
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic enantioselective construction of vicinal quaternary carbon stereocenters
Feng
Zhou
 *a,
Lei
Zhu
*b,
Bo-Wen
Pan
c,
Yang
Shi
c,
Yun-Lin
Liu
d and
Jian
Zhou
*ae
*a,
Lei
Zhu
*b,
Bo-Wen
Pan
c,
Yang
Shi
c,
Yun-Lin
Liu
d and
Jian
Zhou
*ae
aShanghai Key Laboratory of Green Chemistry and Chemical Processes, Shanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, East China Normal University, Shanghai, 200062, P. R. China. E-mail: fzhou@chem.ecnu.edu.cn; jzhou@chem.ecnu.edu.cn
bSchool of Chemistry and Materials Science, Hubei Engineering University, Hubei 432000, P. R. China. E-mail: Lei.zhu@hbeu.edu.cn
cSchool of Pharmaceutical, Guizhou University of Traditional Chinese Medicine, Guiyang, 550002, P. R. China
dSchool of Chemistry and Chemical Engineering, Guangzhou University, Guangzhou, 510006, P. R. China
eState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai, 200032, P. R. China
First published on 28th July 2020
Abstract
This review summarizes the advances in the catalytic enantioselective construction of vicinal quaternary carbon stereocenters, introduces major synthetic strategies and discusses their advantages and limitations, highlights the application of known protocols in the total synthesis of natural products, and outlines the synthetic opportunities.
1. Introduction
Contiguous carbon stereogenic centers are a common feature of complex natural products and drugs.1 They represent a large fraction of three-dimensional space, and the orientation of substituents at carbon stereocenters has an impact on the shape of many structurally complex molecules, which are closely related to the properties of organic molecules.2 Contiguous stereogenic carbon atoms also greatly add complexity and diversity to organic molecules because they encompass a wide range of substructures depending on the types of chiral carbons involved. As shown in Fig. 1, even the simplest subclass, two adjacent stereogenic carbons, can be classified into vicinal tertiary carbon stereocenters (I), vicinal stereocenters containing a fully substituted carbon (II and III), and vicinal fully substituted carbon stereocenters (IV, V, and VI). With both adjacent stereocenters changing from tertiary carbons to tetrasubstituted ones that bear at least one heteroatom substituent, and to quaternary carbon stereocenters, the steric hindrance substantially increases and imposes remarkable difficulties on the synthesis. In particular, sterically congested vicinal quaternary carbon stereocenters (VI) constitute a daunting challenge in synthetic chemistry.3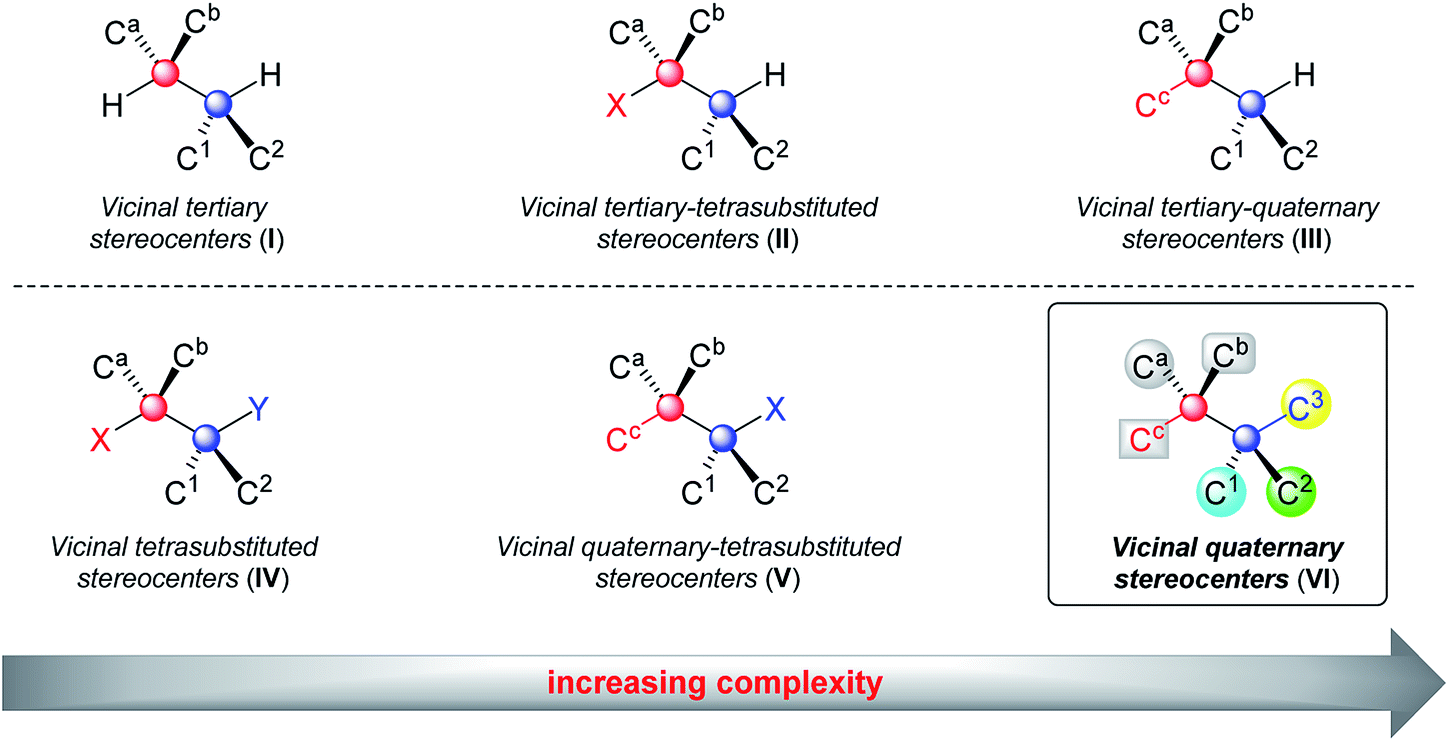 | ||
| Fig. 1 Two adjacent carbon stereocenters. | ||
However, vicinal quaternary carbon stereocenters often feature in various types of fused-ring, spirocyclic, and bridged-ring skeletons of biologically active complex natural products (Fig. 2).4–12 Notably, there are even three or up to four contiguous stereogenic quaternary carbon atoms in architecturally complex molecules, as seen in laurenene, salvileucalin B, sordarin, taiwaniadduct B, and musabalbisiane A.12a–e
 | ||
| Fig. 2 Typical natural products containing vicinal quaternary carbon stereocenters. | ||
Vicinal quaternary carbon stereocenters also exist in drugs, as exemplified by the semisynthetic opioid derivative of thebaine, buprenorphine, a mixed partial agonist opioid receptor modulator to treat opioid addiction.12f Considering that it is a routine strategy in medicinal research to introduce conformational constraints to alleviate the conformational entropy penalty upon binding to the target, the enhanced conformational restriction of vicinal stereogenic quaternary carbons would be helpful to improve the pharmaceutical properties of organic molecules in drug discovery and development.13 Therefore, development of efficient construction of such structural motifs for the synthesis and modification of bioactive compounds is much sought after.
Despite significant achievements in the diastereoselective synthesis of adjacent quaternary carbon stereocenters using chiral substrates or reagents,4 the exploitation of catalytic enantioselective methods is urgent, important, and at the forefront of asymmetric catalysis. However, it is extremely difficult to achieve reasonable reactivity, and control both diastereoselectivity and enantioselectivity in the key catalytic C–C bond-forming step because of the challenge of steric hindrance, such as the steric congestion in the transition state and the diminished steric dissimilarity of carbon substituents on both prochiral carbons.3 In addition, the electronic effect represents another challenge, as it may not only influence the reactivity of reaction reagents but also affect the substrate–catalyst interaction. Therefore, the catalytic enantioselective construction of contiguous stereogenic quaternary carbon atoms constitutes an ideal testing ground for new chiral catalysts and methodologies to exhibit their potency and potential, and reflects some of the latest achievements in asymmetric catalysis.
In the past decade, much progress has been made in catalytic enantioselective creation of two vicinal quaternary carbon stereocenters. A variety of elegant protocols have been developed on the basis of four basic synthetic strategies shown in Fig. 3: (a) the reaction of trisubstituted carbon nucleophiles and electrophiles, (b) difunctionalization of all-carbon tetrasubstituted alkenes, (c) sequential modification of vicinal-activated methines, and (d) the desymmetrization of meso compounds. Despite ongoing progress, a comprehensive review summarizing the latest advances in this important and active research field has not been performed yet, although Overman and coworkers continuously highlighted the importance and challenge of efficient construction of continuous stereogenic quaternary carbon atoms in natural product synthesis.3,4a,b Synthetic efforts to construct contiguous quaternary carbon stereocenters of selected natural products are nicely summarized in several reviews contributed by the groups of Overman,3,4a,b and Gong & Yang;4c however, they are mainly based on diastereoselective methods using chiral starting materials or auxiliaries. Several review articles on catalytic enantioselective construction of quaternary stereocenters have been published by Overman,2,14 Stoltz15 and other groups,16–23 but they only introduced limited methods to construct vicinal quaternary carbon atoms depending on the topic of these reviews. In light of this, we feel it is necessary to present a timely comprehensive review article to summarize the latest advances until now, discuss in depth the advantages and limitations of each strategy, and give the readers some inspiration to develop more useful and excellent methods to construct vicinal quaternary carbon stereocenters.
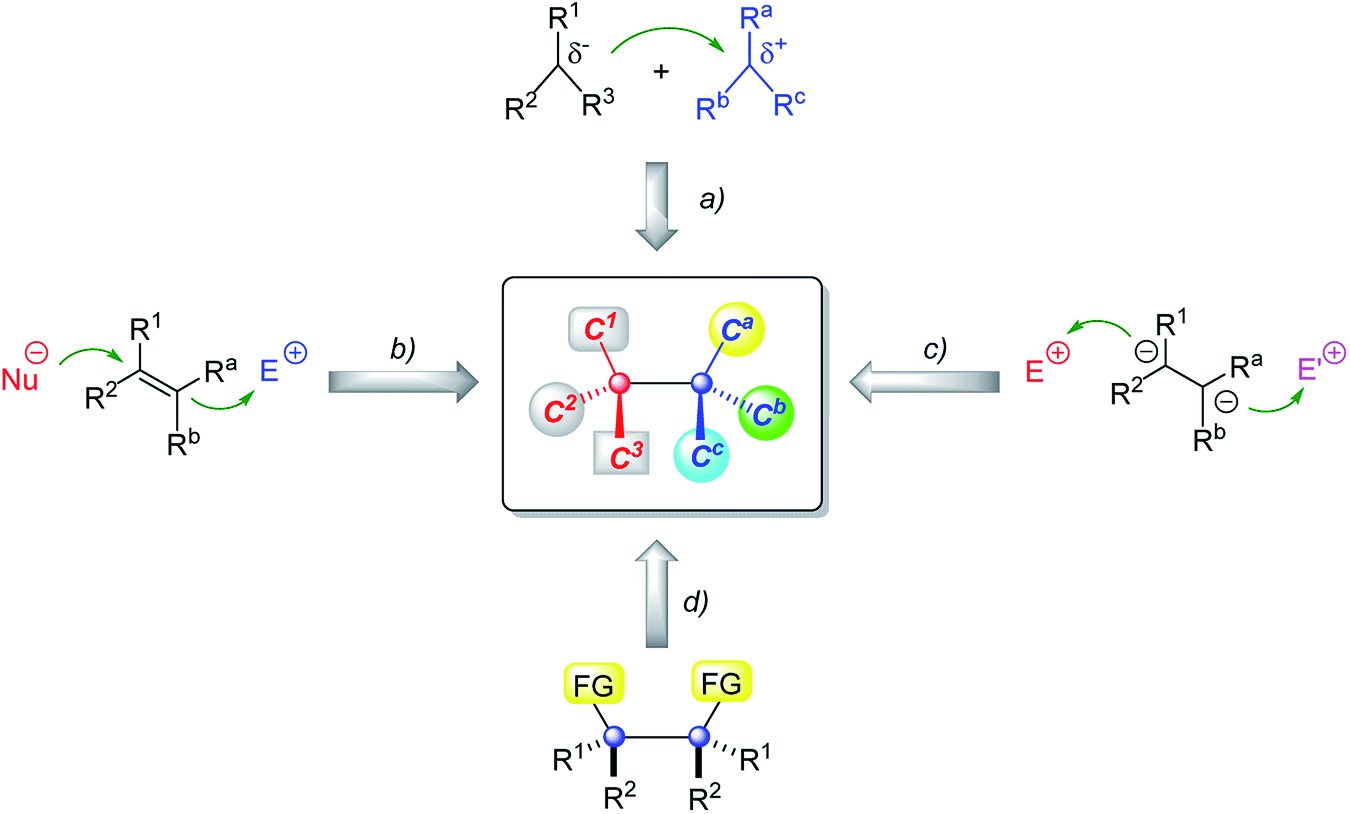 | ||
| Fig. 3 Synthetic strategies for vicinal quaternary carbon stereocenters. | ||
2. The reaction of trisubstituted carbon nucleophiles and electrophiles
The C–C bond-forming reaction between two trisubstituted prochiral carbons, in most cases a nucleophile and an electrophile, is a straightforward strategy for the simultaneous construction of vicinal quaternary carbon stereocenters in one step. Despite the advantages, such as the flexible and diverse synthesis using many types of substrates for reaction design, there are difficult challenges in the reaction development, the low reactivity caused by the steric repulsion and the difficulty in the control of diastereo- and enantioselectivity due to the lesser steric dissimilarity of the substituents on both prochiral carbons. Apart from steric hindrance, the inherent electronic challenges impose extra difficulty for the reaction development. According to the Lapworth–Evans model,24 the all-carbon tertiary anion might be highly unstable and reactive in its own right; meanwhile, the tertiary carbocation should be stable but highly prone to side reactions such as rearrangements and eliminations. In this context, the engineering of adjacent functionality to stabilize these reaction centers and to promote substrate–catalyst interactions plays an important role in utilizing this strategy successfully. Nevertheless, substantial achievements have been made by using highly active substrates or by integrating both prochiral partners into an intramolecular reaction. These elegant advances will be introduced according to reaction types in this section, including cyclopropanation, cycloaddition, cyclization, alkylation, rearrangement, and domino reactions. Notably, there are no clearly defined nucleophiles or electrophiles in some reactions, although they involve the C–C bond formation between two trisubstituted prochiral carbons.2.1 Cyclopropanation reaction
The cyclopropanation is probably the most investigated process for the construction of vicinal quaternary carbon stereocenters. The resulting congested cyclopropanes are interesting targets for medicinal research. In this context, olefin cyclopropanation using diazo reagents, cycloisomerization of enynes and tandem sequences have shown their value.Although the olefin cyclopropanation is the first metal catalyzed asymmetric reaction, pioneered by Nozaki in 1966,25 it was applied to construct vicinal quaternary stereocenters 30 years later, when Doyle reported the reaction of phenyldiazoacetate and α-methylstyrene 2a. The use of catalyst 4 gave 3a in 85% ee.26 To our knowledge, this is probably the first catalytic asymmetric reaction to construct vicinal all-carbon quaternary stereocenters (Scheme 1).
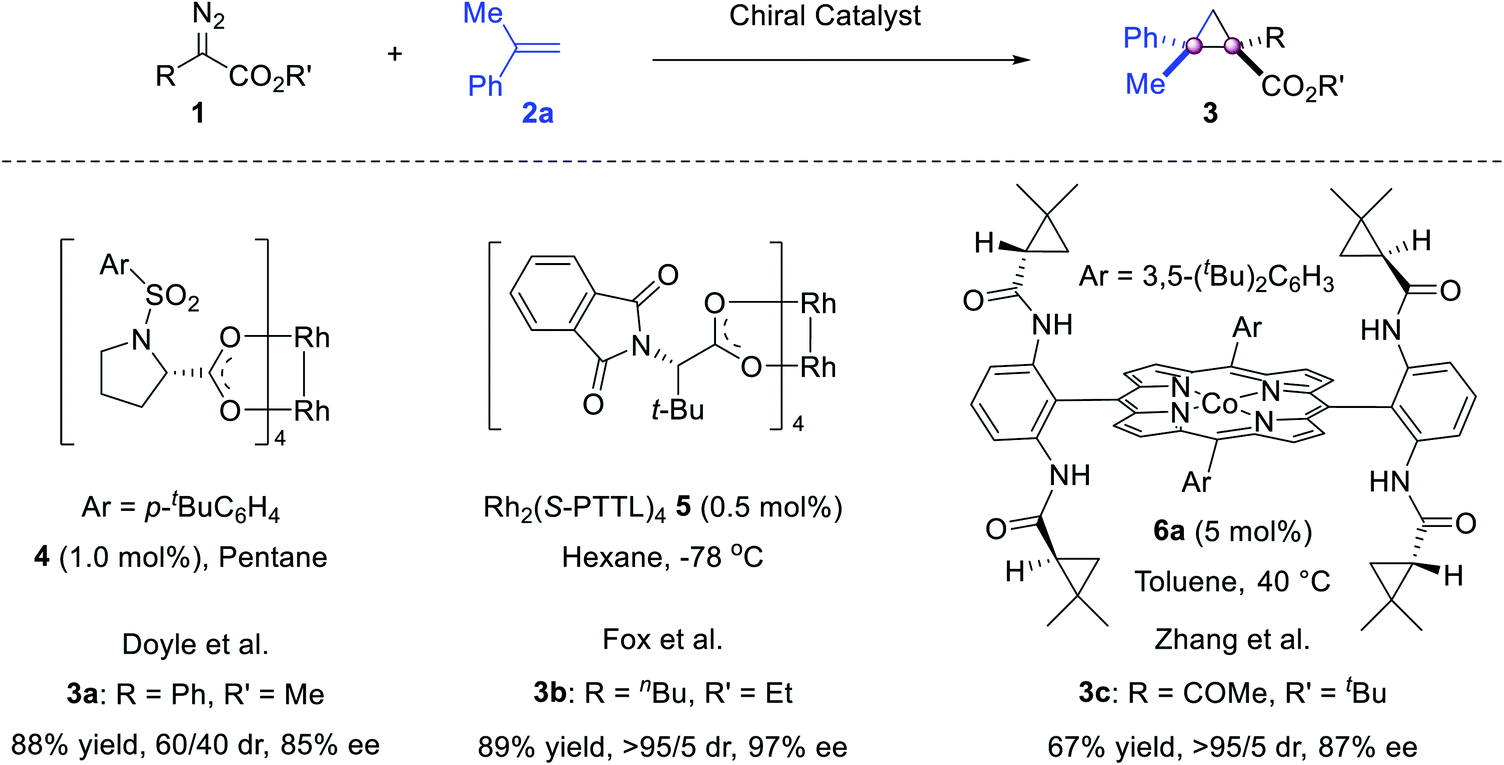 | ||
| Scheme 1 Cyclopropanation of α-methylstyrene with diazoacetates. | ||
Thirteen years later, more studies were reported (Scheme 1). Fox reported a Rh2(S-PTTL)4 mediated highly stereoselective version using α-butyldiazoacetate,27 without competitive intramolecular C–H insertion28 and β-hydride elimination29 by-products. Zhang reported a porphyrin/Co(II) complex 6a catalyzed reaction of α-ketodiazoacetate,30 wherein H-bonding interactions between the amide N–H of the catalyst and two carbonyl acceptors on the carbene intermediate are critical for high catalytic activity.31
The cyclopropanation of cyclic diazo compounds enables the construction of spirocyclic cyclopropanes (Scheme 2). With our efforts in the syntheses of quaternary oxindoles,32 we reported the first enantioselective Hg- and Au-catalyzed olefin cyclopropanation using diazo reagents. While (R)-difluorophos/Hg(PF6)2 catalyzed the reaction of diazooxindole and 2a to give spirocyclopropyloxindole 8a in 83% ee,33 Ding's spiroketal bisphosphine34 (SKP)-derived digold complex 10 was powerful for this reaction, furnishing 8b in excellent drs and ees.35 Later, Qiu and Xu reported a Rh2(S-TBPTTL)211 catalyzed version using N-Boc diazooxindoles, delivering 8c in 82% yield and 95% ee albeit with a moderate dr.36
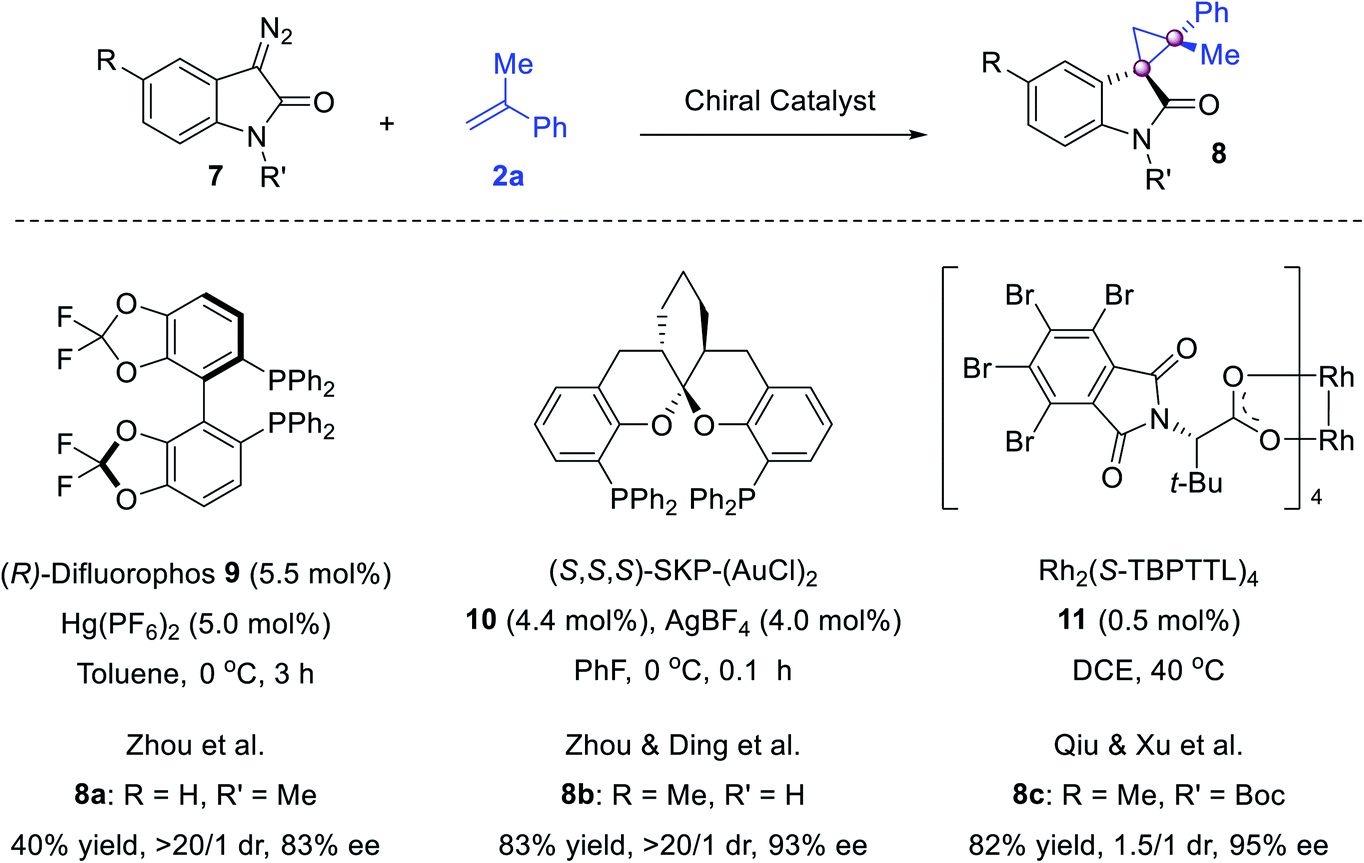 | ||
| Scheme 2 Enantioselective cyclopropanation to spirocyclopropanes. | ||
The cyclopropanation of electron-deficient olefins and diazo compounds to construct vicinal quaternary carbon stereocenters is also achieved. In 2011, Hwang & Ryu reported that the reaction of α,β-unsaturated aldehydes 12 with diazoacetates 1 catalyzed by chiral (S)-oxazaborolidinium 13 gave 14 in high to excellent yield and stereoselectivities (Scheme 3a).37 It was proposed that the Lewis acid-mediated nucleophilic addition of the diazoester from the Si face of the acrolein led to intermediate I, and then the loss of nitrogen delivered the desired product. In 2018, Liu and Feng reported the enantioselective cyclopropanation and C–H insertion reactions of α-alkyldiazoesters 16 with α-substituted vinyl ketones 15 in a one-pot manner (Scheme 3b).38 The chiral N,N′-dioxide 17a/Sc(OTf)3 gave cyclopropanes 18 and E-enone derivatives 19 simultaneously with good yields and excellent ee values. A mono-bonding model was proposed to rationalize the generation of chiral cyclopropanes.
 | ||
| Scheme 3 Lewis acid-catalyzed cyclopropanation of diazo reagents. | ||
The cyclopropanation of diazo reagents and fluoroalkylated olefins paves the way to fluorinated cyclopropanes that are of interest in drug discovery. In 2017, Jubault reported a highly stereoselective synthesis of CF2H-cyclopropanes 21a using difluoromethylated olefins 20a, catalyzed by Rh2(S-BTPCP)422 (Scheme 4).39 One year later, Charette and Jubault further extended the reaction to α-trifluoromethyl olefins 20b.40 We also developed stereoselective syntheses of congested cyclopropanes 21a and 21c and spirocyclopropyl-oxindoles 24 featuring a CF2H or a CFH2 group, via Rh2(R-DOSP)423 mediated cyclopropanation of α-aryl diazoacetates 1 or digold catalyst 10 catalyzed that of diazooxindoles 7.41 A dramatic solvent effect was observed and theoretical calculations suggested that the C–F⋯H–N interaction between PhF and the N–H bond of the diazooxindole-derived Au(I)-carbenoid intermediate effectively lowered the reaction barrier, and tuned both the reactivity and stereoselectivity.42
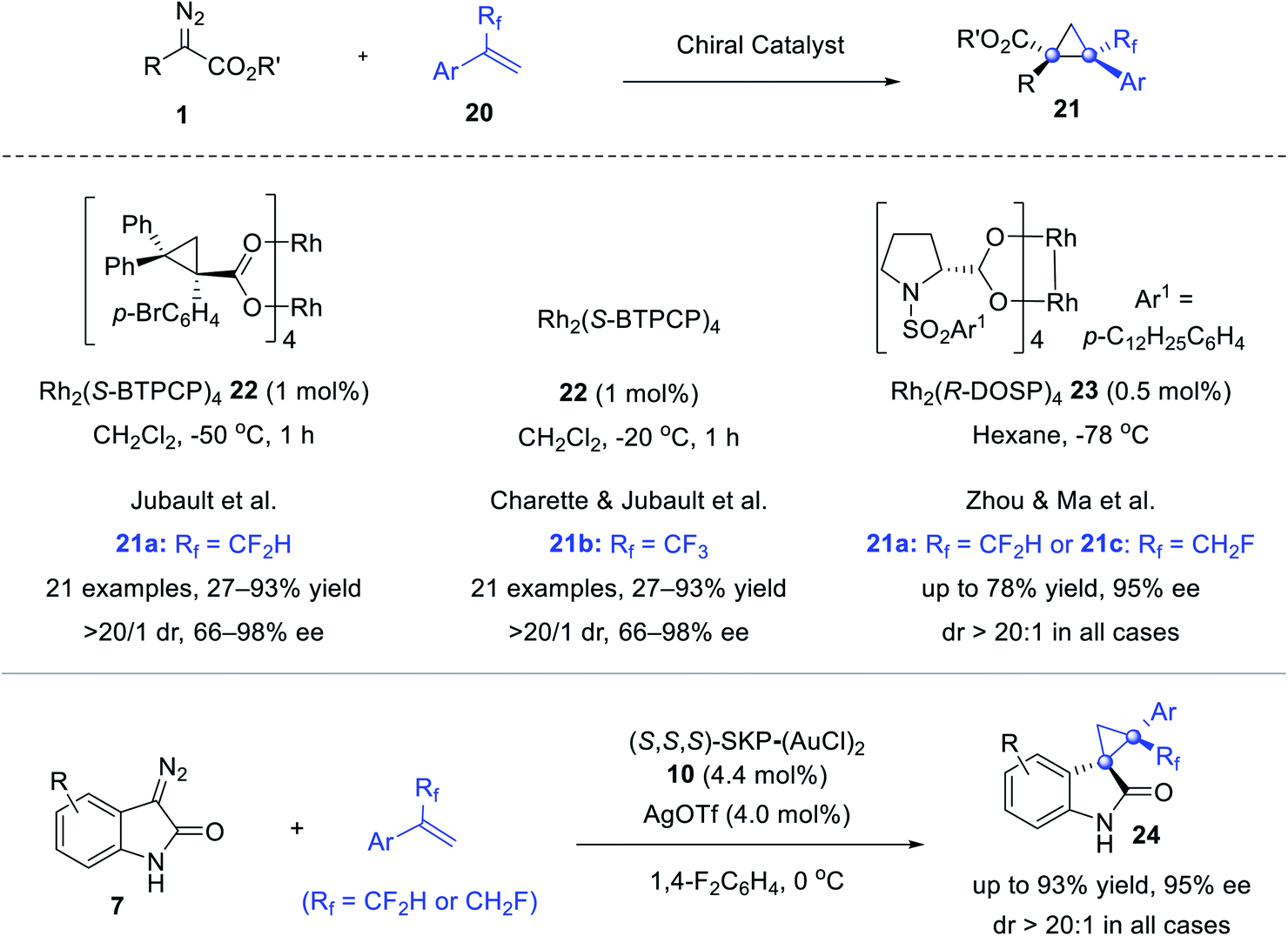 | ||
| Scheme 4 Cyclopropanation of fluoromethylated olefins. | ||
The intramolecular cyclopropanation to cyclopropanes with bicyclic or polycyclic structures is also accomplished. In 2011, Zhang reported a porphyrin/Co(II) catalyst 6b for the intramolecular cyclopropanation of acceptor-substituted diazoacetate 25, giving chiral 3-oxabicyclo[3.1.0]hexan-2-one 26 in 88% yield with 83% ee (Scheme 5a).43 Zhou and Zhu achieved a spirobisoxazoline 28/Fe(II) complex catalyzed version using diazoesters 27, furnishing [3.1.0]bicycloalkanes 29 in high yields with up to 97% ee (Scheme 5b).44 They further applied this strategy to construct indoline-cyclopropanes efficiently (Scheme 5c).45
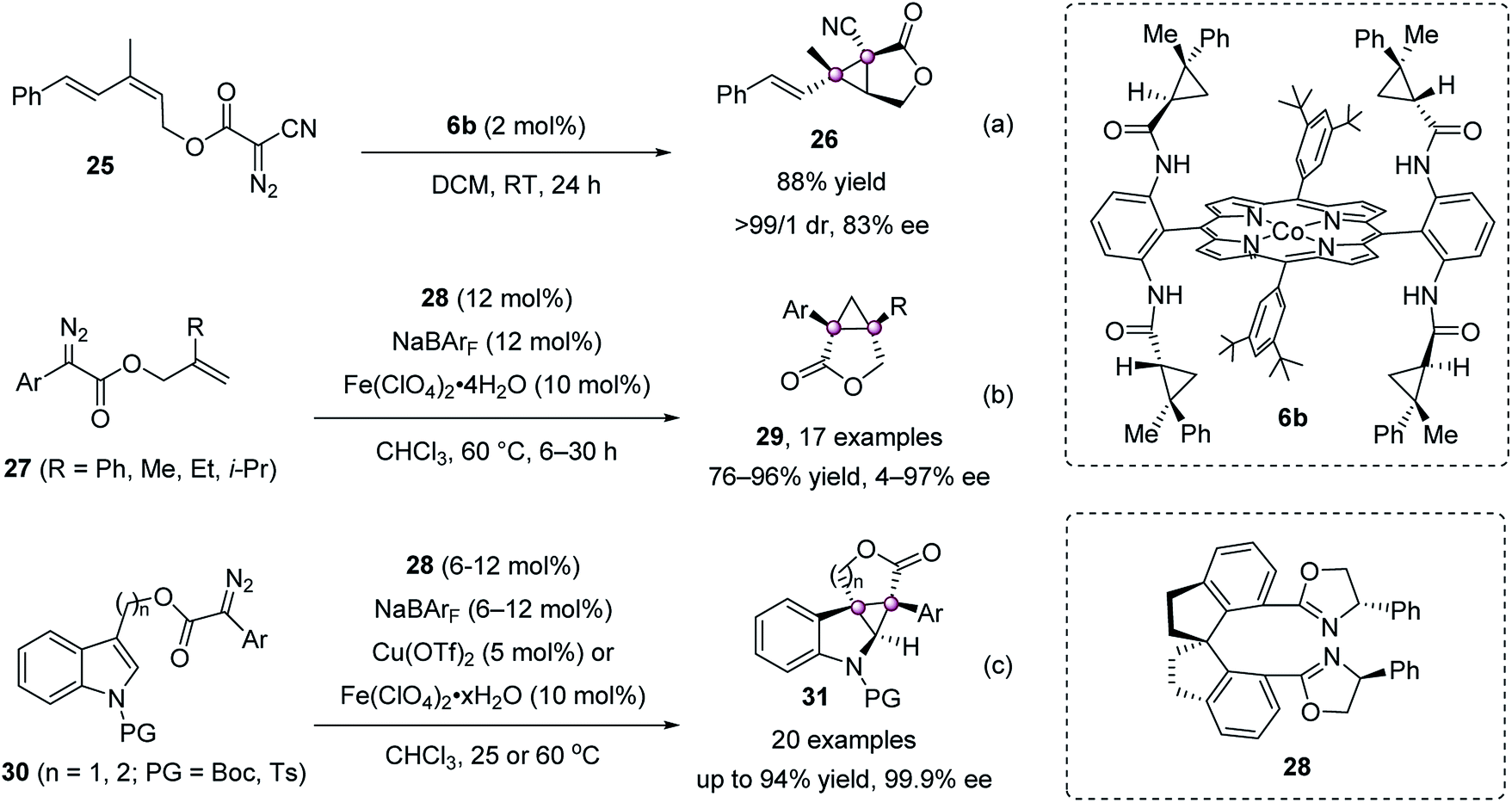 | ||
| Scheme 5 Asymmetric intramolecular cyclopropanation of diazo compounds. | ||
The cyclopropanation shown above requires using acceptor-substituted diazo reagents to inhibit side reactions and achieve high stereoselectivity, so it inevitably affords congested cyclopropanes bearing electron-withdrawing substituents. The cycloisomerization of enynes constitutes a complement to access polysubstituted cyclopropanes with aryl or alkyl groups. In 2005, Shibata first conducted catalytic asymmetric studies and found that the TolBINAP/Ir complex mediated the cycloisomerization of 1,6-enyne 32a well to afford 3-azabicyclo[4.1.0]heptene 33a in 92% yield and 66% ee.46 Later, several groups tried improving the stereoselectivities (Scheme 6). NHC–Pt(II) complex 34 (ref. 47) could afford product 33b in 88% ee, but Au(I) complex 35 (ref. 48) afforded poor enantioselectivity. The use of chiral Ru complex 36 furnished 33c in 81% yield and 68% ee.49 To avoid the dissociation of Ph3P in chiral catalyst 36, a new tridentate ligand-derived Rh complex 37 was developed and afforded higher 98% yield and 88% ee.50 Finally, the chiral phosphoramidite–Au(I) complex 38, developed by Fürstner et al., achieved 96% ee.51 This protocol was applied to the synthesis of anti-depressant agent (−)-GSK 1360707.
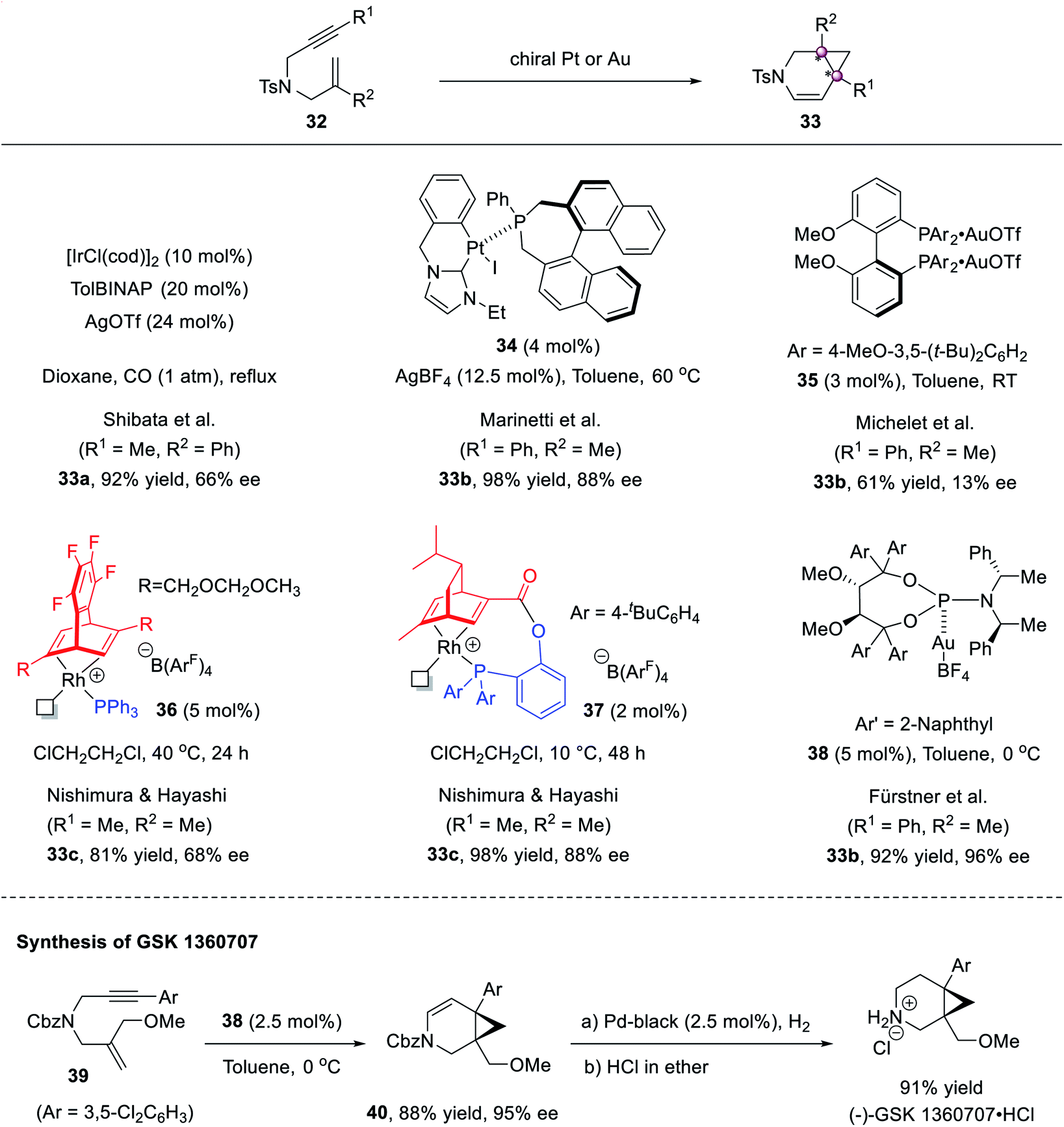 | ||
| Scheme 6 Transition-metal catalyzed cycloisomerization of 1,6-enyne. | ||
In 2014, Trost et al. realized a cycloisomerization of [1,6]- or [1,7]-enynes 41 bearing a propargyl alcohol moiety, affording chiral ketones 43 with a bicyclic backbone in high yield and ee values (Scheme 7a).52 A β-oxo ruthenium carbenoid I, formed via 1,2-hydride migration, was proposed to be involved during the reaction. Pla-Quintana et al. demonstrated that N-tosylhydrazones 44 could deliver metal carbene intermediates II by chiral Rh(I)-catalysis, which then underwent carbene–alkyne metathesis/cyclopropanation to give chiral cyclopropane 45 in 63% yield and 74% ee (Scheme 7b).53 Zhang group reported the gold-catalyzed intramolecular enantioselective cyclopropanation of 1,6-enynes 46 using their chiral sulfonamide–phosphine-type ligand 47 (Scheme 7c).54
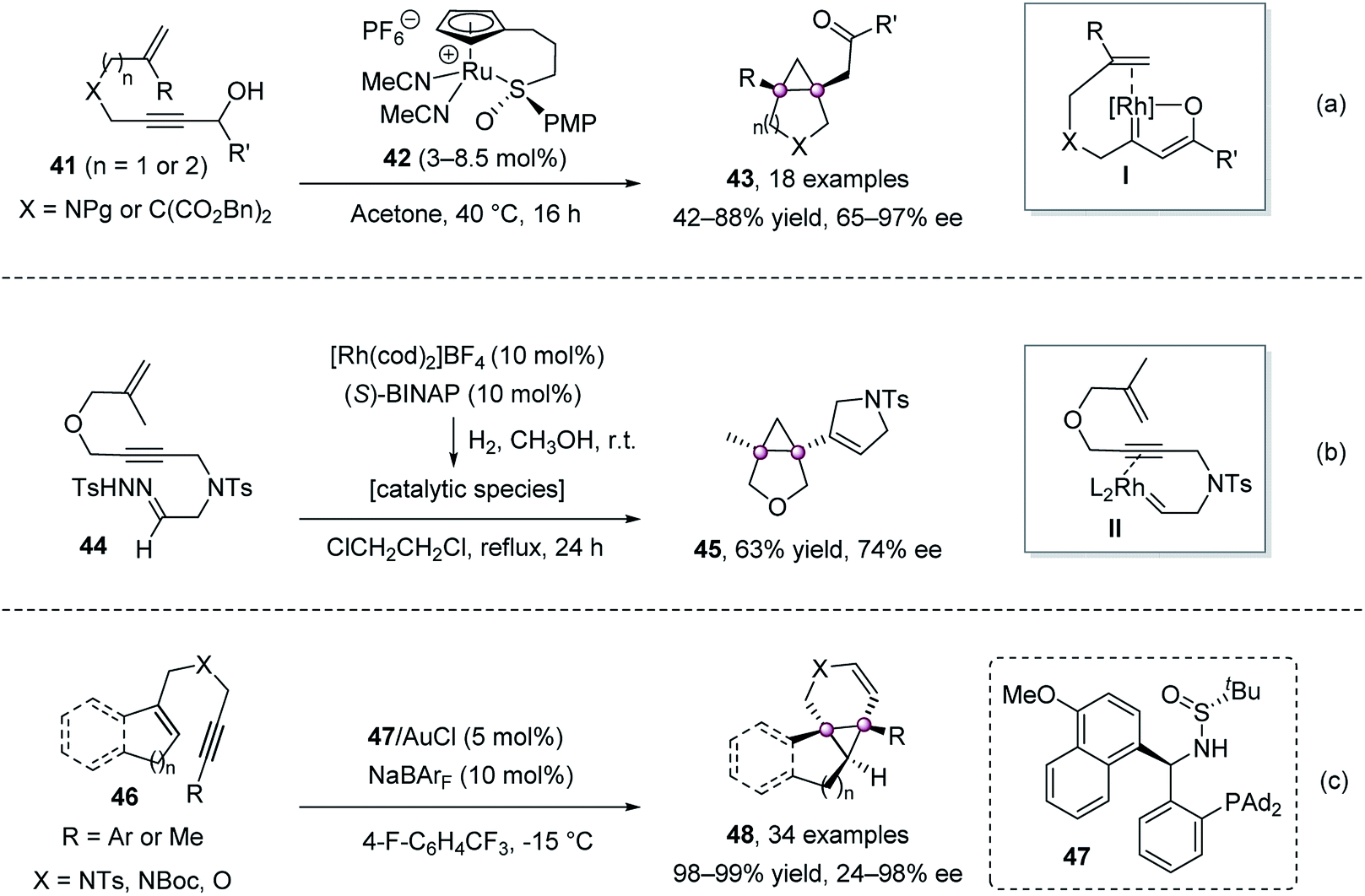 | ||
| Scheme 7 Enantioselective cycloisomerization of [1,6]- or [1,7]-enyne. | ||
Tandem sequences are an effective method to build cyclopropanes with vicinal quaternary stereocenters. Malkov used cinchona alkaloid derived bifunctional catalysts to develop two tandem Michael-cyclization reactions for the synthesis of spirocyclic oxindoles from 3-alkylidene oxindoles 49 with α-chloroacetoacetate 50,55 or with 3-chlorooxindole 53 (Scheme 8a and b).56 Feng et al. achieved a 17b/Mg(OTf)2-catalyzed version using cyclic sulfur ylide 56 with 3-alkylidene oxindole 49a, furnishing chiral 57 in a 57% yield, 95/5 syn/anti-ratio and 68% ee (Scheme 8c).57
 | ||
| Scheme 8 Asymmetric Michael-cyclization reaction. | ||
An elegant cooperative Cu(I)/secondary amine catalysis directed intramolecular radical cyclopropanation of unactivated alkenes to construct bicyclo[3.1.0]hexanes 61 was invented by Liu et al. (Scheme 9).58 The initial single-electron transfer of the enamine I formed in situ with external iodine(III) oxidant DF-BI-OH or Cu(II) gave iminium radical cation II, followed by a sequential 6-endo-trig and 3-exo-trig cyclization to give desired products.
 | ||
| Scheme 9 Radical asymmetric intramolecular α-cyclopropanation. | ||
2.2 Cycloaddition reaction
Cycloaddition is a powerful strategy to construct contiguous carbon stereocenters. In 2007, Yamamoto reported a highly stereoselective Diels–Alder reaction of 1-substituted cyclopentadienes 62 and 2,5-disubstituted benzoquinones 64 catalyzed by oxazaborolidine 65 (Scheme 10).59 Because cyclopentadienes 62 existed as a mixture of 1- and 2-substituted isomers due to the [1,5]-sigmatropic rearrangement, excess ethyl acrylate 63 was employed to consume the 2-substituted 62bvia the formation of 66. Then, the remaining 1-substituted 62a reacted with dienophile 64 to give endo adducts 67 as a single regioisomer in high enantiomeric purity. Because of the strong steric interaction between the phenyl group of catalyst 65 and the substituents of cyclopentadiene 62a, the endo approach of 62a to the double bond of anti-coordinated benzoquinone was favored. | ||
| Scheme 10 Asymmetric Diels–Alder reaction. | ||
In 2013, Ooi and co-workers reported an elegant palladium-catalyzed decarboxylative [3 + 2] cycloaddition of 5-vinyloxazolidinones 68 with activated trisubstituted alkenes 69, enabling the single-step synthesis of multifunctionalized pyrrolidines 71 bearing contiguous tertiary and vicinal quaternary stereocenters in high yields and excellent stereoselectivities (Scheme 11).60 The phosphine ligand 70 bearing a chiral ammonium salt component was critical for the reaction development, assisting in the preferable halide–palladium contact and recognizing the anionic site via facile pairing with the ammonium ion in the transition state, which in turn provided a precise stereoselective control during the plural bond-forming process. The reaction could be amplified to a 10 mmol scale using only 0.5 mol% catalyst, without the erosion of stereoselectivity. The products 71 are of high synthetic value, as shown by the conversion of 71a to the core structure of the thrombin inhibitor analogue, a densely substituted bicyclic lactam 73, as a single stereoisomer.
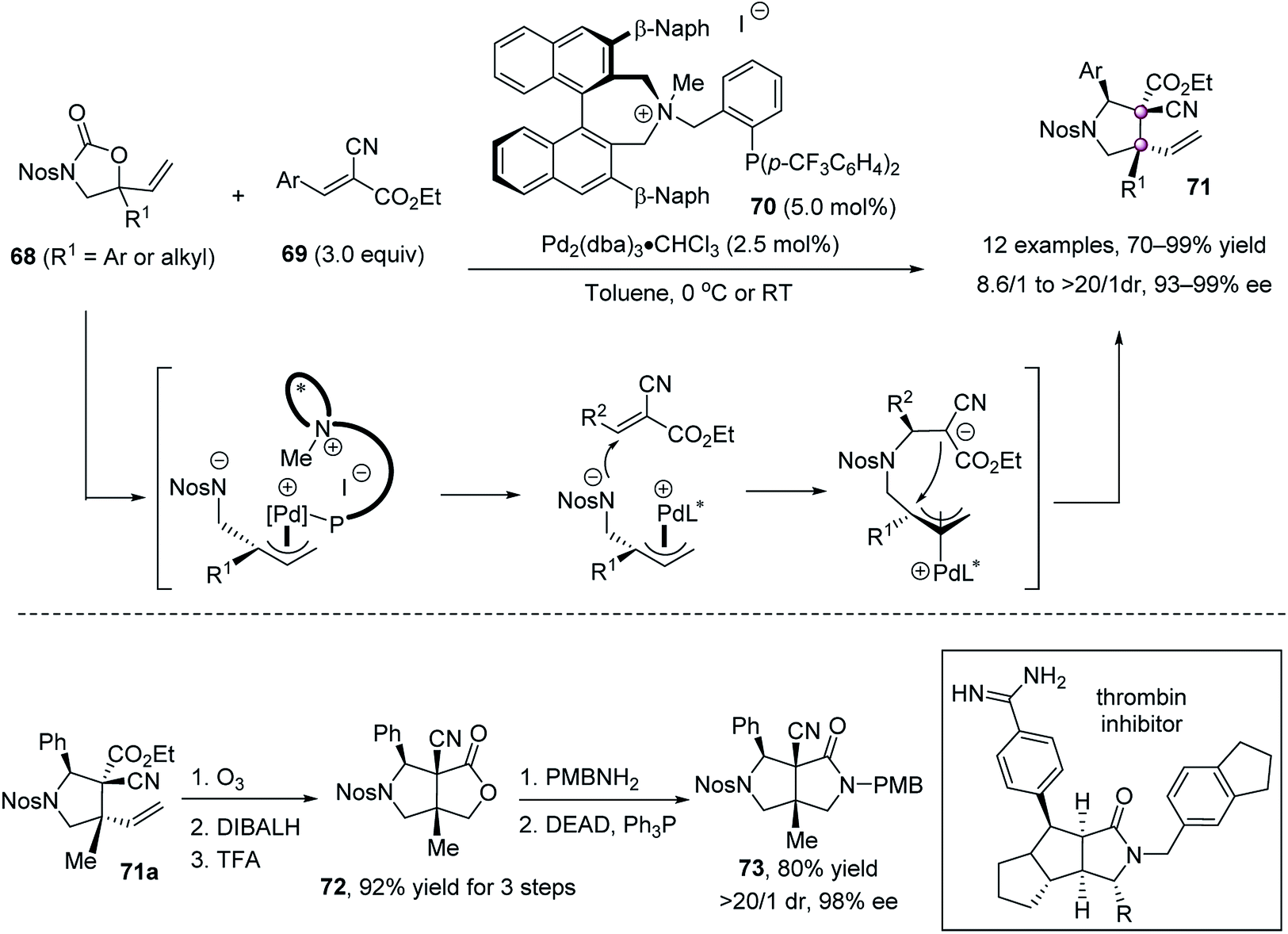 | ||
| Scheme 11 Asymmetric [3 + 2] annulation reaction. | ||
Soon afterward, Zhang et al. reported a highly stereoselective palladium-catalyzed decarboxylative cycloaddition of vinylethylene carbonates 74 and trisubstituted electron-deficient alkenes 75, furnishing densely functionalized tetrahydrofurans 77 in up to 98% yield and 98% ee with a 11/1 dr (Scheme 12).61 It was proposed that cycloaddition is the stereochemistry-determining step, involving a conformationally favored chair-like transition state II.
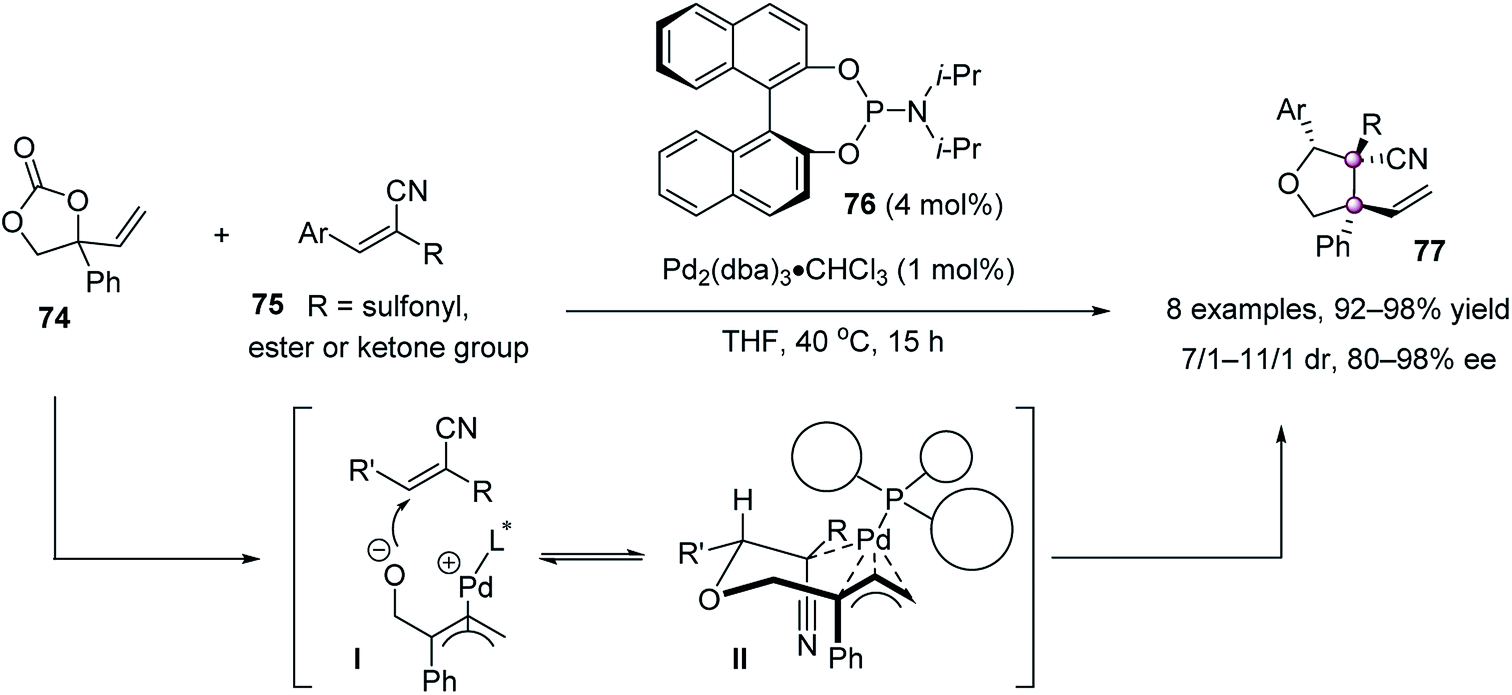 | ||
| Scheme 12 Asymmetric decarboxylative cycloaddition. | ||
Most recently, Song and Gong nicely integrated chiral NHC catalysis with copper catalysis to accomplish an elegant [3 + 3] annulation of isatin-derived enals 78 and ethynylethylene carbonates 79, giving spirooxindole–lactones 81 with excellent stereoselectivities (Scheme 13).62 It was proposed that Cu(CH3CN)4PF6 activated 79 to produce a copper–allenylidene intermediate (II or II′), which reacted with the chiral NHC–homoenolate I to furnish vicinal quaternary stereocenters. The following O-acylation cyclization and protonation produced spirocyclic oxindoles 81.
 | ||
| Scheme 13 NHC-catalyzed [3 + 3] annulation of isatin-derived enals. | ||
Meggers and co-workers reported a visible light-activated asymmetric intermolecular [2 + 2] cycloaddition to construct chiral cyclobutenes (Scheme 14).63 The bis-cyclometalated chiral-at-Rh catalyst 83 played a dual role in both photoredox activation and asymmetric catalysis, allowing the [2 + 2] cycloaddition of α,β-unsaturated 2-acyl imidazole 82 with a wide range of alkenes 2 to work effectively, delivering differently substituted cyclobutanes 84 with up to >99% ee and up to >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr. Mechanism studies suggested the following catalytic cycle. First, the N,O-chelate coordination of α,β-unsaturated 2-acyl imidazole 82 to catalyst 83 gave intermediate I, which was excited by visible light to its lowest singlet state (II). After intersystem crossing (ISC), the excited triplet state III reacted with alkene 2 to afford Rh-bound 1,4-diradical intermediate IV. Then, ISC and cyclization along with the recoordination of another molecule of 82 generated the [2 + 2] cycloaddition product 84 and closed the catalytic cycle.
:1 dr. Mechanism studies suggested the following catalytic cycle. First, the N,O-chelate coordination of α,β-unsaturated 2-acyl imidazole 82 to catalyst 83 gave intermediate I, which was excited by visible light to its lowest singlet state (II). After intersystem crossing (ISC), the excited triplet state III reacted with alkene 2 to afford Rh-bound 1,4-diradical intermediate IV. Then, ISC and cyclization along with the recoordination of another molecule of 82 generated the [2 + 2] cycloaddition product 84 and closed the catalytic cycle.
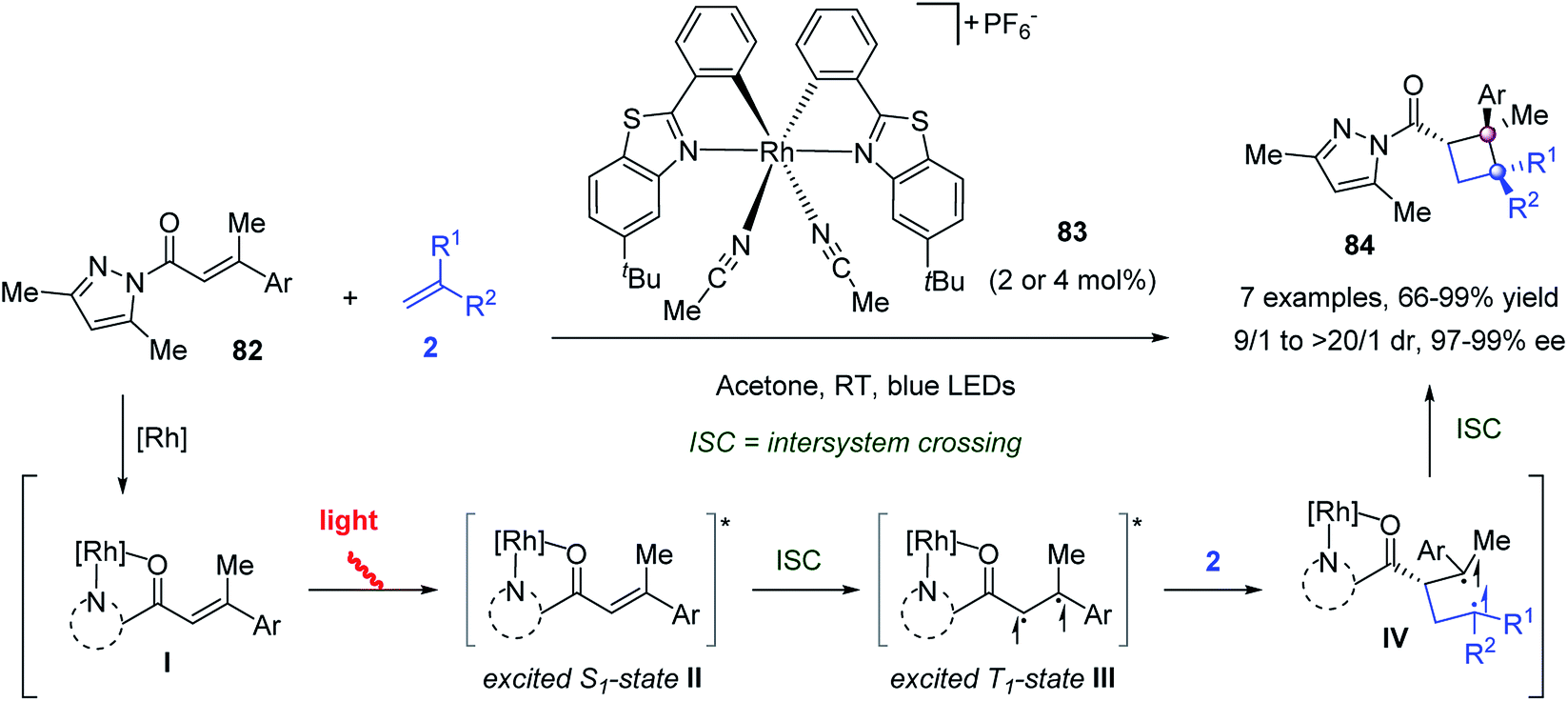 | ||
| Scheme 14 Visible light-activated asymmetric [2 + 2] cycloaddition. | ||
2.3 Cyclization reaction
Cyclization reactions, such as the Nazarov cyclization, enabled the key diastereo- and enantioselective step of forming two adjacent quaternary carbon atoms to be performed in an intramolecular fashion. In light of this, the cyclization strategy is not only helpful to achieve reasonable reactivity but also beneficial for the control of stereoselectivities.In 2014, Tius et al. accomplished a remarkable Nazarov cyclization to construct chiral cyclopentanone with vicinal quaternary stereocenters (Scheme 15).64 In the presence of 10 mol% chiral N-triflyl phosphoramide 86, the Nazarov cyclization of fully substituted dienones 85 proceeded smoothly to furnish optically active cyclopentenones 88 as single diastereoisomers in up to 98% ee. 2-Tert-butylphenol 87 was employed as a cation scavenger to intercept the diphenylmethyl cation generated.
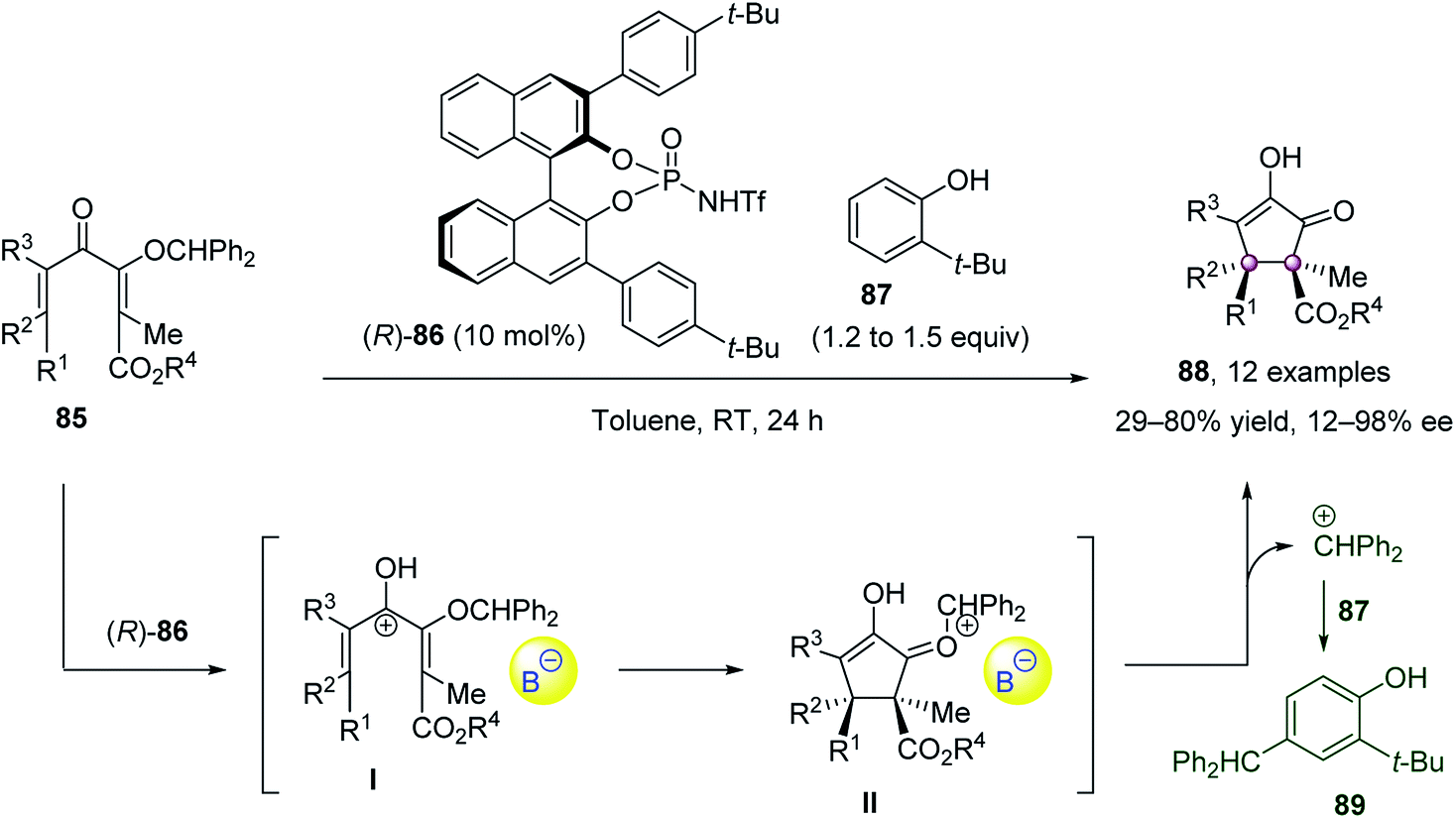 | ||
| Scheme 15 Nazarov cyclization reaction. | ||
2.4 Alkylation reaction
Asymmetric allylic alkylation is one of the most important reactions for the formation of carbon–carbon and carbon–heteroatom bonds and has been widely applied to the fields of both medicinal chemistry and synthesis of natural products.65 Despite intensive studies, the employment of this strategy to construct vicinal quaternary stereogenic carbons is underdeveloped. An impressive example was realized by Trost in 2011. Using a ligand 91 derived chiral palladium catalyst, the reaction of 3-alkyloxindole 90 and racemic linalyl carbonate 92 gave the linalylated quaternary oxindole 94 in 46% yield, >19:1 dr, and 90% ee, along with the formation of geranylated 95 and nerylated 96 (Scheme 16).66 The slow interconversion of syn- and anti-π-allylpalladium complexes leads to the generation of both isomers 94 and 95 in equal amounts. Based on this consideration, the geometrically defined linear carbonate 93 was used, affording the linalylated product 94 in 92% yield with 91% ee and 13:1 selectivity to the nerylated 96.
 | ||
| Scheme 16 Asymmetric allylic alkylation. | ||
Since the pioneering work of Stoltz and co-workers,67 the catalytic enantioselective addition of nucleophiles to indol-2-ones, generated in situ from 3-halooxindoles, has emerged as a fruitful strategy to construct enantioenriched 3,3-disubstituted oxindoles, a privileged scaffold widely distributed in bioactive natural products and pharmaceutically active compounds.68,69 In 2013, Wang and co-workers nicely demonstrated the value of this strategy in constructing oxindoles with vicinal quaternary stereogenic carbons for natural product synthesis. They showed that a chiral diamine 99-derived Ni complex could catalyze the alkylation of 3-bromooxindoles 98 with 3-substituted indoles 97 smoothly, affording indolenines 100 with excellent diastereo- and enantioselectivity (Scheme 17).70 Notably, the stereoselectivity was governed by the formation of chiral catalyst–electrophile complex I, different from previous reports that used chiral catalyst to control the nucleophiles.69 Based on this methodology, the first catalytic enantioselective total synthesis of complex natural product (+)-perophoramidine was accomplished.
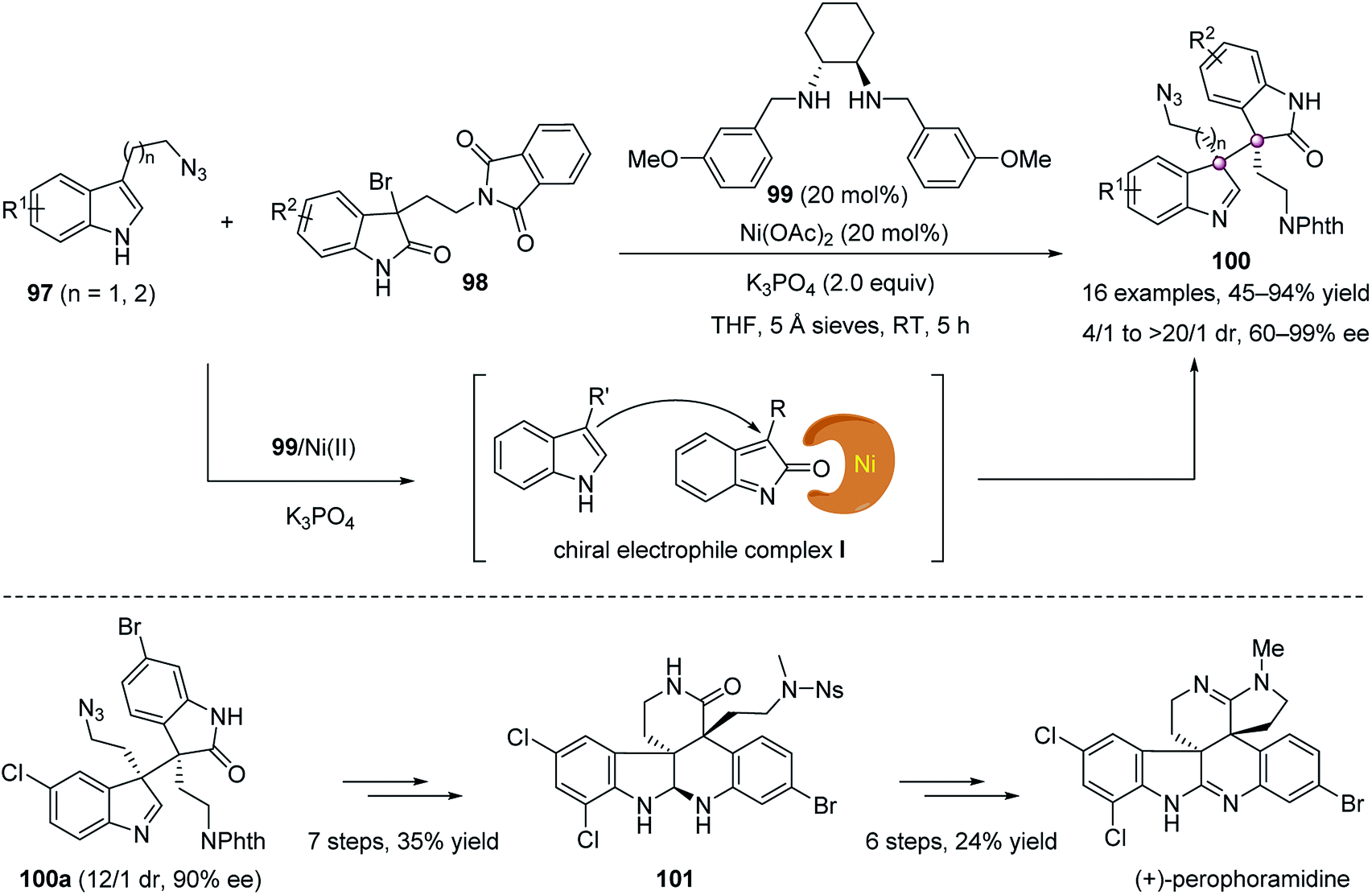 | ||
| Scheme 17 Alkylation of 3-bromooxindoles and its application. | ||
Later, Liu and Feng reported an elegant formal alkylation of 3-bromooxindoles using fully substituted silyl ketene imines 102 (SKI), catalyzed by a chiral N,N′-dioxide 17b/Ni(II) complex (Scheme 18).71 The resulting cyano-substituted quaternary oxindoles 103 were readily accessed in up to 90% yield, 23:1 dr, and 98% ee, which could undergo various diversified reactions. It was believed that the in situ-generated indol-2-one bound to the chiral Ni(II) catalyst, with its Si face blocked by the 2,4,6-triisopropylphenyl group of the ligand, to facilitate the Re-face attack of SKI on the Re face of the electrophile.
 | ||
| Scheme 18 Asymmetric conjugate addition of SKI. | ||
In 2013, Ooi et al. developed an unusual catalytic enantioselective ring-opening alkylation of racemic 2,2-disubstituted aziridines 105 at the tetrasubstituted terminus (Scheme 19).72 In the presence of chiral 1,2,3-triazolium salt 106 and K2CO3, 3-substituted oxindoles 104 preferentially reacted with (S)-105 to give quaternary oxindoles 107 bearing two adjacent quaternary stereocenters in high to excellent yield and dr values, as well as excellent ee values. Control experiments confirmed this kinetic resolution progress because (R)-105a could be recovered as the major enantiomer when using 2.0 equiv. of racemic 105a. On the basis of the absolute configuration of the product with the remaining aziridine, the ring-opening substitution at the tetrasubstituted chiral carbon was believed to proceed in a stereoinvertive manner.
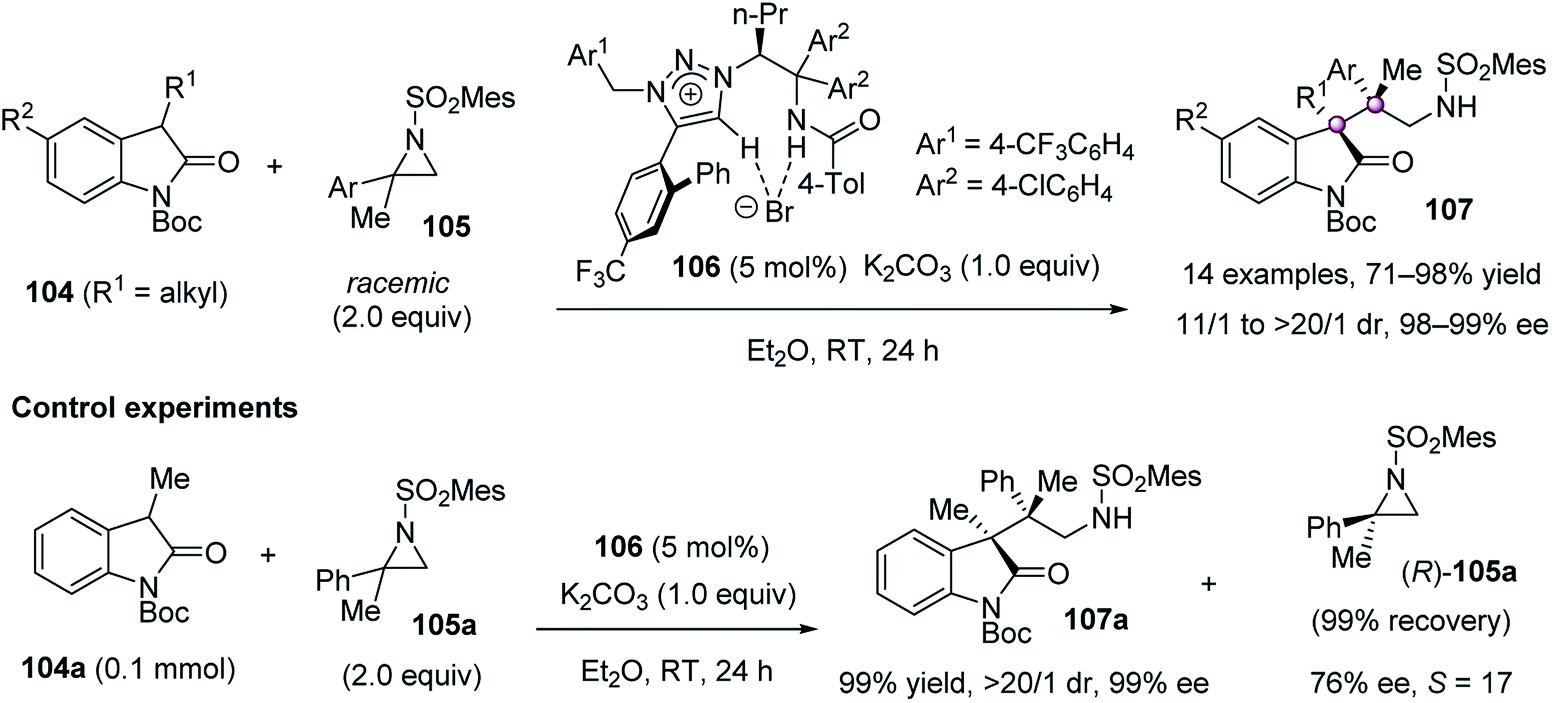 | ||
| Scheme 19 Asymmetric ring-opening alkylation. | ||
Recently, Jørgensen et al. reported a direct diastereo- and enantioselective oxidative homocoupling reaction of α-branched aldehydes (Scheme 20).73 Using secondary amine catalyst 109 in combination with 4-nitrobenzoic acid, together with Ag2CO3 as the oxidant, the succinic 1,4-dialdehydes 110 were obtained in up to 79% yield, 18/1 dr, and 96% ee. It was proposed that a radical cation intermediate I, formed via SET oxidation of enamine by Ag(I), was involved during the reaction process. Mechanistic studies also showed that the reactivity was influenced by radical stability, whereas the stereoselectivity was governed by the cationic character of intermediate I.
 | ||
| Scheme 20 Direct oxidative homocoupling reaction of α-branched aldehydes. | ||
2.5 Rearrangement reaction
The Claisen rearrangement of allyl vinyl ethers74 is a potentially potent approach to forge vicinal quaternary stereocenters because of the predictable and high diastereoselectivity resulting from the concerted nature of the C–O bond-breaking and C–C bond-forming process via a chair-like transition state. The power of this approach was successfully demonstrated by Jacobsen et al. in 2010 by a chiral C2-symmetric guanidinium ion 112-catalyzed [3,3]-sigmatropic rearrangement of cyclic O-allyl β-ketoesters 111 (Scheme 21).75 The allylation products 113 were obtained in high yield and stereoselectivities. The reaction was successfully applied to the construction of the core structure of natural product hyperforin.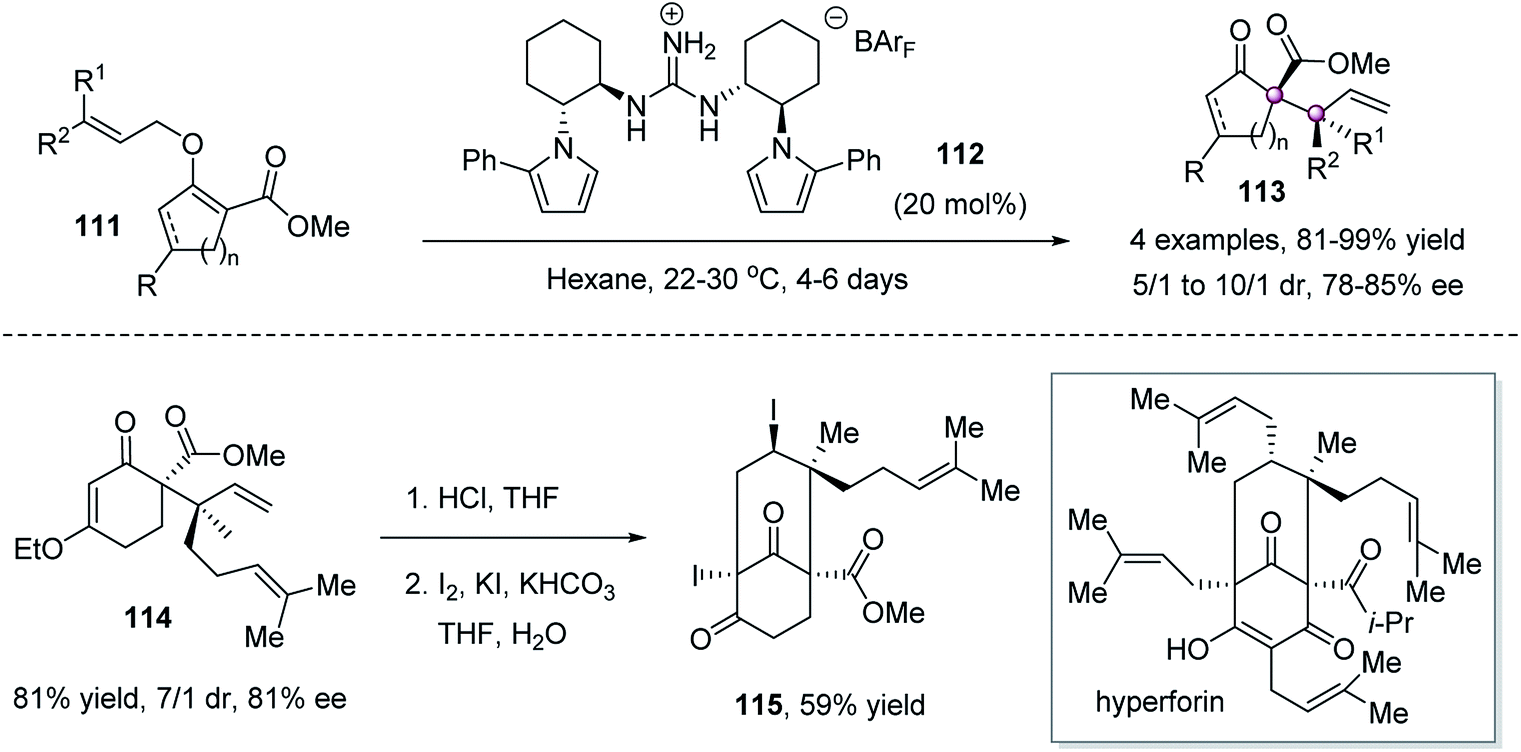 | ||
| Scheme 21 Guanidinium-catalyzed Claisen rearrangement. | ||
2.6 Domino reaction
Domino reactions could merge two or more bond-forming transformations in one-step without the isolation of intermediates, thus providing efficient tactics for the construction of vicinal quaternary stereocenters. In 2009, Melchiorre et al. reported an elegant organocatalytic Michael–Michael sequence to forge spirocyclic oxindoles with structural and stereochemical complexity (Scheme 22).76 The merger of chiral primary amine 118 and ortho-fluorobenzoic acid mediated the double Michael reaction of 3-alkenyloxindole 116 and enone 117 efficiently. The initial intermolecular Michael reaction was facilitated by enamine activation of enone 117, affording iminium intermediate I that underwent the subsequent Michael addition to give highly congested 120 in 19/1 dr and 97% ee. | ||
| Scheme 22 Domino Michael–Michael reaction of alkylidene oxindoles. | ||
Recently, Liu and Zhou designed a novel bifunctional oxindole–chromone 4C synthon 122 as a versatile platform to construct hexahydroxanthones with contiguous stereogenic centers (Scheme 23).77 Takemoto's tertiary amine–thiourea 123 catalyzed the domino Michael–Michael reaction well to give hexahydroxanthones 124 with five stereocenters, including two adjacent spiro quaternary ones, with excellent dr and ee values.
 | ||
| Scheme 23 Domino Michael–Michael reaction to hexahydroxanthones. | ||
3. Difunctionalization of all-carbon tetrasubstituted alkenes
The concerted or stepwise C–C bond-forming reactions of both prochiral carbon atoms of all-carbon tetrasubstituted alkenes constitute an effective strategy to construct adjacent all-carbon stereocenters. Depending on the electronic or steric nature of such alkenes that are significantly influenced by the substituents, this strategy should incorporate a broad range of reactions to construct the target structures with great diversity. However, because of the challenge of functionalization of all-carbon tetrasubstituted alkenes in terms of reactivity and enantiofacial control, this strategy is much less explored, and known examples are limited to highly active fully substituted alkenes with ring fusion to reduce steric hindrance, such as oxindole-based tetrasubstituted alkenes and cyclobutenones. In addition, the known examples all focused on the use of reagents with both nucleophilic/electrophilic dualistic properties for the difunctionalization of all-carbon tetrasubstituted alkenes, allowing the highly stereoselective construction of polycyclic compounds with multiple contiguous stereogenic centers. This not only exhibits the power of this strategy in constructing complex structures but also suggests great possibilities for further exploration.In 2007, Trost reported an enantioselective Pd-catalyzed [3 + 2] trimethylenemethane (TMM) cycloaddition of alkylidene oxindole 125 using cyano-substituted allyl acetate 126 as the TMM precursor (Scheme 24).78 Under the catalysis of 127/Pd complex, the spirocyclopentane oxindole 128 was obtained in 76% yield, 4.2/1 dr, and 90% ee. It was believed that the initially formed Pd–TMM complex I would equilibrate rapidly to the stabilized species II, which then underwent cycloaddition to the double bond to give the final product.
 | ||
| Scheme 24 [3 + 2] TMM cycloaddition. | ||
In 2015, Zhao reported a tandem Michael–Michael reaction of indolylidene cyanoacetate 129 and nitroalkanes 130 catalyzed by quinidine-derived thiourea 131 to afford spirocyclohexane oxindoles 132 in high yields, up to 95/5 dr and 98% ee (Scheme 25).79 It was proposed the intermolecular attack of nitroalkane 130 from the back onto the Re face of 129 led to intermediate II, which then underwent intramolecular addition to give 132.
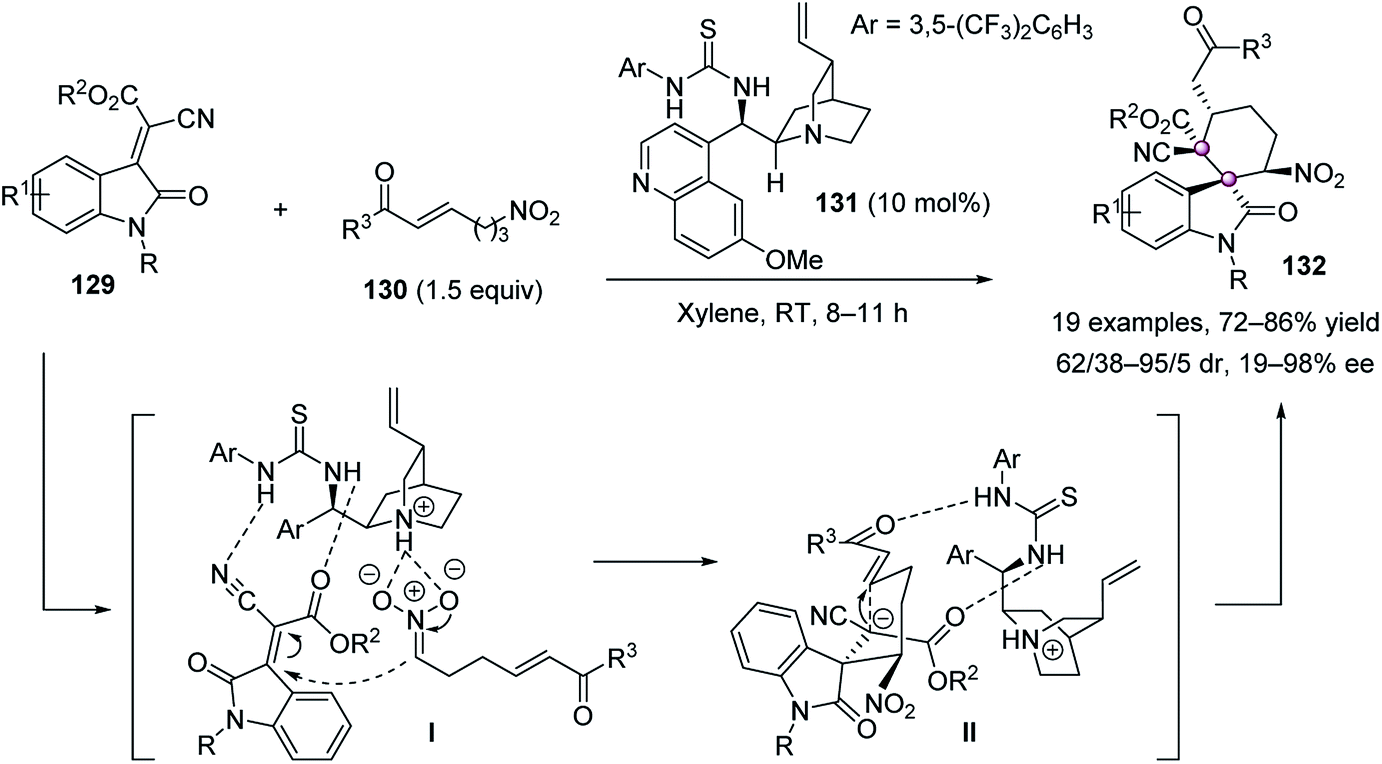 | ||
| Scheme 25 Tandem Michael–Michael reaction. | ||
In 2018, an asymmetric [3 + 2] cycloaddition of azomethine ylides 133 and sterically demanding 2,3-diphenyl cyclobutenone 134 was developed by Adrio and Carretero with a chiral Fesulphos 135/Cu complex as the catalyst, delivering the 3-azabicyclo[3.2.0]heptane 136 in 64% yield and 84% ee (Scheme 26).80 To avoid the steric interaction with the bulky t-Bu group, the cyclobutenone would approach the less hindered face of the tetrahedral chiral Cu complex I, thus affording the desired endo-product in its current configuration.
 | ||
| Scheme 26 [3 + 2] cycloaddition of azomethine ylides and cyclobutenones. | ||
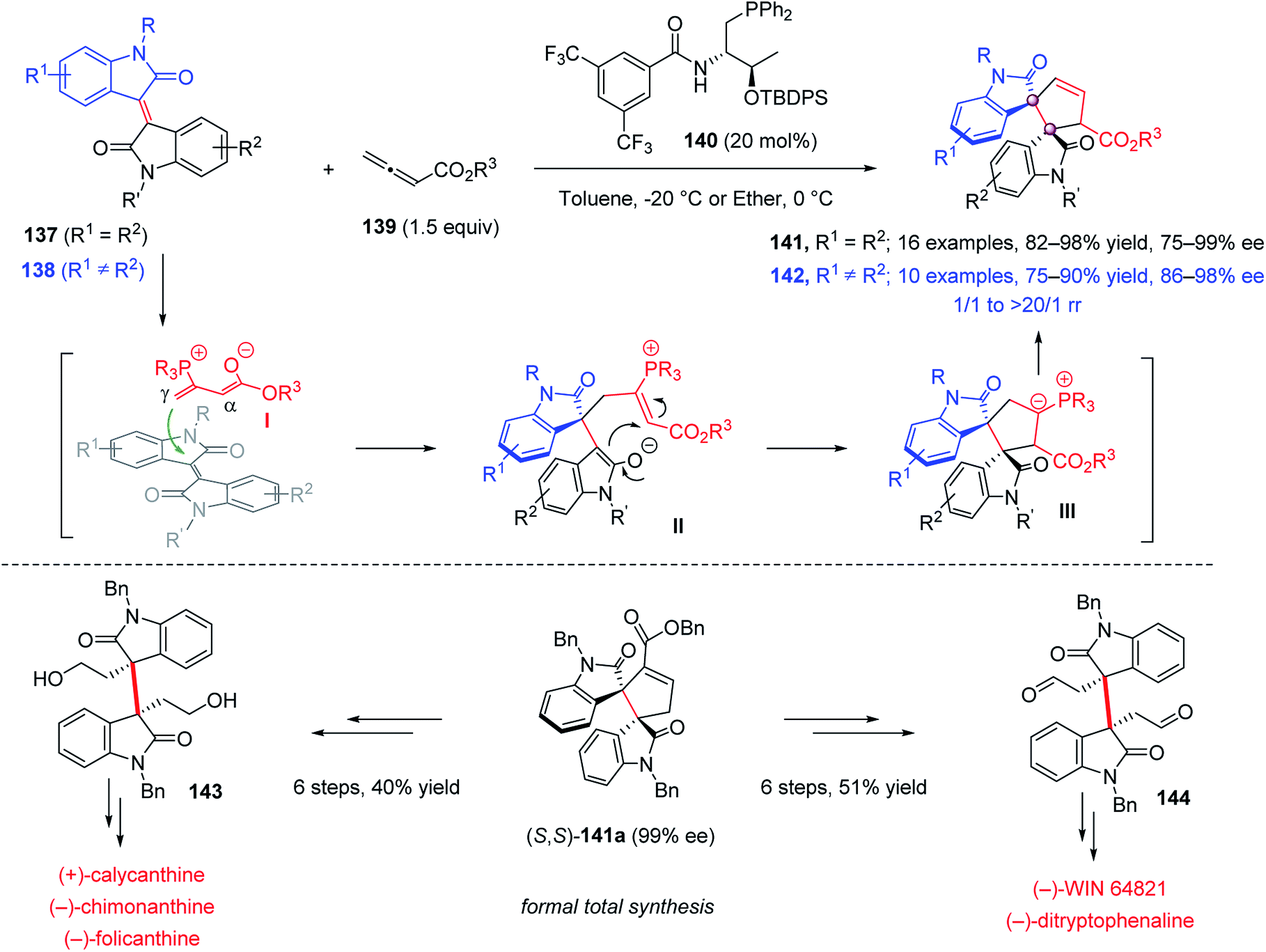
In 2019, Mei and Lu reported a chiral phosphine 140-catalyzed enantioselective [3 + 2] annulation of allenic esters 139 with isoindigos 137, bearing two identical oxindole moieties, for the synthesis of dimeric spirocyclic bisindolines 141 with high yields and excellent ee (Scheme 27).81 The reaction of unsymmetric isoindigos 138 was also performed. Using an electronic/steric-differentiation strategy, the structurally distinct spirocyclic molecules 142 were obtained with high yield and excellent ee. It was proposed that the nucleophilic attack of phosphine 140 on allenes 139 produced the zwitterionic intermediate I, the less hindered γ-attack of which on the structurally encumbered C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond afforded intermediate II. For asymmetric isoindigos, the regioselectivity of this step was governed by the electronic difference between the two oxindole units. The following cyclization and proton transfer gave the final annulation product. The synthetic value of this [3 + 2] annulation was demonstrated by the formal total synthesis of (+)-calycanthine, (−)-chimonanthine, (−)-folicanthine, (−)-WIN64821 and (−)-ditryptophenaline.
C double bond afforded intermediate II. For asymmetric isoindigos, the regioselectivity of this step was governed by the electronic difference between the two oxindole units. The following cyclization and proton transfer gave the final annulation product. The synthetic value of this [3 + 2] annulation was demonstrated by the formal total synthesis of (+)-calycanthine, (−)-chimonanthine, (−)-folicanthine, (−)-WIN64821 and (−)-ditryptophenaline.
 | ||
| Scheme 27 Asymmetric annulation of tetrasubstituted alkenes with allenes. | ||
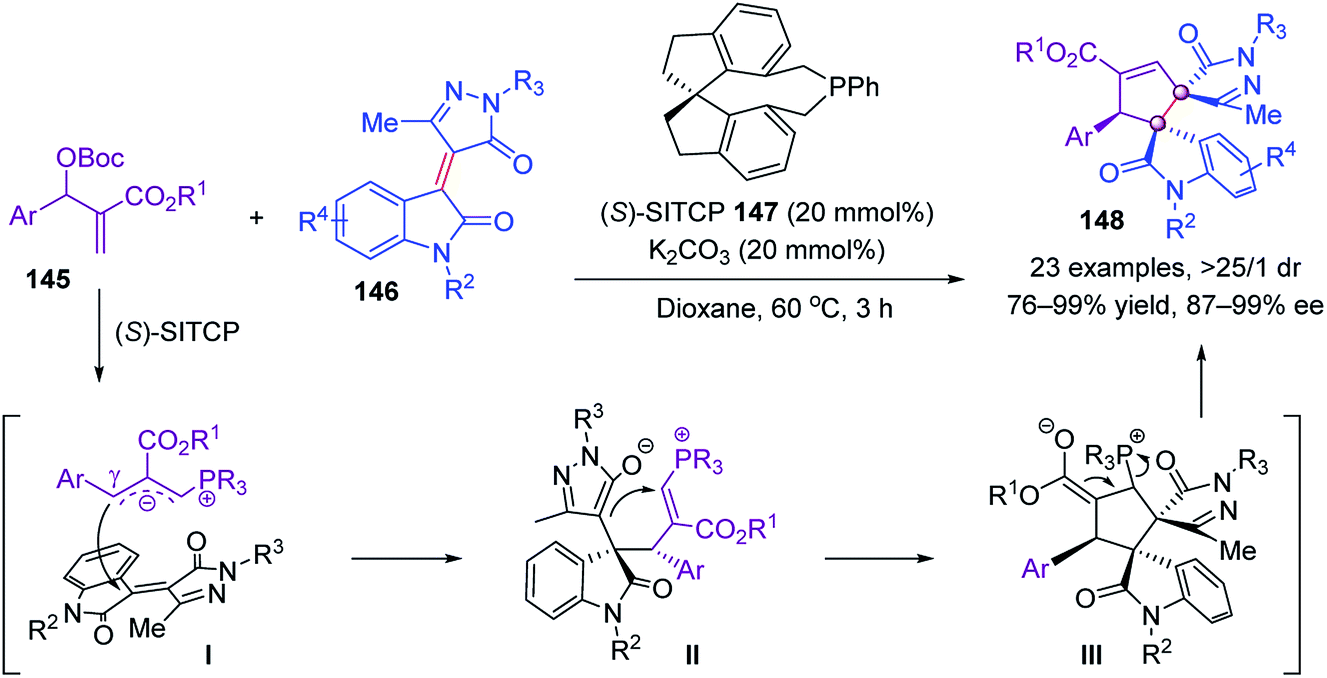
Later, Ullah and Lu further utilized Morita–Baylis–Hillman adducts 145 as C3 synthons to realize another chiral phosphine-catalyzed [3 + 2] annulation with pyrazoloneyldiene oxindoles 146. Under the catalysis of chiral (S)-SITCP 147, bispiro[pyrazolone-3,3′-oxindoles] 148 were obtained in up to 99% yield and 99% ee (Scheme 28).82
 | ||
| Scheme 28 Asymmetric [3 + 2] annulation of pyrazoloneyldiene oxindoles. | ||
4. Sequential modification of vicinal-activated methines
Vicinal trisubstituted carbon nucleophiles such as adjacent methines and enolate equivalents contain two masked adjacent trisubstituted carbanions that can be sequentially released to react with the same or different carbon electrophiles, to furnish vicinal quaternary stereogenic carbons. Because the reaction usually occurred in a stepwise manner, the initial step would produce a chiral quaternary carbon, the matching of which with a chiral catalyst may greatly influence the control of diastereoselectivity in the next step. In addition, when the same carbon electrophile was used to react with symmetric substrates capable of forming two vicinal trisubstituted carbanions, it was possible to generate undesired meso products, which presents another difficulty with this strategy.Dimeric cyclotryptamine alkaloids with vicinal quaternary carbon stereocenters at C3a and C3a′ are present in many plants and animals, showing an array of biological activities.83 The catalytic enantioselective construction of such dimeric structures is difficult due to the lability of the C3a–C3a′ σ-bond84 and the undesired steric hindrance in forging vicinal quaternary stereocenters.85
In 2012, Kanai and Matsunaga achieved an elegant double Michael reaction of bisoxindole 149a with nitroethylene to give quaternary bisoxindole 153 (Scheme 29).86 The chiral 151/Mn(4-F-BzO)2 complex catalyzed the initial Michael addition well, giving monoalkylated adduct 152 in 96% ee; however, its bulky nature prevented it from mediating the next step well, due to the steric hindrance, and side reactions occurred to a severe degree, including the cleavage of the C3a–C3a′ σ-bond. Then, they optimized a sequential procedure that required the removal of the chiral catalyst after the completion of the first step, and a smaller size Mg(OAc)2·4H2O to realize a highly diastereoselective Michael reaction, giving product 153 in 69% yield, >20:1 dr, and 95% ee. Based on chiral synthon 153, the total synthesis of (+)-chimonanthine, (+)-folicanthine, and (−)-calycanthine was realized successfully.
 | ||
| Scheme 29 Sequential Michael reaction. | ||
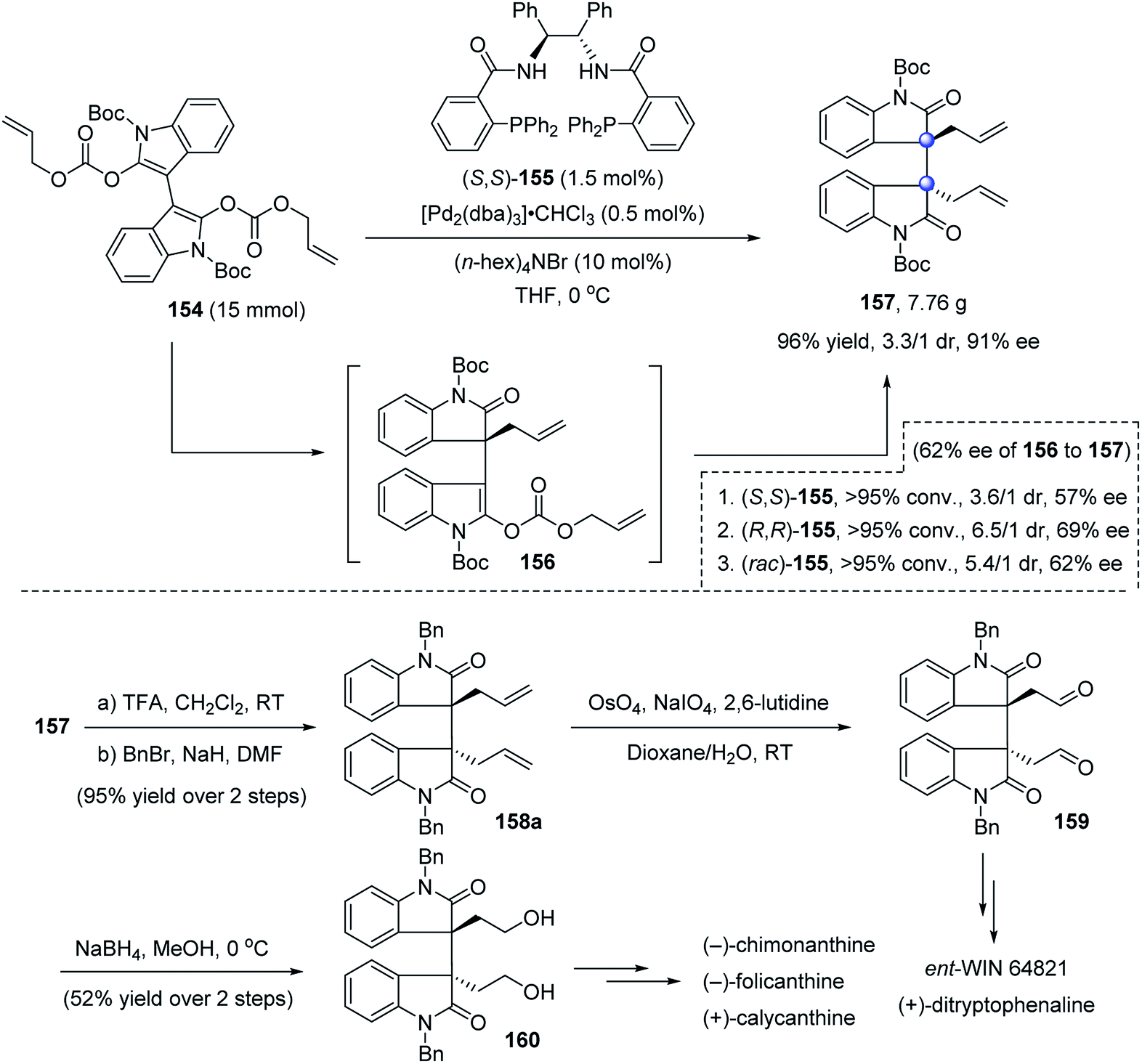
Later, Trost nicely applied decarboxylative asymmetric allylic alkylation (DAAA) to construct such an important bisoxindole structure (Scheme 30).87 In the presence of 0.5 mol% Pd2(dba)3 and 1.5 mol% (S,S)-155, dienol dicarbonate 154 readily converted to the C2-symmetric product 157 on a gram scale in 96% yield, 3.3:1 dr, and 91% ee. The first allylation was the enantiodetermining step, whereas the diastereoselectivity of the second step was more controlled by the substrate than by the ligand, and occurred in a mismatched fashion. The bisallylated oxindole 157 was easily converted to known synthons 159 and 160, enabling the formal total synthesis of cyclotryptamine alkaloids (−)-chimonanthine, (−)-folicanthine, (+)-calycanthine, ent-WIN 64821 and (+)-ditryptophenaline.
 | ||
| Scheme 30 Asymmetric Pd–DAAA of dienol dicarbonate. | ||
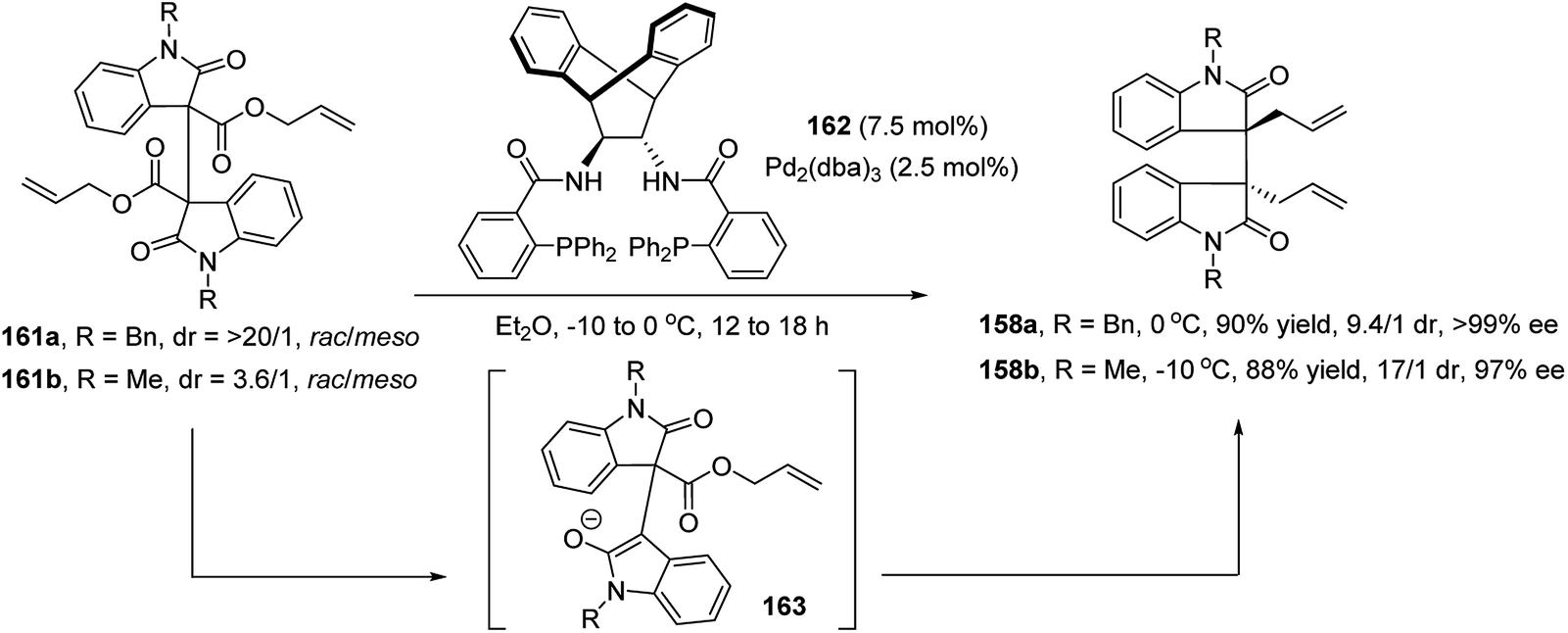
Bisai et al. further developed a similar enantioselective Pd-catalyzed DAAA of dimeric 2-oxindole esters 161 (Scheme 31).88 Trost ligand 162 in combination with Pd2(dba)3 catalyzed the reaction well to afford the N-benzyl and N-methyl protected products 158a and 158b in a high to excellent yield and dr value, with excellent enantioselectivity.
 | ||
| Scheme 31 Asymmetric Pd–DAAA of dimeric 2-oxindole esters. | ||
The above protocols allow facile access to the homo-type alkaloids, but were impotent for hetero-type difunctionalization, which requires the matching of the reactivity and stereocontrol of two different electrophiles. To address this challenge, Tu et al. developed a novel spirocyclic amide–triazolium salt 165 as a remarkable phase-transfer catalyst accomplishing the enantioselective homo- and heterodialkylation of bisoxindoles 149 (Scheme 32).89 In the homodialkylation reaction, benzyl bromoacetates, alkyl bromoacetate and allylic bromides were all viable electrophiles which gave the corresponding homodialkylation products 166 in 41–89% yield, 2.1/1 to >20/1 dr, and 46–99% ee. Notably, this protocol could be extended to a one-pot heterodialkylation. The second electrophile 164′ was added after the completion of the initial monoalkylation using electrophile 164, affording the expected heteroproducts 168 in up to 72% yield, 10.7/1 dr, and 96% ee. The H-bonding between the catalyst amide N–H bond and the enolate intermediate, and the ion-pairing interaction as well as the steric hindrance of the adamantyl group of the chiral catalyst, directed the Re-face nucleophilic attack of the enolate intermediate on electrophiles to afford (S, S)-166 or 168. This elegant protocol paved the way to the formal synthesis of (−)-folicanthine and the first catalytic enantioselective total synthesis of (−)-chimonanthidine.
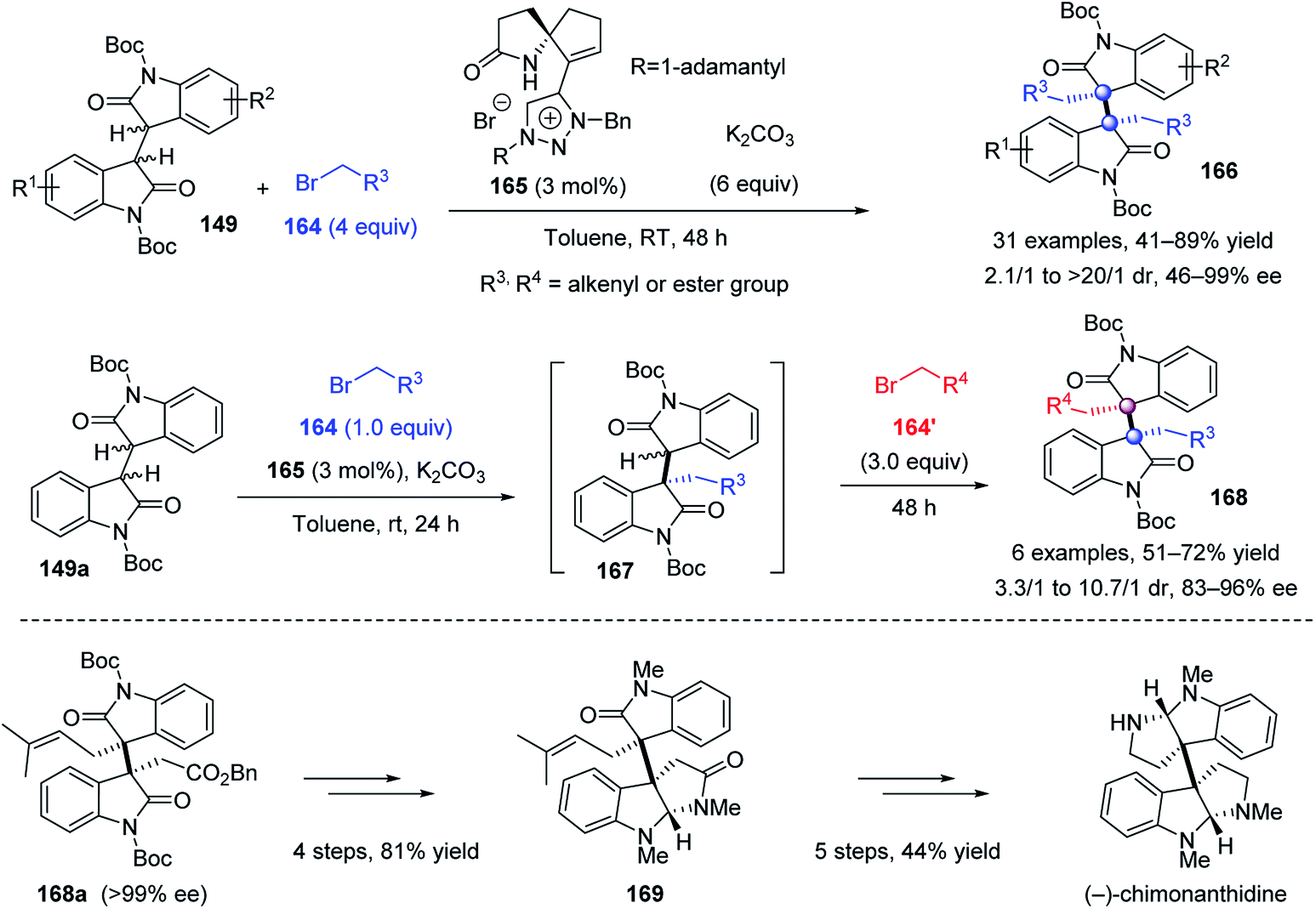 | ||
| Scheme 32 Asymmetric homo- and heterodialkylation of bisoxindoles. | ||
5. The desymmetrization of meso compounds
The catalytic enantioselective desymmetrization of meso compounds with adjacent quaternary prochiral carbons is a powerful strategy to forge vicinal quaternary carbon stereocenters. This strategy has some unique features. First, an important merit is that it can alleviate steric repulsion because the reaction takes place at the tethered functionality that is at least one covalent bond away from the two existing quaternary carbons. However, this also raises the difficulty in the control of stereoselectivities because the chiral environment of the catalyst is farther away from the reaction site. It takes “remote control” to realize excellent stereoselectivities. Second, in principle, all types of catalytic reactions could be utilized to construct vicinal quaternary carbon stereocenters because no matter what kind of reaction takes place at one of the two identical functional groups attached to the preexisting adjacent quaternary carbons, vicinal quaternary stereocenters formed simultaneously. By this strategy, some elegant protocols have been developed successfully.Early in 2002, during the total synthesis of quadrigemine C and psycholeine, Overman et al. showed an elegant desymmetrizing double Heck cyclization to forge the contiguous quaternary stereocenters of the C1-symmetric dioxindole framework (Scheme 33). Under the catalysis of stoichiometric amounts of the chiral (R)-Tol-BINAP/Pd(OAc)2 complex, the desymmetric double Heck cyclization of meso-dibutenanilide 170 afforded C1-symmetric dioxindole 171 in 62% yield and 90% ee, along with the formation of 21% meso-isomer.90 The central contiguous quaternary center and the two peripheral quaternary centers were constructed simultaneously, paving way to the alkaloids quadrigemine C and psycholeine.
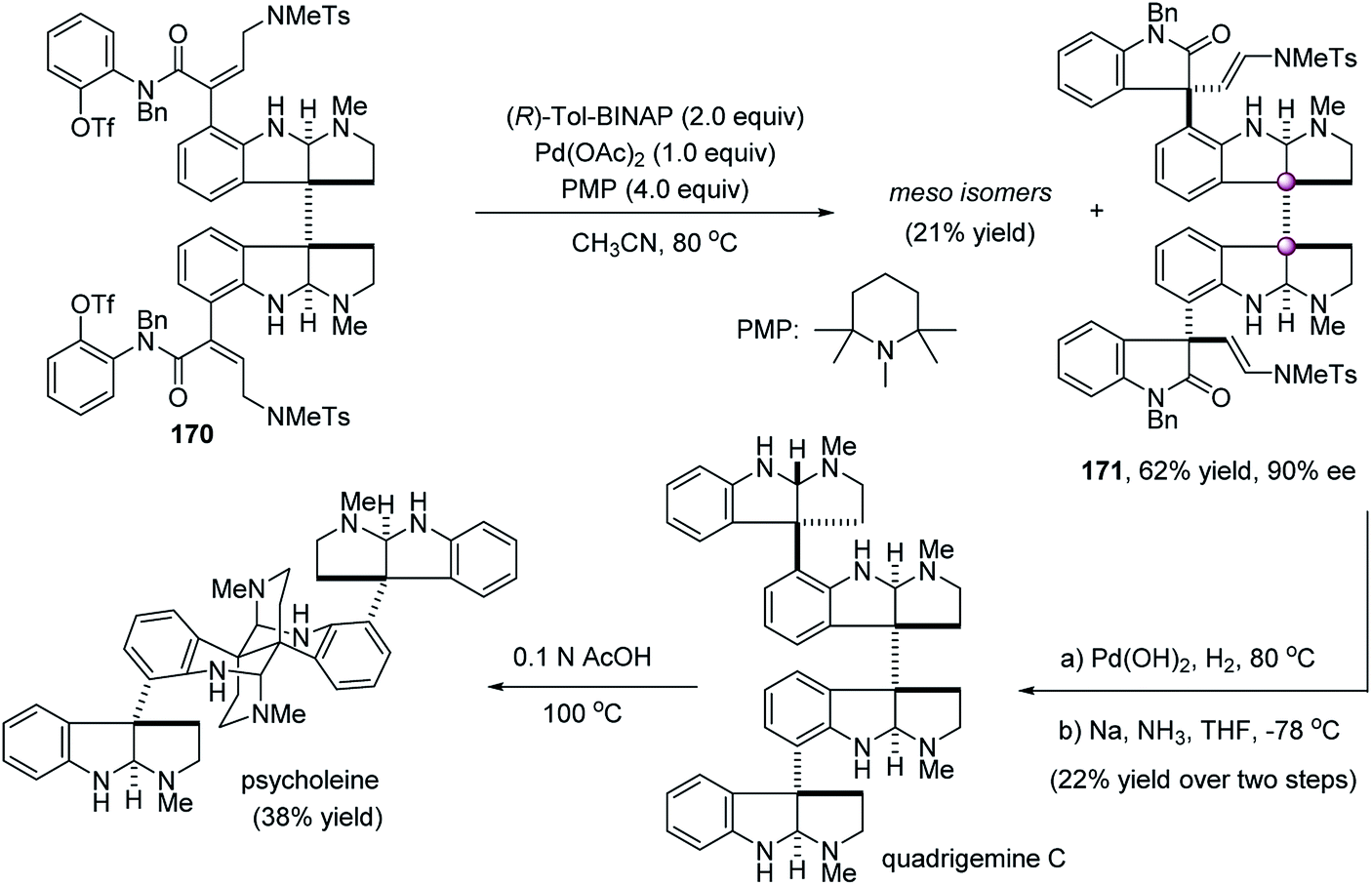 | ||
| Scheme 33 Desymmetric double Heck cyclization. | ||
In 2011, Willis et al. developed a catalytic asymmetric desymmetrization of meso-chimonanthine 172 by means of Trost allylation, enabling the enantioselective total synthesis of hodgkinsine B (Scheme 34).91 The use of a chiral Pd catalyst derived from 173 allowed access to N-allylchimonanthine 174 on a gram scale in high yield with >99% ee. The high efficiency of this reaction was very impressive because meso-chimonanthine 172 was probably the most complex N-nucleophile used in Trost allylation. With dimeric hexahydropyrroloindole 174 as the key intermediate, the total synthesis of hodgkinsine B was accomplished in 14.9% yield over nine steps.
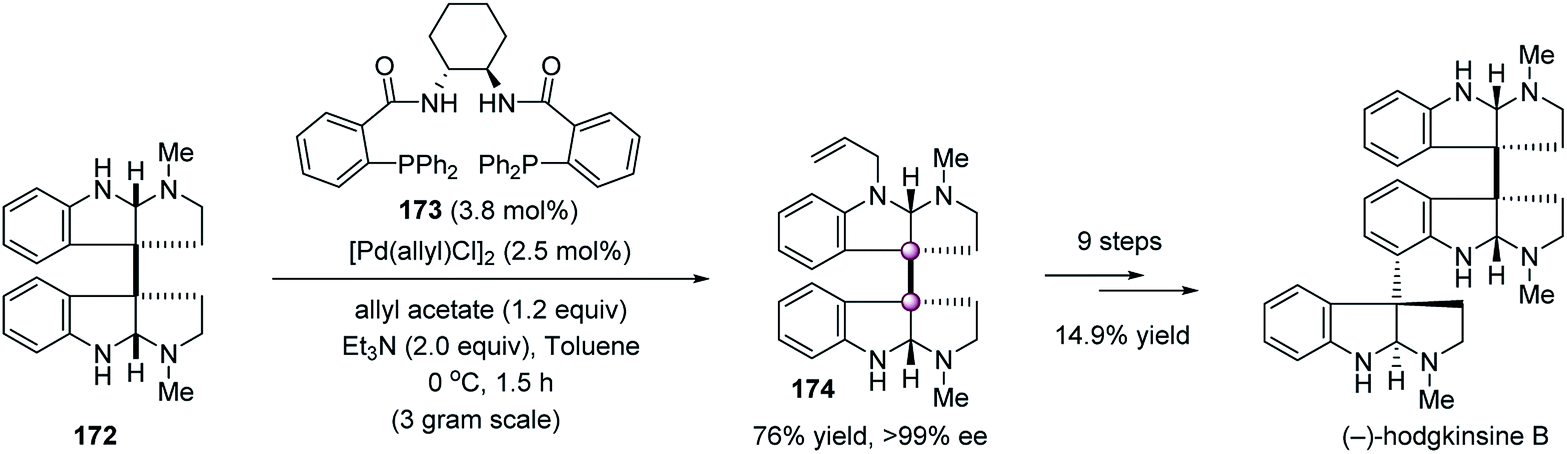 | ||
| Scheme 34 Desymmetric Trost allylation. | ||
In 2009, Stoltz and co-workers first attempted using a catalytic amount of a chiral catalyst to desymmetrize meso compounds to construct vicinal quaternary carbon stereocenters in an effort to construct the challenging carbocyclic core of zoanthenol (Scheme 35).92 They found that in the presence of 10 mol% quinine and 1.0 equiv. pempidine, meso-anhydride 175 reacted with MeOH at −50 °C to afford the half-ester 176 in 88% yield and 70% ee. The key building block half-ester 176 enabled access to the carbocyclic core skeleton of zoanthenol177 in 5.4% yield over 13 steps.
 | ||
| Scheme 35 Desymmetric methanolysis reaction. | ||
Grubbs et al. exploited a novel homochiral stereogenic-at-Ru complex 180 as a powerful catalyst for the Z-selective enantioselective ring-opening/cross-metathesis. They also tried using 1 mol% 180 to desymmetrize norbornene anhydride 178 with allyl acetate 179, affording bicyclic lactone 181 in 65% yield, 96/4 Z/E ratio, and 95% ee (Scheme 36).93 The initial cross-metathesis of 180 with allyl acetate 179 gave the ruthenium methylidene I that underwent the enantioselective ring-opening reaction with the norbornene 178 to give intermediate II. Subsequent cross-metathesis of II with allyl acetate 179 gave the final product 181. It was proposed that the rigidity imparted by the Ru–C chelate was responsible for the high Z selectivity and enantioselectivity.
 | ||
| Scheme 36 Desymmetric ring-opening/cross-metathesis reaction. | ||
Despite intensive studies on enantioselective desymmetrization of diols or triols, the first desymmetrization of tetrols was published as recently as 2017, when Zheng et al. reported a chiral phosphoric acid-mediated oxidative cleavage of benzylidene acetals 182 (Scheme 37).94 Under the catalysis of phosphoric acid 183, the desymmetrization of 182 worked well with 3,3-dimethyldioxirane (DMDO) to give products 184 in 87–96% yield, 4/1–18/1 dr, and >99% ee. It was believed that phosphoric acid 183 served as a proton shuttle to mediate the rate- and enantiodetermining DMDO oxidation step. Meanwhile, the interactions between the PMP group of the substrate and the catalyst were essential to obtain high enantioselectivity.
 | ||
| Scheme 37 Desymmetrization of tetrols. | ||
6. Conclusions
Although the catalytic enantioselective construction of two vicinal quaternary carbon stereocenters is a daunting challenge, remarkable achievements have been made in the past two decades. A series of elegant protocols have been exploited based on four major synthetic strategies as introduced. Some methods have demonstrated their value in facilitating the enantioselective total synthesis of natural products. Research in this field also gives an impetus for the development of new chiral catalysts and synthetic strategies.Despite significant progress, this research is still at its early stage, full of opportunities for future development. First, the discovery of new reactions is the primary task. Although four strategies have been developed for the catalytic enantioselective construction of vicinal quaternary carbon stereocenters, only the strategy using trisubstituted carbon nucleophiles and electrophiles has met with relatively more successful examples, and the other three strategies are less investigated, with only limited examples reported. Second, known protocols have ample room for improvement in terms of stereoselectivities and substrate scope. Many reported reactions are limited in terms of substrate diversity, with only one or several substrates tested. For example, the enantioselective sequential modification of vicinal-activated methines is restricted to active bisoxindole scaffolds, and homo-type reactions dominate, with only one hetero-type reaction known (Scheme 32). In addition, except for the case of the oxidative homocoupling reaction of aldehydes (Scheme 22) and the desymmetrization of tetrols (Scheme 37), the construction of vicinal stereocenters in an acyclic system remains a particularly difficult challenge. Therefore, the development of new reactions using different types of substrates with sufficient structural diversity would be a promising direction. Third, enzyme catalysis has not found its utility in this challenging task, and it is worthwhile to use this powerful tool to realize reactivity and stereoselectivities unattainable by chiral metal catalysis or organocatalysis, which should be a direction that needs to be explored. Nevertheless, as can be expected, with the emergence of new chiral catalysts, more and more efficient enantioselective reactions will be exploited to construct adjacent quaternary carbon stereocenters, and find wide application in natural product and drug synthesis.
Conflicts of interest
The authors declare no competing interests.Acknowledgements
The financial support from the National Natural Science Foundation of China (21725203, 21871090, 21774029), the Ministry of Education (PCSIRT) and the Fundamental Research Funds for the Central Universities is highly appreciated.Notes and references
- (a) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921–2944 CrossRef CAS PubMed; (b) A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945–2963 CrossRef CAS PubMed; (c) K. C. Nicolaou and S. A. Snyder, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11929–11936 CrossRef CAS PubMed; (d) J. Christoffers and A. Baro, Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis, Wiley-VCH, Weinheim, 2005 Search PubMed; (e) K.-S. Yeung and I. Paterson, Chem. Rev., 2005, 105, 4237–4313 CrossRef CAS PubMed; (f) T. Busch and A. Kirschning, Nat. Prod. Rep., 2008, 25, 318–341 RSC; (g) Z.-L. Song, C.-A. Fan and Y.-Q. Tu, Chem. Rev., 2011, 111, 7523–7556 CrossRef CAS PubMed; (h) A. Bauer and M. Bronstrup, Nat. Prod. Rep., 2014, 31, 35–60 RSC.
- K. W. Quasdorf and L. E. Overman, Nature, 2014, 516, 181–191 CrossRef CAS PubMed.
- E. A. Peterson and L. E. Overman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11943–11948 CrossRef CAS PubMed.
- For reviews on the construction of vicinal all–carbon quaternary stereocenters in natural product synthesis: (a) A. Steven and L. E. Overman, Angew. Chem., Int. Ed., 2007, 46, 5488–5508 CrossRef CAS PubMed; (b) M. Büschleb, S. Dorich, S. Hanessian, D. Tao, K. B. Schenthal and L. E. Overman, Angew. Chem., Int. Ed., 2016, 55, 4156–4186 CrossRef PubMed; (c) R. Long, J. Huang, J. Gong and Z. Yang, Nat. Prod. Rep., 2015, 32, 1584–1601 CAS.
- (a) R. M. Lemieux and A. I. Meyers, J. Am. Chem. Soc., 1998, 120, 5453–5457 CrossRef CAS; (b) M. Movassaghi and M. A. Schmidt, Angew. Chem., Int. Ed., 2007, 46, 3725–3728 CrossRef CAS PubMed; (c) L. E. Overman and E. A. Peterson, Angew. Chem., Int. Ed., 2003, 42, 2525–2528 CrossRef CAS PubMed; (d) L. E. Overman and D. V. Paone, J. Am. Chem. Soc., 2001, 123(38), 9465–9467 CAS; (e) A. D. Lebsack, J. T. Link, L. E. Overman and B. A. Stearns, J. Am. Chem. Soc., 2002, 124, 9008–9009 CrossRef CAS PubMed; (f) J. Kim and M. Movassaghi, J. Am. Chem. Soc., 2010, 132, 14376–14378 CrossRef CAS PubMed.
- (a) A. B. Smith III, T. Sunazuka, T. L. Leenay and J. Kingery-Wood, J. Am. Chem. Soc., 1990, 112, 8197–8198 CrossRef; (b) A. B. Smith, N. Kanoh, H. Ishiyama and R. A. Hartz, J. Am. Chem. Soc., 2000, 122, 11254–11255 CrossRef CAS; (c) R. J. Sharpe and J. S. Johnson, J. Am. Chem. Soc., 2015, 137, 4968–4971 CrossRef CAS PubMed; (d) T. Teranishi, T. Murokawa, M. Enomoto and S. Kuwahara, Biosci., Biotechnol., Biochem., 2015, 79, 11–15 CrossRef CAS PubMed; (e) E. J. Corey and S. Lin, J. Am. Chem. Soc., 1996, 118, 8765–8766 CrossRef CAS.
- (a) R. A. Holton, R. M. Kennedy, H. B. Kim and M. E. Krafft, J. Am. Chem. Soc., 1987, 109, 1597–1600 CrossRef CAS; (b) A. Toró, P. Nowak and P. Deslongchamps, J. Am. Chem. Soc., 2000, 122, 4526–4527 CrossRef; (c) L. E. Overman, D. J. Ricca and V. D. Tran, J. Am. Chem. Soc., 1993, 115, 2042–2044 CrossRef CAS; (d) Y. Takahashi, F. Yoshimura, K. Tanino and M. Miyashita, Angew. Chem., Int. Ed., 2009, 48, 8905–8908 CrossRef CAS PubMed; (e) C. P. Ting and T. J. Maimone, J. Am. Chem. Soc., 2015, 137, 10516–10519 CrossRef CAS PubMed; (f) A. K. Miller and D. Trauner, Angew. Chem., Int. Ed., 2003, 42, 549–552 CrossRef CAS PubMed.
- (a) C. He, C. Zhu, Z. Dai, C. -C. Tseng and H. Ding, Angew. Chem., Int. Ed., 2013, 52, 13256–13260 CrossRef CAS PubMed; (b) B. Bradshaw, G. Etxebarria-Jardí and J. Bonjoch, J. Am. Chem. Soc., 2010, 132, 5966–5967 CrossRef CAS PubMed; (c) M. Bian, Z. Wang, X. Xiong, Y. Sun, C. Matera, K. C. Nicolaou and A. Li, J. Am. Chem. Soc., 2012, 134, 8078–8081 CrossRef CAS PubMed; (d) M. A. Corsello, J. Kim and N. K. Garg, Nat. Chem., 2017, 9, 944–949 CrossRef CAS PubMed; (e) V. B. Birman and S. J. Danishefsky, J. Am. Chem. Soc., 2002, 124, 2080–2081 CrossRef CAS PubMed.
- (a) L. A. Paquette, J. Wright, G. J. Drtina and R. A. Roberts, J. Org. Chem., 1987, 52, 2960–2962 CrossRef; (b) J. Belmar and R. L. Funk, J. Am. Chem. Soc., 2012, 134, 16941–16943 CrossRef CAS PubMed; (c) D. Sun, C. Xing, X. Wang, Z. Su and C. Li, Org. Chem. Front., 2014, 1, 956–960 RSC; (d) J. A. Stafford and C. H. Heathcock, J. Org. Chem., 1990, 55, 5433–5434 CrossRef CAS.
- (a) T. Kang, S. B. Song, W.-Y. Kim, B. G. Kim and H.-Y. Lee, J. Am. Chem. Soc., 2014, 136, 10274–10276 CrossRef CAS PubMed; (b) T. Gaich and J. Mulzer, J. Am. Chem. Soc., 2009, 131, 452–453 CrossRef CAS PubMed; (c) M. T. Crimmins, J. M. Pace, P. G. Nantermet, A. S. Kim-Meade, J. B. Thomas, S. H. Watterson and A. S. Wagman, J. Am. Chem. Soc., 2000, 122, 8453–8463 CrossRef CAS; (d) T. Magauer, J. Mulzer and K. Tiefenbacher, Org. Lett., 2009, 11, 5306–5309 CrossRef CAS PubMed; (e) M. T. Crimmins, D. K. Jung and J. L. Gray, J. Am. Chem. Soc., 1993, 115, 3146–3155 CrossRef CAS; (f) D. W. Brooks, P. G. Grothaus and H. Mazdiyasni, J. Am. Chem. Soc., 1983, 105, 4472–4473 CrossRef CAS.
- (a) L. E. Overman, D. V. Paone and B. A. Stearns, J. Am. Chem. Soc., 1999, 121, 7702–7703 CrossRef CAS; (b) K. C. Nicolaou, G. Vassilikogiannakis, W. Mägerlein and R. Kranich, Angew. Chem., Int. Ed., 2001, 40, 2482–2486 CrossRef CAS; (c) C. Liu, R. Chen, Y. Shen, Z. Liang, Y. Hua and Y. Zhang, Angew. Chem., Int. Ed., 2017, 56, 8187–8190 CrossRef CAS PubMed; (d) F.-X. Wang, J.-Y. Du, H.-B. Wang, P.-L. Zhang, G.-B. Zhang, K.-Y. Yu, X.-Z. Zhang, X.-T. An, Y.-X. Cao and C.-A. Fan, J. Am. Chem. Soc., 2017, 139, 4282–4285 CrossRef CAS PubMed; (e) S. M. King, N. A. Calandra and S. B. Herzon, Angew. Chem., Int. Ed., 2013, 52, 3642–3645 CrossRef CAS PubMed; (f) N. Hartrampf, N. Winter, G. Pupo, B. M. Stoltz and D. Trauner, J. Am. Chem. Soc., 2018, 140, 8675–8680 CrossRef CAS PubMed; (g) R. Long, J. Huang, W. Shao, S. Liu, Y. Lan, J. Gong and Z. Yang, Nat. Commun., 2014, 5, 5707 CrossRef CAS PubMed.
- (a) P. A. Wender, T. W. Von Geldern and B. H. Levine, J. Am. Chem. Soc., 1988, 110, 4858–4860 CrossRef CAS; (b) S. Levin, R. R. Nani and S. E. Reisman, J. Am. Chem. Soc., 2011, 133, 774–776 CrossRef CAS PubMed; (c) S. Chiba, M. Kitamura and K. Narasaka, J. Am. Chem. Soc., 2006, 128, 6931–6937 CrossRef CAS PubMed; (d) J. Deng, S. Zhou, W. Zhang, J. Li, R. Li and A. Li, J. Am. Chem. Soc., 2014, 136, 8185–8188 CrossRef CAS PubMed; (e) M. Ali, Phytochemistry, 1992, 31, 2173–2175 CrossRef CAS; (f) R. E. Johnson, M. A. Chutuape, E. C. Strain, S. L. Walsh, M. L. Stitzer and G. E. Bigelow, N. Engl. J. Med., 2000, 343, 1290–1297 CrossRef CAS PubMed.
- (a) I. Sharma and D. S. Tan, Nat. Chem., 2013, 5, 157–158 CrossRef CAS PubMed; (b) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed; (c) A. Harvey, Drug Discovery Today, 2000, 5, 294–300 CrossRef PubMed; (d) P. Hu, H. M. Chi, K. C. DeBacker, X. Gong, J. H. Keim, I. T. Hsu and S. A. Snyder, Nature, 2019, 569, 703–707 CrossRef CAS PubMed.
- C. J. Douglas and L. E. Overman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5363–5367 CrossRef CAS PubMed.
- (a) J. T. Mohr, M. R. Krout and B. M. Stoltz, Nature, 2008, 455, 323–332 CrossRef CAS PubMed; (b) Y. Liu, S.-J. Han, W.-B. Liu and B. M. Stoltz, Acc. Chem. Res., 2015, 48, 740–751 CrossRef CAS PubMed; (c) A. Y. Hong and B. M. Stoltz, Eur. J. Org. Chem., 2013, 2745–2759 CrossRef CAS PubMed.
- (a) I. Marek, Y. Minko, M. Pasco, T. Mejuch, N. Gilboa, H. Chechik and J. P. Das, J. Am. Chem. Soc., 2014, 136, 2682–2694 CrossRef CAS PubMed; (b) D. Pierrot and I. Marek, Angew. Chem., Int. Ed., 2020, 59, 36–49 CrossRef CAS PubMed.
- C. Li, S. S. Ragab, G. Liu and W. Tang, Nat. Prod. Rep., 2020, 37, 276–292 RSC.
- Y. Ping, Y. Li, J. Zhu and W. Kong, Angew. Chem., Int. Ed., 2019, 58, 1562–1573 CrossRef CAS PubMed.
- (a) X.-P. Zeng, Z.-Y. Cao, Y.-H. Wang, F. Zhou and J. Zhou, Chem. Rev., 2016, 116, 7330–7396 CrossRef CAS PubMed; (b) P.-W. Xu, J.-S. Yu, C. Chen, Z.-Y. Cao, F. Zhou and J. Zhou, ACS Catal., 2019, 9, 1820–1882 CrossRef CAS.
- J. Feng, M. Holmes and M. J. Krische, Chem. Rev., 2017, 117, 12564–12580 CrossRef CAS PubMed.
- T. Ling and F. Rivas, Tetrahedron, 2016, 72, 6729–6777 CrossRef CAS.
- C. Hawner and A. Alexakis, Chem. Commun., 2010, 46, 7295–7306 RSC.
- For other early reviews: (a) B. M. Trost and C. Jiang, Synthesis, 2006, 369–396 CrossRef CAS; (b) J. Christoffers and A. Mann, Angew. Chem., Int. Ed., 2001, 40, 4591–4597 CrossRef CAS; (c) E. J. Corey and A. Guzman–Perez, Angew. Chem., Int. Ed., 1998, 37, 388–401 CrossRef.
- F. Serratosa, The Design of Organic Synthesis, Elsevier, Amsterdam, 1990 Search PubMed.
- H. Nozaki, S. Moriuti, H. Takaya and R. Noyori, Tetrahedron Lett, 1966, 7, 5239–5244 CrossRef.
- M. P. Doyle, Q.-L. Zhou, C. Charnsangavej and M. A. Longoria, Tetrahedron Lett, 1996, 37, 4129–4132 CrossRef CAS.
- A. DeAngelis, O. Dmitrenko, G. P. A. Yap and J. M. Fox, J. Am. Chem. Soc., 2009, 131, 7230–7231 CrossRef CAS PubMed.
- (a) D. F. Taber, R. J. Herr, S. K. Pack and J. M. Geremia, J. Org. Chem., 1996, 61, 2908–2910 CrossRef CAS PubMed; (b) D. F. Taber and P. V. Joshi, J. Org. Chem., 2004, 69, 4276–4278 CAS.
- K. Minami, H. Saito, H. Tsutsui, H. Nambu, M. Anada and S. Hashimoto, Adv. Synth. Catal., 2005, 347, 1483–1487 CrossRef CAS.
- X. Xu, S. Zhu, X. Cui, L. Wojtas and X. P. Zhang, Angew. Chem., Int. Ed., 2013, 52, 11857–11861 CrossRef CAS PubMed.
- For mechanism study, see: (a) H. Lu, W. I. Dzik, X. Xu, L. Wojtas, B. de Bruin and X. P. Zhang, J. Am. Chem. Soc., 2011, 133, 8518–8521 CrossRef CAS PubMed; (b) J. L. Belof, C. R. Cioce, X. Xu, X. P. Zhang, B. Space and H. L. Woodcock, Organometallics, 2011, 30, 2739–2746 CrossRef CAS PubMed; (c) W. I. Dzik, X. Xu, X. P. Zhang, J. N. H. Reek and B. de Bruin, J. Am. Chem. Soc., 2010, 132, 10891–10902 CrossRef CAS PubMed.
- Z.-Y. Cao, F. Zhou and J. Zhou, Acc. Chem. Res., 2018, 51, 1443–1454 CrossRef CAS PubMed.
- Z.-Y. Cao, F. Zhou, Y.-H. Yu and J. Zhou, Org. Lett., 2013, 15, 42–45 CrossRef CAS PubMed.
- (a) X. Wang, Z. Han, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2012, 51, 936–940 CrossRef CAS PubMed; (b) X. Wang, F. Meng, Y. Wang, Z. Han, Y. Chen, L. Liu, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2012, 51, 9276–9282 CrossRef CAS.
- Z.-Y. Cao, X. Wang, C. Tan, X.-L. Zhao, J. Zhou and K. Ding, J. Am. Chem. Soc., 2013, 135, 8197–8200 CrossRef CAS.
- Y. Chi, L. Qiu and X. Xu, Org. Biomol. Chem., 2016, 14, 10357–10361 RSC.
- L. Gao, G.-S. Hwang and D. H. Ryu, J. Am. Chem. Soc., 2011, 133, 20708–20711 CrossRef CAS PubMed.
- P. Zhao, S. Wu, C. Ke, X. Liu and X. Feng, Chem. Commun., 2018, 54, 9837–9840 RSC.
- M. Bos, W.-S. Huang, T. Poisson, X. Pannecoucke, A. B. Charette and P. Jubault, Angew. Chem., Int. Ed., 2017, 56, 13319–13323 CrossRef CAS PubMed.
- W.-S. Huang, C. Schlinquer, T. Poisson, X. Pannecoucke, A. B. Charette and P. Jubault, Chem. Eur. J., 2018, 24, 10339–10343 CrossRef CAS PubMed.
- Z.-Y. Cao, W. Wang, K. Liao, X. Wang, J. Zhou and J. Ma, Org. Chem. Front., 2018, 5, 2960–2968 RSC.
- For a review on the influence of these subtle interactions on the reactivity and selectivity of organic reactions, see: Y.-J. Hao, J.-S. Yu, Y. Zhou, X. Wang and J. Zhou, Acta Chim. Sinica, 2018, 76, 925–939 CrossRef CAS.
- X. Xu, H. Lu, J. V. Ruppel, X. Cui, S. L. de Mesa, L. Wojtas and X. P. Zhang, J. Am. Chem. Soc., 2011, 133, 15292–15295 CrossRef CAS PubMed.
- J.-J. Shen, S.-F. Zhu, Y. Cai, H. Xu, X.-L. Xie and Q.-L. Zhou, Angew. Chem., Int. Ed., 2014, 53, 13188–13191 CrossRef CAS PubMed.
- H. Xu, Y.-P. Li, Y. Cai, G.-P. Wang, S.-F. Zhu and Q.-L. Zhou, J. Am. Chem. Soc., 2017, 139, 7697–7700 CrossRef CAS PubMed.
- T. Shibata, Y. Kobayashi, S. Maekawa, N. Toshida and K. Takagi, Tetrahedron, 2005, 61, 9018–9024 CrossRef CAS.
- (a) D. Brissy, M. Skander, H. Jullien, P. Retailleau and A. Marinetti, Org. Lett., 2009, 11, 2137–2139 CrossRef CAS PubMed; (b) H. Jullien, D. Brissy, R. Sylvain, P. Retailleau, J.-V. Naubron, S. Gladiali and A. Marinetti, Adv. Synth. Catal., 2011, 353, 1109–1124 CrossRef CAS.
- A. Pradal, C.-M. Chao, P. Y. Toullec and V. Michelet, Beilstein J. Org. Chem., 2011, 7, 1021–1029 CrossRef CAS PubMed.
- T. Nishimura, T. Kawamoto, M. Nagaosa, H. Kumamoto and T. Hayashi, Angew. Chem., Int. Ed., 2010, 49, 1638–1641 CrossRef CAS PubMed.
- T. Nishimura, Y. Maeda and T. Hayashi, Org. Lett., 2011, 13, 3674–3677 CrossRef CAS PubMed.
- H. Teller, M. Corbet, L. Mantilli, G. Gopakumar, R. Goddard, W. Thiel and A. Fürstner, J. Am. Chem. Soc., 2012, 134, 15331–15342 CrossRef CAS PubMed.
- B. M. Trost, M. C. Ryan, M. Rao and T. Z. Markovic, J. Am. Chem. Soc., 2014, 136, 17422–17425 CrossRef CAS PubMed.
- Ò. Torres, A. Roglans and A. Pla-Quintana, Adv. Synth. Catal., 2016, 358, 3512–3516 CrossRef.
- P.-C. Zhang, Y. Wang, Z.-M. Zhang and J. Zhang, Org. Lett., 2018, 20, 7049–7052 CrossRef CAS PubMed.
- A. Noole, N. S. Sucman, M. A. Kabeshov, T. Kanger, F. Z. Macaev and A. V. Malkov, Chem. Eur. J., 2012, 18, 14929–14933 CrossRef CAS PubMed.
- A. Noole, M. Ošeka, T. Pehk, M. Öeren, I. Järving, M. R. J. Elsegood, A. V. Malkov, M. Lopp and T. Kangera, Adv. Synth. Catal., 2013, 355, 829–835 CrossRef CAS.
- H. Mei, G. Pan, X. Zhang, L. Lin, X. Liu and X. Feng, Org. Lett., 2018, 20, 7794–7797 CrossRef CAS PubMed.
- L. Ye, Q.-S. Gu, Y. Tian, X. Meng, G.-C. Chen and X.-Y. Liu, Nature Commun, 2018, 9, 227 CrossRef PubMed.
- J. N. Payette and H. Yamamoto, J. Am. Chem. Soc., 2007, 129, 9536–9537 CrossRef CAS PubMed.
- K. Ohmatsu, N. Imagawa and T. Ooi, Nature Chem, 2013, 6, 47–51 CrossRef PubMed.
- A. Khan, L. Yang, J. Xu, L. Y. Jin and Y. J. Zhang, Angew. Chem., Int. Ed., 2014, 53, 11257–11260 CrossRef CAS PubMed.
- Z.-J. Zhang, L. Zhang, R.-L. Geng, J. Song, X.-H. Chen and L.-Z. Gong, Angew. Chem., Int. Ed., 2019, 58, 12190–12194 CrossRef CAS PubMed.
- X. Huang, T. R. Quinn, K. Harms, R. D. Webster, L. Zhang, O. Wiest and E. Meggers, J. Am. Chem. Soc., 2017, 139, 9120–9123 CrossRef CAS PubMed.
- A. Jolit, P. M. Walleser, G. P. A. Yap and M. A. Tius, Angew. Chem., Int. Ed., 2014, 53, 6180–6183 CrossRef CAS PubMed.
- For reviews: (a) B. M. Trost, Chem. Pharm. Bull., 2002, 50, 1–14 CrossRef CAS PubMed; (b) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921–2943 CrossRef CAS PubMed; (c) B. M. Trost, J. Org. Chem., 2004, 69, 5813–5837 CrossRef CAS PubMed; (d) B. M. Trost, T. Zhang and J. D. Sieber, Chem. Sci., 2010, 1, 427–440 RSC.
- B. M. Trost, S. Malhotra and W. H. Chan, J. Am. Chem. Soc., 2011, 133, 7328–7331 CrossRef CAS PubMed.
- S. Ma, X. Han, S. Krishnan, S. C. Virgil and B. M. Stoltz, Angew. Chem., Int. Ed., 2009, 48, 8037–8041 CrossRef CAS PubMed.
- (a) C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748–8758 CrossRef CAS PubMed; (b) H. Lin and S. J. Danishefsky, Angew. Chem., Int. Ed., 2003, 42, 36–51 CrossRef CAS PubMed; (c) C. Marti and E. M. Carreira, Eur. J. Org. Chem., 2003, 2209–2219 CrossRef CAS.
- The catalytic enantioselective synthesis of 3,3-disubstituted oxindoles is of current interest, see: (a) F. Zhou, Y.-L. Liu and J. Zhou, Adv. Synth. Catal., 2010, 352, 1381–1407 CrossRef CAS; (b) K. Shen, X. Liu, L. Lin and X. Feng, Chem. Sci., 2012, 3, 327–334 RSC; (c) R. Dalpozzo, Adv. Synth. Catal., 2017, 359, 1772–1810 CrossRef CAS; (d) R. Dalpozzo, Org. Chem. Front., 2017, 4, 2063–2078 RSC.
- H. Zhang, L. Hong, H. Kang and R. Wang, J. Am. Chem. Soc., 2013, 135, 14098–14101 CrossRef CAS PubMed.
- J. Zheng, L. Lin, L. Dai, Q. Tang, X. Liu and X. Feng, Angew. Chem., Int. Ed., 2017, 56, 13107–13111 CrossRef CAS PubMed.
- K. Ohmatsu, Y. Ando and T. Ooi, J. Am. Chem. Soc., 2013, 135, 18706–18709 CrossRef CAS PubMed.
- L. Næsborg, L. A. Leth, G. J. Reyes–Rodríguez, T. A. Palazzo, V. Corti and K. A. Jørgensen, Chem. Eur. J., 2018, 24, 14844–14848 CrossRef PubMed.
- For reviews: (a) A. M. M. Castro, Chem. Rev., 2004, 104, 2939–3002 CrossRef CAS PubMed; (b) M. Hiersemann and U. Nubbemeyer, The Claisen Rearrangement, Wiley-VCH, Weinheim, 2007 CrossRef.
- C. Uyeda, A. R. Rötheli and E. N. Jacobsen, Angew. Chem., Int. Ed., 2010, 49, 9753–9756 CrossRef CAS PubMed.
- G. Bencivenni, L.-Y. Wu, A. Mazzanti, B. Giannichi, F. Pesciaioli, M.-P. Song, G. Bartoli and P. Melchiorre, Angew. Chem., Int. Ed., 2009, 48, 7200–7203 CrossRef CAS PubMed.
- X.-L. Liu, Y. Gong, S. Chen, X. Zuo, Z. Yao and Y. Zhou, Org. Chem. Front., 2019, 6, 1603–1607 RSC.
- B. M. Trost, N. Cramer and S. M. Silverman, J. Am. Chem. Soc., 2007, 129, 12396–12397 CrossRef CAS PubMed.
- S. Abbaraju, N. Ramireddy, N. K. Rana, H. Arman and J. C.-G. Zhao, Adv. Synth. Catal., 2015, 357, 2633–2638 CrossRef CAS.
- J. Corpas, A. Ponce, J. Adrio and J. C. Carretero, Org. Lett., 2018, 20, 3179–3182 CrossRef CAS PubMed.
- W.-L. Chan, X. Tang, F. Zhang, G. Quek, G.-J. Mei and Y. Lu, Angew. Chem., Int. Ed., 2019, 58, 6260–6264 CrossRef CAS PubMed.
- J. Zhang, W.-L. Chan, L. Chen, N. Ullah and Y. Lu, Org. Chem. Front., 2019, 6, 2210–2214 RSC.
- M. A. Schmidt and M. Movassaghi, Synlett, 2008, 313–324 CAS.
- J. T. Link and L. E. Overman, J. Am. Chem. Soc., 1996, 118, 8166–8167 CrossRef CAS.
- C. Guo, J. Song, J. Z. Huang, P. H. Chen, S. W. Luo and L. Z. Gong, Angew. Chem., Int. Ed., 2012, 51, 1046–1050 CrossRef CAS PubMed.
- H. Mitsunuma, M. Shibasaki, M. Kanai and S. Matsunaga, Angew. Chem., Int. Ed., 2012, 51, 5217–5221 CrossRef CAS PubMed.
- B. M. Trost and M. Osipov, Angew. Chem., Int. Ed., 2013, 52, 9176–9181 CrossRef CAS PubMed.
- S. Ghosh, S. Bhunia, B. N. Kakde, S. De and A. Bisai, Chem. Commun., 2014, 50, 2434–2437 RSC.
- S.-K. Chen, W.-Q. Ma, Z.-B. Yan, F.-M. Zhang, S.-H. Wang, Y.-Q. Tu, X.-M. Zhang and J.-M. Tian, J. Am. Chem. Soc., 2018, 140, 10099–10103 CrossRef CAS PubMed.
- A. D. Lebsack, J. T. Link, L. E. Overman and B. A. Stearns, J. Am. Chem. Soc., 2002, 124, 9008–9009 CrossRef CAS PubMed.
- R. H. Snell, R. L. Woodward and M. C. Willis, Angew. Chem., Int. Ed., 2011, 50, 9116–9119 CrossRef CAS PubMed.
- J. L. Stockdill, D. C. Behenna, A. McClory and B. M. Stoltz, Tetrahedron, 2009, 65, 6571–6575 CrossRef CAS.
- J. Hartung and R. H. Grubbs, J. Am. Chem. Soc., 2013, 135, 10183–10185 CrossRef CAS PubMed.
- H. Yang, K.-S. Cao and W.-H. Zheng, Chem. Commun., 2017, 53, 3737–3740 RSC.
| This journal is © The Royal Society of Chemistry 2020 |