
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereospecific nucleophilic substitution at tertiary and quaternary stereocentres
Veeranjaneyulu
Lanke
and
Ilan
Marek
 *
*
Schulich Faculty of Chemistry, Technion – Israel Institute of Technology, Technion City 3200009, Haifa, Israel. E-mail: chilanm@technion.ac.il
First published on 28th July 2020
Abstract
Nucleophilic substitution reactions have always been considered as one of the most powerful reactions for the creation of carbon–carbon or carbon–heteroatom bonds in organic synthesis. In contrast to secondary carbons, the steric shielding of tertiary carbons retards a concerted, stereospecific nucleophilic substitution, and ionizing pathways often lead to nonselective substitution due to ion pair dissociation. In this minireview, we will detail pioneering contributions and more recent achievements emphasizing the feasibility of nucleophilic substitution on tertiary stereocentres under certain conditions, with inversion of configuration. The development of these transformations at tertiary centres are of remarkable added value to practitioners in the field of complex molecule synthesis. A stereoselective substitution at a quaternary carbon stereocentre with inversion of configuration is also discussed in the case of a three-membered ring.
1. Introduction
The mechanism of nucleophilic substitution reactions, originally elucidated by Hughes and Ingold,1 are one of the most fundamental and common transformations in organic chemistry that have found countless applications in synthesis.2 Two distinct mechanistic profiles could be considered. In the first scenario, the nucleophile attacks the electrophilic carbon centre from the back side with concurrent departure of the leaving group. This concerted nucleophilic substitution mechanism in which the rate determining step involves the two initial components, exhibits second order kinetics, symbolized as SN2,3 and proceeds with a Walden inversion4 of configuration (Scheme 1a).5 Experimental observations have contributed to the understanding of the displacement and among several parameters, steric hindrance of the substrate and/or nucleophile play a key-role. The more pronounced is the steric hindrance at the electrophilic carbon centre and less efficient is the transformation (Scheme 1b). In contrast, the alternative nucleophilic substitution scenario in which the rate determining step involves unimolecular dissociation of the electrophile, abbreviated as SN1, tends to be important when the central carbon atom of the substrate is surrounded by bulky groups, both because these groups preclude the SN2 mechanism on steric ground but also because a highly substituted carbon centre tends to form a more stable carbenium ion or ion pair intermediates (Scheme 1c), leading to a partial or total loss of stereochemical integrity. | ||
| Scheme 1 | ||
Over the years, the general understanding regarding the stereochemical outcome of nucleophilic substitution slowly became biased and this misconception, relayed in many text-books, implied that nucleophilic substitution at tertiary carbon centres proceed with racemization (with the exception of stereoinvertive epoxides ring-opening as well as nucleophilic substitution on metallocene derivatives and related cases that will not be discussed in this minireview). Original examples as well as recent achievements indicate that, in some cases, substitution at a tertiary stereocentre proceeds with some conservation of the optical purity. In this minireview, we would like first to reformulate the original reports and then discuss the most recent examples emphasizing the potential that these transformations might have in stereoselective synthesis and in the application to natural product synthesis, hoping that it might change the current paradigm.
2. Historical background
The first hint that a tertiary carbinol could be transformed into its chloro-analogue with some transfer of stereochemical information dates back to the pioneering work of Stevens and McNiven in 1939.6 Following this preliminary investigation, the steric course of methanolysis of enantiomerically enriched tertiary alkyl chloride 1 was then investigated by Ingold, Hughes and coworkers.1a Although the absolute configuration of 1 was unknown at that time, the nucleophilic substitution provided 2 with 34% optical activity (Scheme 2). Similarly, Doering and Zeiss7 solvolyzed optically active 3 in refluxing methanol. The reaction proceeded to give the ether 4 in moderate yield but intriguingly with 54% inversion of configuration (Scheme 2).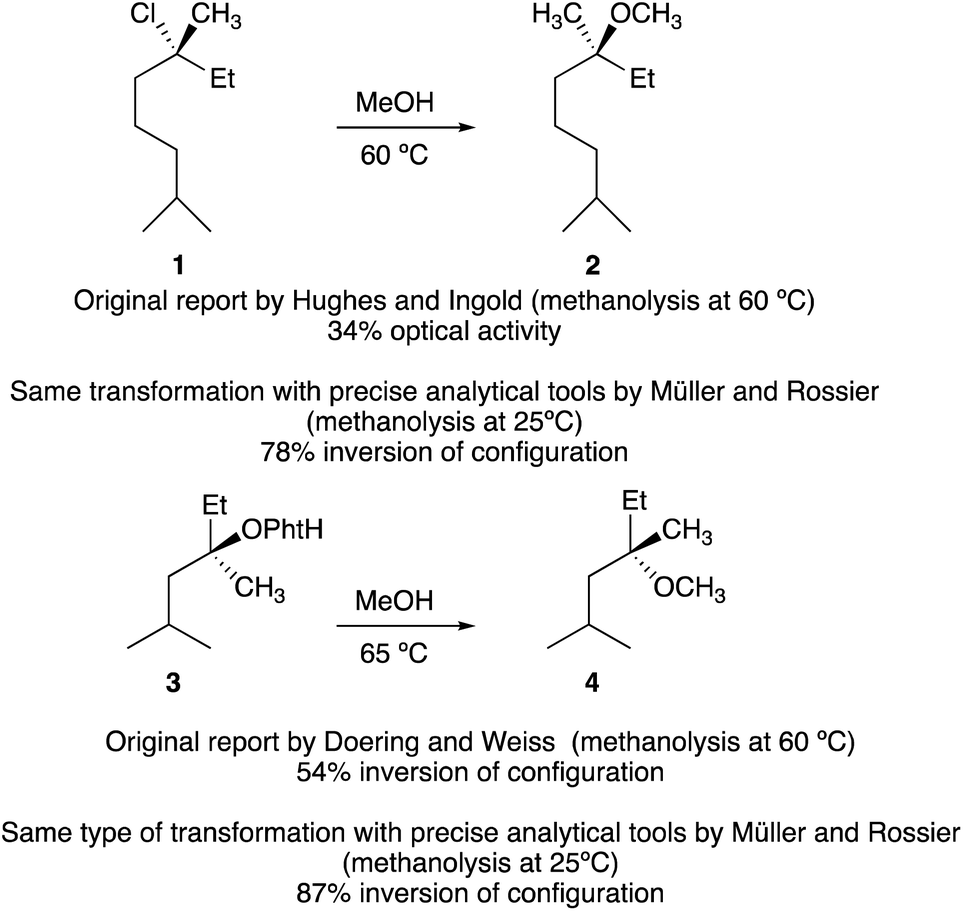 | ||
| Scheme 2 | ||
Since the enantiomeric compositions of the solvolysis products 2 and 4 were determined by comparison of optical rotations of starting materials and products, (both having unfortunately low [α]D values), these initial results were partially ignored by our scientific community until the meticulous study by Müller who irrevocably confirmed that both transformations indeed proceeded with predominantly inversion of configuration.8 For instance, when the methanolysis of (R)-3-chloro-3,7-dimethyloctane 1 was performed at 25 °C, the substitution product 2 was obtained with 78% inversion of configuration. The same holds for the methanolysis of phthalate 3 that proceeded with 87% inversion of configuration.8 Clearly, these reactions do not meet the traditional criteria for simple SN1 processes and a complete understanding of the role of intimate, solvent separated or dissociated ion pairs on the stereochemical outcome of solvolysis and related reactions is of critical importance. The complete ionization (SN1) and direct displacement (SN2) mechanisms can be viewed as the two extremes of a mechanistic continuum. At the SN1 extreme, no covalent interaction exists between the carbocation and the nucleophile in the transition state whereas at the opposite extreme (SN2), the bond formation with the nucleophile is concerted with the bond breaking step. In between these two extreme cases lies a network of intermediates separated by equilibria governed by low energy barriers called ion pairs with partial degrees of interactions between the nucleophile and the reactant. Based on kinetics, salt effects and stereochemistry of solvolysis reactions, the concept of ion pairs involved in nucleophilic substitution, was introduced and developed by S. Winstein.9 This scenario could be divided into three distinct parts: the process of ionization initially generates an intimate ion-pair (or contact ion-pair) where the carbocation and counteranion are in close proximity to each other (Fig. 1). This species can proceed to a solvent-separated ion pair, in which one or more solvent molecules have inserted between the carbocation and the leaving group but in which the ions have not yet diffused apart. Finally, the “free”carbocation is formed by diffusion away from the anion, which is the dissociative step.10
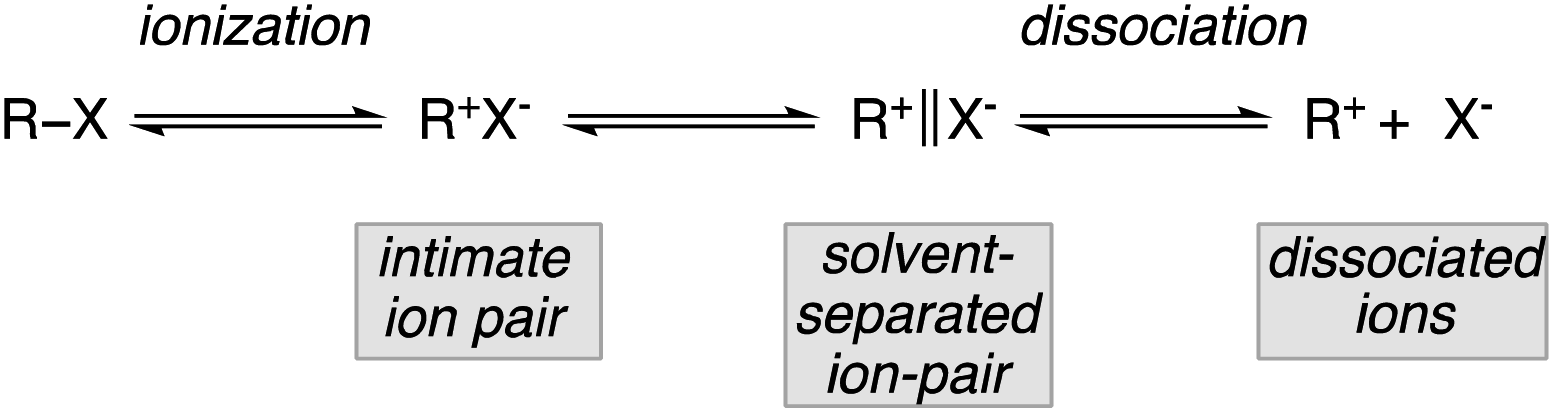 | ||
| Fig. 1 Network of intermediates with partial degrees of interactions. | ||
To understand that nucleophilic substitution might occur on a tertiary stereocentre with some preservation of the optical purity, one has to realize that the nucleophile can attack at either of the ion pairs. If the nucleophilic reaction proceeds on the intimate ion pair, the leaving group still shields the front side of the carbocation and an inversion of configuration is expected. In contrast, at the solvent-separated ion pair stage, the nucleophile might approach from either face, particularly in the case where the solvent is the nucleophile (solvolysis). When the nucleophile is different from the solvent (and not attach to the solvent), the nucleophilic substitution reaction on solvent separated ion pair is also expected to result in inversion of configuration. Obviously, reactions by capture of dissociated ions should lead to the formation of a complete racemate. This is an outstanding area of research and although the initial report is now more than 80 years old, the stereoselective substitution of tertiary alcohols and tert-alkyl leaving groups remains a significant problem as one needs to intercept the reaction at the intimate ion or solvent separated ions-pair levels.
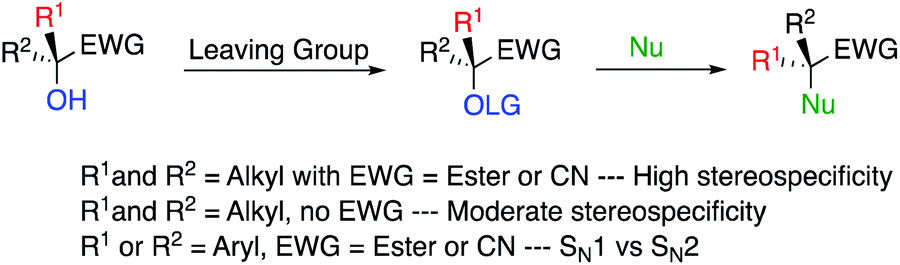
On the other hand, a relevant strategy for a stereospecific SN2 reaction at a tertiary centre would be to convert an alcohol into a corresponding good leaving group, being at the α-position of an ester (or nitrile) to impede the formation of a potential carbocation. In such case, the steric hindrance at the tertiary alcohol centre is slightly decreased due to the planar nature of the ester (nitrile) functionalities (Scheme 3). Obviously, one needs to avoid the presence of any stabilizing groups at the tertiary alcohol centre (i.e. R1 and R2 ≠ aromatic) to inhibit potential dissociation into a SN1 process.
 | ||
| Scheme 3 | ||
Below are few recent examples illustrating the state of the art in this field.
3. Stereospecific SN2 reaction of tertiary alcohol derivatives
Based on the pioneering work of Mayer,11 Cohen has reported a ZnCl2-mediated intramolecular cyclization of hydroquinone 5 into chroman-2-carboxylic ester 6, a key intermediate in the synthesis of α-tocopherol (vitamin E), with inversion of configuration.12 This example of a displacement at a tertiary centre was further corroborated by the nucleophilic substitution of a tertiary mesylate, easily obtained from 7. The subsequent reduction into hydroquinone is in situ treated with a base to provide virtually optically pure 6 with a complete inversion of configuration (Scheme 4). These reports on nucleophilic displacement on a tertiary alcohol- or mesylate- with an inversion of configuration proceed rather easily most probably due to the combination of an intramolecular reaction with the electronic effect of the carbomethoxy group generating a better electrofuge towards the nucleophilic displacement.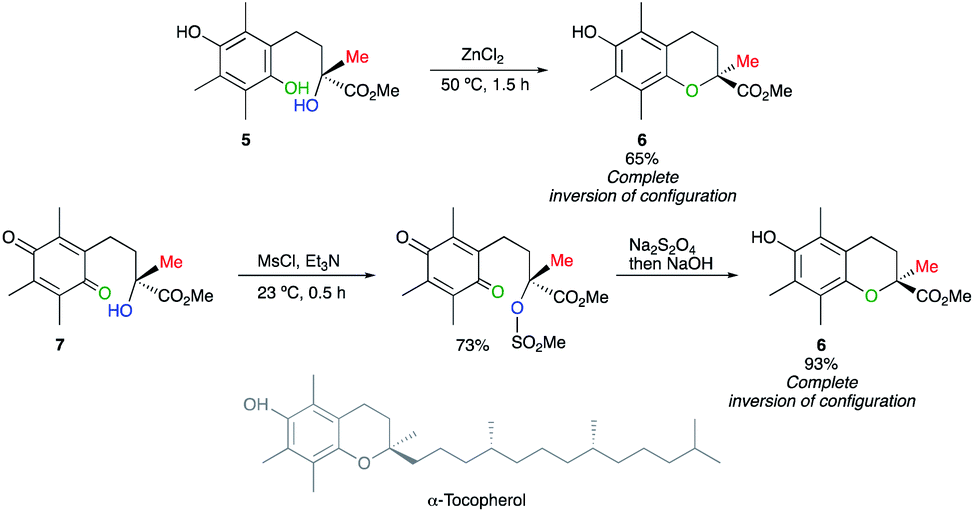 | ||
| Scheme 4 | ||
One elegant illustration of this strategy is the synthesis of unnaturally (R)-(+)-spirobrassinin from naturally occurring (S)-(−)-dioxibrassinin (Scheme 5).13 The absolute configuration was determined by chiroptical techniques such as vibrational circular dichroism (VCD) and electronic circular dichroism (ECD).
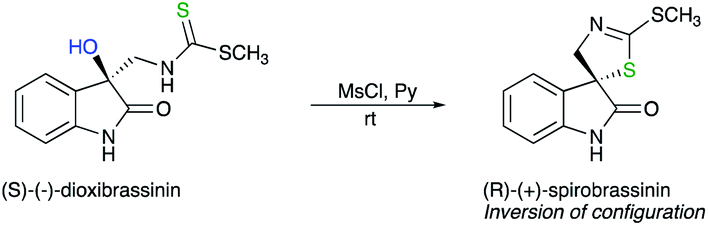 | ||
| Scheme 5 | ||
Activation of tertiary alcohols could also be achieved by using the Mitsunobu reaction.14,15 In this context, Shi developed a protocol to transform chiral tertiary alcohol 8 into ether 9 under Mitsunobu conditions (Scheme 6a).16 As the reaction now proceeds intermolecularly, the required reaction conditions were harsher than mentioned earlier (100 °C). Interestingly, slight increase of the steric bulk around the tertiary alcohol (R1 and R2), completely shut down the transformation (Scheme 6b).
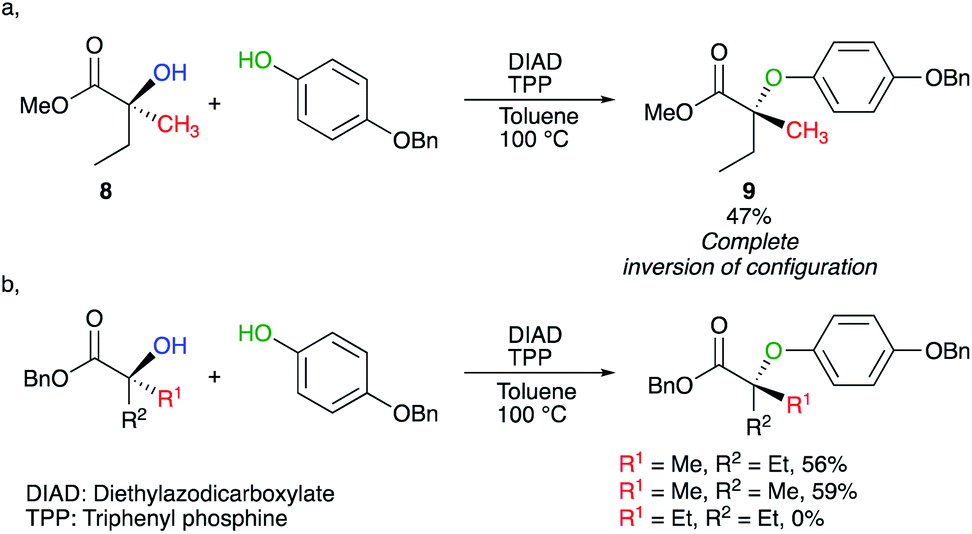 | ||
| Scheme 6 | ||
The preparation of quaternary amino acids has been similarly reported by using the Mitsunobu reaction conditions, by transforming chiral tertiary alcohols with HN3 to tertiary azides with a complete inversion of configuration (Scheme 7).17 It should be noted that classical Mitsunobu conditions using diisopropyl azodicarboxylate (DIAD) and triphenyl phosphine (TPP) led to a poor transformation and only the combination of ADDP (1,1-(azodicarbonyl)dipiperidine) with the less bulky PMe3 led to the expected transformation at the exception of the latest example which resulted in >99% elimination.18 Simple reduction of the azide function and ester hydrolysis provide the expected quaternary amino acids with 99% enantiospecificity (abbreviated as es in Scheme 7).
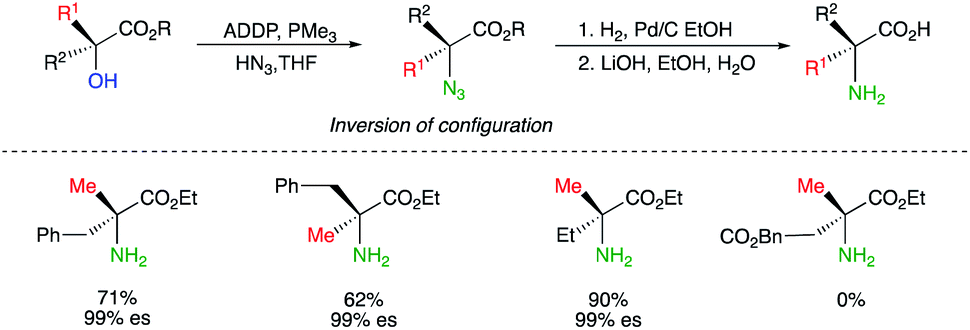 | ||
| Scheme 7 | ||
An alternative and mild activation of tertiary alcohols was also reported for nucleophilic substitution of tertiary alcohol derivatives by Mukaiyama by using a new combination of reductant and oxidant.19 The method is based on the interaction of alkoxydiphenylphosphine 10 with weak oxidants such as quinone to provide a key intermediate for the substitution. The reaction proceeds from an in situ basic activation of the alcohol with chlorodiphenylphosphine to afford alkoxydiphenylphosphines 10. Then, the addition of quinone provides the phosphonium salt 11, which is a key intermediate in the course of the reaction, and act as deprotonating agent of the nucleophile to provide a reactive species that allows the nucleophilic substitution to provide 12 with a complete inversion of configuration (Scheme 8).20 However, to the best of our knowledge, this strategy has never been used successfully by any other research groups.
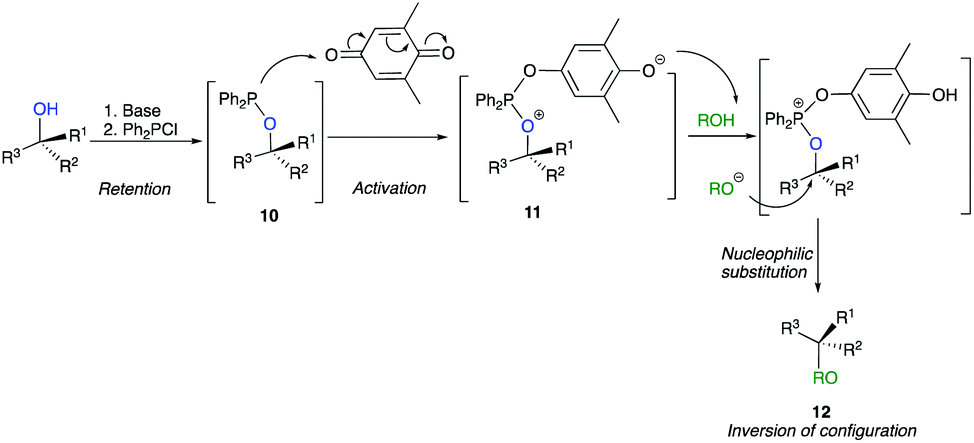 | ||
| Scheme 8 | ||
Although ring-opening of substituted epoxides are not treated in this minireview, the SN2 displacement at a tertiary carbon centre of a 3-fold tertiary alkyl oxonium salt is worth to be mentionned.21 When 1,4,7-trimethyloxatriquinane 13 was treated with tetrabutylammonium azide in CHCl3, bicyclic azide 14 was obtained as the sole product (Scheme 9). This SN2 pathway is in sharp contrast to the absence of reaction under classic solvolysis conditions (refluxing ethanol) or to the elimination products obtained with basic nucleophiles such as methoxide, cyanide or acetate.
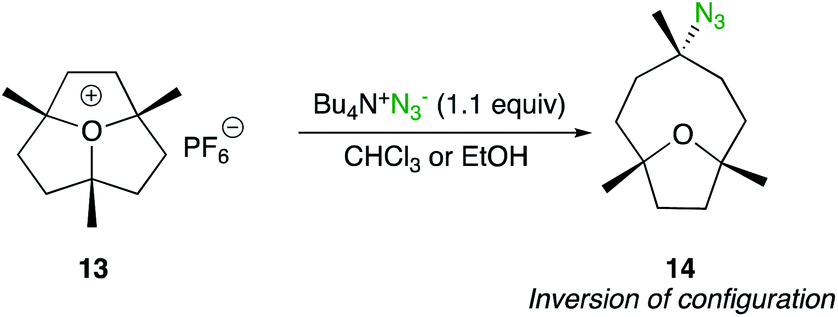 | ||
| Scheme 9 | ||
4. Stereospecific nucleophilic displacement via carbenium ions
The alternative mechanism for a nucleophilic displacement of a leaving group on a tertiary carbon centre proceeds though potentially a network of intermediates separated by equilibria governed by low energy barriers leading finally to the formation of a dissociated ions and ultimately to the formation of a racemic product.22 To avoid racemization, the interception of the intermediate by the nucleophile must occur at the level of intimate or solvent separated ion pairs.23 In a pioneering report, by the research group of Shenvi, chiral tertiary alcohols were stereochemically inverted by addition of a nitrogeneous nucleophile via Lewis acid-catalysed solvolysis.24 The transformation starts by an acylation of the alcohol with trifluoroacetic anhydride followed by the Lewis acid-catalysed solvolysis with trimethylsilyl cyanide (TMSCN) as a nitrogen nucleophile (Scheme 10). The strategy shows a wide tolerance of functional groups such as esters, nitriles, alkenes, alkynes and primary alcohols. An additional potential advantage of this protocol is the straightforward conversion of isocyanides to different functionalities. Although the complete mechanistic picture has not yet been fully addressed, the authors convincingly proposed an active species B, generated from mixing Sc(OTf)3, TMSCN and trifluoroacetate. Once B is formed, ionization could lead to the contact ion pair C, that would undergo a nucleophile attack of TMSCN on the planar carbocation at the opposite side of the large and electron-rich counter anion. Very subtle changes in the concentration of contact ion pair towards solvated cation might lead to reduced selectivity of the process. It should be noted that the direct SN2 displacement of TMSCN on the active species B could not be completely excluded even though all experimental evidence are consistent with the existence of the ion-pair pathway.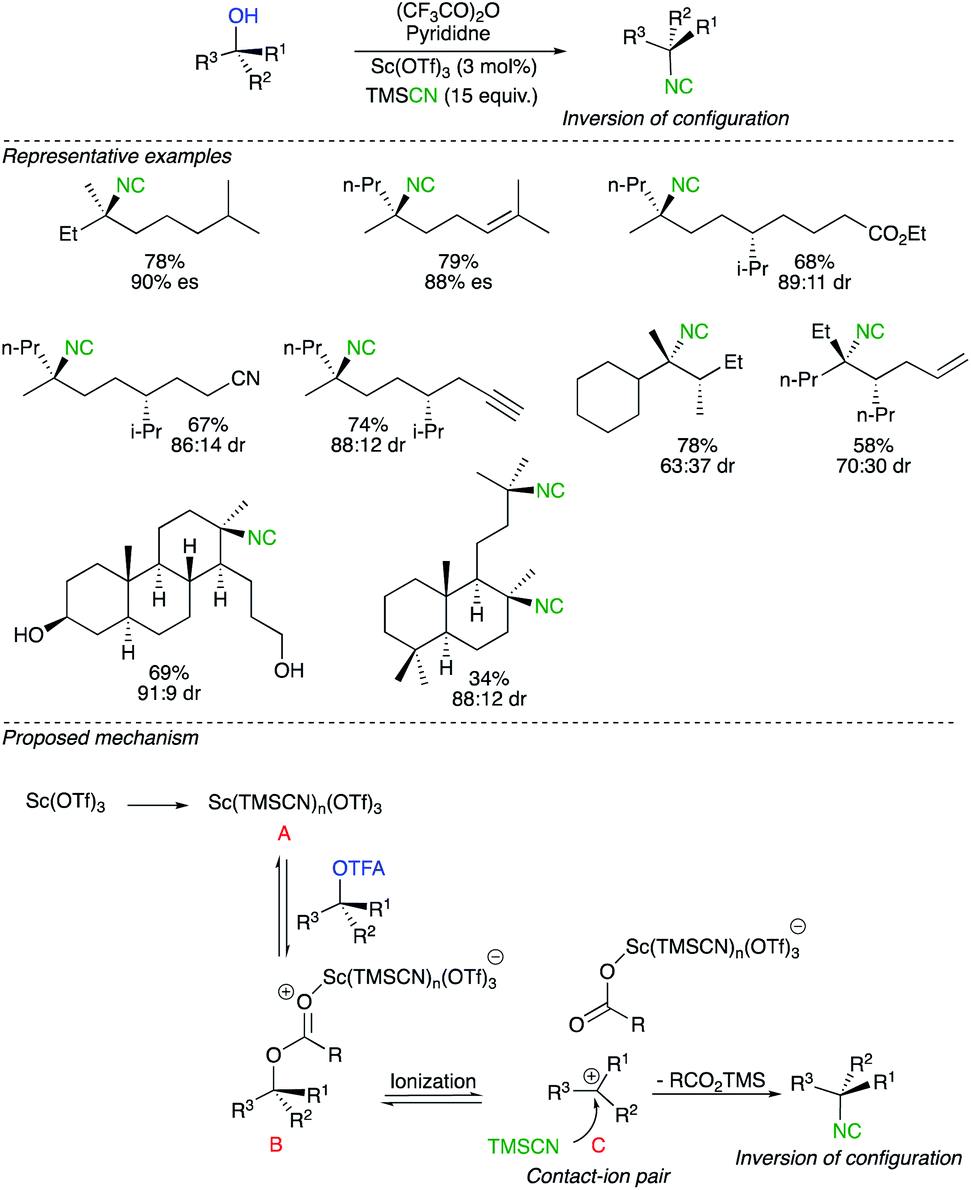 | ||
| Scheme 10 | ||
Recently, Cook reported an iron-catalysed conversion of tertiary alcohols to sulfonamides (Scheme 11).25 Enantioenriched indolines were prepared from enantioenriched tertiary alcohols by intramolecular nucleophilic substitution with a complete inversion of configuration. Although the scope of the reaction presents a few limitations, the method enables effective access to enantioenriched 2,2-disubstituted indolines. The proposed mechanism proceeds through the formation of tight Fe ion-pairs, at low temperature and with a solvent of low dielectric constant. Reaction of the nucleophile from the opposite face of the Fe-protected carbenium ion leads to the products with inversion of configuration and high enantiospecificity.
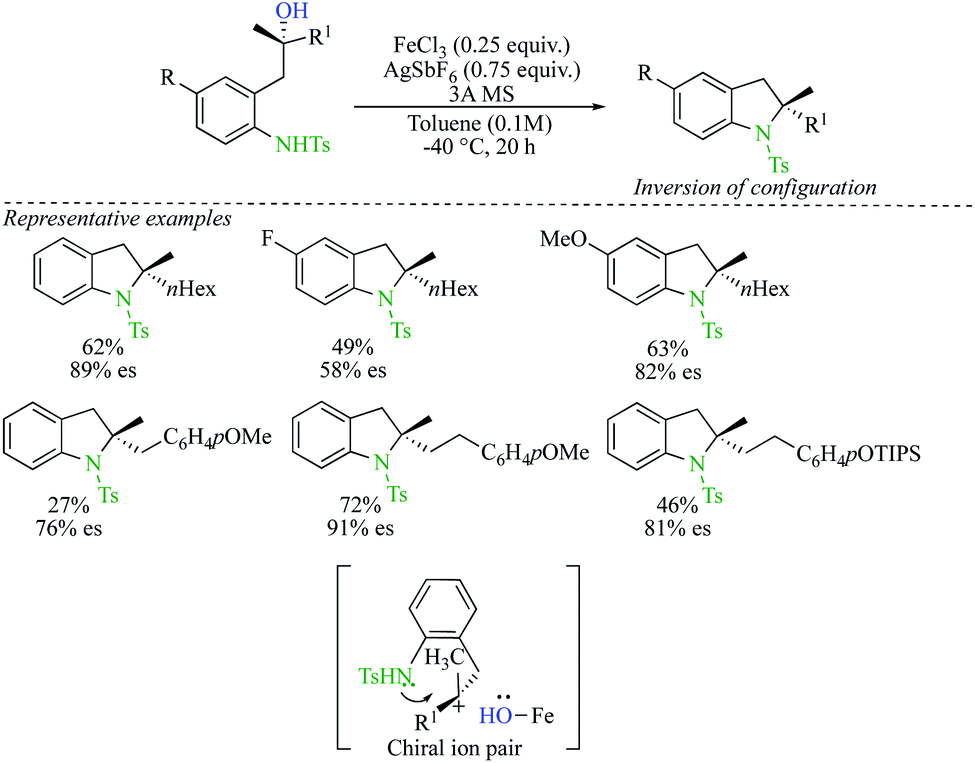 | ||
| Scheme 11 | ||
Six-membered heterocyclic compounds 16 could also be prepared by a Fe-catalysed substitution reaction of underivatized tertiary alcohol 15. The key to achieving high chirality transfer turned out to be decreasing the polarity of the solvent system. By using a mixture of n-hexane and DCE at either −15 °C or room temperature (depending of the nucleophilicity of the heteroatom), various O-, N- and S-substituted heterocycles were prepared with a complete chirality transfer (Scheme 12).26 It is interesting to note that for the substrate with X = O and R = alkyl, the combination of a tertiary alcohol prone to undergo elimination with a weak nucleophile still generates the product 16c with high chirality transfer. Experimental results suggests that the coordination of iron to the nucleofuge would lead to C–O bond cleavage to generate a tight ion pair intermediate. The use of non-polar solvent can therefore be rationalized as it “tightens” the ion pair whereas more polar solvent would loosen the ion pairing and nucleophile addition could occur from either side (in pure DCE, only 9% of enantiospecificity was observed).
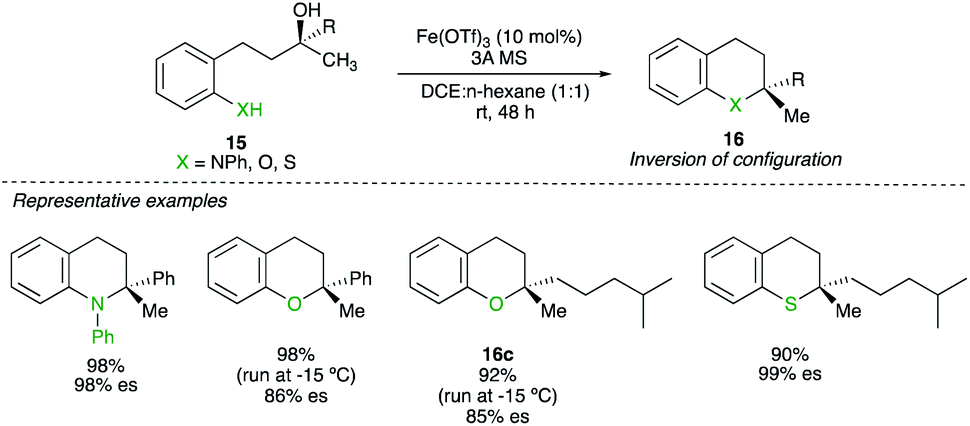 | ||
| Scheme 12 | ||
When the equilibrium between intimate ion-pair versus solvated cation is shifted towards the latter, a planar carbocation is formed. If the nucleophile can be intercepted on only one face of the carbocation, a non-racemic product could eventually be formed. Therefore, despite the intrinsic instability and high reactivity of carbocation intermediate, this alternative approach might theoretically lead to the formation of enantiomerically enriched quaternary carbon stereocentre from a racemic starting material. A rare example of this concept of enantioconvergent catalytic SN1 reaction of racemic tertiary alcohols was reported by Jacobsen and relies on the synergistic action of a chiral hydrogen-bond-donor catalyst with a strong Lewis-acid promoter.27 The reaction of propargyl acetate with allyltrimethylsilane in the presence of a squaramide catalysts L1 and TMSOTf involves the formation of a carbocation intermediate that subsequently reacts with a nucleophile to produce the propargylic quaternary carbon stereocentre 17 with high enantiomeric excess (Scheme 13).
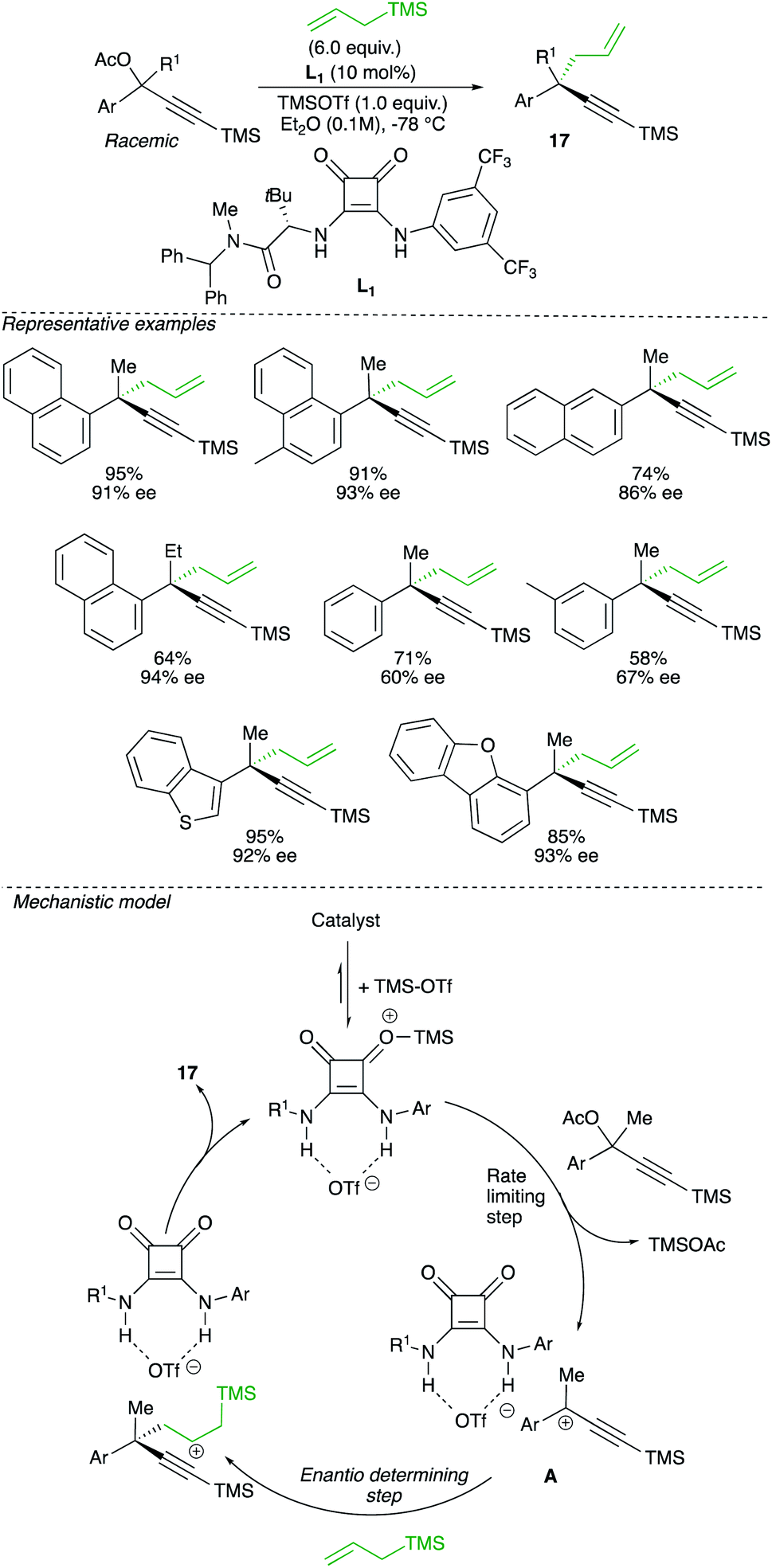 | ||
| Scheme 13 | ||
An interesting correlation was observed between polarizability values and enantioselectivities, suggesting that stabilizing aromatic interactions are likely to contribute in the enantiodifferentiation step. A comprehensive mechanistic study, including cross-over experiments and detailed kinetic studies, have been performed to probe the mechanism. The first step of the proposed mechanism involves the generation of charged active catalyst from squaramide and TMSOTf. This active catalyst dissociates the propargyl acetate to generate the tertiary carbocation, interacting with the catalyst to lead to A. One face of the carbocation complex A is thus shielded, the nucleophile approaching from the opposite side, inducing the enantio-discriminating step (Scheme 13).
5. Nucleophilic substitution at quaternary carbon stereocentres
All of the above transformations formally result from the displacement of a leaving group by a nucleophile. The contra-intuitive nucleophilic displacement at a quaternary carbon centre where a carbon–carbon bond would be cleaved was not yet fully exploited in the literature. This peculiar transformation would be possible if one could take into account the release of ring strain as driving force to promote the reaction. In this context, we have recently reported an intermolecular regio- and stereoselective nucleophilic substitution reaction at quaternary stereocentres of cyclopropyl carbinol derivatives with various nucleophiles to provide the corresponding acyclic products as a single diastereomer (Scheme 14).28 The strategy capitalizes on rapid and efficient access to diastereomerically pure and enantiomerically enriched cyclopropyl carbinol 19, from enantioenriched cyclopropenes 18, enabling the straightforward preparation of diastereomerically pure tertiary alkyl bromide, chloride, ester and fluoride derivatives 20. In all cases, the substitution occurs at the most substituted carbon centre C2 with excellent diastereoselectivity and a complete inversion of configuration. The robustness of the protocol can be evaluated in Scheme 14 for a large variety of substrates and nucleophiles. Several catalytic systems allowed the transformation but CuBr2 was used as the most convenient choice. The reaction is stereospecific as both diastereomers 21 and 22 at the quaternary carbon centre (Scheme 14, Nu = HBF4) could be equally prepared by simply permuting the two substituents on the cyclopropyl ring (R and R2). To rationalize the observed selectivity for the nucleophilic substitution at the most substituted carbon centre, one has first to consider that the reaction is independent of the stereochemistry at the carbinol centre C4 (Scheme 14, mechanistic insight), suggesting that the reaction might proceed through the formation of a cyclopropyl carbocation A, best represented as the hybrid form B.29 As the transition state has a positive charge spread all over the molecular backbone, the reaction of B with a nucleophile should occur at the site bearing the highest density of positive charge leading to the homoallyl product with inversion of configuration (Scheme 14). The starting material 18 can easily be prepared enantiomerically enriched as a single diastereomer at the three cyclopropyl carbon centres in only two catalytic steps from commercially available alkynes.30 Therefore, the nucleophilic substitution of cyclopropylcarbinol 19 allows the easy and straightforward access to functionalized acyclic homoallyl species possessing for instance, a tertiary alkyl fluoride or ester functionalities in excellent diastereo- and enantiomeric ratios (see 23 and 24, Scheme 14).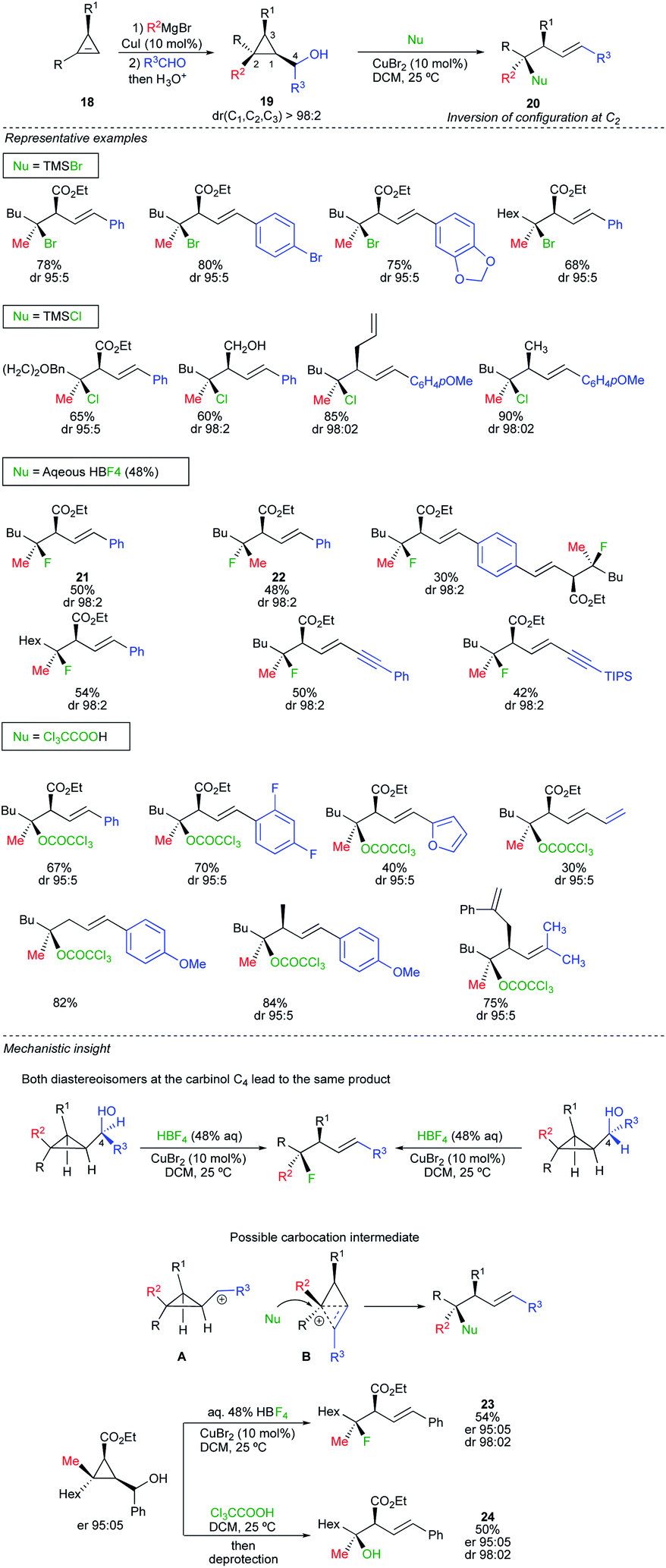 | ||
| Scheme 14 | ||
Although the nucleophilic substitution at a quaternary carbon stereocentre of a three-membered ring is less intuitive than the more traditional nucleophilic displacement on an tertiary alcohol or other tertiary alkyl leaving groups, the potential preservation of the optical activity for the former transformation is higher due to the particular structure of the cyclobutonium species B.31 In this case, as one face of the transient carbocation is shielded by the molecular backbone, the diastereo- and enantiospecificity of the reaction is easier to control, even using only stoichiometric amount of nucleophiles (not solvolysis), than through the formation of intimate and solvent separated ion pairs.
Nucleophilic substitution reactions have always been considered as one of the most powerful reactions for the creation of carbon–carbon or carbon–heteroatom bonds in organic synthesis. However, our scientific community was generally considering that the steric shielding of tertiary carbons retards a concerted, stereospecific nucleophilic substitution, and ionizing pathways often lead to nonselective substitution due to ion pair dissociation. In this minireview, we underlined the fascinating and rich area that stereoselective or stereospecific nucleophilic substitution at fully-substituted carbon centres represents through the original contributions of leading scientists that demonstrated, several decades ago, that stereoselective nucleophilic substitution can proceed equally well at tertiary carbon centre via either via a SN2 or SN1 mechanism with inversion of configuration.
More recently, we have witnessed a renaissance of the field by the development of impressive transformations at tertiary centres providing remarkable added values to practitioners in the field of stereoselective synthesis and synthesis of natural products. Even more fascinating is the stereoselective substitution at a quaternary carbon stereocentre where a selective cleavage of a carbon–carbon bond occurs. A single set of examples has been reported where the release of the ring-strain allows the substitution at a quaternary carbon centres with an inversion of configurations. Further development of selective nucleophilic substitution at sterically encumbered carbon centres as well as utilization of this concept in unexplored territories will surely lead to exciting surprises, and opportunities for innovation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement no. 786976.Notes and references
- (a) C. A. Bunton, E. D. Hughes, C. K. Ingold and D. F. Meigh, Nature, 1950, 166, 679 CrossRef PubMed . For original reviews, see:; (b) E. D. Hughes, Trans. Faraday Soc., 1941, 37, 603 RSC; (c) E. D. Hughes, Quart. Rev., 1951, 5, 145 RSC.
- (a) G. W. Wheland, Structure and Mechanism in Organic Chemistry, ed. C. K. Ingold and N. Y. Ithaca, Cornell Univ. Press, 1953, p. 828 Search PubMed; (b) S. R. Hartshorn, Aliphatic Nucleophilic Substitution, Cambridge University Press, London, 1973 Search PubMed; (c) M. B. Smith and J. March, Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Wiley-Interscience, New York, 6th edn, 2007, ISBN 978-0-471-72091-1 Search PubMed.
- E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, University Science Books, Sausalito, CA, 2006, ch. 1 Search PubMed.
- P. Walden, Ber. Dtsch. Chem. Ges., 1896, 29, 133 CrossRef.
- T. A. Hamlin, M. Swart and F. M. Bickelhaupt, ChemPhysChem, 2018, 19, 1315 CrossRef CAS PubMed.
- P. G. Stevens and N. L. McNiven, J. Am. Chem. Soc., 1939, 61, 1295 CrossRef CAS.
- W. v. E. Doering and H. H. Zeiss, J. Am. Chem. Soc., 1953, 75, 4733 CrossRef CAS.
- P. Müller and J.-C. Rossier, J. Chem. Soc., Perkin Trans. 2, 2000, 2232 RSC.
- S. Winstein, E. Clippinger, A. H. Fainberg, R. Heck and G. C. Robinson, J. Am. Chem. Soc., 1956, 78, 328 CrossRef CAS.
- (a) D. J. Raber, J. M. harris and P. v. R. Schleyer, in Ion Pairs, ed. M. Szware, John Wiley & Sons, New York, 1974, ch. 3 Search PubMed; (b) T. W. Bentley and P. v. R. Schleyer, Adv. Phys. Org. Chem., 1977, 14, 1 CrossRef CAS; (c) J. P. Richard, Adv. Carbocation Chem., 1989, 1, 121 CAS; (d) Y. Marcus and G. Hefter, Chem. Rev., 2006, 106, 4585 CrossRef CAS PubMed; (e) J. P. Richard, M. M. Toteva and T. L. Amyes, Org. Lett., 2001, 3, 2225 CrossRef CAS PubMed.
- P. Schudel, H. Mayer, J. Metzger, R. Ruegg and O. Isler, Helv. Chim. Acta, 1963, 46, 333 CrossRef CAS.
- N. Cohen, R. J. Lopresti and C. Neukom, J. Org. Chem., 1981, 46, 2445 CrossRef CAS.
- K. Monde, T. Taniguchi, N. Miura, S.-I. Nishimura, N. Harada, R. K. Dukor and L. A. Nafie, Tetrahedron Lett., 2003, 44, 6017 CrossRef CAS.
- O. Mitsunobu, Synthesis, 1981, 1 CrossRef CAS.
- K. C. Kumara Swamy, N. N. Bhuvan Kumar, E. Balaraman and K. V. P. Pavan Kumar, Chem. Rev., 2009, 109, 2551 CrossRef PubMed.
- Y.-J. Shi, D. L. Hughes and J. M. McNamara, Tetrahedron Lett., 2003, 3609 CrossRef CAS.
- J. E. Green, D. M. Bender, S. Jackson, M. J. O'Donnell and J. R. McCarthy, Org. Lett., 2009, 11, 807 CrossRef CAS PubMed.
- T. Tsunoda, Y. Yamamiya and S. Ito, Tetrahedron Lett., 1993, 34, 1639 CrossRef CAS.
- T. Mukaiyama, T. Shintou and K. Fukumoto, J. Am. Chem. Soc., 2003, 125, 10538 CrossRef CAS PubMed.
- (a) T. Mukaiyama and H. Yamabe, Chem. Lett., 2007, 36, 1 Search PubMed; (b) T. Shintou and T. Mukaiyama, J. Am. Chem. Soc., 2004, 126, 7359 CrossRef CAS PubMed; (c) T. Mukaiyama, K. Kuroda and Y. Maruyama, Heterocycles, 2010, 80, 63–81 CrossRef CAS; (d) T. Mukaiyama, T. Shintou and K. Fukumoto, J. Am. Chem. Soc., 2003, 125, 10538 CrossRef CAS PubMed; (e) K. Kuroda, Y. Hayashia and T. Mukaiyama, Tetrahedron, 2007, 63, 6358 CrossRef CAS; (f) T. Mukaiyama and K. Ikegai, Chem. Lett., 2004, 33, 1522 CrossRef CAS; (g) K. Ikegai, W. Pluempanupat and T. Mukaiyama, Chem. Lett., 2005, 34, 638 CrossRef CAS; (h) K. Ikegai, W. Pluempanupat and T. Mukaiyama, Bull. Chem. Soc. Jpn., 2006, 79, 780 CrossRef CAS; (i) K. Kuroda, Y. Maruyama, Y. Hayashi and T. Mukaiyama, Chem. Lett., 2008, 37, 836 CrossRef CAS.
- M. Mascal, N. Hafezi and M. D. Toney, J. Am. Chem. Soc., 2010, 132, 10662 CrossRef CAS PubMed.
- A. Streitwieser, Chem. Rev., 1956, 56, 571 CrossRef CAS.
- Stereocontrol imparted by cyclic scaffolds will not be discussed..
- (a) S. V. Pronin and R. A. Shenvi, J. Am. Chem. Soc., 2012, 134, 19604 CrossRef CAS PubMed; (b) S. V. Pronin, C. A. Reiher and R. A. Shenvi, Nature, 2013, 501, 195 CrossRef CAS PubMed.
- P. T. Marcyk, L. R. Jefferies, D. I. AbuSalim, M. Pink, M.-H. Baik and S. P. Cook, Angew. Chem., Int. Ed., 2019, 58, 1727 CrossRef CAS PubMed.
- R. A. Watile, A. Bunrit, J. Margalef, S. Akkarasamiyo, R. Ayub, E. Lagerspets, S. Biswas, T. Repo and J. S. M. Samec, Nat. Commun., 2019, 10, 3826 CrossRef PubMed.
- E. Wendlandt, P. Vangal and E. N. Jacobsen, Nature, 2018, 556, 447 CrossRef PubMed.
- V. Lanke and I. Marek, J. Am. Chem. Soc., 2020, 142, 5543 CrossRef CAS PubMed ; Correction: V. Lanke and I. Marek, J. Am. Chem. Soc., 2020, 142, 7710 CrossRef PubMed.
- (a) J. Wang, M. Sanchez-Rosello, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed; (b) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422 CrossRef CAS PubMed.
- (a) D. Didier, P. Delaye, M. Simaan, B. Island, G. Eppe, H. Eijsberg, A. Kleiner, P. Knochel and I. Marek, Chem.–Eur. J., 2014, 20, 1038 CrossRef CAS PubMed; (b) L. Dian and I. Marek, Chem. Rev., 2018, 118, 8415 CrossRef CAS PubMed; (c) D. S. Müller and I. Marek, J. Am. Chem. Soc., 2015, 137, 15414 CrossRef PubMed; (d) L. Dian, D. S. Müller and I. Marek, Angew. Chem., Int. Ed., 2017, 56, 6783 CrossRef CAS PubMed; (e) H. Sommer and I. Marek, Chem. Sci., 2018, 9, 6503 RSC; (f) D. S. Müller, V. Werner, S. Akyol, H.-G. Schmalz and I. Marek, Org. Lett., 2017, 19, 3970 CrossRef PubMed; (g) H. Zhang, W. Huang, T. Wang and F. Meng, Angew. Chem., Int. Ed., 2019, 58, 11049 CrossRef CAS PubMed; (h) L. Dian and I. Marek, Angew. Chem., Int. Ed., 2018, 57, 3682 CrossRef CAS PubMed.
- H.-U. Siehl, in Advances in Physical Organic Chemistry, Elsevier Ltd, 2018, vol. 52, pp. 1–47 Search PubMed.
| This journal is © The Royal Society of Chemistry 2020 |