
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Switchable foldamer ion channels with antibacterial activity†
Anna D.
Peters
ab,
Stefan
Borsley
ac,
Flavio
della Sala
 ab,
Dominic F.
Cairns-Gibson
c,
Marios
Leonidou
ab,
Jonathan
Clayden
d,
George F. S.
Whitehead
a,
Iñigo J.
Vitórica-Yrezábal
a,
Eriko
Takano
ab,
John
Burthem
ef,
Scott L.
Cockroft
c and
Simon J.
Webb
*ab
ab,
Dominic F.
Cairns-Gibson
c,
Marios
Leonidou
ab,
Jonathan
Clayden
d,
George F. S.
Whitehead
a,
Iñigo J.
Vitórica-Yrezábal
a,
Eriko
Takano
ab,
John
Burthem
ef,
Scott L.
Cockroft
c and
Simon J.
Webb
*ab
aDepartment of Chemistry, University of Manchester, Oxford Road, Manchester M13 9PL, UK. E-mail: S.Webb@manchester.ac.uk
bManchester Institute of Biotechnology, University of Manchester, 131 Princess St, Manchester M1 7DN, UK
cEaStCHEM School of Chemistry, University of Edinburgh, Joseph Black Building, David Brewster Road, Edinburgh EH9 3FJ, UK
dSchool of Chemistry, University of Bristol, Cantock's Close, Bristol BS8 1TS, UK
eDepartment of Haematology, Manchester Royal Infirmary, Manchester University NHS Foundation Trust, Manchester M13 9WL, UK
fDivision of Cancer Sciences, School of Medical Sciences, University of Manchester, Manchester, UK
First published on 4th June 2020
Abstract
Synthetic ion channels may have applications in treating channelopathies and as new classes of antibiotics, particularly if ion flow through the channels can be controlled. Here we describe triazole-capped octameric α-aminoisobutyric acid (Aib) foldamers that “switch on” ion channel activity in phospholipid bilayers upon copper(II) chloride addition; activity is “switched off” upon copper(II) extraction. X-ray crystallography showed that CuCl2 complexation gave chloro-bridged foldamer dimers, with hydrogen bonds between dimers producing channels within the crystal structure. These interactions suggest a pathway for foldamer self-assembly into membrane ion channels. The copper(II)-foldamer complexes showed antibacterial activity against B. megaterium strain DSM319 that was similar to the peptaibol antibiotic alamethicin, but with 90% lower hemolytic activity.
Introduction
Natural ion channels carry out key functions in cellular membranes, including chemical and electrical signal transduction, control of cell volume, and maintenance of internal ion concentrations.1 Ion channel function is necessarily tightly regulated, as unregulated function can result in a disease state. Many diseases, such as epilepsy, myotonia, ataxia cardiac, arrhythmia, and cystic fibrosis, arise because of ion channel dysfunctions (channelopathies).2,3Artificial ion channels are less structurally complex than natural protein ion channels,4 so their behaviour can be more easily analysed. Furthermore, synthetic ionophores could potentially alleviate the symptoms of channelopathies5,6 or provide new antibiotic classes able to overcome the growing problem of resistance.7 Of the wide range of artificial ion channels developed and studied,8 several show either cation selectivity9 or anion selectivity.10 However, a significant challenge in the field is to “switch” any channel activity,11 so that, like natural examples, activity can be controlled by external stimuli, such as light or chemical messengers. Closing channels by blocking the lumen with an added ligand is known, but examples of ligand-induced channel opening are less common.12 These have typically involved ligand-mediated self-assembly of channels in a membrane; for example, dialkoxynaphthalene addition to rigid-rod p-octiphenyl staves gave weakly anion selective channels.13 Metal ions, such as palladium(II), have also been used to assemble channels within membranes.10a,14,15,16 Alternatively, multivalent ligands outside the membrane can drive the lateral assembly of individual membrane-spanning staves. This was elegantly shown by Matile and co-workers, who used coordination of external polyhistidine to copper(II)(iminodiacetate)-terminated p-septiphenyls to open K+-selective channels.17
α-Aminoisobutyric acid (Aib) foldamers have recently shown promise for synthetic ion channel18 and signal transduction activities.19 These highly hydrophobic foldamers contain high proportions of Aib, a residue that favours folding into 310 helices.20 Simple Aib foldamers in phospholipid bilayers show length-dependent ion channel and antibiotic activity,18 with properties that mimic the ionophoric behaviour of the Aib-rich antimicrobial peptide alamethicin, the archetypical peptaibol.21 These easy-to-modify Aib foldamers make an interesting platform for developing switchable ionophores that may also show antibacterial activity. For example, Cu(II)-capped Aib foldamers might cluster upon binding to an external polyvalent ligand (e.g. polyhistidine),22 perhaps “switching on” ion channel activity that is reversible by ligand sequestration. Alternatively, Cu(II) complexation might itself promote self-assembly into active ion channels.15
The design of the Cu(II) complexing Aib foldamers 1 and 2 (Fig. 1) combines structural elements from previous studies. Since the ionophoric activity of Aib foldamers is strongly length-dependent, an octameric (Aib8) foldamer was used as the core, which had been found to be the minimum length needed to give good activity.18b This hydrophobic octamer would fold into a ∼1.6 nm long 310 helix, which would be capped at the N-terminus by an N,N-bis(pyridin-2′-ylmethyl)-N-((1,2,3-triazol-4-yl)methyl)amine (BPTA) chelator unit. The tetrameric Aib foldamer 7 would provide a shorter control compound. We have shown that the structurally related N,N-bis(quinolin-2-ylmethyl)-N-((5′-carboxypyridyl)methyl)amine (BQPA) motif at the N-terminus of foldamers complexes tightly to Cu(II) and Zn(II).19b,23 Much like related N,N,N-tris(pyridin-2-ylmethyl)amine (TPA) complexes,24 the BPTA group should adopt a trigonal pyramidal geometry around the metal ion with at least one potential coordination site for external ligands. It should also be accessible in one synthetic step from readily available Aib foldamers with an N-terminal azido group but different C-terminal groups. Since hydrophobic groups at the C-terminus generally enhance the activity of foldamers in phospholipid vesicles and bacteria,18a foldamers 1 and 2 bear respectively a tBu and a CH2CH2SiMe3 terminus.
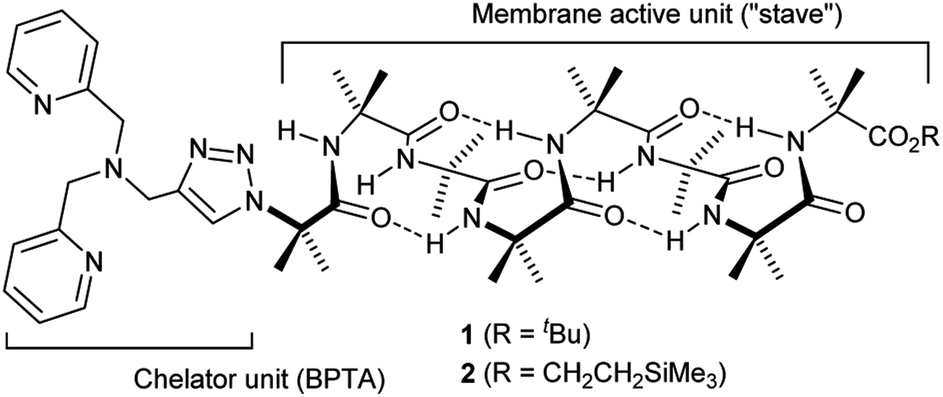 | ||
| Fig. 1 Structure of Aib foldamers 1 and 2. | ||
Results and discussion
Synthesis
Octameric Aib foldamers 3 and 4 were produced by a modification of previous synthetic procedures.25 Copper-catalysed azide–alkyne cycloaddition chemistry26 was then used to produce the BPTA chelator unit at the N-terminus and give target compounds 1 and 2 (Scheme 1). Tetrameric Aib foldamer 7 was produced from 5 using a similar procedure, providing the control compound with a much shorter membrane-active unit (the “stave”).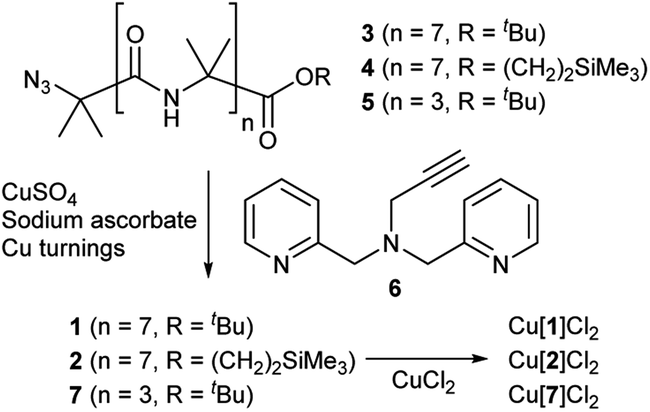 | ||
| Scheme 1 Synthesis of Aib foldamers 1, 2 and 7 and their corresponding copper(II) chloride complexes. | ||
Mixing 1 or 2 with CuCl2 or CuCl provided green or blue complexes respectively. The green complexes with CuCl2, proposed to be Cu[1]Cl2 and Cu[2]Cl2, displayed relatively high solubility in water that was unlike other octameric Aib foldamers. The blue complexes from the addition of CuCl gave very broad NMR spectra even after preparing fresh samples, which was ascribed to in situ oxidation of Cu(I); Cu(I)(TPA) complexes are very efficient one electron donors and will rapidly reduce alkyl halides27 and oxygen.28 Furthermore these blue products gave green solids when dried (see the ESI, Fig. S2†).
X-ray crystallography
Crystal structures were obtained for three compounds: foldamer 2, the green complex from the addition of CuCl2 to 2 and the blue complex from the addition of CuCl to 2. The solid state structure of 2 shows a 310 helical structure in the foldamer body (Fig. 2a) and a head-to-tail distance of 2.1 nm, which is too short to easily span the hydrophobic width of a typical bilayer (e.g. for a typical EYPC/cholesterol bilayer, ca. 2.8 nm).29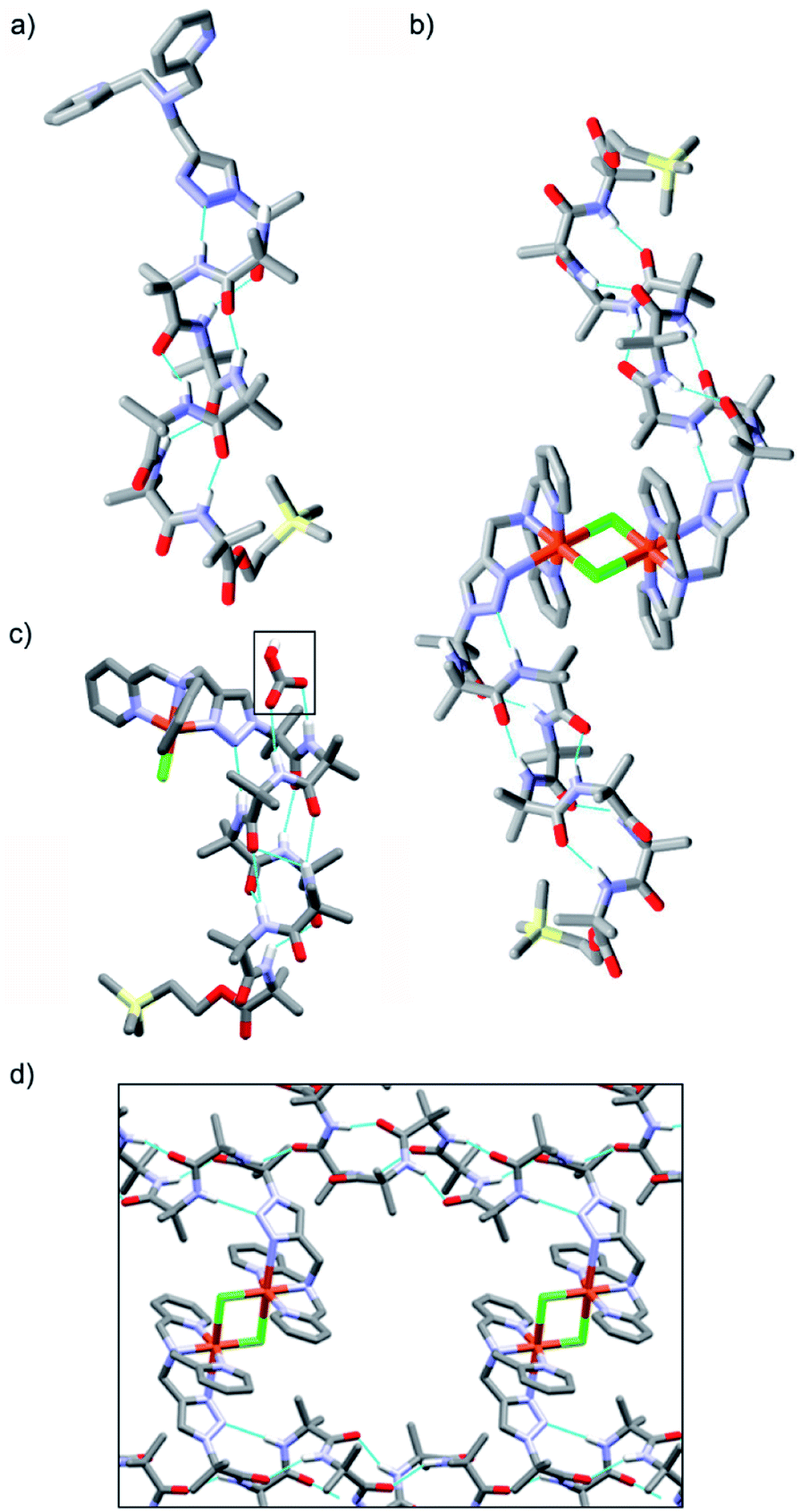 | ||
| Fig. 2 (a and b) X-ray crystal structures of (a) 2, (b) [Cu(II)[2](μ-Cl)]2Cl2 and (c) [Cu(II)[2]Cl]·HCO3, with bicarbonate ion indicated (boxed). (d) Channels within the X-ray crystal structure of [Cu(II)[2](μ-Cl)]2Cl2. | ||
The green complex, Cu[2]Cl2, that resulted from the addition of CuCl2 to 2 crystallized as a dimer of foldamers (Fig. 2b). Two octahedral metal centres share two chloride ligands to give a dimer with a 3.5 nm end-to-end length between the C-termini of the linked 310 helices. The screw sense of the Aib helix inverts at the dicopper centre. The copper ions show a Jahn–Teller distortion that is consistent with Cu(II), with elongation of one Cu–Cl bond (2.836(2) Å) compared to the other (2.241(2) Å); this distortion around the Cu(II) centres is similar to that reported in a [Cu(II)[Ph-TPA](μ-Cl)]2Cl2 complex.30 Electron paramagnetic resonance (EPR) data also showed that unpaired electrons were present (consistent with Cu(II)). The Cu(BPTA) interacts with the foldamer body through a hydrogen bond between the triazole N2 and the NH of Aib(3). A head-to-tail intermolecular hydrogen bond between dimers (from the NH of Aib(2) to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of Aib(7)) along with side-to-side packing of the dimers in the solid state produces channels (Fig. 2d) that run through the crystal and were filled with disordered electron density. The counterions of the dimeric Cu(II) complex were not located crystallographically, but an extra chloride was identified by elemental analysis. It is proposed that these chloride anions are located (along with solvent) in a region of disordered electron density that lies in the channels behind the cationic headgroups.
O of Aib(7)) along with side-to-side packing of the dimers in the solid state produces channels (Fig. 2d) that run through the crystal and were filled with disordered electron density. The counterions of the dimeric Cu(II) complex were not located crystallographically, but an extra chloride was identified by elemental analysis. It is proposed that these chloride anions are located (along with solvent) in a region of disordered electron density that lies in the channels behind the cationic headgroups.
The solid state structure was also determined for crystals obtained from the pale blue product that resulted from CuCl addition to 2. The bond lengths around the metal ion indicate that a copper(II) centre and not a copper(I) centre is present (see ESI†). The copper complex has a trigonal bipyramidal geometry, with the copper displaced outwards from the pocket by 0.322 Å, a Cu–Cl bond length of 2.203(3) Å and a copper to central nitrogen distance of 2.055(8) Å. In this structure, the counterion is located (boxed, Fig. 2c). Bicarbonate is found in a region of disordered electron density behind the N-terminal headgroup, where it is hydrogen bonded to both the NH of Aib(2) and the NH of Aib(3) in the foldamer body. This counterion is proposed to result from hydroxide formation during aerial oxidation of an intermediate Cu(I) complex,28 followed by sequestration of CO2. The foldamer body shows a hydrogen bond between the triazole N2 and the NH of Aib(4). The helix displays a mixture of a 310- and α-helical structure that leads to a shorter end-to-end length of 1.9 nm compared the parent foldamer 2.31 The observation of this monomeric species suggests that the formation of the μ-Cl bridged dimer occurs through a reversible process, which would be influenced by the surrounding environment.
Ionophoric activity
The membrane activity of 1, 2, Cu[1]Cl2 and Cu[2]Cl2 was assessed in phospholipid vesicle membranes using 8-hydroxypyrene-1,3,6-trisulfonate (HPTS) assays. HPTS assays can show ionophoric activity that occurs via any combination of M+/H+ antiport, X−/OH− antiport, M+/OH− symport and X−/H+ symport.The HPTS assays of 1, 2, Cu[1]Cl2 and Cu[2]Cl2 used 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 cholesterol:egg yolk phosphatidylcholine (EYPC) vesicles in MOPS buffer (pH 7.4) with 100 mM of an appropriate salt (e.g. KCl, KBr, NaCl), following previously reported procedures.16 The resulting normalised data were fitted to pseudo first-order rate equations as an approximation (see the ESI†).32 Although the change in fluorescence after the “burst phase” is likely to arise from multiple processes, including inter-vesicle transfer of foldamers,33 this fitting allows the relative effectiveness of each compound to be compared.
:4 cholesterol:egg yolk phosphatidylcholine (EYPC) vesicles in MOPS buffer (pH 7.4) with 100 mM of an appropriate salt (e.g. KCl, KBr, NaCl), following previously reported procedures.16 The resulting normalised data were fitted to pseudo first-order rate equations as an approximation (see the ESI†).32 Although the change in fluorescence after the “burst phase” is likely to arise from multiple processes, including inter-vesicle transfer of foldamers,33 this fitting allows the relative effectiveness of each compound to be compared.
At 10 μM foldamer, ion transport by the parent foldamers 1 and 2 was just above the leakage rate cause by addition of the methanol control (an observed constant rate constant, kobs, of 1 × 10−3 s−1, Fig. 3a and b). For example, in the presence of KCl, kobs was 3 × 10−3 s−1 for compound 1 and 3.3 × 10−3 s−1 for compound 2. The kobs values for 1 and 2 in the presence of KBr, KNO3 and NaCl were similar to, or less than, kobs for KCl (see the ESI†). The CH2CH2OSiMe3 C-terminus (in 2) led to higher activity than the tBu C-terminus (in 1), an effect that has previously been noted for other Aib foldamers.18a
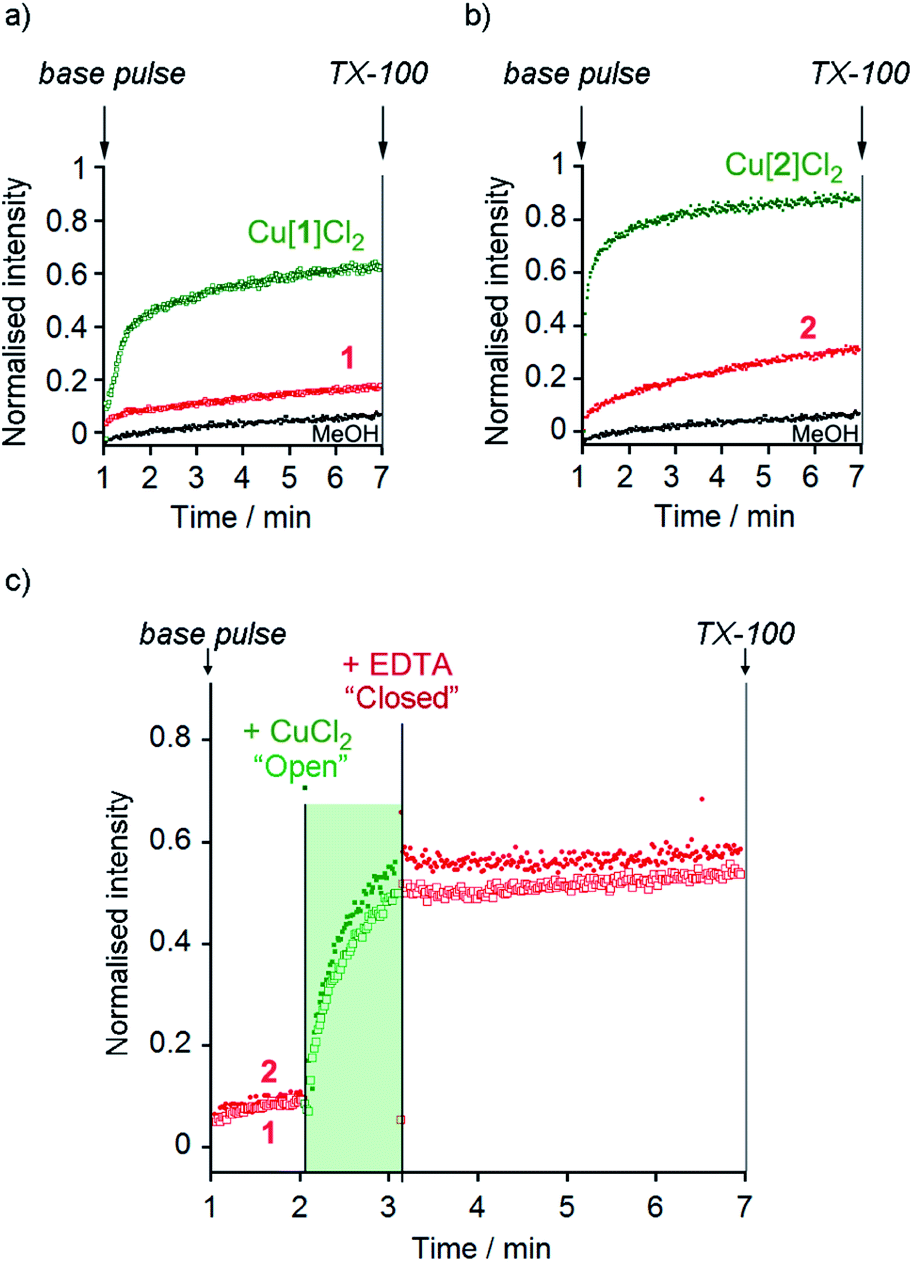 | ||
Fig. 3 (a and b) HPTS assays of (a) MeOH (•), 1 ( ), Cu(II)[1]Cl2 ( ), Cu(II)[1]Cl2 ( ) and (b) MeOH (•), 2 ( ) and (b) MeOH (•), 2 ( ) and Cu(II)[2]Cl2 ( ) and Cu(II)[2]Cl2 ( ) (all 10 μM except Cu(II)[2]Cl2 6 μM) in phospholipid vesicles (760 μM lipid) in the presence of KCl (100 mM). Compounds added at 0 min, base pulse at 1 min, TX-100 added at 7 min allows data normalisation. (c) Switching of ionophoric activity for 1 (10 μM) and 2 (6 μM) in the presence of KCl (100 mM); CuCl2 addition (2 eq.) at 120 s followed by EDTA addition (2.2 eq.) at 180 s. Compounds added at 0 min, base pulse at 1 min, TX-100 added at 7 min allows data normalisation. The corresponding data for the addition of MeOH (20 μL) has been subtracted from these data in (c). ) (all 10 μM except Cu(II)[2]Cl2 6 μM) in phospholipid vesicles (760 μM lipid) in the presence of KCl (100 mM). Compounds added at 0 min, base pulse at 1 min, TX-100 added at 7 min allows data normalisation. (c) Switching of ionophoric activity for 1 (10 μM) and 2 (6 μM) in the presence of KCl (100 mM); CuCl2 addition (2 eq.) at 120 s followed by EDTA addition (2.2 eq.) at 180 s. Compounds added at 0 min, base pulse at 1 min, TX-100 added at 7 min allows data normalisation. The corresponding data for the addition of MeOH (20 μL) has been subtracted from these data in (c). | ||
The Cu(II) complexes displayed markedly higher activity than the parent foldamers; Cu(II)[1]Cl2 (10 μM) showed ca. 5-fold higher ionophoric activity for KCl transport (kobs = 10.8 × 10−3 s−1) than 1 after accounting for background leakage. Cu(II)[2]Cl2 was too active to be accurately measured at 10 μM, therefore the concentration was reduced to 6 μM. Even at this lower concentration, Cu(II)[2]Cl2 showed ca. 8-fold higher ionophoric activity for KCl transport (kobs = 16.5 × 10−3 s−1) than 2 at 10 μM. Ionophoric activity was dependent on oligomer length, with the shorter Cu(II)-Aib tetrameric analogue, Cu(II)[7]Cl2, showing a >50% drop in rate compared to Cu(II)[1]Cl2 (see the ESI†).
Switching of ionophoric activity
The marked difference in activity between 1 and 2 and their corresponding Cu(II) species Cu(II)[1]Cl2 and Cu(II)[2]Cl2 provides the opportunity to “switch” ion transport on and off. “Switching on” of activity should be induced by addition of CuCl2 to compounds 1 and 2, producing an active Cu(II) species in situ. “Switching off” ionophoric activity might then be possible by subsequent extraction of the Cu(II) from foldamer 1 or 2. To demonstrate the switching of ionophoric activity, the HPTS assay was modified so that 2 eq. CuCl2 was added at 2 min, followed by addition of 2.2 eq. EDTA at 3 min. As a control, the addition of CuCl2 to a methanol-containing vesicle suspension (no foldamer present) showed CuCl2 did not significantly affect the HPTS assay or produce leakage (see the ESI†).Both compounds 1 and 2 were “switched on” by CuCl2 addition (Fig. 3c), with a rapid burst of activity that suggests fast ion transport through pores or channels.10e Subsequent EDTA addition promptly “switched off” ion transport, only ten seconds after EDTA addition (Fig. 3c). The rapid “switch off” of activity upon the addition of EDTA suggests that the Cu(II)-foldamer complexes are not fixed at a location deep in the membrane, as EDTA would not be expected to be able to partition deep into the hydrophobic region.
Characterisation of ionophoric activity
Comparison of the rates of cation transport by Cu(II)[2]Cl2 (Li+, Na+, K+ and Rb+, Fig. 4a) and by Cu(II)[1]Cl2 (Na+ and K+, see the ESI†) showed some difference in transport rates for different M+, e.g. kobs for NaCl was 63% the value of kobs for KCl for Cu(II)[2]Cl2. However larger rate differences were observed for the transport of anions (Cl−, Br−, I−, NO3− and SO42−) by Cu(II)[2]Cl2, e.g. kobs for KNO3 was 6% of the value for KCl (Fig. 4b). Cu(II)[1]Cl2 shows similar differences between anions, with kobs for KCl double that of KBr and 5-fold greater than that of KNO3 (see ESI†).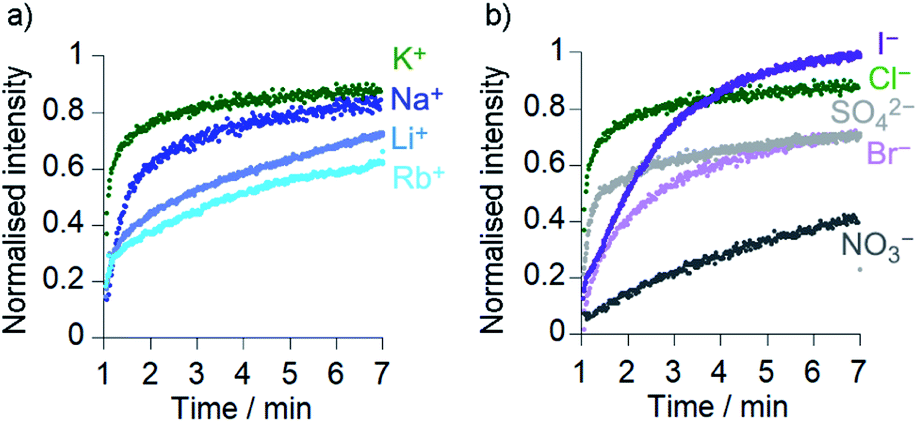 | ||
Fig. 4 Ionophoric activity of Cu(II)[2]Cl2 (6 μM) determined through HPTS assays in the presence of different salts (all 100 mM): (a) KCl ( ), NaCl ( ), NaCl ( ), LiCl ( ), LiCl ( ), RbCl ( ), RbCl ( ); (b) KCl ( ); (b) KCl ( ), KI ( ), KI ( ), K2SO4 ( ), K2SO4 ( ), KBr ( ), KBr ( ), KNO3 (•). Compounds added at 0 min, base pulse at 1 min, TX-100 addition at 7 min allows data normalisation. ), KNO3 (•). Compounds added at 0 min, base pulse at 1 min, TX-100 addition at 7 min allows data normalisation. | ||
The addition of a selective proton transporter, carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) only gave a very small increase in the rate of transport by Cu(II)[1]Cl2 and Cu(II)[2]Cl2 (although significant rate increases were observed for transport by 1 and 2; see the ESI†). The small effect of FCCP on Cu(II)[1]Cl2 and Cu(II)[2]Cl2 activity suggests proton transport is not rate limiting.34
To confirm that chloride could pass through the bilayers, lucigenin assays5a were performed for 2, Cu(II)[1]Cl2 and Cu(II)[2]Cl2 (Fig. 5 and ESI, Fig. S10†). The same lipids were used as for the HPTS assays and lucigenin was encapsulated with NaNO3 in the vesicles. Both Cu(II)[1]Cl2 and Cu(II)[2]Cl2 showed faster Cl− transport than 2, albeit with a smaller difference between 2 and Cu(II)[2]Cl2 than in the HPTS assays (Fig. 5, cf.Fig. 3). The chloride transport activities are lower than those of the thioureas developed by Gale, Davis and co-workers35 but comparable to those of Talukdar and co-workers fumaramides (up to 40% transport after 3 minutes at 30 μM fumaramide).36
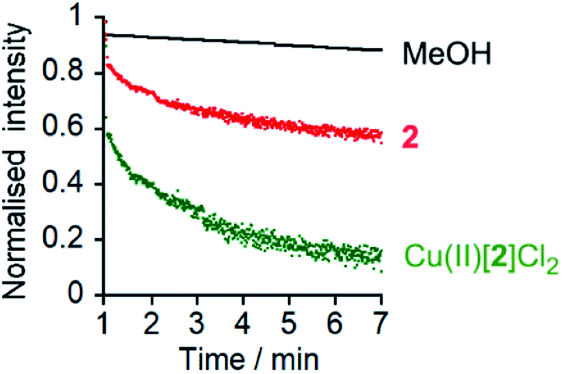 | ||
Fig. 5 Lucigenin assays of Cl− transport by MeOH (•), 2 (10 μM,  ) and Cu(II)[2]Cl2 (10 μM, ) and Cu(II)[2]Cl2 (10 μM,  ) with interior NaNO3 (200 mM) and exterior NaCl (2 M). Compounds added at 0 min, NaCl added at 1 min, TX-100 addition at 7 min allows data normalization. ) with interior NaNO3 (200 mM) and exterior NaCl (2 M). Compounds added at 0 min, NaCl added at 1 min, TX-100 addition at 7 min allows data normalization. | ||
Mechanism of ionophoric activity
Cation/anion carrier,37 ion channel/pore,21a,38 and membrane disruption mechanisms39 have all been observed for Aib-rich ionophores, so a series of assays were performed to determine which mechanism predominated for these foldamers.To assess if these compounds can act as cation carriers, U-tube experiments for sodium picrate transport were performed on 1, Cu(II)[1]Cl2 and Cu(II)[2]Cl2. No transport across the bulk phase was observed for any of these foldamers after 24 h, unlike the dibenzo-18-crown-6 positive control (see the ESI, Fig. S11†). Similarly, a U-tube experiment for chloride transport was performed using lucigenin.4,40 This showed no chloride was carried through the organic phase by 2, Cu(II)[1]Cl2 and Cu(II)[2]Cl2, unlike the 2-aminopentane positive control (see the ESI, Fig. S12†). These assays suggest that these Aib foldamers do not predominantly act via an ion carrier mechanism.
A 5/6-carboxyfluorescein (5/6-CF) release assay18b was then performed to assess if Cu(II)[1]Cl2 and Cu(II)[2]Cl2 disrupt the membrane or form very large pores able to accommodate the passage of 5/6-CF (∼10 Å diameter from molecular modelling).41 Despite high activity in the HPTS assays at 10 μM foldamer, dye release was insignificant in both cases (see the ESI, Fig. S13†), indicating that membrane disruption is not extensive.
To assess if the most active compound, Cu(II)[2]Cl2, and its parent foldamer 2 form channels, planar bilayer conductance (PBC) studies were carried out. The observation of discrete conductance events can indicate channel formation and provide information on the nature of these channels.
PBC experiments were performed in a custom-built cell, with either 2 or Cu(II)[2]Cl2 (5–10 μL of a 1 mM solution in MeOH) added to the ground well. Characterisation sweeps were continued for 2 h, or until substantial channel-forming activity was observed. Single channel experiments were conducted under an applied potential of +100 or −100 mV in 50 s sweeps. Under these conditions, parent foldamer 2 did not display any ion channel behaviour after 2 h under these conditions (see the ESI, Fig. S16†). In contrast, discrete channel-forming behaviour was observed for Cu(II)[2]Cl2 (Fig. 6a), with current levels only slightly higher in KCl than in NaCl. Large current levels and reproducible well-defined quantized steps from 0.5 to 5 ms in duration (similar to the “flicker” behaviour described by Chui and Fyles42) were measured (Fig. 6). Greater increases in macroscopic current value were observed under a negative applied potential rather than a positive applied potential, which could suggest a positive charge is being driven into (or through) the membrane by the applied negative potential; analogous behaviour has been observed for other channel-forming compounds bearing positive charges.43 This could imply [Cu(II)[2]Cl]+ cations44 are involved in the channel-forming species. Channel events have multilevel conductances, both at +100 mV (∼0.08 nS, ∼0.14 nS) and at −100 mV potentials (∼0.08 nS, ∼0.15 nS).
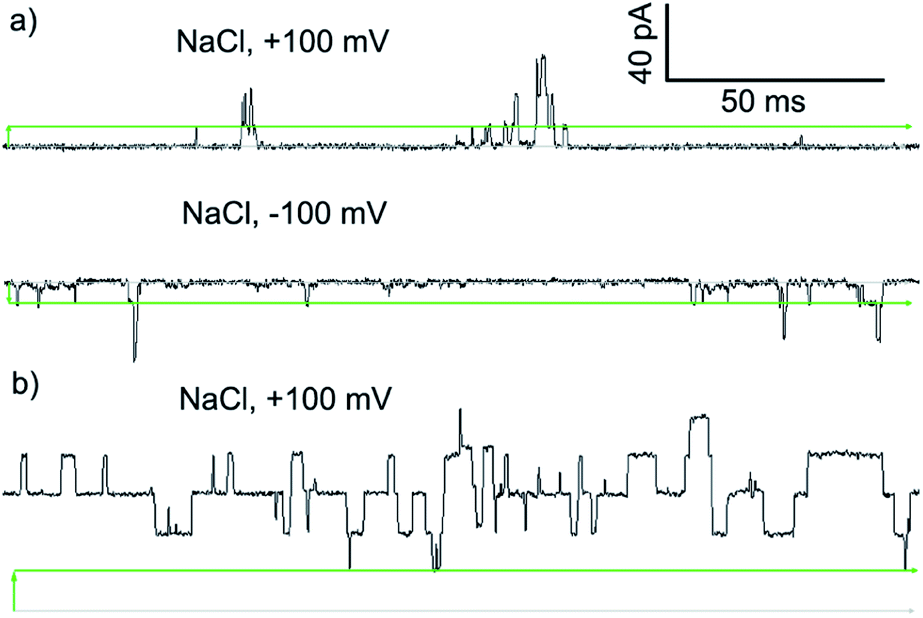 | ||
| Fig. 6 Step changes in conductance induced by Cu(II) complexes of 2 added to cis side of the membrane. Membranes were formed from EYPC lipid/cholesterol (4:1, w/w) in MOPS buffer at room temperature (20 mM MOPS, 100 mM NaCl, pH 7.4). Change from 0 pA (grey bars) indicated by green arrows. Green bars mark the approximate level of a single quantized current-step. (a) Green complex ([Cu(II)[2](μ-Cl)]2Cl2, final concentration 8.3 μM). (b) Blue complex ([Cu(II)[2]Cl]·HCO3, final concentration 8.3 μM). | ||
The I–V curve for Cu(II)[2]Cl2 in KCl is not linear, and shows sharp increases in conductance as the potential is increased to +100 mV or −40 mV with a progressive increase in current after repeated sweeps between these potentials (see the ESI, Fig. S17†). The increase in conductance over time may be due to slow insertion of the foldamer into the membrane, a process that is accelerated as the potential difference increases (especially for negative potentials).45
The blue product from the addition of CuCl to 2 was also assessed using PBC experiments. Under the same conditions used for Cu(II)[2]Cl2, Cu(II)[2]Cl·HCO3 showed a mixture of behaviours. Short-lived “flicker”-type openings could be observed, which displayed conductance levels (0.07 ± 0.01 nS, 0.16 ± 0.01 nS, see the ESI†) similar to those observed for Cu(II)[2]Cl2. In addition, much longer lived “square topped” openings could also be observed (Fig. 6b). These very regular well-defined quantized current steps were open for up to 20 ms at +100 mV, with multiple stepwise increases in conductance with ∼0.18 nS increments.46 A linear increase in the conductance of these current steps was observed with increasingly positive applied potential, suggesting channels with a symmetrical charge distribution are formed (see the ESI†).7b
Application of Hille's equation47 to estimate the inner channel diameters for both Cu(II)[2]Cl2 and Cu(II)[2]Cl·HCO3 provided approximate pore diameters of 1.5 nm, 2.1 nm and 2.3 nm for the different conductance levels (see the ESI†). These diameters are comparable to those proposed for different alamethicin pores (1.1 nm for hexameric pores; 1.8 nm for octameric pores).38,48
Antibiotic activity
Several synthetic ion channels have been shown to exhibit biological activity, most commonly in the form of antibiotic activity, which correlates with ion channel activity in non-biological membranes.8b,10h,14f,49 Long Aibn foldamers (n > 10) show good activity against the Gram-positive B. megaterium strain DSM319.18a N-terminal functionalisation of such Aib foldamers with BPTA will provide a new type of amino terminal Cu(II)- and Ni(II)-binding (ATCUN) motif, which may improve antimicrobial properties.50 Like other ATCUN motifs, the Cu(II) will be tightly complexed in the BPTA pocket of 1 and 2 (the stability constant for Cu(II) with TPA is >1016 M−1),50a,51 preventing the release of potentially toxic free Cu(II) ions into the blood.The antibiotic activities of 1, 2, Cu(II)[1]Cl2 and Cu(II)[2]Cl2 were measured against B. megaterium strain DSM319 (see the ESI†). The complexes Cu(II)[1]Cl2 and Cu(II)[2]Cl2 showed much lower minimum inhibitory concentrations (MICs) than 1 and 2 (Fig. 7a), which correlates inversely with the relative ionophoric activities measured by HPTS assays. Interestingly, the MIC for Cu(II)[2]Cl2 is the same within error as that for alamethicin under the same conditions against this Gram-positive bacterial strain (alamethicin showed a MIC of 6 ± 2 μM, see ESI†).
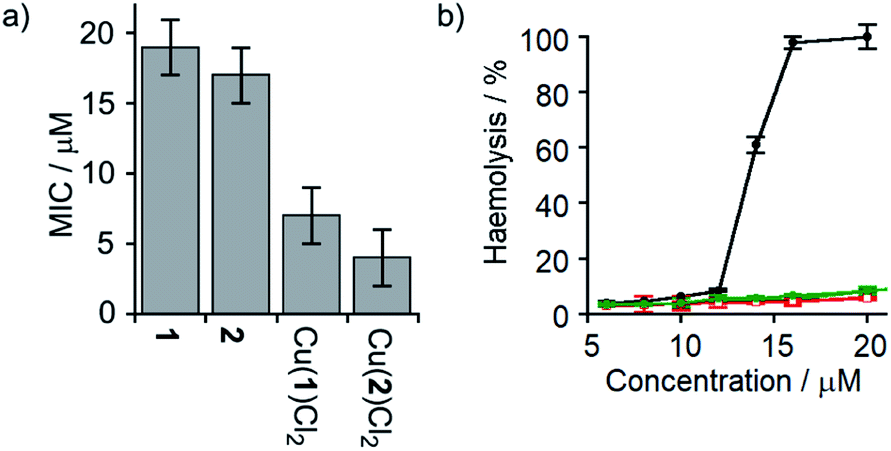 | ||
Fig. 7 (a) MIC against B. megaterium strain DSM319. (b) Haemolysis of human erythrocytes caused by alamethicin (•) and 1 ( ), Cu(II)[1]Cl2 ( ), Cu(II)[1]Cl2 ( ), 2 ( ), 2 ( ) and Cu(II)[2]Cl2 ( ) and Cu(II)[2]Cl2 ( ). ). | ||
Although alamethicin is a very active channel former, its use in the clinic is hampered by its high haemolytic activity.52 Since Cu(II)[2]Cl2 shows similar antibiotic activity to alamethicin and also bears a cationic centre (which often produce haemolysis),53 its haemolytic activity against human erythrocytes was assessed. The dose response curve for haemoglobin release gave an EC50 value for haemolysis by alamethicin of 1.4 μM, similar to its reported EC50 value (1.45 μM in phosphate buffer after 1 h incubation).54 Interestingly, all foldamers showed much lower haemolytic activity under these conditions than alamethicin, although no EC50 values could be determined for the Aib foldamers due to their solubility limits (20 μM for 1 and 2; the more water soluble Cu(II) complexes produced only 10% haemolysis at 25 μM). It is clear however that Cu(II)[1]Cl2 and Cu(II)[2]Cl2 both produce low haemolysis below 20 μM, despite their positively charged centres55 and high ionophoric/antibiotic activity at this concentration. It is known that the haemolytic activity of amphipathic peptides, including alamethicin, decreases substantially on a reduction of peptide hydrophobicity and hydrophobic moment.56 We suggest that the lower haemolytic activity of Cu(II)[1]Cl2 and Cu(II)[2]Cl2 compared to alamethicin is due to their lower hydrophobicity, arising from the shorter length and the cationic N-termini of the Cu(II) complexes of 1 and 2, yet ionophoric activity is maintained due to stronger interactions between foldamers.
Conclusions
The chelating Aib foldamers described here show remarkably efficient “switching” of ion transport activity. As observed for unfunctionalised Aib octamers,18b BPTA-capped Aib octamers showed measurable ionophoric activity at micromolar concentrations. However complexation to CuCl2 markedly increased this ionophoric activity, with the Cu(II) complexes showing 5- to 8-fold increases in activity compared to the parent octamers. There was also clear length dependence, with a shorter Aib tetramer showing markedly lower activity. Complexation of Cu(II) was reversible, allowing the ionophoric activity of the Aib octamers 1 and 2 to be activated in situ by addition of CuCl2 and deactivated by extraction of the Cu(II) by EDTA. The ionophoric activity of the CuCl2 complex of 2 approaches that measured for the archetypical peptaibol alamethicin.18b Channels formed by these CuCl2/Aib octamer complexes appear to transport both cations and anions, and showed good chloride transport activity in lucigenin assays. PBC studies showed square-topped conductance profiles for [Cu(II)[2](μ-Cl)]2Cl2 and [Cu(II)[2]Cl]·HCO3 but no conductance for the parent foldamer 2, consistent with multimeric ion channel formation by the Cu(II) complexes.The observation of hydrogen bonds and Cu–Cl–Cu bridges between foldamers in the solid state (Fig. 2b) indicates the Cu(II) complexes of the foldamers can form multiple strong intermolecular interactions. These interactions will promote self-assembly of monomeric foldamers into multimeric channels within the membrane, perhaps aided by the absence of competing ligands like water and the high effective concentrations that result after partitioning into the membrane.57 The solid-state structure of [Cu(II)[2]Cl]·HCO3 also suggests the Aib foldamer body has a role in facilitating the passage of ions through the bilayer, and may favour one ion over another. This structure shows that the bicarbonate counterion is not bound to the Cu(II) centre but is located behind the headgroup, where it is hydrogen-bonded to the Aib foldamer body.
The antibiotic activities of 1, 2, Cu(II)[1]Cl2 and Cu(II)[2]Cl2 correlate well with their relative ion channel activity. The MICs of Cu(II)[1]Cl2 and Cu(II)[2]Cl2 were similar to that of alamethicin. However, these ionophores did not show the high haemolytic activity of alamethicin, a significant barrier to the adoption of peptaibols in the clinic.
These Aib foldamers show remarkably efficient switching of channelling activity upon Cu(II) complexation/decomplexation. This activity switching in synthetic membranes, although not easily applied in vivo, may indicate a pathway towards switchable non-haemolytic peptaibol antibiotics and switchable drugs for treating the symptoms of channelopathies.
Author contributions
ADP and SJW conceived the study. ADP and ML performed the synthesis with advice from JC. GFSW and IJV-Y carried out the crystallography. ADP and FdS performed the ionophoric activity assays in vesicles. SB, DFC-G and SLC performed and analysed the PBC assays. ADP and ET carried out the antibiotic studies. ADP and JB carried out the haemolysis assays. ADP, FdS and SJW wrote the manuscript with input from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a Colloid Science PhD studentship from the School of Chemistry (University of Manchester, UK), the EPRSC (grants EP/P027067/1, EP/N009134/1 and EP/K039547), and an Erasmus+ Traineeship. We thank Prof. D. Collison and Dr A. Baldansuren from the EPSRC National Service for Electron Paramagnetic Resonance Spectroscopy for use of facilities and support. We thank Mrs R. Sung for her guidance in developing HPLC protocols. We also thank Dr R. Spiess and the Mass Spectrometry Service in the School of Chemistry (University of Manchester) for mass spectrometry support. Prof. Takano was funded by the European Union's Horizon 2020 Research and Innovation Programme (Grant Agreement No. 720793, H2020 TOPCAPI-thoroughly Optimised Production Chassis for Advanced Pharmaceutical Ingredients project). Prof. Cockroft was funded by the Leverhulme Trust (Philip Leverhulme Prize) and the European Research Council (ERC) under the European Union's Seventh Framework Programme (Grant agreement No. 336935 TransPoreT). We would like to thank Dr K. A. P. Payne and Prof. D. Leys for providing B. megaterium strain DSM319 and Dr S. Y. Huang and Ms. K. Baker for training in antibiotic assays and use of facilities.References
- (a) N. Unwin, Neuron, 1989, 3, 665–676 CrossRef CAS PubMed; (b) C. A. Hübner and T. J. Jentsch, Hum. Mol. Genet., 2002, 11, 2435–2445 CrossRef PubMed.
- L. J. Ptáček, Neuromuscular Disord., 1997, 7, 250–255 CrossRef.
- D. C. Gadsby, P. Vergani and L. Csanády, Nature, 2006, 440, 477–483 CrossRef CAS PubMed.
- S. Matile, N. Sakai and A. Hennig, Transport Experiments in Membranes, in Supramolecular Chemistry: From Molecules to Nanomaterials, John Wiley & Sons, Ltd, 2012 Search PubMed.
- (a) A. P. Davis, D. N. Sheppard and B. D. Smith, Chem. Soc. Rev., 2007, 36, 348–357 RSC; (b) N. Busschaert, S.-H. Park, K.-H. Baek, Y. P. Choi, J. Park, E. N. W. Howe, J. R. Hiscock, L. E. Karagiannidis, I. Marques, V. Félix, W. Namkung, J. L. Sessler, P. A. Gale and I. Shin, Nat. Chem., 2017, 9, 667–675 CrossRef CAS PubMed; (c) P. A. Gale, J. T. Davis and R. Quesada, Chem. Soc. Rev., 2017, 46, 2497–2519 RSC; (d) H. Li, H. Valkenier, L. W. Judd, P. R. Brotherhood, S. Hussain, J. A. Cooper, O. Jurček, H. A. Sparkes, D. N. Sheppard and A. P. Davis, Nat. Chem., 2016, 8, 24–32 CrossRef CAS PubMed; (e) I. Alfonso and R. Quesada, Chem. Sci., 2013, 4, 3009–3019 RSC; (f) H. Behera and N. Madhavan, J. Am. Chem. Soc., 2017, 139, 12919–12922 CrossRef CAS PubMed.
- K. A. Muraglia, R. S. Chorghade, B. R. Kim, X. X. Tang, V. S. Shah, A. S. Grillo, P. N. Daniels, A. G. Cioffi, P. H. Karp, L. Zhu, M. J. Welsh and M. D. Burke, Nature, 2019, 567, 405–408 CrossRef CAS PubMed.
- (a) E. Martens and A. L. Demain, J. Antibiot., 2017, 70, 520–526 CrossRef CAS PubMed; (b) T. M. Fyles, Chem. Soc. Rev., 2007, 36, 335–347 RSC.
- (a) P. Reiss and U. Koert, Acc. Chem. Res., 2013, 46, 2773–2780 CrossRef CAS PubMed; (b) G. W. Gokel and S. Negin, Acc. Chem. Res., 2013, 46, 2824–2833 CrossRef CAS PubMed; (c) N. Sakai and S. Matile, Langmuir, 2013, 29, 9031–9040 CrossRef CAS PubMed; (d) W. Si, P. Xin, Z.-T. Li and J.-L. Hou, Acc. Chem. Res., 2015, 48, 1612–1619 CrossRef CAS PubMed; (e) W.-X. Feng, Z. Sun and M. Barboiu, Isr. J. Chem., 2018, 58, 1209–1218 CrossRef.
- (a) N. Rodríguez-Vázquez, A. Fuertes, M. Amorín and J. R. Granja, Met. Ions Life Sci., 2016, 16, 485–556 Search PubMed; (b) C. Lang, X. Deng, F. Yang, B. Yang, W. Wang, S. Qi, X. Zhang, C. Zhang, Z. Dong and J. Liu, Angew. Chem., Int. Ed., 2017, 56, 12668–12671 CrossRef CAS PubMed.
- (a) B. P. Benke, P. Aich, Y. Kim, K. L. Kim, M. R. Rohman, S. Hong, I.-C. Hwang, E. H. Lee, J. H. Roh and K. Kim, J. Am. Chem. Soc., 2017, 139, 7432–7435 CrossRef CAS PubMed; (b) V. Sidorov, F. W. Kotch, G. Abdrakhmanova, R. Mizani, J. C. Fettinger and J. T. Davis, J. Am. Chem. Soc., 2002, 124, 2267–2278 CrossRef CAS PubMed; (c) X. Li, B. Shen, X.-Q. Yao and D. Yang, J. Am. Chem. Soc., 2007, 129, 7264–7265 CrossRef CAS PubMed; (d) V. Gorteau, G. Bollot, J. Mareda, A. Perez-Velasco and S. Matile, J. Am. Chem. Soc., 2006, 128, 14788–14789 CrossRef CAS PubMed; (e) V. Gorteau, G. Bollot, J. Mareda and S. Matile, Org. Biomol. Chem., 2007, 5, 3000–3012 RSC; (f) A. Hennig, L. Fischer, G. Guichard and S. Matile, J. Am. Chem. Soc., 2009, 131, 16889–16895 CrossRef CAS PubMed; (g) M. Vidal and A. Schmitzer, Chem.–Eur. J., 2014, 20, 9998–10004 CrossRef CAS PubMed; (h) T. Saha, A. Gautam, A. Mukherjee, M. Lahiri and P. Talukdar, J. Am. Chem. Soc., 2016, 138, 16443–16451 CrossRef CAS PubMed; (i) L. Yuan, J. Shen, R. Ye, F. Chen and H. Zeng, Chem. Commun., 2019, 55, 4797–4800 RSC.
- (a) R. Kawano, ChemPhysChem, 2018, 19, 359–366 CrossRef CAS PubMed; (b) T. Muraoka, K. Umetsu, K. V. Tabata, T. Hamada, H. Noji, T. Yamashita and K. Kinbara, J. Am. Chem. Soc., 2017, 139, 18016–18023 CrossRef CAS PubMed; (c) T. Muraoka, T. Endo, K. V. Tabata, H. Noji, S. Nagatoishi, K. Tsumoto, R. Li and K. Kinbara, J. Am. Chem. Soc., 2014, 136, 15584–15595 CrossRef CAS PubMed; (d) S.-P. Zheng, L.-B. Huang, Z. Sun, and M. Barboiu, Angew. Chem., Int. Ed. DOI:10.1002/anie.201915287.
- J.-Y. Chen and J.-L. Hou, Org. Chem. Front., 2018, 5, 1728–1736 RSC.
- (a) P. Talukdar, G. Bollot, J. Mareda, N. Sakai and S. Matile, J. Am. Chem. Soc., 2005, 127, 6528–6529 CrossRef CAS PubMed; (b) P. Talukdar, G. Bollot, J. Mareda, N. Sakai and S. Matile, Chem.–Eur. J., 2005, 11, 6525–6532 CrossRef CAS PubMed.
- (a) U. Devi, J. R. D. Brown, A. Almond and S. J. Webb, Langmuir, 2011, 27, 1448–1456 CrossRef CAS PubMed; (b) M. Boccalon, E. Iengo and P. Tecilla, J. Am. Chem. Soc., 2012, 134, 20310–20313 CrossRef CAS PubMed; (c) M. Jung, H. Kim, K. Baek and K. Kim, Angew. Chem., Int. Ed., 2008, 47, 5755–5757 CrossRef CAS PubMed; (d) L. J. Lalgee, L. Grierson, R. A. Fairman, G. E. Jaggernauth, A. Schulte, R. Benz and M. Winterhalter, Biochim. Biophys. Acta, Biomembr., 2014, 1838, 1247–1254 CrossRef CAS PubMed; (e) C. J. E. Haynes, J. Zhu, C. Chimerel, S. Hernández-Ainsa, I. A. Riddell, T. K. Ronson, U. F. Keyser and J. R. Nitschke, Angew. Chem., Int. Ed., 2017, 56, 15388–15392 CrossRef CAS PubMed; (f) J. Kempf and A. R. Schmitzer, Chem.–Eur. J., 2017, 23, 6441–6451 CrossRef CAS PubMed.
- C. P. Wilson and S. J. Webb, Chem. Commun., 2008, 4007–4009 RSC.
- C. P. Wilson, C. Boglio, L. Ma, S. L. Cockroft and S. J. Webb, Chem.–Eur. J., 2011, 17, 3465–3473 CrossRef CAS PubMed.
- M. M. Tedesco, B. Ghebremariam, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 1999, 38, 540–543 CrossRef CAS PubMed.
- (a) C. Adam, A. D. Peters, M. G. Lizio, G. F. S. Whitehead, V. Diemer, J. A. Cooper, S. L. Cockroft, J. Clayden and S. J. Webb, Chem.–Eur. J., 2018, 24, 2249–2256 CrossRef CAS PubMed; (b) J. E. Jones, V. Diemer, C. Adam, J. Raftery, R. E. Ruscoe, J. T. Sengel, M. I. Wallace, A. Bader, S. L. Cockroft, J. Clayden and S. J. Webb, J. Am. Chem. Soc., 2016, 138, 688–695 CrossRef CAS PubMed.
- (a) M. De Poli, W. Zawodny, O. Quinonero, M. Lorch, S. J. Webb and J. Clayden, Science, 2016, 352, 575–580 CrossRef CAS PubMed; (b) F. G. A. Lister, B. A. F. Le Bailly, S. J. Webb and J. Clayden, Nat. Chem., 2017, 9, 420–425 CrossRef CAS.
- (a) C. Toniolo, M. Crisma, F. Formaggio and C. Peggion, Biopolymers, 2001, 60, 396–419 CrossRef CAS PubMed; (b) J. Venkatraman, S. C. Shankaramma and P. Balaram, Chem. Rev., 2001, 101, 3131–3152 CrossRef CAS PubMed; (c) M. De Poli, M. De Zotti, J. Raftery, J. A. Aguilar, G. A. Morris and J. Clayden, J. Org. Chem., 2013, 78, 2248–2255 CrossRef CAS PubMed; (d) S. J. Pike, T. Boddaert, J. Raftery, S. J. Webb and J. Clayden, New J. Chem., 2015, 39, 3288–3294 RSC; (e) M. G. Lizio, V. Andrushchenko, S. J. Pike, A. D. Peters, G. F. S. Whitehead, I. J. Vitórica-Yrezábal, S. T. Mutter, J. Clayden, P. Bouř, E. W. Blanch and S. J. Webb, Chem.–Eur. J., 2018, 24, 9399–9408 CrossRef CAS PubMed; (f) C. M. Venkatachalam, Biopolymers, 1968, 6, 1425–1436 CrossRef CAS PubMed.
- (a) S. Futaki, D. Noshiro, T. Kiwada and K. Asami, Acc. Chem. Res., 2013, 46, 2924–2933 CrossRef CAS PubMed; (b) G. A. Woolley and B. A. Wallace, J. Membr. Biol., 1992, 129, 109–136 CAS.
- X. Wang, R. J. Mart and S. J. Webb, Org. Biomol. Chem., 2007, 5, 2498–2505 RSC.
- N. Eccles, B. A. F. Le Bailly, F. della Sala, I. J. Vitórica-Yrezábal, J. Clayden and S. J. Webb, Chem. Commun., 2019, 55, 9331–9334 RSC.
- (a) W. Xu, J. A. Craft, P. R. Fontenot, M. Barens, K. D. Knierim, J. H. Albering, F. A. Mautner and S. S. Massoud, Inorg. Chim. Acta, 2011, 373, 159–166 CrossRef CAS; (b) K. D. Karlin, J. C. Hayes, S. Juen, J. P. Hutchinson and J. Zubieta, Inorg. Chem., 1982, 21, 4106–4108 CrossRef CAS.
- D. Leibfritz, E. Haupt, N. Dubischar, H. Lachmann, R. Oekonomopulos and G. Jung, Tetrahedron, 1982, 38, 2165–2181 CrossRef CAS.
- V. D. Bock, H. Hiemstra and J. H. Van Maarseveen, Eur. J. Org. Chem., 2006, 51–68 CrossRef CAS.
- A. Kaur, T. G. Ribelli, K. Schröder, K. Matyjaszewski and T. Pintauer, Inorg. Chem., 2015, 54, 1474–1486 CrossRef CAS PubMed.
- K. D. Karlin, N. Wei, B. Jung, S. Kaderli, P. Niklaus and A. D. Zuberbühler, J. Am. Chem. Soc., 1993, 115, 9506–9514 CrossRef CAS.
- (a) N. Kučerka, S. Tristram-Nagle and J. F. Nagle, J. Membr. Biol., 2006, 208, 193–202 CrossRef PubMed; (b) J. Gallová, D. Uhríková, N. Kučerka, M. Svorková, S. S. Funari, T. N. Murugova, L. Almásy, M. Mazúr and P. Balgavý, J. Membr. Biol., 2011, 243, 1–13 CrossRef PubMed.
- N. A. C. dos Santos, F. Lorandi, E. Badetti, K. Wurst, A. A. Isse, A. Gennaro, G. Licini and C. Zonta, Polymer, 2017, 128, 169–176 CrossRef.
- The complex of Zn(ClO4)2 with 2 has a similar structure but with water coordinated in the place of chloride. See: A. D. Peters, G. F. S. Whitehead and S. J. Webb, CCDC 1969243, CSD Communication, 2019, DOI:10.5517/ccdc.csd.cc2434zd.
- A. F. DiGiorgio, S. Otto, P. Bandyopadhyay and S. L. Regen, J. Am. Chem. Soc., 2000, 122, 11029–11030 CrossRef CAS.
- S. M. Elvington and J. W. Nichols, Biochim. Biophys. Acta, Biomembr., 2007, 1768, 502–508 CrossRef CAS PubMed.
- N. Sakai and S. Matile, J. Phys. Org. Chem., 2006, 19, 452–460 CrossRef CAS.
- S. J. Edwards, H. Valkenier, N. Busschaert, P. A. Gale and A. P. Davis, Angew. Chem., Int. Ed., 2015, 54, 4592–4596 CrossRef CAS PubMed.
- A. Roy, A. Gautam, J. A. Malla, S. Sarkar, A. Mukherjee and P. Talukdar, Chem. Commun., 2018, 54, 2024–2027 RSC.
- H. Duclohier, C. F. Snook and B. A. Wallace, Biochim. Biophys. Acta, Biomembr., 1998, 1415, 255–260 CrossRef CAS.
- S. Qian, W. Wang, L. Yang and H. W. Huang, Biophys. J., 2008, 94, 3512–3522 CrossRef CAS PubMed.
- M. Eid, S. Rippa, S. Castano, B. Desbat, J. Chopineau, C. Rossi and L. Béven, J. Biophys., 2010, 179641 Search PubMed.
- J. Gravel and A. R. Schmitzer, J. Supramol. Chem., 2015, 27, 364–371 CrossRef CAS.
- R. Pajewski, R. Ferdani, J. Pajewska, N. Djedovič, P. H. Schlesinger and G. W. Gokel, Org. Biomol. Chem., 2005, 3, 619–625 RSC.
- J. K. W. Chui and T. M. Fyles, Chem. Soc. Rev., 2012, 41, 148–175 RSC.
- (a) R. U. Muller and C. S. Peskin, J. Gen. Physiol., 1981, 78, 201–229 CrossRef CAS PubMed; (b) W. Si, Z.-T. Li and J.-L. Hou, Angew. Chem., Int. Ed., 2014, 53, 4578–4581 CrossRef CAS PubMed.
- W. T. Eckenhoff and T. Pintauer, Inorg. Chem., 2007, 46, 5844–5846 CrossRef CAS PubMed.
- R. U. Muller and O. S. Andersen, J. Gen. Physiol., 1982, 80, 427–449 CrossRef CAS PubMed.
- We speculate that this complex may be more soluble in buffer than Cu(II)[2]Cl2, leading to higher membrane loadings that produce longer-lived openings of larger, higher nuclearity pores.
- (a) B. Hille, Ionic Channels of Excitable Membranes, Sinauer, Sunderland, MA, 3rd edn, 2001 Search PubMed; (b) O. S. Smart, J. Breed, G. R. Smith and M. S. P. Sansom, Biophys. J., 1997, 72, 1109–1126 CrossRef CAS PubMed.
- P. Pieta, J. Mirza and J. Lipkowski, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 21223–21227 CrossRef CAS PubMed.
- (a) C. Ren, X. Ding, A. Roy, J. Shen, S. Zhou, F. Chen, S. F. Y. Li, H. Ren, Y. Y. Yang and H. Zeng, Chem. Sci., 2018, 9, 4044–4051 RSC; (b) J. W. Meisel, M. B. Patel, E. Garrad, R. A. Stanton and G. W. Gokel, J. Am. Chem. Soc., 2016, 138, 10571–10577 CrossRef CAS PubMed; (c) M. Zhang, P.-P. Zhu, P. Xin, W. Si, Z.-T. Li and J.-L. Hou, Angew. Chem., Int. Ed., 2017, 56, 2999–3003 CrossRef CAS PubMed.
- (a) C. Harford and B. Sarkar, Acc. Chem. Res., 1997, 30, 123–130 CrossRef CAS; (b) M. D. Libardo, J. L. Cervantes, J. C. Salazar and A. M. Angeles-Boza, ChemMedChem, 2014, 9, 1892–1901 CAS; (c) J. L. Alexander, Z. Thompson and J. A. Cowan, ACS Chem. Biol., 2018, 13, 844–853 CrossRef CAS PubMed.
- G. Anderegg, E. Hubmann, N. G. Podder and F. Wenk, Helv. Chim. Acta, 1977, 60, 123–140 CrossRef CAS.
- (a) B. Leitgeb, A. Szekeres, L. Manczinger, C. Vágvölgyi and L. Kredics, Chem. Biodiversity, 2007, 4, 1027–1051 CrossRef CAS PubMed; (b) A. Taylor, Proc. N. S. Inst. Sci., 1986, 36, 27–58 Search PubMed; (c) P. Dong, Y. Zhou, W. He and D. Hua, Chem. Commun., 2016, 52, 896–899 RSC.
- T. Kondo and M. Tomizawa, J. Pharm. Sci., 1969, 58, 1378–1381 CrossRef CAS PubMed.
- G. Irmscher and G. Jung, Eur. J. Biochem., 1977, 80, 165–174 CrossRef CAS PubMed.
- E. J. Helmerhorst, I. M. Reijnders, W. van't Hof, E. C. I. Veerman and A. V. Nieuw Amerongen, FEBS Lett., 1999, 449, 105–110 CrossRef CAS PubMed.
- M. Dathe, C. Kaduk, E. Tachikawa, M. F. Melzig, H. Wenschuh and M. Bienert, Biochim. Biophys. Acta, 1998, 1370, 175–183 CrossRef CAS.
- E. L. Doyle, C. A. Hunter, H. C. Phillips, S. J. Webb and N. H. Williams, J. Am. Chem. Soc., 2003, 125, 4593–4599 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and compound characterisation. HPTS and PBC procedures and data. Antibacterial and haemolysis procedures. X-ray crystallography data. CCDC 1999499–1999501. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02393k |
| This journal is © The Royal Society of Chemistry 2020 |