
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCobalt-catalyzed aminocarbonylation of (hetero)aryl halides promoted by visible light†
Alexander M.
Veatch
and
Erik J.
Alexanian
 *
*
Department of Chemistry, The University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, USA. E-mail: eja@email.unc.edu
First published on 22nd June 2020
Abstract
The catalytic aminocarbonylation of (hetero)aryl halides is widely applied in the synthesis of amides but relies heavily on the use of precious metal catalysis. Herein, we report an aminocarbonylation of (hetero)aryl halides using a simple cobalt catalyst under visible light irradiation. The reaction extends to the use of (hetero)aryl chlorides and is successful with a broad range of amine nucleophiles. Mechanistic investigations are consistent with a reaction proceeding via intermolecular charge transfer involving a donor–acceptor complex of the substrate and cobaltate catalyst.
Introduction
Carbonylative transformations of (hetero)aryl halides are widely utilized across chemical synthesis for the preparation of functionalized aromatics.1 The aminocarbonylation of (hetero)aryl halides is a prototypical example in the class, first reported by Heck in 1974 using palladium catalysis.2 The significant value of this transformation in amide synthesis has since led to the development of a range of novel palladium-catalyzed aminocarbonylations of broad scope.3 While often efficient, the use of expensive palladium catalysts and phosphine ligands in these reactions is suboptimal. The continued lack of catalytic systems for aminocarbonylation featuring inexpensive, first-row metals hampers further application of this important synthetic process.4Metal carbonyl complexes (e.g., Co2(CO)8) have been shown to catalyze carbonylative transformations of (hetero)aryl halides using phase-transfer catalysis or visible light promotion, most notably in alkoxycarbonylations.5 In these reactions, the strongly basic reaction conditions are proposed to generate tetracarbonyl cobaltate (Co(CO)4−) in situ as the active catalyst.6 However, the requirement of alkoxides presents a challenge in the use of other nucleophiles in this manifold.5 Indeed, there is scant precedent for intermolecular aminocarbonylation using cobalt catalysis (Fig. 1).5a Moreover, the mechanistic details concerning the activation of the (hetero)aryl substrate by the cobaltate catalyst remain unknown.5c
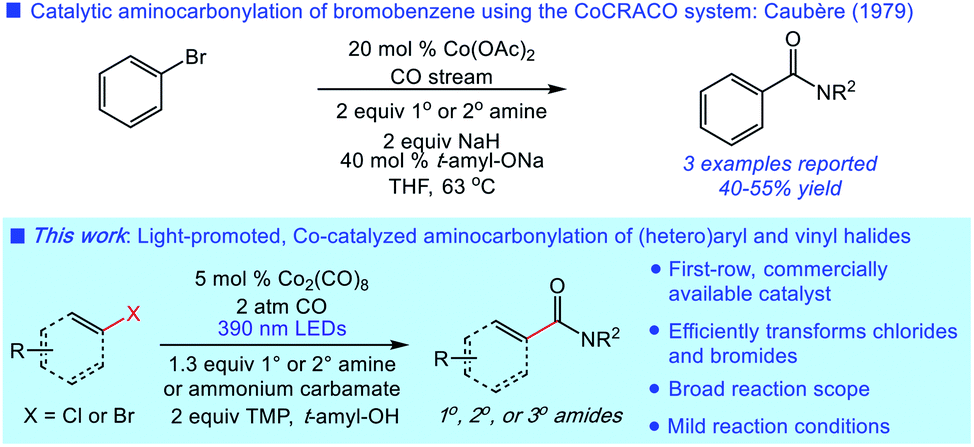 | ||
| Fig. 1 Cobalt-catalyzed aminocarbonylations of sp2 C–X electrophiles. | ||
We have recently developed a mild system for the generation of cobaltate ion from Co2(CO)8 for the catalytic carbonylative functionalization of alkyl electrophiles.7 We hypothesized that this system could also unlock a general, cobalt-catalyzed aminocarbonylation of (hetero)aryl electrophiles. Herein, we report the successful development of such a process using a broad range of electrophiles-including (hetero)aryl chlorides – and diverse amine nucleophiles. Initial mechanistic studies support a visible light-promoted electron-transfer of a donor–acceptor complex involving the substrate and cobaltate in the critical activation step.
Results and discussion
Our studies commenced with the aminocarbonylation of 1-bromo-4-methoxybenzene (1) using piperidine as the nucleophile. The use of simple carbonyl complex Co2(CO)8 in conjunction with 2 equiv. of tetramethylpiperidine (TMP) under visible light irradiation (390 nm LEDs) delivered the desired aminocarbonylation product in virtually quantitative isolated yield (Table 1, entry 1). As expected, the substitution of 10 mol% of a cobaltate catalyst was equally effective (entry 2). The use of either Mn2(CO)10 or CoCl2 instead of Co2(CO)8 was not effective in the aminocarbonylation (entries 3 and 4). Switching from violet LEDs (390 nm) to either blue LEDs (427 nm), ambient light, or dark conditions significantly lowered the yield (entries 5–7). The use of a balloon of CO (1 atm) instead of 2 atm CO was suboptimal, although the reaction proceeded in 58% yield (entry 8). Decreasing the reaction time to 20 h led to incomplete conversion (entry 9). Control experiments indicated that removing TMP lowered reaction efficiency (entry 10), and no reaction took place in the absence of Co2(CO)8 (entry 11).|
|
||
|---|---|---|
| Entry | Variation from standard conditions above | Yieldb (%) |
| a Reactions were performed with [1]0 = 0.40 M. b Yields determined by 1H NMR spectroscopy of crude reaction mixture using an internal standard. c Isolated yield. | ||
| 1 | None | 99c |
| 2 | 10 mol% K Co(CO)4 instead of Co2(CO)8 | 99 |
| 3 | 10 mol% Mn2(CO)10 instead of Co2(CO)8 | <2 |
| 4 | 10 mol% CoCl2 instead of Co2(CO)8 | <2 |
| 5 | 427 nm LEDs instead of 390 nm LEDs | 10 |
| 6 | Ambient light, 50 °C instead of 390 nm LEDs | <2 |
| 7 | No light instead of 390 nm LEDs | <2 |
| 8 | 1 atm CO (balloon) instead of 2 atm CO | 58 |
| 9 | 20 h instead of 48 h | 73 |
| 10 | No TMP | 69 |
| 11 | No Co2(CO)8 | <2 |
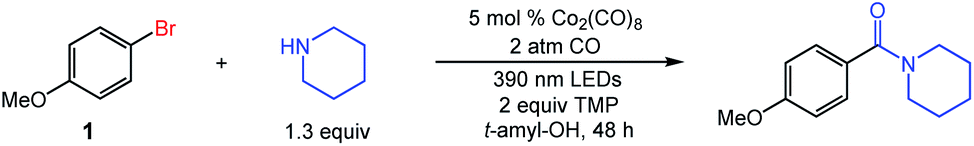
A diverse array of (hetero)aryl and vinyl electrophiles are excellent substrates for the aminocarbonylation, as demonstrated in Fig. 2 using piperidine as a representative nucleophile. Aminocarbonylations of electron-rich and electronically neutral aryl electrophiles proceed in good to quantitative yields across a range of substitution patterns, as demonstrated by the reactions of substrates 1–14. Aryl triflates react comparably to aryl bromides, as indicated by the quantitative aminocarbonylation of substrate 8. For this substrate and a few others, irradiation at 370 nm slightly improved reaction efficiency. Electron-poor aryl bromides are also good coupling partners in the aminocarbonylation (substrates 15–18), with the decreased yield of aryl nitrile 18 attributed to undesired reactivity of the nitrile functionality. Cyclooctenyl bromide 19 and (E)-(2-bromovinyl)benzene 20 delivered aminocarbonylation products in 94% and 50% yield (76![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :24 E:Z), respectively, indicating the viability of vinyl halide substrates.
:24 E:Z), respectively, indicating the viability of vinyl halide substrates.
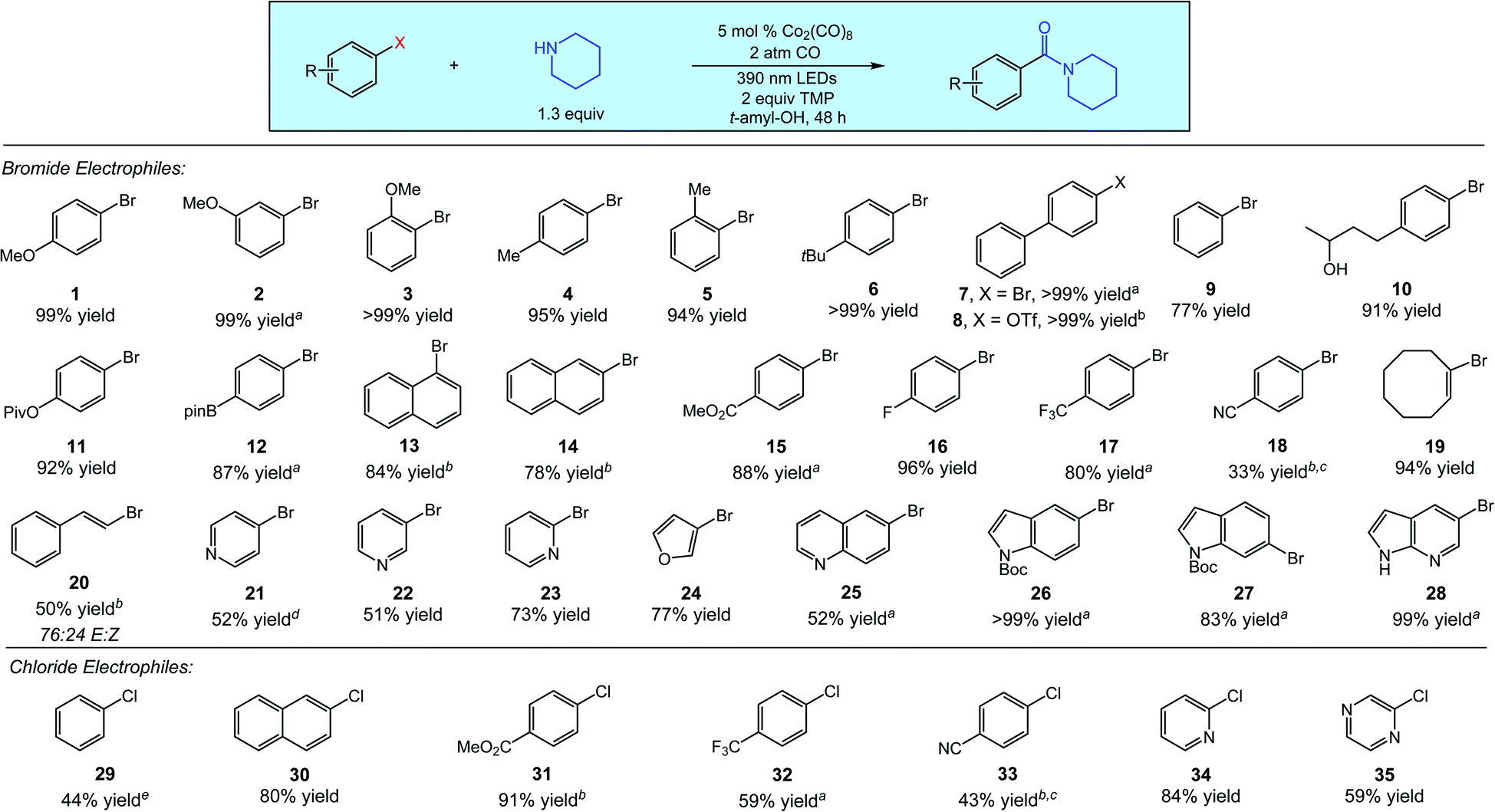 | ||
| Fig. 2 Aminocarbonylation of diverse (hetero)aryl electrophiles. a10 mol% Co2(CO)8 used as catalyst. b10 mol% Co2(CO)8 used as catalyst under 370 nm LEDs. cCyclohexylamine used as the nucleophile, reaction time of 24 h. dSubstrate HCl salt used, 3 equiv. of TMP used as base. e20 mol% Co2(CO)8 used as catalyst, reaction time of 144 h. | ||
We additionally surveyed several classes of common heterocyclic bromides, with the aminocarbonylations proceeding in good to excellent yields. Bromopyridines (substrates 21–23), 3-bromofuran (24), and 6-bromoquinoline (25) all delivered the corresponding amide products in good yield. We also examined the reactions of a set of bromo(aza)indoles (substrates 26–28) which were similarly successful. We additionally performed the aminocarbonylation of 1-bromo-4-methoxybenzene (1) on a one-gram scale in 96% isolated yield with no change in reaction conditions. In short, Fig. 2 (top) clearly indicates that the cobalt-catalyzed aminocarbonylations of (hetero)aryl and vinyl bromides proceed with notable efficiency across a broad range of important substrates.
Owing to their wider availability and lower cost than other electrophiles, (hetero)aryl chlorides are attractive substrates for carbonylative processes. These substrates also pose significant limitations in palladium-catalyzed carbonylations, as the presence of CO is known to present significant challenges in the catalytic activation of these substrates.1a,3a We next explored the use of (hetero)aryl chlorides in the aminocarbonylation (Fig. 2, bottom). While the overall levels of reactivity were lower than that of bromides, promising results were obtained across several substrates. The aminocarbonylation of chlorobenzene (29) proceeded in moderate yield (44%) after an extended reaction time and an increase in catalyst loading, while the reaction of 2-chloronaphthalene (30) proceeded under standard conditions in good yield (80%). The aminocarbonylations of electron-poor aryl chlorides were similar to their bromide counterparts (substrates 31–33), whereas reactions of electron-rich aryl chlorides gave poor conversions and are a current limitation likely owing to higher reduction potentials. As expected, heteroaryl chlorides also reacted efficiently, as 2-chloropyridine (34) and 2-chloropyrazine (35) delivered aminocarbonylation products in 84% and 59% yield, respectively.
We continued by evaluating the scope of the amine nucleophiles permissible in the aminocarbonylation, which was also quite broad. Cyclic and acyclic secondary amines afforded benzamides 36–38 in good to excellent yield. Primary amines were also useful coupling partners, as demonstrated by the synthesis of products 39–44 using a diverse set of common amine nucleophiles (Fig. 3). We have also obtained a promising preliminary result indicating the potential for the preparation of primary amides using our system. Aminocarbonylation using ammonium carbamate as an in situ source of ammonia produced amide 45 in moderate yield (46%). Notably, the catalytic carbonylative coupling of (hetero)aryl electrophiles with ammonia remains a significant challenge in catalysis, with few successful systems to date even involving precious palladium catalysis with tuned phosphine ligands.3a,8 Finally, we also achieved preliminary results for alkoxycarbonylation and carboxylation using substrate 1 with only minor changes. The alkoxycarbonylation proceeded in 69% yield using 2,2,2-trifluoroethanol with Ph3SiCo(CO)4 as catalyst, and the carboxylation used KOH as the nucleophile with KCo(CO)4 as catalyst to deliver benzoic acid 46 in 82% yield (see ESI† for further details).
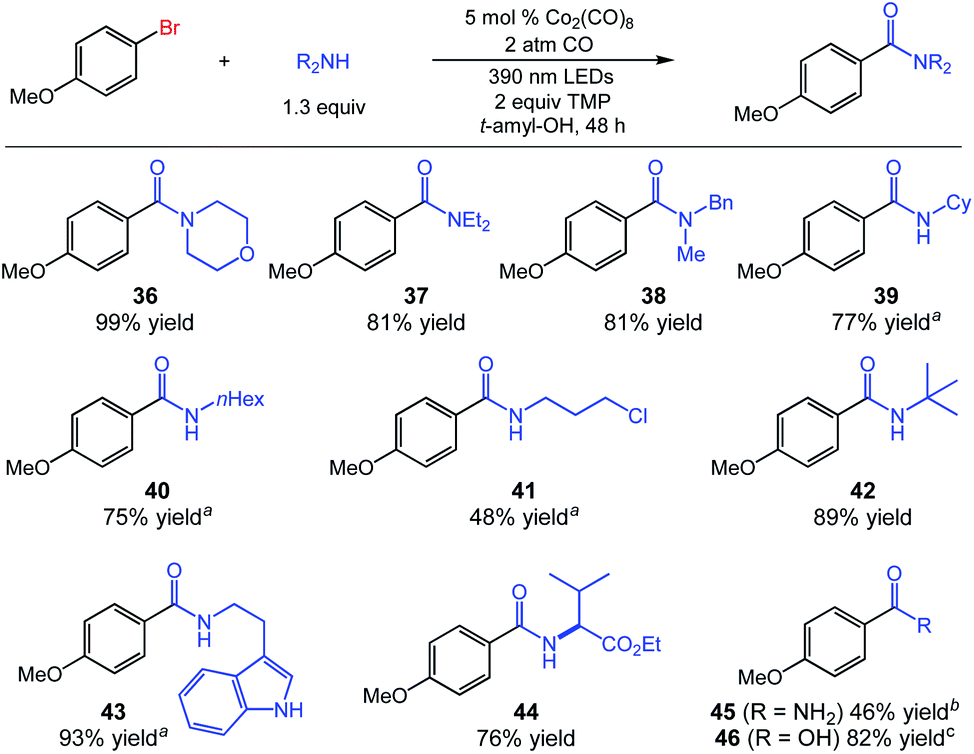 | ||
| Fig. 3 Cobalt-catalyzed aminocarbonylation of 1-bromo-4-methoxybenzene using diverse amine nucleophiles. aReaction time of 24 h. b10 mol% Co2(CO)8 used as catalyst with ammonium carbamate as nucleophile in the presence of 4 Å molecular sieves. c10 mol% KCo(CO)4 used as catalyst with 4.0 equiv. of KOH as nucleophile. | ||
Despite the clear potential for cobalt catalysis in the carbonylative functionalization of sp2 C–X electrophiles, the mechanistic details of this class of transformations remain obscure. While previous studies have speculated that photochemical charge transfer of a donor–acceptor complex involving the electrophile and Co(CO)4− could be involved, there is no experimental evidence for such a step.5c In order to gain insight into this key step, we performed UV-vis spectroscopic measurements on bromobenzene, KCo(CO)4 (used as a proxy for cobaltate formed in situ under our reaction conditions, and also a viable catalyst – see Table 1, entry 2), and the combination of the two in THF at 10 μM of the cobaltate ion. As clearly indicated in Fig. 4, we observed an increase in absorbance (λmax = 275 nm) upon mixing the two species; this peak is proposed to arise from the absorption of a donor–acceptor complex formed from the bromobenzene and the cobaltate, providing support for such a complex in the light-promoted activation via cobaltate catalysis.
 | ||
| Fig. 4 UV-vis absorption spectra of bromobenzene and KCo(CO)4 in THF with [10 μM]. | ||
A mechanistic proposal for the cobalt-catalyzed aminocarbonylation is depicted in Scheme 1. Octacarbonyldicobalt first disproportionates to the cobaltate anion by addition of the amine or TMP.9 The active cobaltate next coordinates to the (hetero)aryl or vinyl electrophile, followed by a reversible light-promoted charge transfer of the donor–acceptor complex. Loss of the halide (or triflate) through an SRN1 mechanism leads to a radical pair which recombines in the solvent cage to form a (hetero)aryl or vinyl cobalt species.5b,10 This intermediate undergoes CO migratory insertion to generate an acylcobalt which is then substituted by the amine nucleophile to deliver the product and regenerate the catalyst.
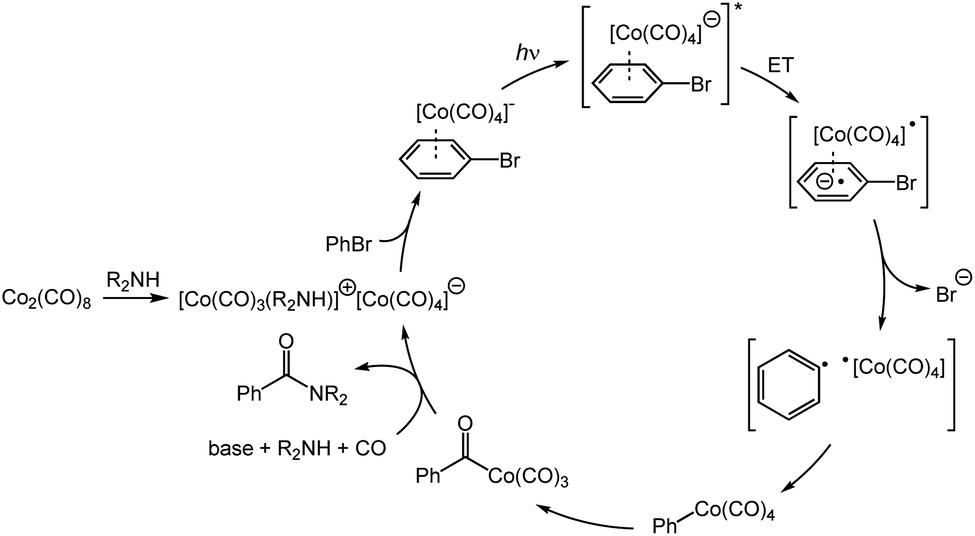 | ||
| Scheme 1 Plausible catalytic cycle for the cobalt-catalyzed aminocarbonylation. | ||
Conclusions
In conclusion, we have developed a visible-light promoted, cobalt-catalyzed aminocarbonylation of diverse sp2 C–X electrophiles. The broad scope of the reaction extends to the use of (hetero)aryl and vinyl bromides, chlorides, and triflates with a range of amine nucleophiles, including an ammonia surrogate. The overall scope of the cobalt-catalyzed aminocarbonylation rivals that of the common phosphine-ligated, palladium-catalyzed methods, and proceeds at low CO pressure, ambient temperatures, and uses visible light LEDs. Our mechanistic studies provide evidence for the important role of a donor–acceptor complex between cobaltate and (hetero)aryl electrophiles with implications beyond the present work. We anticipate that the notable practicality of this inexpensive cobalt-based catalytic system will prove enabling in carbonylative catalysis. Ongoing efforts will continue to develop carbonylative transformations for synthesis featuring anionic metal catalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Award No. R35 GM131708 from the National Institute of General Medical Sciences. We thank the UNC Department of Chemistry Mass Spectrometry Core Laboratory for assistance with MS analysis, supported by the National Science Foundation under award number CHE 1726291. We thank Brendon Sargent (University of North Carolina—Chapel Hill) for assistance with optimization and helpful discussions. We also thank Carla Morton (UNC—CH) for assistance with UV-vis spectroscopic measurements and Xander Deetz (UNC—CH) for assistance with calculation of the donor–acceptor binding constant.Notes and references
- (a) A. Brennführer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114–4133 CrossRef PubMed; (b) C. L. Allen and J. M. Williams, Chem. Soc. Rev., 2011, 40, 3405–3415 RSC; (c) S. T. Gadge and B. M. Bhanage, RSC Adv., 2014, 4, 10367–10389 RSC; (d) L. W. Hernandez, J. Pospech, U. Klöckner, T. W. Bingham and D. Sarlah, J. Am. Chem. Soc., 2017, 139, 15656–15659 CrossRef CAS PubMed; (e) R. J. Atkins, A. Banks, R. K. Bellingham, G. F. Breen, J. S. Carey, S. K. Etridge, J. F. Hayes, N. Hussain, D. O. Morgan and P. Oxley, et al. , Org. Process Res. Dev., 2003, 7, 663–675 CrossRef CAS.
- A. Schoenberg and R. F. Heck, J. Org. Chem., 1974, 39, 3327–3331 CrossRef CAS.
- (a) J. Y. Wang, A. E. Strom and J. F. Hartwig, J. Am. Chem. Soc., 2018, 140, 7979–7993 CrossRef CAS PubMed; (b) J. R. Martinelli, D. A. Watson, D. M. M. Freckmann, T. E. Barder and S. L. Buchwald, J. Org. Chem., 2008, 73, 7102–7107 CrossRef CAS PubMed; (c) C. F. J. Barnard, Organometallics, 2008, 27, 5402–5422 CrossRef CAS; (d) S. D. Friis, T. Skrydstrup and S. L. Buchwald, Org. Lett., 2014, 16, 4296–4299 CrossRef CAS PubMed; (e) S. N. Gockel and K. L. Hull, Org. Lett., 2015, 17, 3236–3239 CrossRef CAS PubMed.
- For aminocarbonylations not involving simple amine coupling partners (aryl carbamoylation), see: (a) X.-P. Wen, Y.-L. Han and J.-X. Chen, RSC Adv., 2017, 7, 45107–45112 RSC; (b) C. M. Lindsay and D. A. Widdowson, J. Chem. Soc., Perkin Trans. 1, 1988, 3, 569 RSC; (c) N. Alandini, L. Buzzetti, G. Favi, T. Schulte, L. Candish, K. D. Collins and P. Melchiorre, Angew. Chem., Int. Ed., 2020, 59, 5248–5253 CrossRef CAS PubMed.
- (a) J.-J. Brunet, C. Sidot, B. Loubinoux and P. Caubère, J. Org. Chem., 1979, 44, 2199–2202 CrossRef CAS; (b) J.-J. Brunet, C. Sidot and P. Caubère, Tetrahedron Lett., 1981, 22, 1013–1016 CrossRef CAS; (c) J.-J. Brunet, C. Sidot and P. Caubère, J. Org. Chem., 1983, 48, 1166–1171 CrossRef CAS; (d) J. Marchal, J. Bodiguel, Y. Fort and P. Caubère, J. Org. Chem., 1995, 60, 8336–8340 CrossRef CAS; (e) R. Vanderesse, J. Marchal and P. Caubère, Synth. Commun., 1993, 23, 1361–1370 CrossRef CAS.
- J.-J. Brunet, C. Sidot and P. Caubère, J. Organomet. Chem., 1981, 204, 229–241 CrossRef.
- B. T. Sargent and E. J. Alexanian, Angew. Chem., Int. Ed., 2019, 58, 9533–9536 CrossRef CAS PubMed.
- (a) X. Qi, H.-J. Ai, C.-X. Cai, J.-B. Peng, J. Ying and X.-F. Wu, Eur. J. Org. Chem., 2017, 7222–7225 CrossRef CAS; (b) A. Schnyder, M. Beller, G. Mehltretter, T. Nsenda, M. Studer and A. F. Indolese, J. Org. Chem., 2001, 66, 4311–4315 CrossRef CAS PubMed.
- (a) P. L. Pauson, J. P. Stambuli, T.-C. Chou and B.-C. Hong, Octacarbonyldicobalt, in Encyclopedia of Reagents for Organic Synthesis, 2014, pp. 1–26 Search PubMed; (b) J. Guo, H. D. Pham, Y.-B. Wu, D. Zhang and X. Wang, ACS Catal., 2020, 10, 1520–1527 CrossRef CAS.
- J. F. Bunnett, Acc. Chem. Res., 1978, 11, 413–420 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization data. See DOI: 10.1039/d0sc02178d |
| This journal is © The Royal Society of Chemistry 2020 |