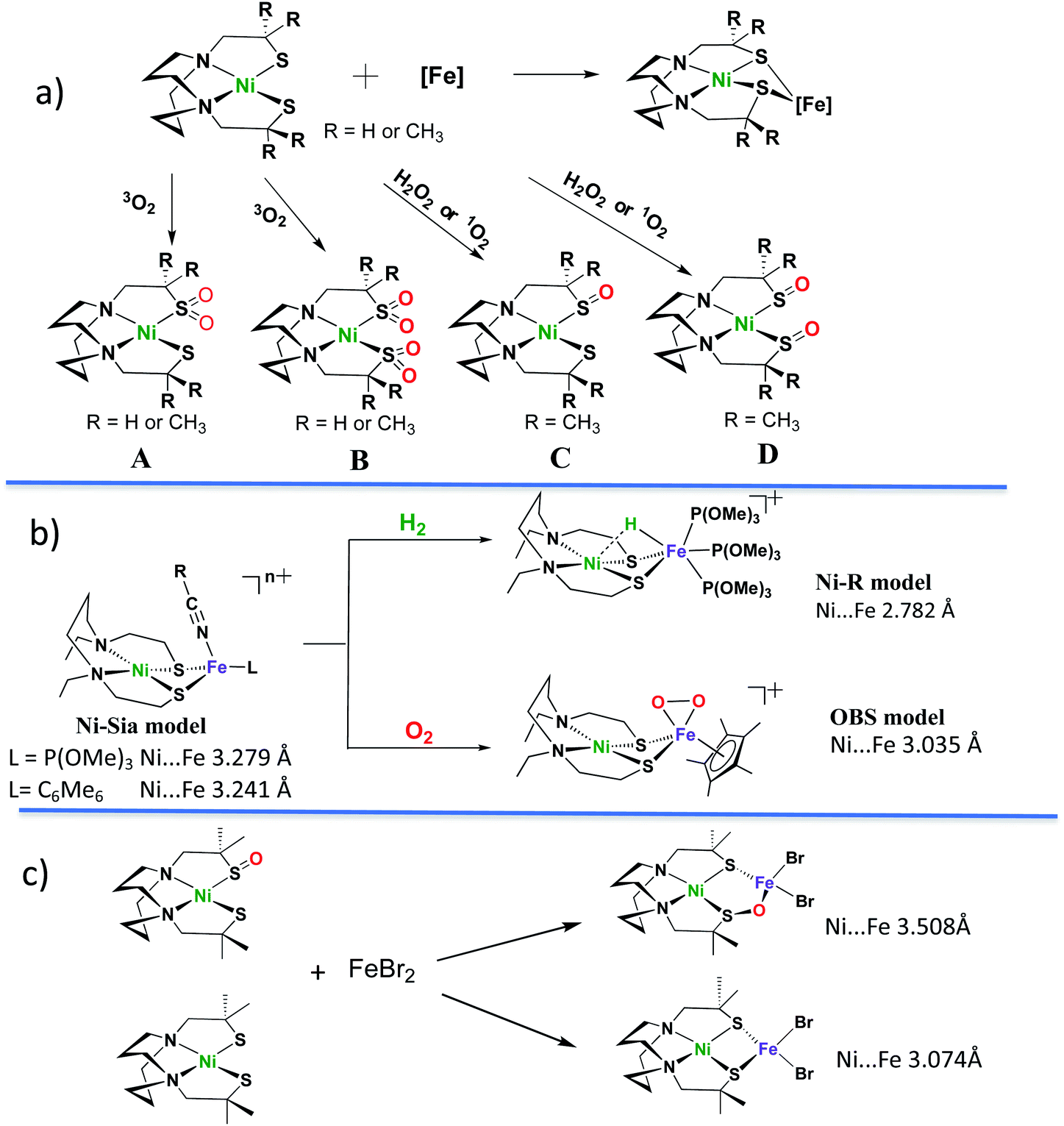 | ||
| Fig. 4 Sulfoxygenates from monomeric NiN2S2 and examples of NiN2S2 in NiFe complexes. Descriptions of (a)–(c) are in text.29–32,36–43 | ||
Oxygen reactivity at sulfur was also discovered in monomeric nickel complexes developed from cleavage of [NS2Ni]2 and [NSe2Ni]2 complexes by cyanide.35 Sulfinato (2-oxy) species resulted on addition of O2 to the [NS2Ni(CN)]− anion, however the analogous selenium analogue was said to be stable to oxygen for up to 4 days. Sulfoxygenation occurs on a single S, and isotopic labeling suggests the oxygen uptake from O2 occurs by a concerted mechanism.33 This study provides a rare opportunity to explore the chemical differences of S vs. Se when bound to nickel.35
As indicated in panel (a) of Fig. 4, the rich nucleophilic chemistry at the non-oxygenated, reduced nickel-dithiolate includes the ability to serve as metallodithiolate ligands to a variety of Lewis acid, transition metal receivers, notably, iron.36,37 From such reactions the Ni(μ-SR)2Fe butterfly core of the [NiFe]–H2ase active site was obtained; some examples of these demonstrated electrocatalytic ability in the proton reduction, Hydrogen Evolution Reaction (HER).
3.2 O-damaged Ni–Fe heterobimetallics bridged by sulfurs
Panel (b) in Fig. 4 displays features of H2 uptake by one of hydrogenase biomimetics as well as an O2 binding study at iron.38–42 The former, with hydride located between Ni and Fe is an approximate model of the Ni-R state (Fig. 4b) of the [NiFe]–H2ase which has been shown by crystallography to contain H2 in “arrested” heterolytic cleavage.5 The latter is the first reported example of a side-on FeIV peroxo complex, which is a biomimetic for a postulated oxygen-bound species in [NiFe]–H2ase.38 It is derived from bubbling O2 gas into a propionitrile solution at −80 °C or acetonitrile at −40 °C in its reduced form (the Ni-Sia model in the Fig. 4b) with a 30% yield.38 The oxygen uptake shortens the Ni⋯Fe distance as compared to the reduced form by ca. 0.2 Å (3.2325(6) Å in reduced and 3.0354(7) Å in the oxidized form). The iron peroxide species can be returned to the reduced form by supplying an electron source, nBu4NBH4, and EtOH as proton source.38No sulfur oxidation was reported in the Ogo et al. studies of NiN2S2·(η5-C5H5)Fe complexes. In fact, to our knowledge no S-oxygenation of any Ni(μ-SR)2Fe complexes in which the dithiolates are connected into the tetradentate N2S2 ligand has been thus far observed. The chelated backbone in the ligand and sulfur bridging between metals likely evades O2 attack on sulfur; instead, the labile solvent binding on iron provided a reaction site for O2.38
Panel (c) of Fig. 4 summarizes a report from Driess and coworkers where structures derived from reaction of NiN2S2 and NiN2S(SO), i.e., the reduced and the isolated nickel mono-sulfoxygenate from Panel (a), with FeBr2, were compared.43 This first model complex of an oxygen-damaged [NiFe]–H2ase active site was assembled from pre-formed components, and its structure shows a ∼0.4 Å expansion of the NiFe core from that of the reduced form. That is, the distance between Ni and Fe (3.508 Å) within the 5-membered NiSOFeS ring contrasts to the 4-membered NiSFeS “reduced form” (3.074 Å), and, differing from the Ogo study, there are no oxidation state changes on the metals. Although further explorations on conversions between the two forms were not reported, this study suggested an approach to model complexes for sulfur-oxygenation and O-damaged [NiFe]–H2ase.
3.3 O-uptake in complexes related to [NiFeS]– and [NiFeSe]–H2ases active sites
The pursuit of [NiFe]–H2ase synthetic analogues designed to contrast the Cys–S and Cys–Se analogues would appear to be a valuable endeavor. While the biological and electrochemical/biological studies that query these differences are rather mature, there are few reports of model compounds that might be used for this purpose. Nature's design for the [FeFe]–H2ase is clear regarding the role of a pendant base for the final steps of proton transfer into reduced iron, to generate an iron hydride.6 The [FeFe]–H2ase pendant base is a secondary amine nitrogen positioned just over an open site on the reactive iron—the site destined to become Fe–H or Fe–(η2-H2) with added electrons and a proton or with capture of H2, respectively.6 The well-engineered open site on the distal iron, needed for proton uptake or H2 bonding in the productive chemistry, also accounts for the greater air-sensitivity of the [FeFe]–H2ase.The Ni-R state of [NiFeS]–H2ase, Fig. 2, indicates a terminal thiolate S performs the role of base in the [NiFe]–H2ase mechanism.5 Hence it is not surprising, in view of the superior properties of [NiFeSe]–H2ases that the analogous position, the terminal cysteine that plays a critical role as “pendant” base in the final steps of H+ delivery to the NiFe assembly or alternately in the opposite direction holds the proton in Ni-R, is precisely where SeCysteine resides.2,3 The superior properties of [NiFeSe]– over [NiFeS]–H2ases include better HER catalytic ability, reduced H2 inhibition and rapid reactivation from O2 damage.44 Protein film voltammetry studies of the [NiFeSe]–hydrogenase by Armstrong et al. have found that the enzyme retains partial catalytic activity for H2 production even in the presence of 1% O2 in the atmosphere.45 We point out that the immediate outersphere “canopy” described earlier provides a nearby base that might perform this function, however further distanced from the eventual proton lodging site than the terminal cysteinyl S or Se.7
The key experiments that demonstrated the lower catalytic activity and higher oxygen sensitivity (poorer tolerance) of [NiFeS]–, as compared to [NiFeSe]–H2ase, derived from the single site mutagenesis of Se–cysteine to cysteine from identical bacterial sources, the enzyme in D. vulgaris Hildenborough.14 If the reasons for these superior properties of [NiFeSe]–H2ase lie solely with the elemental differences in chalcogens (rather than some as yet not established subtle differences in protein residues or folding), there are obvious explanations. The larger size of Se, with more electrons and higher polarizability results in better nucleophilicity of the RSe−, higher acidity, of RSeH, and lower redox potential than sulfur.26 Higher acidity is proposed to be the reason for better “H+” shuttling and higher H2 production of [NiFeSe]–H2ases.44 Concomitantly, the larger size means that Se–O bonds, once formed because of the better nucleophilicity, are weaker than S–O. Thus facile Se–O bond release accounts for high oxygen-tolerance of [NiFeSe]–H2ases.14,26,44
The synthesis of appropriate synthetic analogues of enzyme active sites requires precursors that contain similar core features and sufficient stability to be chemically assayed in some way that relates to the reactivity of the enzyme function. Such concomitant requirements are not always easily met as the protein matrix stabilizes structures that are slightly distorted from the thermodynamic first-coordination sphere choices of ligand and metal that are seen in the “free world” of solution/molecular chemistry. Lubitz and coworkers generated a complex ligand system as an apt model of the sulfur-rich nickel subsite in [NiFe]–H2ase, and one that could be modified to include selenium, Fig. 5.46,47 Importantly, these models have a terminal chalcogenide, which could be viewed as a pendant base for proton shuttling to reduced iron. With E = S, the NiFe complex is capable of electrochemical catalysis of H2 production in aprotic solvents; however, in the selenium analogue, which was explored by Reisner, et al., electrocatalytic H2 production derives from species that deposit on the electrode.47 Studies of these compounds were carried out under air-free conditions, and, to our knowledge, they have not been explored for oxygen uptake.
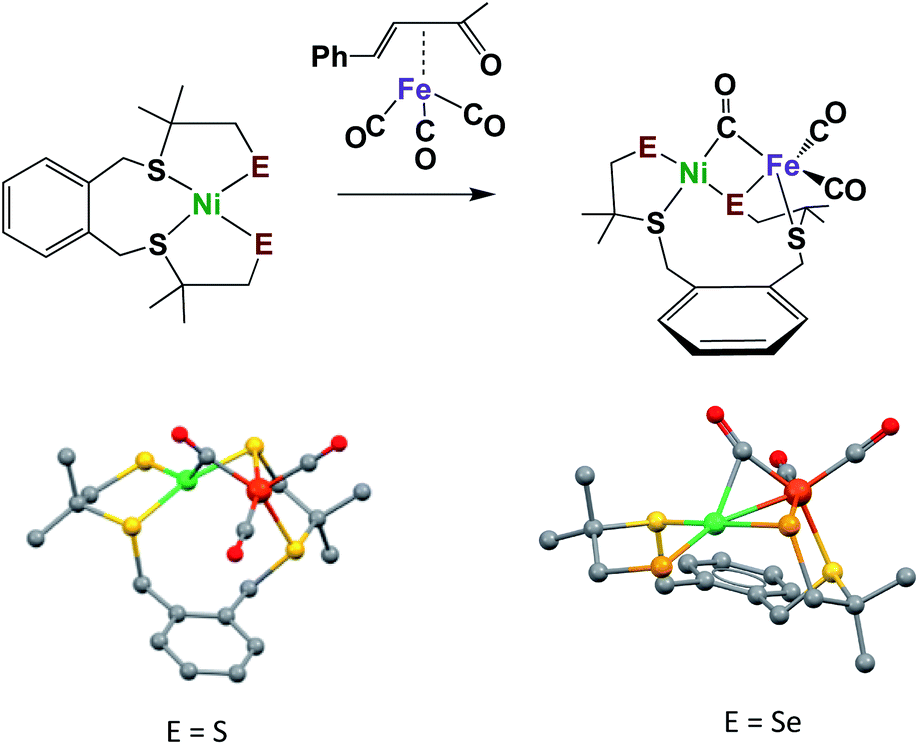 | ||
| Fig. 5 Model complexes for [NiFe]–H2ase active sites with terminal S or Se.46,47 | ||
A somewhat related dinickel complex, [NE2Ni]2, with bridging and terminal thiolates, was the synthetic precursor to the NS2Ni(CN) complex described above. Reactions with O2 finds uptake at the terminal thiolate S for the [NS2Ni]2 yielding the terminal sulfinate [N(μ-S)SO2Ni]2. However no such reaction was observed with the analogous [NSe2Ni]2 complex having selenium as terminal chalcogen.48
With the goal of pursuing heterobimetallics, and heterochalcogenates, for their potential as more stable electrocatalysts, we have adopted dimeric [N2SNi]22+ as synthon, Fig. 6. The simple dinickel butterfly complex precursor may be split by various nucleophiles including carbenes, imidazoles, thiolates, and selenoates.49–51 With the chalcogenates, entry to NiFe bimetallics is possible making use of the (η5-C5H5)Fe(CO)(NCCH3)2+ receiver species outlined in Fig. 6. Although less sophisticated than the Lubitz/Reisner models, and lacking the EAS requirement for a terminal chalcogenide as pendant base, the products from the −SC6H4X and −SeC6H4X nucleophiles are nevertheless informative. Specifically, it is possible to correlate the response of irreversible Ec reduction potentials for both the monomeric NiN2S(SAr) and the derivative [NiFe]-heterobimetallics, as well as ν(CO)IR values for the latter, according to the remote effects of X in the −EC6H4X via Hammett parameters. The shifts in the ν(CO) positions are small, yet they systematically vary within the series with electron-donor ability of X, indicating the polarization of electron richness from the S or Se to Fe is further transmitted via π-backbonding to CO. From ν(CO) values it is also concluded, as expected, that the aryl selenoates are more donating to iron than the analogous aryl thiolates. In fact, a type of synthetic alchemy is seen to convert S into Se in terms of the equivalence of the electron-donating ability of p-−SC6H4NH2 and −SeC6H5.50
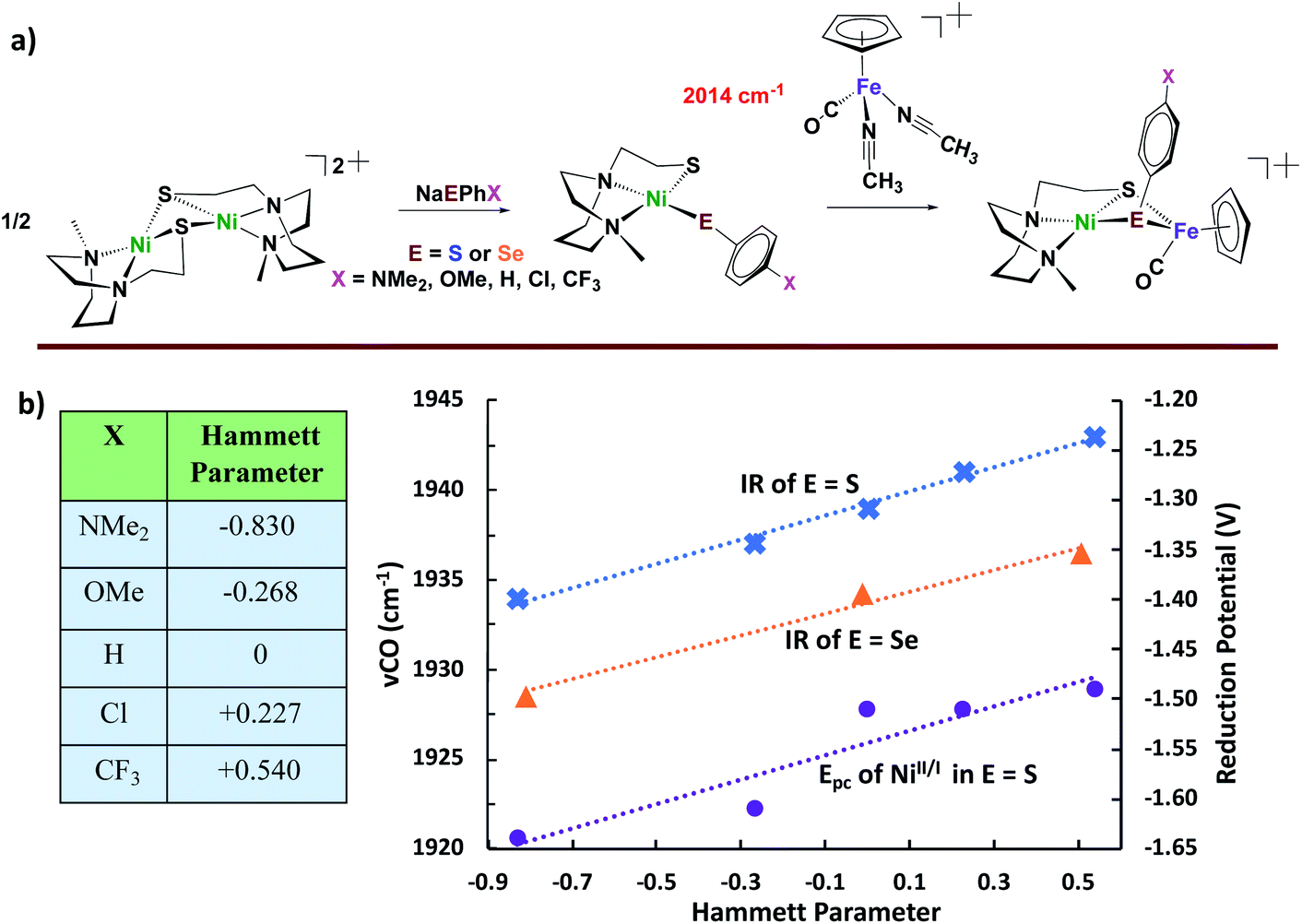 | ||
| Fig. 6 (a) Synthetic approach for Ni2 dimer-splitting leading to NiFe bimetallic analogues of [NiFeS]– and [NiFeSE]–H2ase active sites; and (b) correlation of redox potentials and ν(CO) IR data with Hammett parameters.49–51 | ||
Included in the detailed characterization of the new complexes, is their reactivity with O2, Scheme 3. Unlike the tetradentate NiN2S2 monomeric complexes which form oxygenated sulfur species, exposure of the Ni(N2S)(EPh) to O2 results in degradation. Nevertheless, as metallo-dichalcogenide ligands in NiFe complexes, O2 uptake occurs at the monodentate bridging chalcogenide, leading to the conversion of the butterfly bimetallic, Ni(μ-EPh)(μ-SN2)Fe (E = S or Se), to a stable Ni–O–EPh–Fe–SN2 5-membered-ring arrangement, where an O atom has inserted between E and Ni in both the 1-oxy species as well as the 2-oxy or sulfinate form. Fig. 7 displays representative structures highlighting the NiFe core. As indicated the Ni⋯Fe distance in the 4-membered Ni(μS)(μE)Fe core is only slightly larger for E = Se vs. E = S. Expansion to 3.568 Å occurs in the 5-membered Ni–S–Fe–Se–O ring and to 3.395 Å for the analogous the Ni–S–Fe–S′–O ring in the 2-oxy species. The 5-membered rings in the selenium and sulfur forms are largely the same as the additional oxygen in the latter projects outside of the 5-membered ring.49,50 Notably the 2-oxy species and the 1-oxy sulfur species have been detected by mass spectroscopy but are not isolated sufficiently pure for XRD determinations. Interesting features in these reactions and products include (a) the more rapid reaction of O2 with the Se as compared to the S analogues; (b) oxygen partitioning in S vs. Se analogues; (c) the position of the oxygen-inserted products between E and Fe, or E and Ni; and (d) the mechanism of O2 addition.
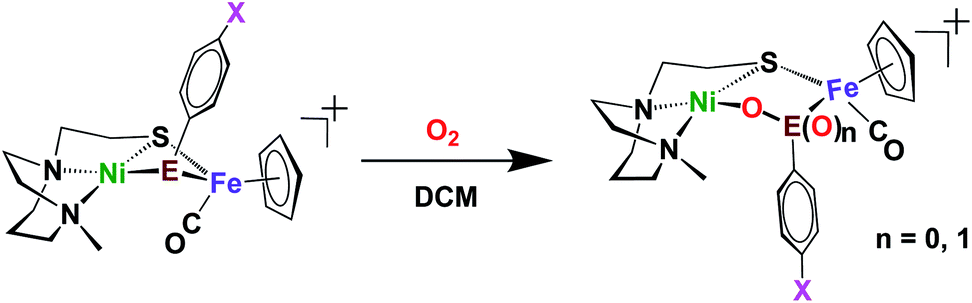 | ||
| Scheme 3 Oxygenation reaction of Ni(μ-ER)Fe complexes.50 | ||
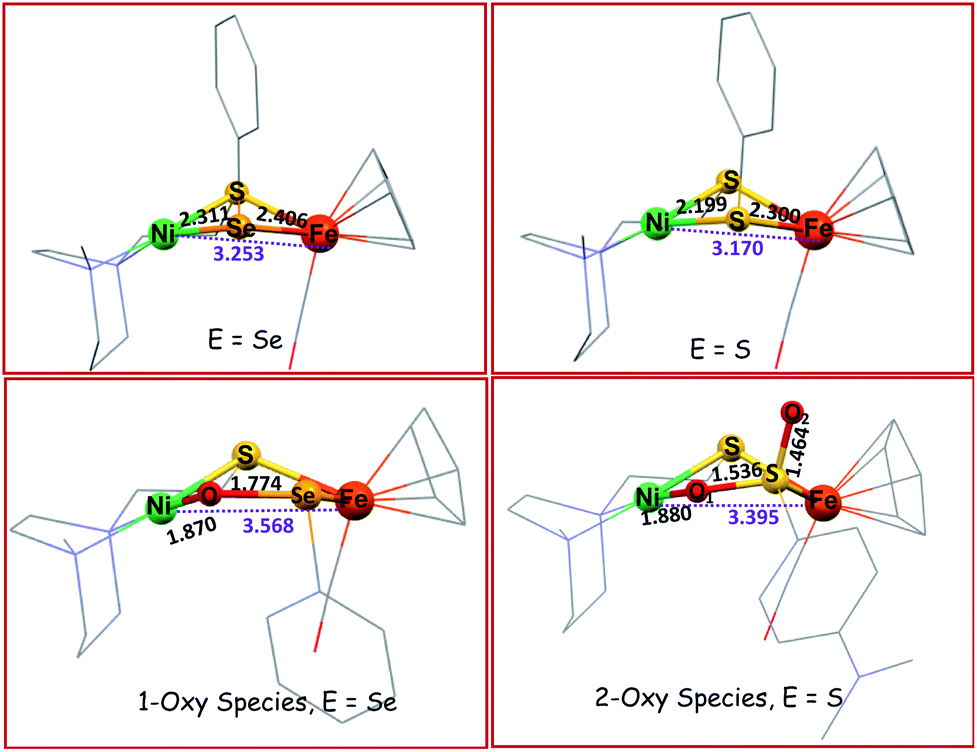 | ||
| Fig. 7 Solid state XRD molecular structures of NiFe complexes.49,50 | ||
Analysis of the protein crystal structures described in the introduction to this Perspective finds, at this time, only one case of a 2-oxy selenocysteine within an oxygen-damaged [NiFeSe]–H2ase active site, Scheme 2. In the natural system, the two oxygen atoms bridge between Fe and Se. In contrast, our biomimetic study finds primarily 1-oxy products for the selenium complex, but faster reactions as compared to the sulfur analogue that generates both 1- and 2-oxy species.50
To reiterate, several 2-oxy sulfinato species have been discovered in various oxygen-damaged [NiFeS]–H2ase structures, but 2-oxy selenoates are rare. A computational study in collaboration with M. B. Hall and L. C. Elrod pursued a thermodynamic approach to address this dichotomy.50Scheme 4 shows that for both [NiFeS]– and [NiFeSe]–H2ases model complexes that explore differences for S vs. Se, the 2-oxy species are thermodynamically favored, for both S and Se, the difference between the 1-oxy and 2-oxy species is around 10 kcal in Se analogue and 20 kcal in the S version. However, comproportionation of the 2-oxy with the 0-oxy selenium species to generate two 1-oxy products is favored by −6 kcal. The analogous comproportionation for the sulfur species is thermodynamically disfavored, as the reaction is uphill by +6.9 kcal. That is, the 2-oxy sulfinate more readily forms and is likely to remain in this form.50
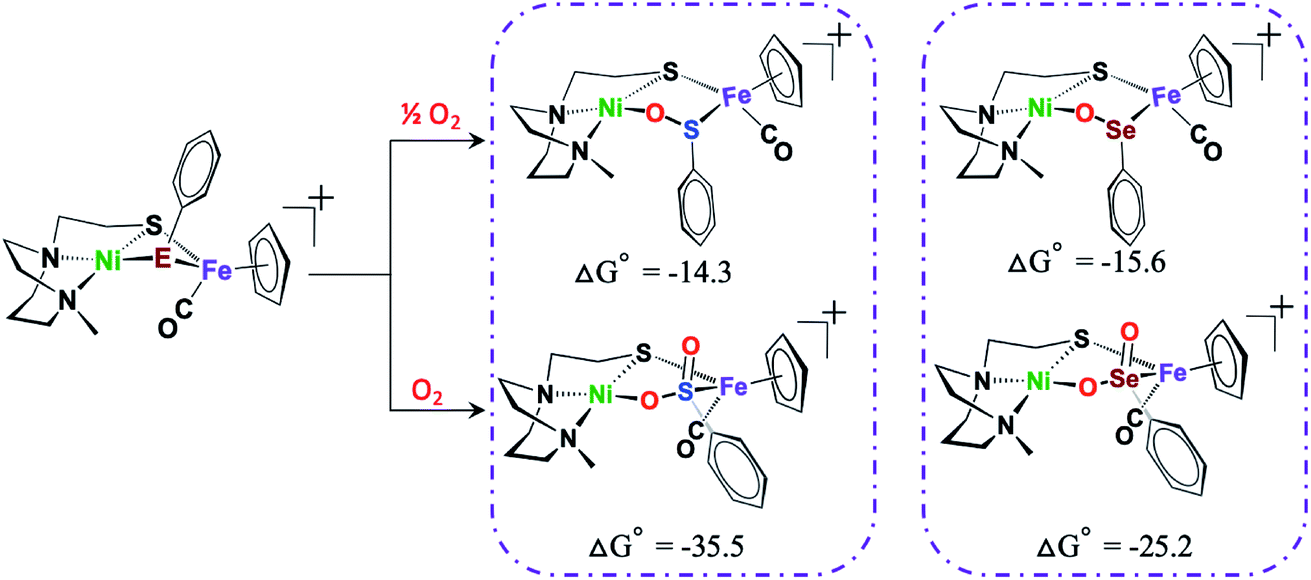 | ||
| Scheme 4 DFT calculated free energy values, ΔG°, for comparison of oxygen-uptake reactions of Ni(μ-EPhH)(μ-S′N2)Fe complexes, E = S and Se, in kcal mol−1.50 | ||
Computations also agree the observation that the bridging-S that is tethered to the N of the diazacycle are disfavored to become oxygenated. A computational cleavage of the tether results in a 15 kcal mole−1 lowering of the oxygenated sulfur, and a preference for it over the S of the PhS.49
While various ratios of 1- and 2-oxy species are found in the oxygenation reactions of most −E–Ph–X derivatives in this family of complexes, the thiolate of greater basicity, –SC6H4NH2, shows selectivity for the 2-oxy species.50 Such a result provided opportunity to explore one aspect of the mechanism of O2 addition via isotope distribution reactions, as presaged in O2 addition studies of the monomeric NiN2S2 species.32,33 An isotopic labeling/mass spectroscopy experiment analyzing products from a mixture of 32O2/36O2 found the two oxygens on −S(O)2C6H4NH2 are from the same O2 molecule, consistent with a concerted dioxygen addition reaction.50 If analogous to the concerted addition of 32O2/36O2 earlier studied, this result suggests O2 binding at Ni as precursor to its transfer to the chalcogen. However the nuclearity of the complex that takes up the O2 is as unknown as is the fate of the second oxygen in the 1-oxy species.
The possibility of reversal of oxygenated damage was explored using both chemical deoxygenation, O-abstracting agents, PR3 (R = Me, or o-tolyl), and the “electrochemical” deoxygenation, by using CpCo2 as reductant and HBF4 as proton source. Both methods realize oxygen removal from the 1-oxy species giving spectroscopic yield of 60%, however, the 2-oxy compounds are inert.49,50 Mass spectra and infrared spectra confirmed the oxygen removal from 1-oxy species; and proton NMR spectroscopy indicated the oxygen was removed as H2O (Scheme 5).
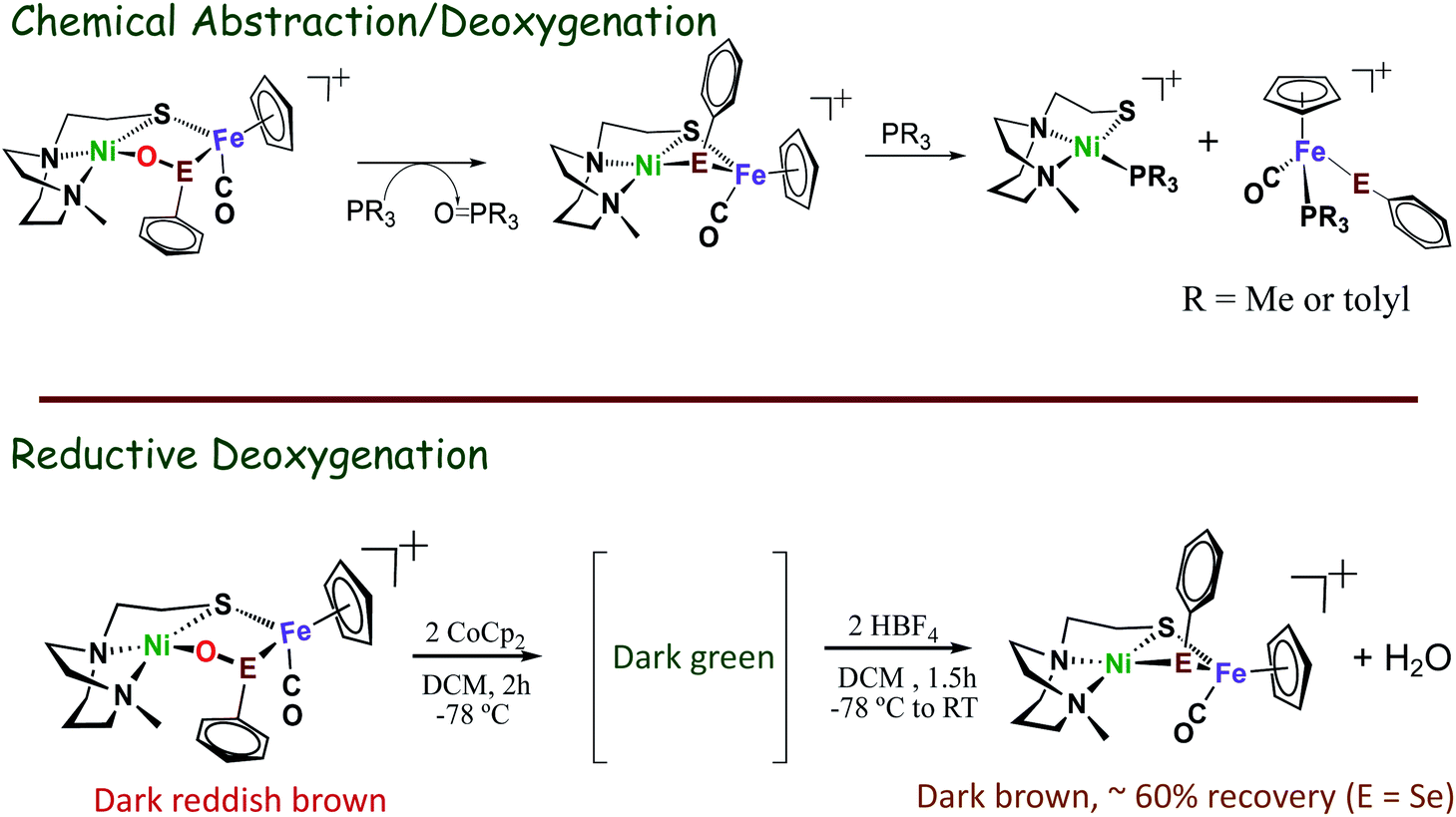 | ||
| Scheme 5 Oxygen removal from oxy-species by O-abstracting agents, PR3 (R = Me, or o-tolyl), or when using CoCp2 as reductant and HBF4 as proton source.49,50 | ||
4. O2-damage studies on models for [FeFe]–H2ases active sites
While the [NiFe]–H2ases and the model complexes show reversible O-uptake and removal at S and Se, no S-oxy species have been found in structures of the [FeFe]–H2ase. Compared to [NiFe]–H2ases, the [FeFe]–H2ases are much less oxygen tolerant.52 Adventitious O2 diffuses through the gas channel in the protein and appears to first land on the vacant site at the distal Fed, which is remote from the 4Fe4S cubane. The ROS forms from the attack and further results in the diffusion and destruction of the 4Fe4S subcluster.52,53 Recent crystallographic studies on [FeFe]–H2ases from the Happe group show the Fed dissociation and loss on exposure to O2.54Although there is no evidence of oxygen protection by S in [FeFe]–H2ases, biomimetics based on the ubiquitous diiron complexes, (μ-pdt)[Fe(CO)2L][Fe(CO)2L′] (L′/L = CO, PPh3, or PMe3), form sulfenates at the propanedithiolate sulfurs on reaction with O-atom sources such as H2O2, or meta-chloroperoxybenzoic acid, as shown in Scheme 6a.55 Deoxygenation with reclaimation of the binding dithiolate occurs with addition of Cp*2Co and H2O. More details can be found in a 2011 microreview by Darensbourg and Weigand in Eur. J. Inorg. Chem.56
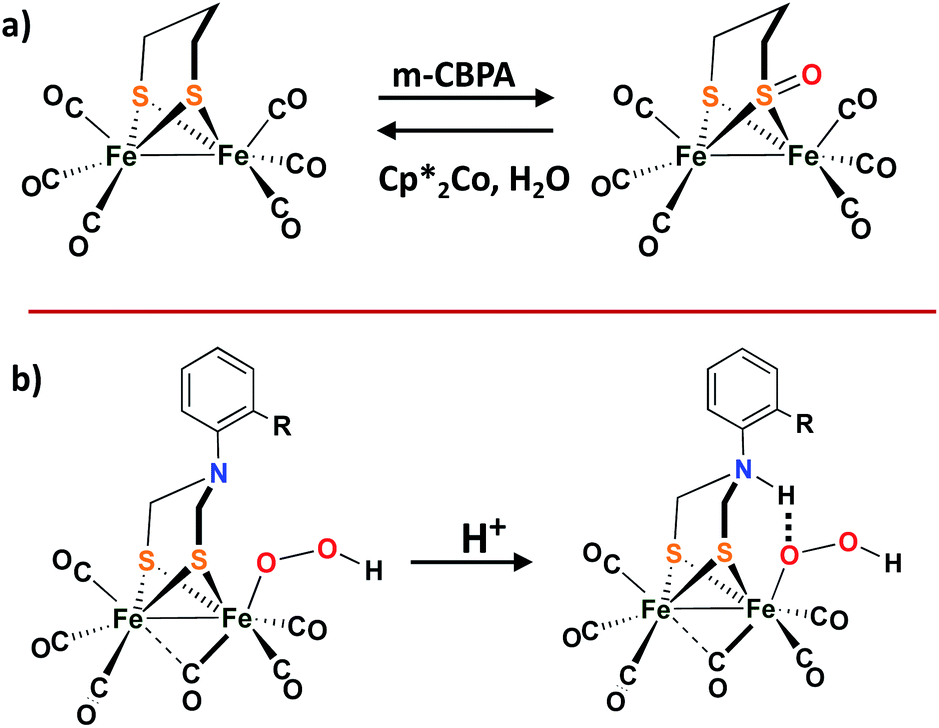 | ||
| Scheme 6 (a) The sulfur oxygenation in model complex of [FeFe]–H2ases; (b) Proposed protonated bridging nitrogen reduction by ortho-substituted ADT-bridged [FeFe]–H2ases mimics.57,58 | ||
Dey, et al., reported an interesting role for the μ-SRS cofactor in the complex (μ-S2(CH2)2NAr)[Fe(CO)3]2. On replacement of the –CH2– group in (μ-pdt)[Fe(CO)2L][Fe(CO)2L′] with a secondary amine bridgehead, H2 production was possible in the presence of dissolved O2 in water (pH = 5.5).57,58 It is proposed that the bridgehead N-protonation and subsequent H-bonding capability assists SO–O bond cleavage which accounts for O2 tolerance, as shown in the Scheme 6b.58
5. Advances in technological development: O2-protection provided by protective polymer films layer in films
Clearly the protein matrix offers much more than the physical protection of the active sites of H2ase's from O2 that was simplistically described early in this manuscript. The ability to maintain activity in the presence of O2 exists for many enzymes and the importance of understanding such a desirable characteristic is vital for practical applications both of enzymes and of biomimetics. Artificial methods to increase oxygen tolerance and extend the lifetimes of such hydrogenases and synthetic analogues of their active sites, while using them in H2 production/oxidation have long been sought and a full description of this field is beyond the scope of our Perspective. Recent developments addressing such challenges describe devices designed both for practicality and for interrogating best approaches to their improvement. As example, redox polymers have been employed as a protective film over [NiFe]–H2ase, isolated from D. vulgaris Miyazaki F by an collaborative efforts of Plumere, Rudiger, Schuhmann, Lubitz et al.59 By adding a redox hydrogel film, consisting of a viologen-functionalized polymer over the O2-sensitive catalyst layer on the electrode surface, the device is able to efficiently work and produce hydrogen under ∼5% O2. Subsequently, a paradigm shift was made on observing improved efficiency when the size of the thick film (>100 μm) was reduced to thin films (down to as much as 3 μm).60 That is, they uncovered the precise conditions that balanced the competition for electrons needed for current generation and those needed for the O2 protection process. The contributions from kinetics of H2 oxidation and for O2 consumption were fit to a mathematical model that included diffusion of the oxidized enzyme back to the electrode within regimes in the film, explaining the advantages of the thin film. The basis of such protection is the ability of the redox viologen-functionalized polymer to rapidly divert electrons from the inner catalyst layer and use them for O2 reduction, in this way protecting the inner catalyst film from oxygen attack. The role of the extra redox protecting matrix is actually “similar” to the [4Fe3S] cluster adopted in membrane-bound hydrogenase (MBH); the latter provides extra two electrons to reduce oxygen.Electrode disposition also plays a role in oxygen protection of model complexes for [NiFe] and [NiFeSe]–H2aes. The Ni site models, [Ni(LE)(MesE)]− (E = S or Se), reported by Reisner, are decomposed under oxygen and form disulfides/diselenides.8 However, they are precursors for oxygen-tolerant catalysts for heterogeneous H2 production by depositing on the electrode.61
6. Concluding remarks
While systematic changes in ligand properties hoping to distinguish first coordination sphere contributions from “sterics vs. electronics” are widely desirable in accounting for the efficacy of a homogeneous metallo-catalyst, rarely are chemists presented with such a controlled coordination environment as is found in the two enzyme active sites described above. The modification of a single atom in the [NiFeS]– vs. [NiFeSe]–H2ase active site, resulted in profound differences in activity, in oxygen tolerance, and in structures of O-damaged active sites can be largely rationalized based on fundamental differences between sulfur and selenium. From our perspective the few NiFe models of those sites have informed on the value of Nature's well-designed active sites comprised of two metals, which permit various levels of O2 uptake because of the varying O-affinity of the S and Se based ligand, preventing untenable metal oxidation. Such M–Se–O–M′ and M–S–O–M′ species appear to be transient O-atom depositories which provide a chance for repair or O-atom removal, without drastic damage to the protein. Clearly the biomimetic complexes are limited at this time and they are flawed, i.e., their structures lack features assumed to be critical to the efficient functioning of the active sites.Selenium wins in both the hydrogen-processing catalytic ability and in recovery from O-damage. Yet it has downsides to wider use in nature because of (1) geographic availability (the ratio of S over Se is as low as 6000![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and as high as 55500:126); and (2) the ease – or lack thereof—of biosynthetic machinery for the incorporation of selenocysteine into proteins. For example, to recode the stop codon UGA to a sense codon for Selenocysteine in the Eukaryotic Sec-insertion machinery, several accessory proteins and ATP units are required26. We stress that the critical studies from Portugal that compared [NiFeS]– and [NiFeSe]–H2ase were based on single site mutations of the latter that essentially converted the [NiFeSe]–H2ase in D. vulgaris Hildenborough into the identical protein except with Se replaced by S. The reverse process (S → Se), cannot be readily performed. The significance of this work is that the only difference in the hydrogenase studied was the active sites; everything else was the same. Hence any differences were due to the single chalcogen atom. Those differences are comfortably explained by the physical properties of the elements themselves, although subtle changes in protein structure could arguably propagate into a different controlling feature.
:1 and as high as 55500:126); and (2) the ease – or lack thereof—of biosynthetic machinery for the incorporation of selenocysteine into proteins. For example, to recode the stop codon UGA to a sense codon for Selenocysteine in the Eukaryotic Sec-insertion machinery, several accessory proteins and ATP units are required26. We stress that the critical studies from Portugal that compared [NiFeS]– and [NiFeSe]–H2ase were based on single site mutations of the latter that essentially converted the [NiFeSe]–H2ase in D. vulgaris Hildenborough into the identical protein except with Se replaced by S. The reverse process (S → Se), cannot be readily performed. The significance of this work is that the only difference in the hydrogenase studied was the active sites; everything else was the same. Hence any differences were due to the single chalcogen atom. Those differences are comfortably explained by the physical properties of the elements themselves, although subtle changes in protein structure could arguably propagate into a different controlling feature.
A final note is directed towards the protective, anti-oxidant, value of selenium. The studies we described all involve the chalcogens in the first coordination sphere of the NiFe moieties. Studies that explore differences induced by placing selenium in second or third coordination spheres have yet to be done. Could selenium be a watch dog or guard for O2 invasion in general air-sensitive molecular catalysts? At what distance would such a guard need to be placed? This is an intriguing question for future synthetic or post-translational designs.
The very specific interactions of residues in the protein polymer that account for the slight distortion in the coordination geometry of the active sites structures from solution forms also permit changes that occur with substrate binding and activation. This role of the protein matrix is beginning to be understood with more clarity for the hydrogenases as these organometallic active sites, especially the [FeFe]–H2ase, have fewer restrictions from covalent attachments. The matrix provided by the redox-active polymer films, discussed in the last part of this manuscript holds much interest for the possibility of practical application both of enzymes and of fragile biomimetics of the active sites.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financial supported by the National Science Foundation (CHE-1665258) and the Robert A. Welch Foundation (A-0924). The authors express appreciation to colleagues and coworkers through the years, especially Professor M. B. Hall of TAMU.Notes and references
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, Science, 1998, 282, 1853–1858 CrossRef CAS PubMed
.
- A. Volbeda, M. H. Charon, C. Piras, E. C. Hatchikian, M. Frey and J. C. Fontecilla-Camps, Nature, 1995, 373, 580–587 CrossRef CAS PubMed
- E. Garcin, X. Vernede, E. C. Hatchikian, A. Volbeda, M. Frey and J. C. Fontecillla-Camps, Structure, 1999, 7, 557–566 CrossRef CAS
- S. Shima and R. K. Thauer, Chem. Rec., 2007, 7, 37–46 CrossRef CAS PubMed
- H. Ogata, K. Nishikawa and W. Lubitz, Nature, 2015, 520, 571–574 CrossRef PubMed
- W. Lubitz, H. Ogata, O. Rudiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed
- R. M. Evans, E. J. Brooke, S. A. Wehlin, E. Nomerotskaia, F. Sargent, S. B. Carr, S. E. Phillips and F. A. Armstrong, Nat. Chem. Biol., 2016, 12, 46–50 CrossRef CAS PubMed
- C. Wombwell, C. A. Caputo and E. Reisner, Acc. Chem. Res., 2015, 48, 2858–2865 CrossRef CAS PubMed
- D. M. Heinekey, J. Organomet. Chem., 2009, 694, 2671–2680 CrossRef CAS
- H. Ogata, P. Kellers and W. Lubitz, J. Mol. Biol., 2010, 402, 428–444 CrossRef CAS PubMed
- H. Ogata, S. Hirota, A. Nakahara, H. Komori, N. Shibata, T. Kato, K. Kano and Y. Higuchi, Structure, 2005, 13, 1635–1642 CrossRef CAS PubMed
- T. Buhrke, O. Lenz, N. Krauss and B. Friedrich, J. Biol. Chem., 2005, 280, 23791–23796 CrossRef CAS PubMed
- S. Zacarias, A. Temporão, M. d. Barrio, V. Fourmond, C. Léger, P. M. Matias and I. A. C. Pereira, ACS Catal., 2019, 9, 8509–8519 CrossRef CAS
- M. C. Marques, C. Tapia, O. Gutierrez-Sanz, A. R. Ramos, K. L. Keller, J. D. Wall, A. L. De Lacey, P. M. Matias and I. A. C. Pereira, Nat. Chem. Biol., 2017, 13, 544–550 CrossRef CAS PubMed
- P. Rodríguez-Maciá, L. M. Galle, R. Bjornsson, C. Lorent, I. Zebger, Y. Yoda, S. P. Cramer, S. DeBeer, I. Span and J. A. Birrell, Angew. Chem., Int. Ed. Engl., 2020, 59, 2–11 CrossRef
- Y. Shomura, K. S. Yoon, H. Nishihara and Y. Higuchi, Nature, 2011, 479, 253–256 CrossRef CAS PubMed
- A. Volbeda, P. Amara, C. Darnault, J. M. Mouesca, A. Parkin, M. M. Roessler, F. A. Armstrong and J. C. Fontecilla-Camps, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5305–5310 CrossRef CAS PubMed
- J. Fritsch, P. Scheerer, S. Frielingsdorf, S. Kroschinsky, B. Friedrich, O. Lenz and C. M. Spahn, Nature, 2011, 479, 249–252 CrossRef CAS PubMed
- C. Gebner, O. Trofanchuk, K. Kawagoe, Y. Higuchi, N. Yasuoka and W. Lubitz, Chem. Phys. Lett., 1996, 256, 518–524 CrossRef
- S. Foerster, M. Stein, M. Brecht, H. Ogata, Y. Higuchi and W. Lubitz, J. Am. Chem. Soc., 2003, 125, 83–93 CrossRef CAS PubMed
- A. Volbeda, L. Martin, C. Cavazza, M. Matho, B. W. Faber, W. Roseboom, S. P. Albracht, E. Garcin, M. Rousset and J. C. Fontecilla-Camps, J. Biol. Inorg Chem., 2005, 10, 239–249 CrossRef CAS PubMed
- A. Volbeda, L. Martin, E. Barbier, O. Gutiérrez-Sanz, A. L. D. Lacey, P. P. Liebgott, S. b. Dementin, M. Rousset and J. C. Fontecilla-Camps, J. Biol. Inorg Chem., 2014, 20, 11 CrossRef PubMed
- A. Volbeda, P. Amara, M. Iannello, A. L. De Lacey, C. Cavazza and J. C. Fontecilla-Camps, Chem. Commun., 2013, 49, 7061–7063 RSC
- M. C. Marques, R. Coelho, A. L. De Lacey, I. A. Pereira and P. M. Matias, J. Mol. Biol., 2010, 396, 893–907 CrossRef CAS PubMed
- M. J. Maroney and R. J. Hondal, Free Radicals Biol. Med., 2018, 127, 228–237 CrossRef CAS PubMed
- H. J. Reich and R. J. Hondal, ACS Chem. Biol., 2016, 11, 821–841 CrossRef CAS PubMed
- A. C. McQuilken and D. P. Goldberg, Dalton Trans., 2012, 41, 10883–10899 RSC
- T. Arakawa, Y. Kawano, S. Kataoka, Y. Katayama, N. Kamiya, M. Yohda and M. Odaka, J. Mol. Biol., 2007, 366, 1497–1509 CrossRef CAS PubMed
- V. E. Kaasjager, E. Bouwman, J. Reedijk, C. A. Grapperhaus, J. H. Reibenspies, J. J. Smee, M. Y. Darensbourg, A. Derecskei-Kovacs and L. M. Thomson, Inorg. Chem., 2002, 41, 1837 CrossRef CAS PubMed
- C. A. Grapperhaus and M. Y. Darensbourg, Acc. Chem. Res., 1998, 31, 451 CrossRef CAS
- P. J. Farmer, J. N. Verpeaux, C. Amatore, M. Y. Darensbourg and G. Musie, J. Am. Chem. Soc., 1994, 116, 9355 CrossRef CAS
- C. A. Grapperhaus, M. Y. Darensbourg, L. W. Sumner and D. H. Russell, J. Am. Chem. Soc., 1996, 118, 1791–1792 CrossRef CAS
- S. A. Mirza, M. Kumar, M. A. Pressler, R. O. Day and M. J. Maroney, Inorg. Chem., 1993, 32, 977–987 CrossRef CAS
- M. J. Maroney, S. B. Choudhury, P. A. Bryngelson, S. A. Mirza and M. J. Sherrod, Inorg. Chem., 1996, 35, 1073–1076 CrossRef CAS PubMed
- S. B. Choudhury, M. A. Pressler, S. A. Mirza, R. O. Day and M. J. Maroney, Inorg. Chem., 1994, 33, 4831–4839 CrossRef CAS
- S. Ding, P. Ghosh, A. M. Lunsford, N. Wang, N. Bhuvanesh, M. B. Hall and M. Y. Darensbourg, J. Am. Chem. Soc., 2016, 138, 12920–12927 CrossRef CAS PubMed
- P. Ghosh, M. Quiroz, N. Wang, N. Bhuvanesh and M. Y. Darensbourg, Dalton Trans., 2017, 46, 5617–5624 RSC
- T. Kishima, T. Matsumoto, H. Nakai, S. Hayami, T. Ohta and S. Ogo, Angew. Chem., Int. Ed. Engl., 2016, 55, 724–727 CrossRef CAS PubMed
- T. Matsumoto, T. Kishima, T. Yatabe, K.-S. Yoon and S. Ogo, Organometallics, 2017, 36, 3883–3890 CrossRef CAS
- K. Kim, T. Matsumoto, A. Robertson, H. Nakai and S. Ogo, Chem.–Asian J., 2012, 7, 1394–1400 CrossRef CAS PubMed
- S. Ogo, Chem. Rec., 2014, 14, 397–409 CrossRef CAS PubMed
- S. Ogo, Coord. Chem. Rev., 2017, 334, 43–53 CrossRef CAS
- N. J. Lindenmaier, S. Wahlefeld, E. Bill, T. Szilvasi, C. Eberle, S. Yao, P. Hildebrandt, M. Horch, I. Zebger and M. Driess, Angew. Chem., Int. Ed. Engl., 2017, 56, 2208–2211 CrossRef CAS
- C. S. A. Baltazar, M. C. Marques, C. M. Soares, A. M. DeLacey, I. A. C. Pereira and P. M. Matias, Eur. J. Inorg. Chem., 2011, 2011, 948–962 CrossRef
- A. Parkin, G. Goldet, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2008, 130, 13410–13416 CrossRef CAS PubMed
- K. Weber, T. Kramer, H. S. Shafaat, T. Weyhermuller, E. Bill, M. van Gastel, F. Neese and W. Lubitz, J. Am. Chem. Soc., 2012, 134, 20745–20755 CrossRef CAS PubMed
- C. Wombwell and E. Reisner, Chem.–Eur. J., 2015, 21, 8096–8104 CrossRef CAS PubMed
- S. A. Mirza, R. O. Day and M. J. Maroney, Inorg. Chem., 1996, 35, 1992–1995 (
Inorg. Chem.
, 1996
, 35
, 4
) CrossRef CAS
- X. Yang, L. C. Elrod, J. H. Reibenspies, M. B. Hall and M. Y. Darensbourg, Chem. Sci., 2019, 10, 1368–1373 RSC
- X. Yang, L. C. Elrod, T. Le, V. S. Vega, H. Naumann, Y. Rezenom, J. H. Reibenspies, M. B. Hall and M. Y. Darensbourg, J. Am. Chem. Soc., 2019, 141, 15338–15347 CrossRef CAS PubMed
- R. M. Jenkins, M. L. Singleton, L. A. Leamer, J. H. Reibenspies and M. Y. Darensbourg, Inorg. Chem., 2010, 49, 5503–5514 CrossRef CAS PubMed
- C. Lambertz, N. Leidel, K. G. Havelius, J. Noth, P. Chernev, M. Winkler, T. Happe and M. Haumann, J. Biol. Chem., 2011, 286, 40614–40623 CrossRef CAS PubMed
- A. Kubas, C. Orain, D. De Sancho, L. Saujet, M. Sensi, C. Gauquelin, I. Meynial-Salles, P. Soucaille, H. Bottin, C. Baffert, V. Fourmond, R. B. Best, J. Blumberger and C. Leger, Nat. Chem., 2017, 9, 88–95 CrossRef CAS PubMed
- J. Esselborn, L. Kertess, U. P. Apfel, E. Hofmann and T. Happe, J. Am. Chem. Soc., 2019, 141, 17721–17728 CrossRef CAS PubMed
- T. Liu, B. Li, M. L. Singleton, M. B. Hall and M. Y. Darensbourg, J. Am. Chem. Soc., 2009, 131, 8296–8307 CrossRef CAS PubMed
- M. Y. Darensbourg and W. Weigand, Eur. J. Inorg. Chem., 2011, 2011, 994–1004 CrossRef
- S. Dey, A. Rana, D. Crouthers, B. Mondal, P. K. Das, M. Y. Darensbourg and A. Dey, J. Am. Chem. Soc., 2014, 136, 8847–8850 CrossRef CAS PubMed
- M. E. Ahmed, S. Dey, M. Y. Darensbourg and A. Dey, J. Am. Chem. Soc., 2018, 140, 12457–12468 CrossRef CAS PubMed
- N. Plumere, O. Rudiger, A. A. Oughli, R. Williams, J. Vivekananthan, S. Poller, W. Schuhmann and W. Lubitz, Nat. Chem., 2014, 6, 822–827 CrossRef CAS PubMed
- H. Li, D. Buesen, S. Dementin, C. Leger, V. Fourmond and N. Plumere, J. Am. Chem. Soc., 2019, 141, 16734–16742 CrossRef CAS PubMed
- C. Wombwell and E. Reisner, Dalton Trans., 2014, 43, 4483–4493 RSC
| This journal is © The Royal Society of Chemistry 2020 |