
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Prediction of NHC-catalyzed chemoselective functionalizations of carbonyl compounds: a general mechanistic map†
Xue
Li
a,
Jun
Xu
b,
Shi-Jun
Li
 a,
Ling-Bo
Qu
a,
Zhongjun
Li
a,
Yonggui Robin
Chi
c,
Donghui
Wei
*a and
Yu
Lan
*ad
a,
Ling-Bo
Qu
a,
Zhongjun
Li
a,
Yonggui Robin
Chi
c,
Donghui
Wei
*a and
Yu
Lan
*ad
aCollege of Chemistry, Institute of Green Catalysis, Zhengzhou University, 100 Science Avenue, Zhengzhou, Henan 450001, China. E-mail: donghuiwei@zzu.edu.cn
bCollege of Pharmacy, Guizhou University of Traditional Chinese Medicine, Guiyang, China
cDivision of Chemistry & Biological Chemistry, School of Physical & Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore
dCollege of Chemistry and Chemical Engineering, Chongqing University, Chongqing 400044, China. E-mail: lanyu@cqu.edu.cn
First published on 22nd June 2020
Abstract
Generally, N-heterocyclic carbene (NHC) complexed with carbonyl compounds would transform into several important active intermediates, i.e., enolates, Breslow intermediates, or acylazolium intermediates, which act as either a nucleophile (Nu) or an electrophile (E) to react with the other E/Nu partner. Hence, the key to predicting the origin of chemoselectivity is to compute the activity (i.e., electrophilic index ω for E and nucleophilic index N for Nu) and stability of the intermediates and products, which are suggested in a general mechanistic map of these reactions. To support this point, we selected and studied different cases of the NHC-catalyzed reactions of carbonyl compounds in the presence of a base and/or an oxidant, in which multiple possible pathways involving acylazolium, enolate, Breslow, and α,β-unsaturated acylazolium intermediates were proposed and a novel index ω + N of the E and Nu partners was employed to exactly predict the energy barrier of the chemoselective step in theory. This work provides a guide for determining the general principle behind organocatalytic reactions with various chemoselectivities, and suggests a general application of the reaction index in predicting the chemoselectivity of the nucleophilic and electrophilic reactions.
Introduction
The combination of a nitrogen atom with a lone pair of electrons and a 6e carbon atom in N-heterocyclic carbenes (NHCs) provides a conjugation between the two atoms, which can significantly stabilize the unoccupied orbital of the carbon atom to achieve a typical singlet carbene.1 In this kind of chemistry, NHCs usually act as nucleophiles by using the occupied sp2 orbital of the carbon atom to activate polarized unsaturated substrates, which can reversibly form a covalent bond with active molecules.2 Further transformations of the forming intermediates can reverse the electrophilic substrates to nucleophiles by the electron-donation of the conjugative nitrogen atoms. Therefore, NHCs can be used as powerful catalysts in organocatalysis due to the special characteristics of nucleophilicity,3 good leaving group ability,4 and tunable electronic5 and steric properties.6 As an example (Scheme 1), the reaction of NHCs with aldehydes can generate a formal enol, namely Breslow intermediates, which can be used as a nucleophile to react with electrophilic partners for the synthesis of a variety of functional molecules including biologically active natural products,7 organic materials,8 pharmaceuticals,9 agrochemicals, etc.10 Indeed, the other electrophiles, such as ketenes, enals, olefins, or imines, can also be activated by NHCs according to their nucleophilicity, which significantly increases the complexity of carbene chemistry.11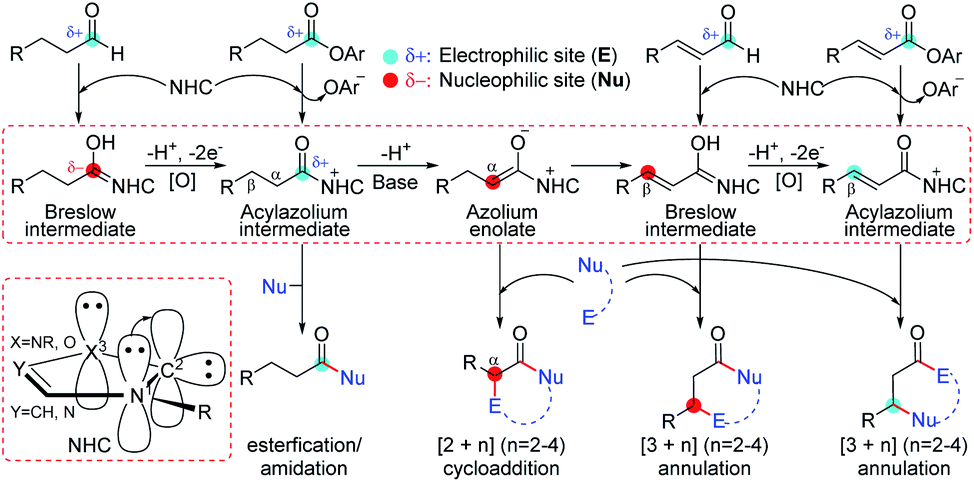 | ||
| Scheme 1 NHC-catalyzed transformation modes of carbonyl compounds. | ||
In NHC-mediated transformations, the introduction of an external base or oxidant increases the complication of the reaction mechanism because the additives can probably reverse the electronic properties of NHC-involved active intermediates. As a contrast, the mechanism for the NHC-catalyzed cycloaddition of ketenes only involves three steps of adsorption, cycloaddition, and dissociation in the absence of external additives. Alternatively, when a Brønsted base participates in the NHC-catalyzed functionalization of aldehydes,12 enals,13 or esters,14 the transformations would involve more steps due to the extra proton transfer in those reactions. Furthermore, the situation becomes much more complex in the presence of an external oxidant,15 because the key step of hydrogen transfer to oxidants might take place through hydride transfer to oxygen or carbon (HTO/HTC),16 single electron transfer-hydrogen atom transfer (SET-HAT),17 or even HAT-SET18 processes. However, the lack of a universal principle to disclose the mechanism of the NHC-catalyzed carbonyl compound transformations restricts the rational design of both NHC-catalysts and reaction types. Hence, predicting the chemoselectivity and designing a map for the general mechanisms of the NHC-catalyzed transformations of carbonyl compounds in the presence of an external base and/or oxidant are still challenging and highly desirable in this field.
In NHC-catalysis, carbonyl compounds are one of the most commonly used electrophiles that can be activated by the adsorption of NHCs. When external additives are added, further deprotonation and/or oxidation greatly increase the complexity of the reaction mechanism. For instance, an aldehyde can be activated by the NHC catalyst to afford the Breslow intermediate, which can be oxidized to the acylazolium intermediate by an oxidant (Scheme 1). The deprotonation of the acylazolium assisted by a base can generate azolium enolate, which can isomerize to an α,β-unsaturated Breslow intermediate through proton transfer. Further oxidation can provide the α,β-unsaturated acylazolium species. Previous experiments have demonstrated that all the above mentioned intermediates can react with specific nucleophiles or electrophiles to achieve the functionalization of carbonyl compounds.13a–i,14,15 However, the origin of chemoselectivity for the competitive transformation of corresponding intermediates still remains a challenge for synthetic chemists and could be solved by the exploration of reaction mechanisms. In this work, a series of NHC-catalyzed carbonyl compound transformation reactions will be considered theoretically, and we will try to predict the origin of chemoselectivity from a general perspective. We hope that the understanding of the reaction mechanism and origin of chemoselectivity would be helpful for the design of new transformations in NHC catalysis.
Computational details
The Gaussian09 program19 and M06-2X (ref. 20) functional were employed to perform all the calculations, which have been widely used to study the mechanisms and selectivities21 of various catalytic reactions.22 All the geometries were optimized at the 6-31G(d,p) basis set in THF and other solvents such as toluene, in which the solvent effect was simulated by the appropriate integral equation formalism polarizable continuum model (IEF-PCM).23 The harmonic vibrational frequency calculations were performed at the same computational level as that of the optimization, which ensures no imaginary frequency in the intermediate and only one imaginary frequency in the transition state. The discussed energies of all the optimized geometries were gained by adding the single-point energies and the Gibbs free energy corrections at the M06-2X-D3/6-31++G(2df,2pd)/IEF-PCMTHF//M06-2X/6-31G(d,p)/IEF-PCMTHF computational level (L1).In addition, the global reactivity index (GRI) analysis was performed on the active intermediates to evaluate their nucleophilic (nucleophilic index N)24 or electrophilic (electrophilic index ω)25 reactivities on the basis of the equations: N = EH(R) − EH(TCNE) (EH(TCNE) = −0.38586 a.u.), ω = μ2/2η, μ = (EH + EL)/2, and η = (EL − EH),26 while the local reactivity index (electrophilic (P+k) and nucleophilic (P−k) Parr functions)27 analysis was also performed to uncover the nucleophilic or electrophilic sites. To confirm the reliability of the computational level (L1), other density functional theory (DFT) methods (i.e., B3LYP-D3,28 CAM-B3LYP-D3,29 ωB97X-D,30 and MP2) at different levels (L2–6) were carried out on the crucial stereoselective step. Furthermore, other configurations of the stereoselective transition states have been additionally considered, to confirm that all the discussed geometries had the lowest energy. More computational results and details can be found in the ESI.†
Results and discussion
General mechanistic map for NHC-catalyzed chemoselective functionalizations of esters
As a selected model reaction (Scheme 2), when the NHC precursor Pre-NHC was used as a catalyst, ester R1 can react with imine R2 to afford the [3 + 3] oxidative annulation product P with up to 76% yield and a 60% enantiomeric excess (ee) value in the presence of the base DBU and oxidant 3,3′,5,5′-tetra-tert-butyl diphenoquinone (DQ).31 The Chi group also found that the additive HOBt can significantly improve the yield (from 76% to 94%) and enantioselectivity (from 60% to 94% ee).31 According to our proposed transformation model in Scheme 1, when acylazolium intermediate M02 is formed, it can be deprotonated to form an enolate intermediate, M3. Moreover, its Breslow type isomer M4 can be oxidized to an α,β-unsaturated acylazolium species, M05. As shown in Scheme 2, all these intermediates can react with nucleophile R2− or electrophile R2 to afford the corresponding amidation/ketolation, [2 + 2] cycloaddition, [3 + 2] annulation, and [3 + 3] annulation products. However, only the [3 + 3] oxidative annulation product P was observed experimentally, while the origin of chemoselectivity for this transformation still remains unclear. Moreover, the enantioselectivity for [3 + 3] oxidative annulation with additive effects should also be studied. Therefore, this transformation was selected in our theoretical calculation as a model reaction to reveal the common model for the NHC-catalyzed carbonyl compound transformations. | ||
| Scheme 2 Possible reaction pathways of NHC-catalyzed reactions between saturated carboxylic esters and imines. | ||
Among the proposed pathways, the [3 + 3] annulation pathway was mainly discussed in detail for the NHC-catalyzed reactions, while the other four possible pathways were divorced from the key active intermediates. Both the non-free-carbene pathway and carbene generation pathway were considered, and the computed results indicated that the DBU-assisted deprotonation of azolium catalyst Pre-NHC to afford free NHC can be easily happen (Fig. S1 of the ESI†).32 The [3 + 3] annulation pathway contains nine steps, as shown in Fig. 1–3.
 | ||
| Fig. 1 Gibbs free energy profiles of the NHC-catalyzed oxidative α,β-C(sp3)–H functionalization of the ester. | ||
 | ||
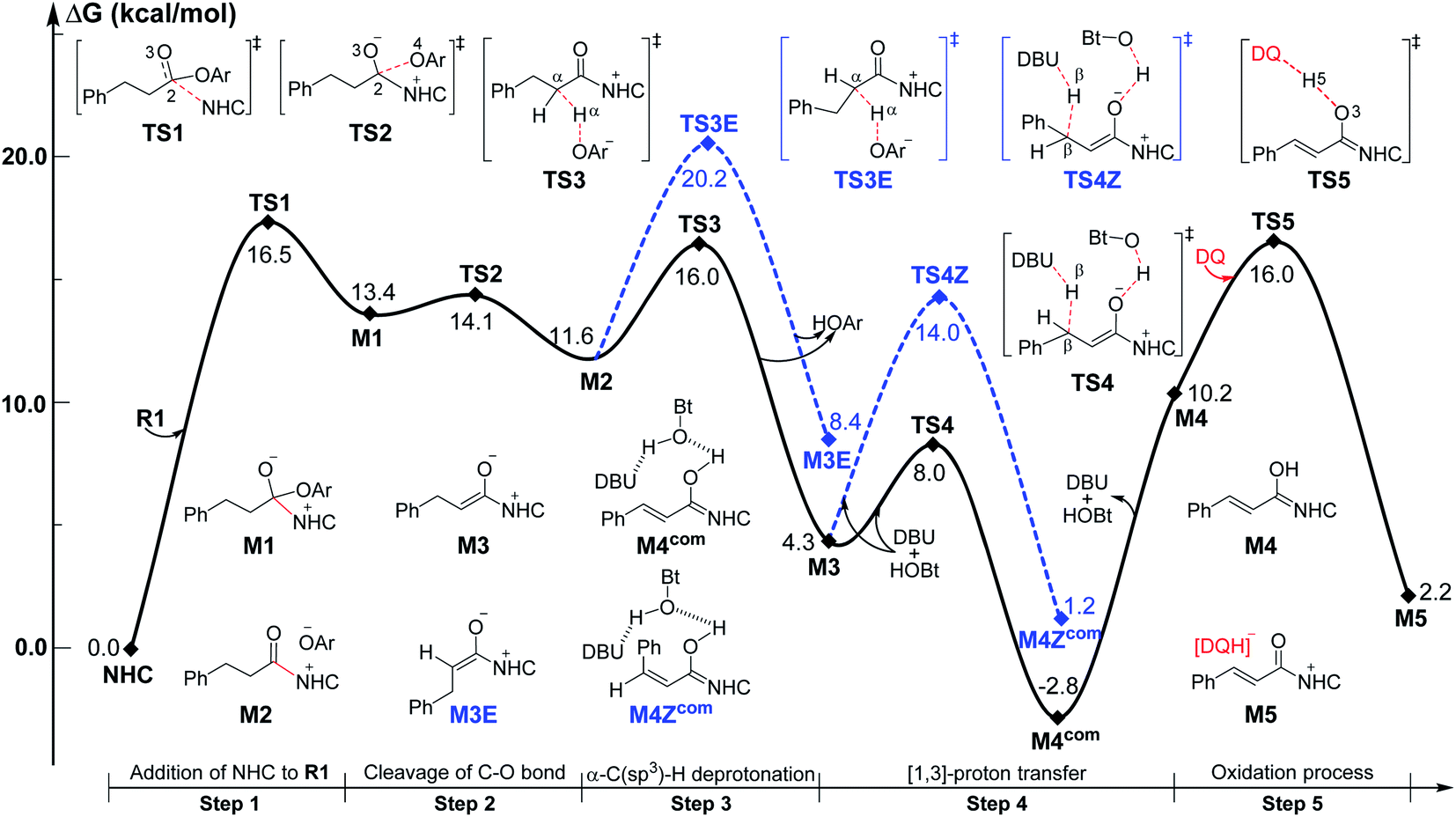
| Fig. 2 Optimized geometries of transition states in the [3 + 3] annulation pathway with HOBt; the hydrogen atoms that are not involved in the reaction have been omitted (distances: Å). | ||
 | ||
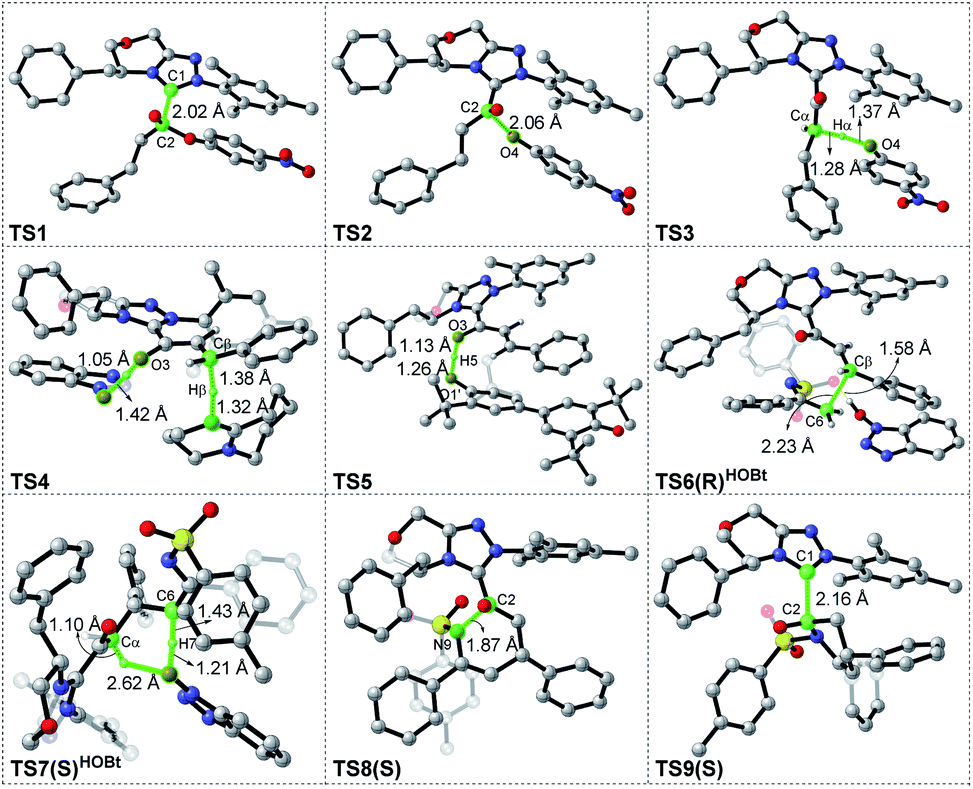
| Fig. 3 Gibbs free energy profiles for NHC-catalyzed oxidative [3 + 3] annulation of the ester with imine in the presence of HOBt. | ||
As shown in Fig. 1, the active site C1 of the NHC nucleophilically attacks the carbonyl carbon C2 of ester R1 initially through transition state TS1, forming zwitterionic intermediate M1 with an energy barrier of 16.5 kcal mol−1. The structural transformation from NHC and aryl ester to the zwitterionic (tetrahedral) intermediate needs to overcome an energy barrier of 16.5 kcal mol−1, which is close to that reported in ref. 33. Subsequently, the dissociation of OAr− takes place via a C2–O4 bond cleavage transition state TS2 with an energy barrier of only 0.7 kcal mol−1, affording intermediate M2 (the complexation of the acylazolium intermediate M02 and OAr−). Then, an α-C(sp3)–H deprotonation of M2 is assisted by anionic OAr− through transition states TS3/TS3E to generate the corresponding enolate intermediates M3/M3E. The relative free energy of transition state TS3 is 4.2 kcal mol−1 lower than that of TS3E; hence, the pathway associated with TS3/M3 is preferred. In addition, the base DBU-assisted α-C(sp3)–H deprotonation pathway (Fig. S2 and S3 of the ESI†) was also considered theoretically; however, much higher activation barriers were observed via transition states TS3DBU/TS3EDBU.
After the α-C(sp3)–H deprotonation of M2, intermediate M3 undergoes a DBU and HOBt cooperatively assisted [1,3]-proton transfer process via transition states TS4/TS4Z (ΔG‡ = 3.7/9.7 kcal mol−1, Fig. 1), affording the related complexes M4com/M4Zcom. These complexes subsequently dissociate into Breslow intermediates M4/M4Z and DBU + HOBt. The energies of TS4/M4com are 6.0/4.0 kcal mol−1 lower than those of TS4Z/M4Zcom, implying that the pathway associated with TS4 is energetically preferred. Then, intermediate M4 is oxidized into a complex M5, which consists of the unsaturated acylazolium intermediate M05 and anion [DQH]−. The oxidation occurs via transition state TS5 (ΔG‡ = 5.8 kcal mol−1, Fig. 1) through the most favorable HTO pathway, which is determined after computing and comparing the four proposed oxidative pathways, namely, HTO, HTC, HAT-SET, and SET-HAT.34 More information on the natural population analysis (NPA) charge and other possible oxidative pathways can be found in the ESI.†
For the Michael addition process, the two reacting parts are generally nucleophilic (Nu) and electrophilic (E). Obviously, intermediates M5 or M05 are the electrophilic parts as their electrophilic indexes (ω) equal to 3.093 and 2.979 eV, respectively, so the other reacting part related to R2 should be nucleophilic. Thus, imine R2 must be first deprotonated by a base (i.e., OAr−, DBU, or [DQH]−) to afford the nucleophilic R2− anion. As revealed by the computed results in Fig. S9 of the ESI,† the deprotonation of R2 by base [DQH]− through transition state TS0DQH− (ΔG‡ = 12.5 kcal mol−1, Fig. S9 and S10 of the ESI†) is the most favorable pathway among the three possible deprotonation pathways, affording the stable nucleophilic intermediate R2−HOBt by interacting with HOBt and dissociating DQH2. As depicted in Scheme 3, the nucleophilic site C6 (with P−k(C6) = 0.61) of anionic R2−HOBt can attack the Re-/Si-faces of the electrophilic site Cβ (with P+k(Cβ) = 0.24) of M5 through transition state TS6(R/S)HOBt (ΔG‡ = 9.9/12.3 kcal mol−1, Fig. 3) in the process of forming intermediate M6(R/S)HOBt and releasing anion [DQH]−, in which a Cβ–C6 bond is formed. The energy difference of 2.4 kcal mol−1 between 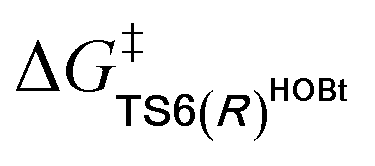 and
and 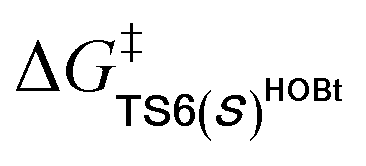 corresponds to the calculated 96.6% ee value, which is close to the 94% ee value in the presence of HOBt observed in the experiment.31 The letters “R/S” in M6(R/S)HOBt represent the chirality of the Cβ center. In addition, since the energies of TS6(R)HOBt and M6(R)HOBt locate below those of TS6(S)HOBt and M6(S)HOBt, the processes that follow M6(S)HOBt are unnecessary to discuss in detail.
corresponds to the calculated 96.6% ee value, which is close to the 94% ee value in the presence of HOBt observed in the experiment.31 The letters “R/S” in M6(R/S)HOBt represent the chirality of the Cβ center. In addition, since the energies of TS6(R)HOBt and M6(R)HOBt locate below those of TS6(S)HOBt and M6(S)HOBt, the processes that follow M6(S)HOBt are unnecessary to discuss in detail.
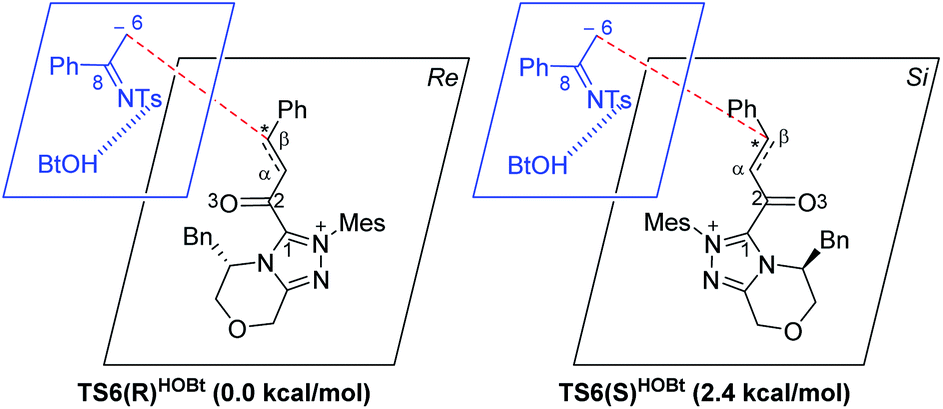 | ||
| Scheme 3 Stereochemical possibilities for the catalytic Michael addition process (unit in kcal mol−1). | ||
Next, M6(R)HOBt conducts the other [1,3]-proton transfer process with the help of the protic media HOBt through transition state TS7(S)HOBt (ΔG‡ = 12.0 kcal mol−1), generating intermediate M7(S) and dissociating HOBt. It should be noted that the R-chirality of the Cβ atom in TS6/M6 is apparently converted to the S-chirality in TS7/M7, since the unsaturated bond is changed from the C2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Cα bond in TS6/M6 to the C6C8 bond in TS7/M7. Hereafter, M7(S) undergoes a ring closure process via a six-membered ring transition state TS8(S) (ΔG‡ = 3.7 kcal mol−1) to produce M8(S). Eventually, the catalyst NHC is recycled and a six-membered ring main product of lactam P(S) is produced through transition state TS9(S) (ΔG‡ = 3.2 kcal mol−1). The energy of P(S) is found to be 23.7 kcal mol−1 below the energy of the reactant, suggesting that the entire reaction is exothermic. In addition, the difference between the [3 + 3] annulation pathways with or without HOBt is only reflected in steps 6 and 7. Fig. S11 of the ESI† shows that the energy barriers of steps 6 and 7 via transition states TS6(R/S) and TS7(S/R)HOAr are 13.6/14.4 and 13.2/10.4 kcal mol−1 in the [3 + 3] annulation pathway without HOBt, respectively. Apparently, the energy barrier difference between ΔGTS6(R)‡ and ΔGTS6(S)‡ is only 0.8 kcal mol−1 and can be theoretically converted to a 59% ee value, which is very close to the observed 60% ee value in the experiment,31 indicating that HOBt is indeed able to advance the stereoselectivity.
Cα bond in TS6/M6 to the C6C8 bond in TS7/M7. Hereafter, M7(S) undergoes a ring closure process via a six-membered ring transition state TS8(S) (ΔG‡ = 3.7 kcal mol−1) to produce M8(S). Eventually, the catalyst NHC is recycled and a six-membered ring main product of lactam P(S) is produced through transition state TS9(S) (ΔG‡ = 3.2 kcal mol−1). The energy of P(S) is found to be 23.7 kcal mol−1 below the energy of the reactant, suggesting that the entire reaction is exothermic. In addition, the difference between the [3 + 3] annulation pathways with or without HOBt is only reflected in steps 6 and 7. Fig. S11 of the ESI† shows that the energy barriers of steps 6 and 7 via transition states TS6(R/S) and TS7(S/R)HOAr are 13.6/14.4 and 13.2/10.4 kcal mol−1 in the [3 + 3] annulation pathway without HOBt, respectively. Apparently, the energy barrier difference between ΔGTS6(R)‡ and ΔGTS6(S)‡ is only 0.8 kcal mol−1 and can be theoretically converted to a 59% ee value, which is very close to the observed 60% ee value in the experiment,31 indicating that HOBt is indeed able to advance the stereoselectivity.
To ensure the enantioselective transition states associated with the lowest energy configurations, we have performed a conformational study via constructing different configurations of the TS6(R/S) and TS6(R/S)HOBt by rotating 90° per time of the dihedral Φ1(N–C1–C2–Cα) (see Schemes 3 and S5 of the ESI†). Therefore, eight (2 × 4) possible conformations were constructed and computed for either TS6(R/S) or TS6(R/S)HOBt, which can be named TS6(R/S), TS6(R/S)′, TS6(R/S)′′, TS6(R/S)′′′, and TS6(R/S)HOBt, TS6(R/S)HOBt′, TS6(R/S)HOBt′′, TS6(R/S)HOBt′′′, respectively. The computed results in Table S3 of the ESI† indicate that the energies of the TS6(R/S) and TS6(R/S)HOBt are the lowest among the distinctive conformers.
According to the recent work reported by Singleton and Plata, the calculated proton transfer barrier might be unreliable and has a large error in the alcohol-mediated Morita–Baylis–Hillman (MBH) reactions using the most popular DFT methods, i.e., B3LYP and M06-2X,35 which would be due to the absence of explicit solvents in the models.36 Therefore, to test whether the same problem exists in the NHC-catalyzed reactions, we have additionally constructed the models with 100 explicit THF solvents, and computed the energy barriers of the proton transfer processes (steps 4 and 5) at the ONIOM(B3LYP/6-31G(d,p):UFF) and ONIOM(M06-2X/6-31G(d,p):UFF) levels.
As summarized in Table 1, the calculated results indicate that the free energy barriers ΔΔGtot (with the normal Gibbs free energy correction), ΔΔG50% (with 50% of the Gibbs free energy correction), and ΔΔGexplicit (calculated in the explicit solvents without the implicit model) are close, and the free energy barriers obtained by the two DFT methods have tiny differences, which is remarkably different from the computed results reported in Singleton's work. As mentioned above, the computational errors in this system should not be significant and the calculated results using the M06-2X method are consistent with the experiment. More details on the intrinsic reaction coordinate (IRC) results of transition states TS4 and TS5, which were calculated by the different DFT methods in both implicit and explicit models, have been provided in Fig. S18 and S19 of the ESI.†
| Method | kcal mol−1 |
|---|---|
| a The transition state TS4/5 and intermediate IM3/4 obtained from the IRC calculations were calculated at the level of M06-2X/6-31G(d,p)/IEF-PCMTHF or B3LYP/6-31G(d,p)/IEF-PCMTHF. b The transition state TS4/5explicit was first located in the sphere with a radius of 15 Å of the explicit solvents at the ONIOM(M06-2X/6-31G(d,p):UFF) or ONIOM(B3LYP/6-31G(d,p):UFF) levels. Then IRC calculation was performed to locate the corresponding intermediate IM3/4explicit. | |
| M06-2X(ΔΔGtot[TS4-IM3])a | 13.4 |
| M06-2X(ΔΔG50%[TS4-IM3]) | 13.0 |
| M06-2X(ΔΔG[TS4explicit-IM3explicit])b | 11.3 |
| B3LYP(ΔΔGtot[TS4B3LYP-IM3B3LYP])a | 12.1 |
| B3LYP(ΔΔG50%[TS4B3LYP-IM3B3LYP]) | 13.0 |
| B3LYP(ΔΔG[TS4explicit-IM3explicit])b | 10.9 |
| M06-2X(ΔΔGtot[TS5-IM4])a | 6.8 |
| M06-2X(ΔΔG50%[TS5-IM4]) | 7.9 |
| M06-2X(ΔΔG[TS5explicit-IM4explicit])b | 6.4 |
| B3LYP(ΔΔGtot[TS5B3LYP-IM4B3LYP])a | 7.0 |
| B3LYP(ΔΔG50%[TS5B3LYP/IM4B3LYP]) | 5.6 |
Roles of HOBt and DBU
To explore the roles of the additive HOBt and base DBU, we have provided more evidence in both experiment and theory. As shown in Fig. 4, the DBU and HOBt cooperatively assisted [1,3]-proton transfer via transition state TS4 (ΔG‡ = 3.7 kcal mol−1) is the most energetically favorable pathway among all the possible [1,3]-proton transfer pathways, including DBU and DBU·H+ cooperatively assisted [1,3]-proton transfer pathway viaTS4DBU–DH+ (ΔG‡ = 15.2 kcal mol−1) and DBU or DBU·H+ separately assisted [1,3]-proton transfer pathway viaTS4DBU (ΔG‡ = 25.2 kcal mol−1)/TS4DH+ (ΔG‡ = 24.6 kcal mol−1), implying that DBU serves as a base and the participation of HOBt can significantly lower the energy barrier of the key 1,3-proton transfer (i.e., β-C–H deprotonation for the transformation from the Cα–Cβ single bond of saturated ester to the CαCβ double bond), which thus causes the reaction to happen faster and improves the yield of the reaction. In order to prove this point, we have collaborated with Chi's group and the experimental results indicate that the yield indeed can be improved from 76% to 94% by adding the HOBt in the reaction (see Scheme 2).31 In contrast, when unsaturated ester is used as the reactant in the NHC-catalyzed [3 + 3] annulation for the formation of the same product,14a the DFT calculations in Scheme S6 and Fig. S22 of the ESI† demonstrated that the addition of HOBt or other alcohols should be not necessary for improving reaction rate or yield, since the β-C–H deprotonation is not involved in the reaction, and the experimental observations also confirmed this conclusion. Hence, the participation of HOBt for the reaction is critical in promoting the reaction yield and enantioselectivity.
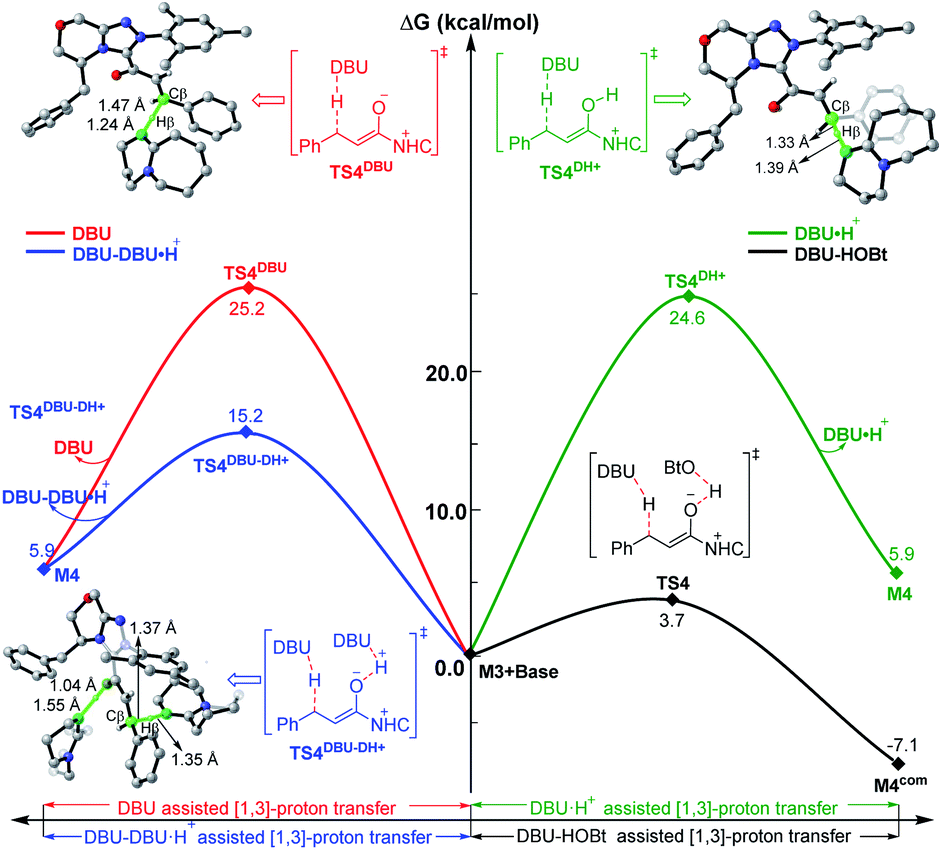 | ||
| Fig. 4 Relative Gibbs free energy profiles of different types of base-assisted [1,3]-proton transfer pathways, and the energies of the minima are relative to the energy of M3 + base (0.0 kcal mol−1). | ||
Origin of chemoselectivity
For the other four proposed pathways divorced from the active intermediates M2, M3, and M4, the detailed discussions of their related energy profiles have been provided in the ESI† for the production of SPA/SPB, SPC, and SPD, as conjectured in Scheme 2. To pursue the origin of the chemoselectivity, we just need to compare the energy barriers involved in the several possible pathways: the possible amidation, ketolation, [2 + 2] cycloaddition, [3 + 2] annulation, and [3 + 3] annulation pathways.Both of the amidation and ketolation pathways are divorced from M2, and the energy profile of the ketolation pathway lies below that of the amidation pathway, which is shown in Fig. S12 of the ESI.† Subsequently, we compared the other four possible pathways. As shown in Fig. 5, since M2 (ω = 1.548 eV, P+k(C2) = 0.18) and R2− (N = 4.459 eV, P−k(C6) = 0.64) are separately electrophilic and nucleophilic, the nucleophilic addition of R2− onto M2 through transition state TS3B is likely to happen. However, the much higher energy of TS3B in the ketolation pathway compared to that of TS3 indicates that it is impossible to occur under the experimental conditions. In addition to the isomerization from enolate M3 to Breslow intermediate M4, since enolate M3 (N = 4.116 eV, P−k(Cα) = 0.62) and R2 (ω = 1.709 eV, P+k(C8) = 0.40) are nucleophilic and electrophilic, respectively, the [2 + 2] cycloaddition between M3 and R2via transition state TS4C(SS) is likely to occur. However, the much higher energy of TS4C(SS) in the [2 + 2] cycloaddition pathway compared to that of TS4 demonstrates the unfavorability of the corresponding pathway. In contrast to the oxidation of Breslow intermediate M4, since M4 (N = 4.962 eV, P−k(Cβ) = 0.62) and R2 (ω = 1.709 eV, P+k(C8) = 0.40) are nucleophilic and electrophilic, respectively, we have located a transition state TS5D(RS) for the nucleophilic addition of M4 onto R2 combined with a proton transfer. The energy barrier of the single-step reaction viaTS5D(RS) in the [3 + 2] annulation pathway is 11.5 kcal mol−1 and not high. However, as summarized in Table S9 of the ESI,† the energy barriers of the four possible reactions (ΔG‡total) viaTS3B, TS4C(SS), TS5D(RS), and TS6(R)HOBt are 31.0, 28.0, 24.5, and 9.9 kcal mol−1, hence, only the product P can be formed in theory, which is consistent with the experimental observations.31
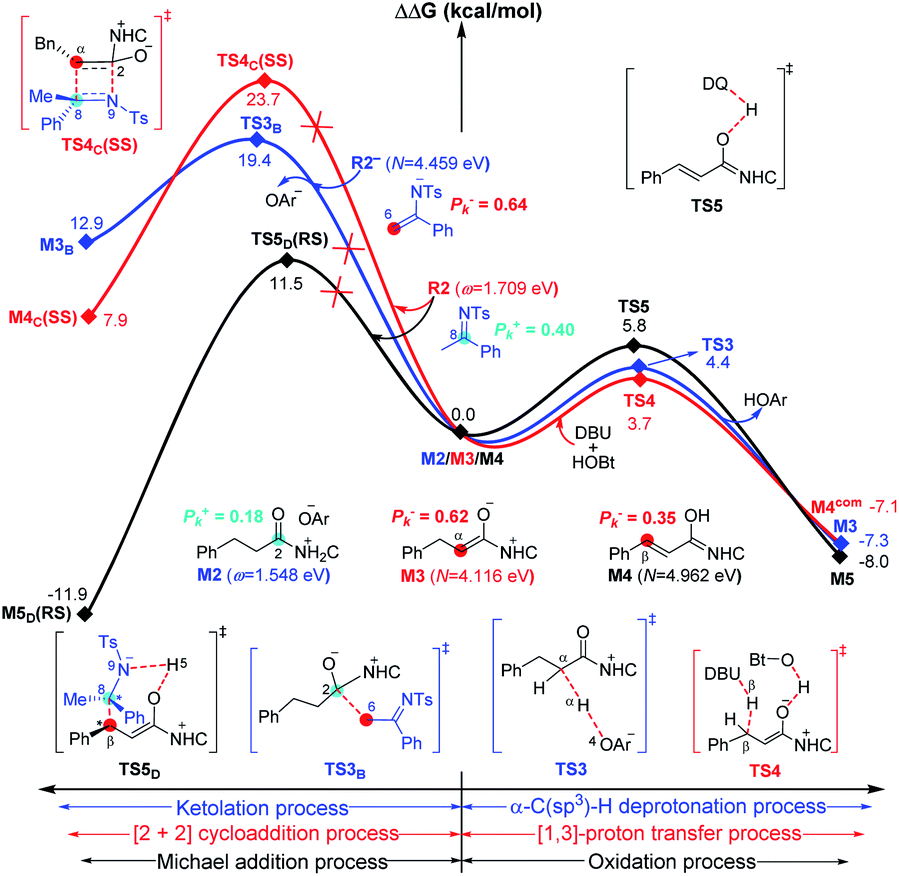 | ||
| Fig. 5 Gibbs free energy profiles for the possible processes between ketolation and α-C(sp3)–H deprotonation (blue line), [2 + 2] cycloaddition and [1,3]-proton transfer (red line), and Michael addition and oxidation (black line) processes involved in the four competing pathways. | ||
As is well accepted by chemists, for the nucleophilic or electrophilic addition processes, if the two reacting partners have much stronger nucleophilicities or electrophilicities, their corresponding transition states would be related to lower energy barriers. Therefore, based on the empirical point, to uncover the origin of this reaction chemoselectivity, we assumed that the larger electrophilic (ω) and nucleophilic (N) indexes ω + N of the stable nucleophile (Nu) → electrophile (E) partners (R2− → M2, M4 → R2, and R2−HOBt → M5) would lead to lower energy barriers, ΔG‡, of the nucleophilic or electrophilic addition processes associated with the transition states (TS3B, TS5D(RS), and TS6(R)HOBt) involved in the three pathways. As summarized in Table S9 of the ESI,† the decreasing single-step energy barriers, ΔG‡, are presented a linear relationship with the corresponding increasing ω + N indexes as shown in Fig. 6. It should be noted that the energy barrier via a four-membered ring transition state TS4C(SS) does not completely correspond to the activity of the reaction partners (M3 → R2), which is due to the large ring strain of the four-membered ring involved in TS4C(SS); thus we did not consider its data for the linear relationship depicted in Fig. 6.
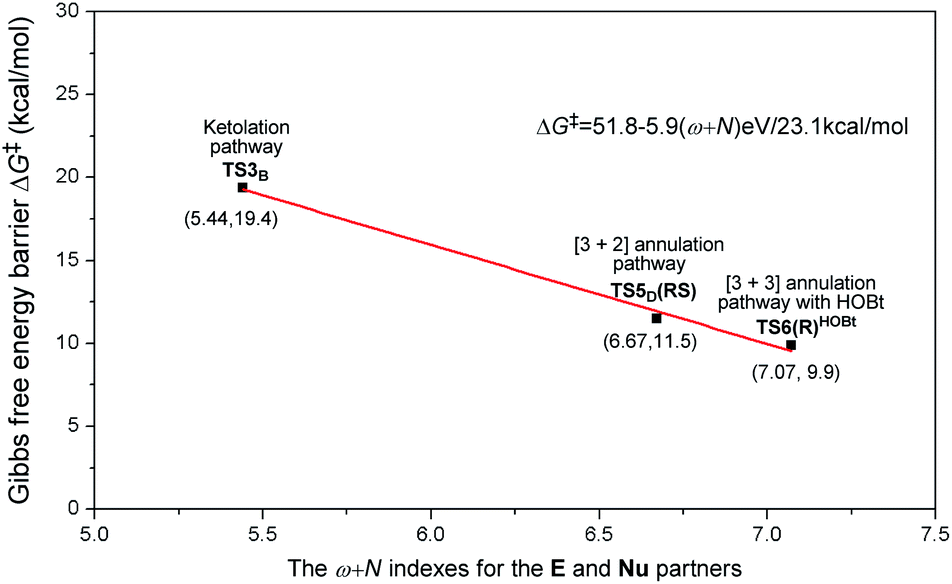 | ||
| Fig. 6 The linear relationship between the single-step energy barriers, ΔG‡, (unit: kcal mol−1) of chemoselective transition states and their corresponding ω + N indexes (unit: eV) of the E and Nu partners. | ||
Considering the above, we suggested a new application of the ω + N index from the reacting ability between the Nu and E to simply and quickly predict the energy barrier of the chemoselective step (ΔG‡p = 51.8–5.9(ω + N) eV/23.1 kcal mol−1), which would be one of the key factors for exploring the origin of chemoselectivity of the possible reactions commonly involved in a large amount of NHC-catalyzed reactions of saturated or unsaturated carbonyl compounds.
General principle for predicting the chemoselectivity of NHC-mediated reactions of carbonyl compounds
To ensure that the ω + N index of Nu and E partners can be used to generally predict the single-step energy barriers and even the chemoselectivity in these types of reactions based on our proposed general mechanistic map, we selected three additional NHC-mediated reactions of carbonyl compounds presented in Fig. 7–9 as valuable cases for testing.13j,14b,17a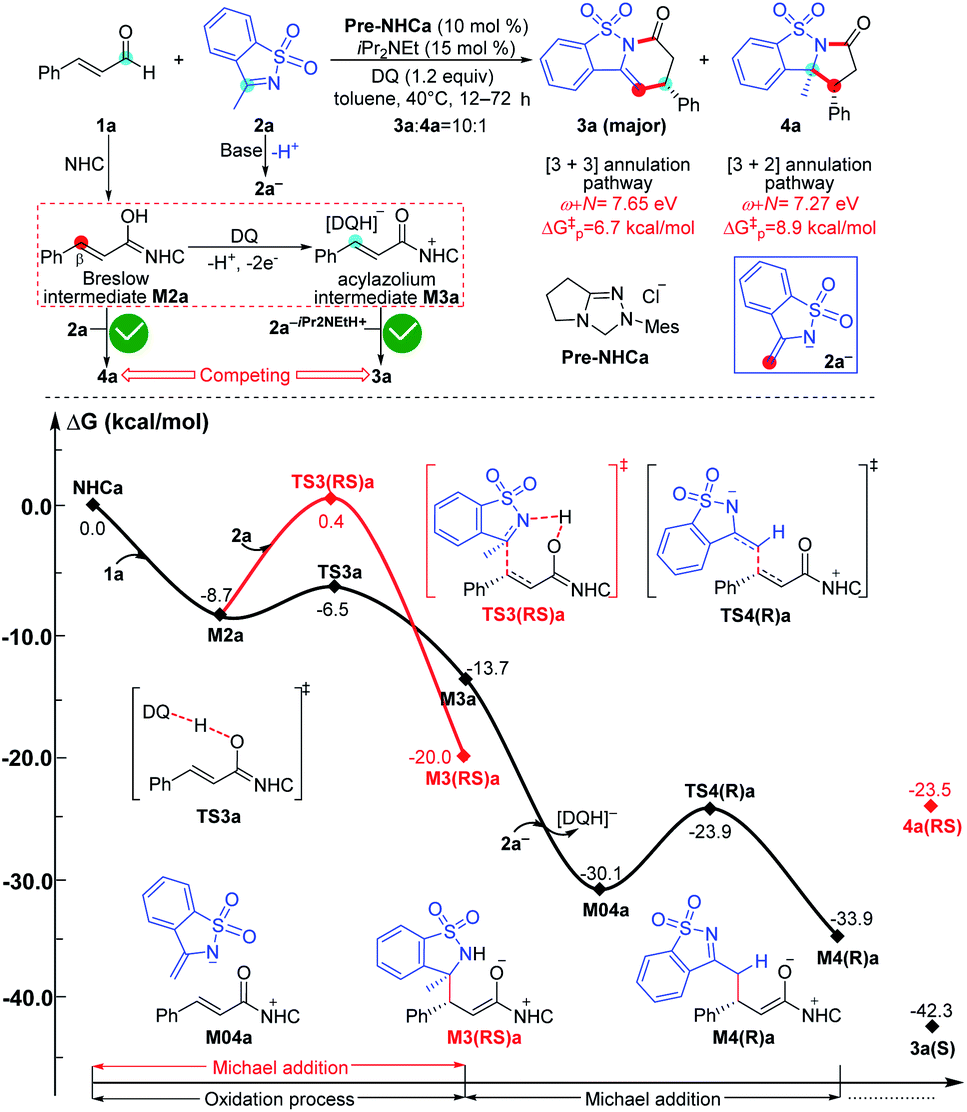 | ||
| Fig. 7 Relative Gibbs free energy profiles of NHC-catalyzed chemoselective [3 + n] (n = 2, 3) annulation of enals with imines. (“ΔG‡p” is calculated by the correlation ΔG‡ = 51.8–5.9(ω + N) eV/23.1 kcal mol−1 and represents the predicted energy barrier of nucleophilic or electrophilic addition.) | ||
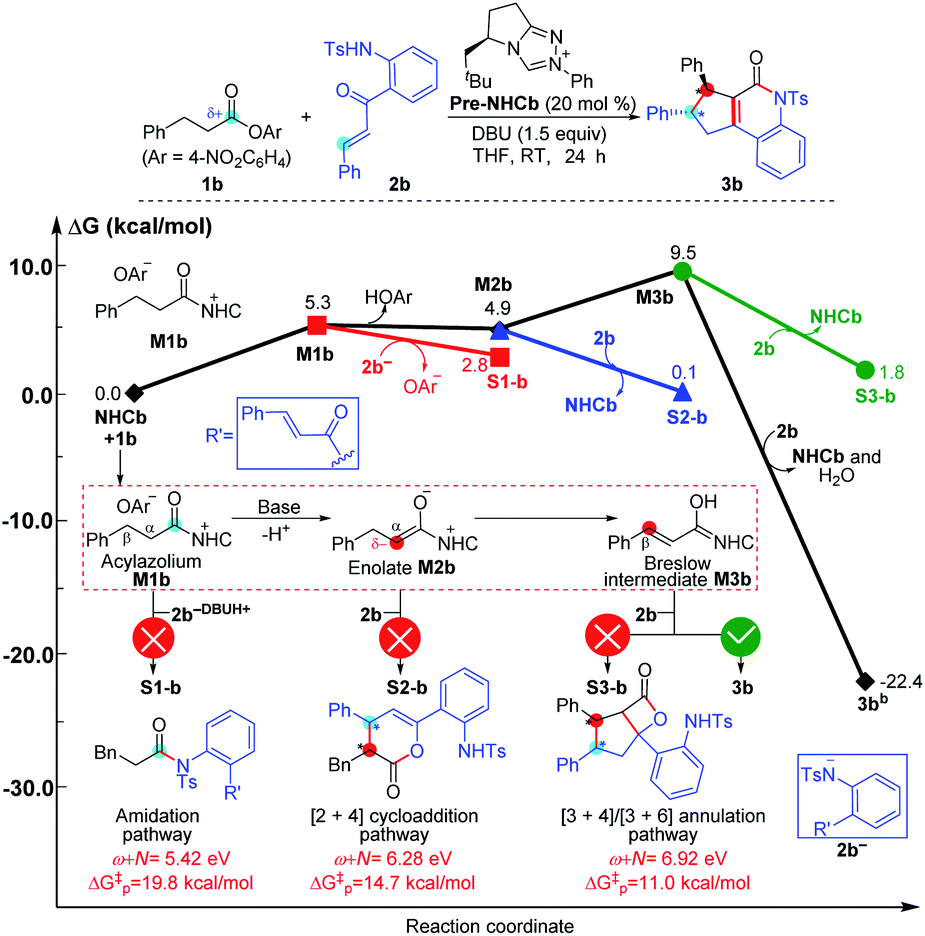 | ||
| Fig. 8 Relative Gibbs free energy profiles of NHC-catalyzed reactions of saturated carboxylic esters with o-tosylamino enones. | ||
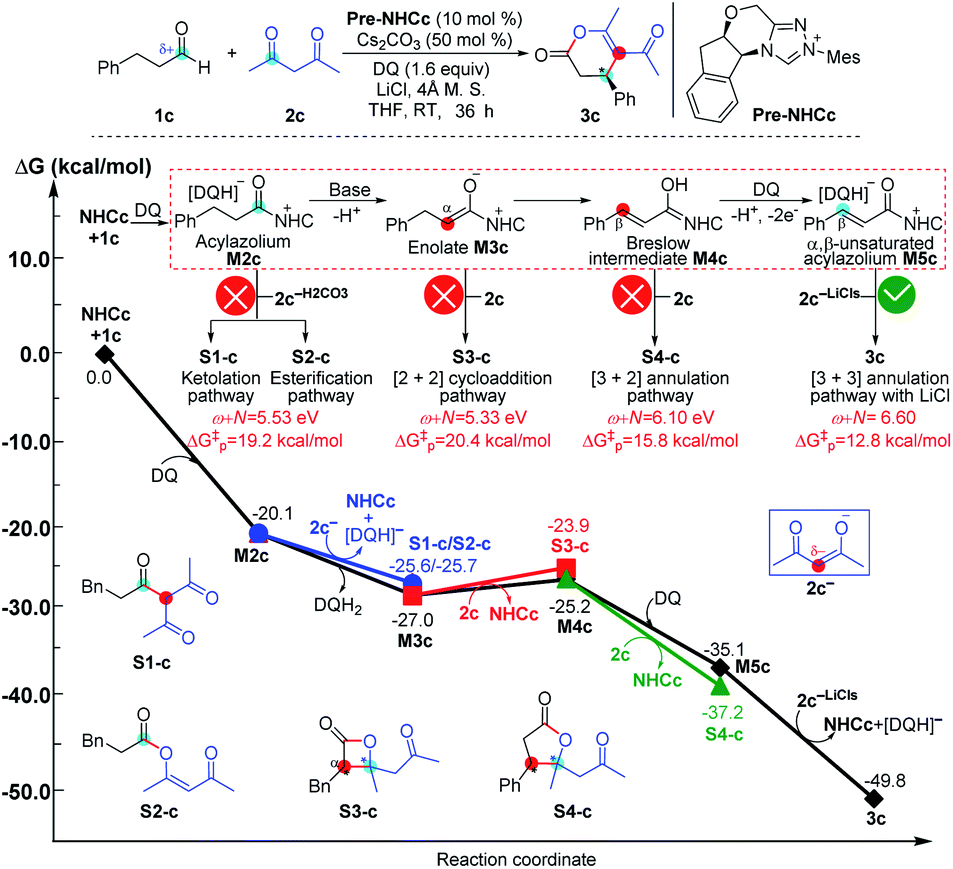 | ||
| Fig. 9 Relative Gibbs free energy profiles of NHC-catalyzed reactions of simple aldehydes with acetylacetones. | ||
As shown in Fig. 7, the NHC-catalyzed chemoselective [3 + n] (n = 2, 3) annulation of enal with imine was selected as one special case,13j and two competing pathways including the [3 + 2] and [3 + 3] annulation pathways were considered according to the general mechanistic map suggested in Scheme 1. The relevant ω + N indexes of the Nu and E partners are 7.27 and 7.65 eV, which correspond to the predicted energy barriers of 8.9 and 6.7 kcal mol−1, respectively, based on the correlation ΔG‡p = 51.8–5.9(ω + N) eV/23.1 kcal mol−1 depicted in Fig. 6. Apparently, the predicted energy barriers are close to the calculated energy barriers (9.1 and 6.2 kcal mol−1, Fig. 7) for the Michael addition process in producing products 3a and 4a. Both of the two transformations should be irreversible, so the chemoselectivity mainly be controlled by kinetics. In kinetics, the [3 + 3] annulation pathway for affording 3a is much more energetically favorable than the [3 + 2] annulation pathway for generating 4a, which is in agreement with the experimental results that the ratio of 3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4a is 10:1.13j
:4a is 10:1.13j
As depicted in Fig. 8, the NHC-catalyzed reactions of saturated carboxylic esters with o-tosylamino enones probably proceed through the following four possible pathways: the amidation, [2 + 4] cycloaddition, [3 + 4], and [3 + 6] annulation pathways. The corresponding ω + N indexes of the Nu and E partners in the four pathways are equal to 5.42, 6.28, 6.92, and 6.92 eV, which indicates that the corresponding ΔG‡ would be relatively lower in the [3 + 4]/[3 + 6] annulation pathway. In thermodynamics, the energy of 3b is much lower than those of S1-b, S2-b, and S3-b, and only the transformation to product 3b is irreversible at room temperature. In summary, we can predict that the formation of 3b through the [3 + 6] annulation pathway is more favorable in both kinetics and thermodynamics. Hence, only the product 3b can be formed in theory, which is in agreement with the experimental observations.14b
As shown in Fig. 9, the NHC-catalyzed reaction of simple aldehydes with acetylacetones was selected as another case, and five pathways, including the ketolation/esterification, [2 + 2] cycloaddition, [3 + 2] annulation, and [3 + 3] annulation pathways with LiCl, were proposed based on the general mechanistic map (Scheme 1). The relevant ω + N indexes of the Nu and E partners correspond to 5.53, 5.33, 6.10, and 6.60 eV, and the energy of main product 3c was found to be much lower than those of S1-c, S2-c, S3-c, and S4-c. The above analyses imply that the [3 + 3] annulation pathway with LiCl for affording 3c is more energetically favorable than the other four pathways for generating S1-c, S2-c, S3-c, and S4-c in both kinetics and thermodynamics. Hence, the main product should be 3c in theory, which is still in agreement with the results observed in experiments.17a All the side products were proposed according to the experimental references,13j,15a,37 and the justifications of the side products were provided in Schemes S7–S9 of the ESI.†
The reactivity indices could be useful for catalyst screening, but we still suggest that the searching of the transition state should be necessary to explore the origin of chemo- and stereo-selectivities. Noteworthy, we have also computed the key transition states involved in the multiple pathways for generating different products in the reaction models depicted in Fig. 8 and 9. As shown in Fig. S23 of the ESI,† the order of the calculated Gibbs free energy barrier (i.e., ΔG‡(TS4b) = 11.2 kcal mol−1 < ΔG‡(S2-TS3b) = 18.3 kcal mol−1 < ΔG‡(S1-TS2b) = 32.6 kcal mol−1) is the same with that of their predicted values (i.e., ΔG‡p ([3 + 4]/[3 + 6] annulation pathway) = 11.0 kcal mol−1 < ΔG‡p ([2 + 4] cycloaddition pathway) = 14.7 kcal mol−1 < ΔG‡p (amidation pathway) = 19.8 kcal mol−1), indicating that the conclusion on the origin of chemoselectivity should be reliable. Meanwhile, a similar conclusion can be obtained from the calculated results in Fig. S24 of the ESI;† the computed energy barriers of the transition states can be used to explain the chemoselectivity well.
Conclusions
In summary, the introduction of an external base or oxidant indeed increases the complication of the reaction mechanism for the four NHC-mediated reactions of carbonyl compounds in theory, as the additives can reverse the electronic properties of NHC-involved active intermediates, i.e., acylazolium intermediate, enolate, Breslow intermediate, or α,β-unsaturated acylazolium intermediate.As revealed by the DFT calculations of the NHC-mediated transformations, the nucleophilic enolate is obtained from the base-assisted deprotonation of the electrophilic acylazolium intermediate. The nucleophilic Breslow intermediate is generated from a base and protic media cooperatively assisted isomerization of a nucleophilic enolate, while the electrophilic α,β-unsaturated acylazolium intermediate is produced from the oxidation of a nucleophilic Breslow intermediate by an oxidant. These active intermediates could probably undergo multiple competing pathways, including the amidation/ketolation, [2 + n] (n = 2, 4) cycloaddition, and [3 + n] (n = 2, 4, 6) annulation pathways, when they react with other nucleophilic (Nu) or electrophilic (E) partners. This could lead to the generation of chemoselective products and greatly increases the number of theoretical calculations. Hence, we suggested an exact mechanistic map and a simple rule by merely computing the ω + N indexes of the stable E and Nu partners and relative energies of intermediates and products to predict the energy barriers via the chemoselective transition states, and even predict the origin of chemoselectivity to significantly reduce the number of theoretical computations.
Notably, this simple rule has been successfully used in several cases of NHC-mediated reactions of saturated/unsaturated esters or aldehydes. Finally, we hope that the obtained insights will facilitate rational design according to the prediction of organocatalytic reactions with special chemoselectivities. Therefore, this work provides a theoretical method for searching and identifying the active intermediates, possible pathways, and even main products in NHC chemistry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the Excellent Youth Fund of the Natural Science Fund of China (No. 21822303), the National Natural Science Foundation of China (No. 21903071, 21772020, and 21773214), the China Postdoctoral Science Foundation (No. 2015T80776), and the Outstanding Young Talent Research Fund of Zhengzhou University (No. 1521316001).Notes and references
- (a) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, An Overview of N-Heterocyclic Carbenes, Nature, 2014, 510, 485 CrossRef CAS PubMed; (b) S. J. Ryan, L. Candish and D. W. Lupton, Acyl Anion Free N-Heterocyclic Carbene Organocatalysis, Chem. Soc. Rev., 2013, 42, 4906–4917 RSC.
- (a) X.-Y. Chen, Q. Liu, P. Chauhan and D. Enders, N-Heterocyclic Carbene Catalysis Via Azolium Dienolates: An Efficient Strategy for Remote Enantioselective Functionalizations, Angew. Chem., Int. Ed., 2018, 57, 3862–3873 CrossRef CAS PubMed; (b) L. C. Morrill and A. D. Smith, Organocatalytic Lewis Base Functionalisation of Carboxylic Acids, Esters and Anhydrides Via C1-Ammonium or Azolium Enolates, Chem. Soc. Rev., 2014, 43, 6214–6226 RSC; (c) R. S. Menon, A. T. Biju and V. Nair, Recent Advances in Employing Homoenolates Generated by N-Heterocyclic Carbene (NHC) Catalysis in Carbon–Carbon Bond-Forming Reactions, Chem. Soc. Rev., 2015, 44, 5040–5052 RSC; (d) V. Nair, S. Vellalath and B. P. Babu, Recent Advances in Carbon–Carbon Bond-Forming Reactions Involving Homoenolates Generated by NHC Catalysis, Chem. Soc. Rev., 2008, 37, 2691–2698 RSC.
- D. Enders and T. Balensiefer, Nucleophilic Carbenes in Asymmetric Organocatalysis, Acc. Chem. Res., 2004, 37, 534–541 CrossRef CAS PubMed.
- W. Schrader, P. P. Handayani, C. Burstein and F. Glorius, Investigating Organocatalytic Reactions: Mass Spectrometric Studies of a Conjugate Umpolung Reaction, Chem. Commun., 2007, 716–718 RSC.
- M. Huang, Y. Li, J. Liu, X.-B. Lan, Y. Liu, C. Zhao and Z. Ke, A Bifunctional Strategy for N-Heterocyclic Carbene-Stabilized Iridium Complex-Catalyzed N-Alkylation of Amines with Alcohols in Aqueous Media, Green Chem., 2019, 21, 219–224 RSC.
- K. Zeitler, Extending Mechanistic Routes in Heterazolium Catalysis–Promising Concepts for Versatile Synthetic Methods, Angew. Chem., Int. Ed., 2005, 44, 7506–7510 CrossRef CAS PubMed.
- (a) J. L. Koniarczyk, J. W. Greenwood, J. V. Alegre-Requena, R. S. Paton and A. McNally, A Pyridine-Pyridine Cross-Coupling Reaction Via Dearomatized Radical Intermediates, Angew. Chem., Int. Ed., 2019, 58, 14882–14886 CrossRef CAS PubMed; (b) C. T. Lohans, H. T. H. Chan, T. R. Malla, K. Kumar, J. J. A. G. Kamps, D. J. B. McArdle, E. van Groesen, M. de Munnik, C. L. Tooke, J. Spencer, R. S. Paton, J. Brem and C. J. Schofield, Non-Hydrolytic β-Lactam Antibiotic Fragmentation by L,D-Transpeptidases and Serine β-Lactamase Cysteine Variants, Angew. Chem., Int. Ed., 2019, 58, 1990–1994 CrossRef CAS PubMed.
- (a) A. Maji, Y. Reddi, R. B. Sunoj and D. Maiti, Mechanistic Insights on Orthogonal Selectivity in Heterocycle Synthesis, ACS Catal., 2018, 8, 10111–10118 CrossRef CAS; (b) H. K. Kisan and R. B. Sunoj, Insights on the Origin of Regiodivergence in the Parallel Kinetic Resolution of Rac-Aziridines Using a Chiral Lanthanum–Yttrium Bimetallic Catalyst, ACS Catal., 2018, 8, 7633–7644 CrossRef CAS.
- M. C. Hilton, X. Zhang, B. T. Boyle, J. V. Alegre-Requena, R. S. Paton and A. McNally, Heterobiaryl Synthesis by Contractive C–C Coupling Via P(V) Intermediates, Science, 2018, 362, 799 CrossRef CAS PubMed.
- (a) X. Bugaut and F. Glorius, Organocatalytic Umpolung: N-Heterocyclic Carbenes and Beyond, Chem. Soc. Rev., 2012, 41, 3511–3522 RSC; (b) Q. Ren, M. Li, L. Yuan and J. Wang, Recent Advances in N-Heterocyclic Carbene Catalyzed Achiral Synthesis, Org. Biomol. Chem., 2017, 15, 4731–4749 RSC; (c) K. J. R. Murauski, A. A. Jaworski and K. A. Scheidt, A Continuing Challenge: N-Heterocyclic Carbene-Catalyzed Syntheses of γ-Butyrolactones, Chem. Soc. Rev., 2018, 47, 1773–1782 RSC.
- (a) P. H.-Y. Cheong, C. Y. Legault, J. M. Um, N. Çelebi-Ölçüm and K. N. Houk, Quantum Mechanical Investigations of Organocatalysis: Mechanisms, Reactivities, and Selectivities, Chem. Rev., 2011, 111, 5042–5137 CrossRef CAS PubMed; (b) K. Zhong, C. Shan, L. Zhu, S. Liu, T. Zhang, F. Liu, B. Shen, Y. Lan and R. Bai, Theoretical Study of the Addition of Cu–Carbenes to Acetylenes to Form Chiral Allenes, J. Am. Chem. Soc., 2019, 141, 5772–5780 CrossRef CAS PubMed.
-
(a) S. S. Sohn, E. L. Rosen and J. W. Bode, N-Heterocyclic Carbene-Catalyzed Generation of Homoenolates: γ-Butyrolactones by Direct Annulations of Enals and Aldehydes, J. Am. Chem. Soc., 2004, 126, 14370–14371 CrossRef CAS PubMed;
(b) Q. Ni, H. Zhang, A. Grossmann, C. C. J. Loh, C. Merkens and D. Enders, Asymmetric Synthesis of Pyrroloindolones by N-Heterocyclic Carbene Catalyzed [2+3] Annulation of α-Chloroaldehydes with Nitrovinylindoles, Angew. Chem., Int. Ed., 2013, 52, 13562–13566 CrossRef CAS PubMed.
-
(a) C. Burstein and F. Glorius, Organocatalyzed Conjugate Umpolung of α,β-Unsaturated Aldehydes for the Synthesis of γ-Butyrolactones, Angew. Chem., Int. Ed., 2004, 43, 6205–6208 CrossRef CAS PubMed;
(b) S. P. Lathrop and T. Rovis, Asymmetric Synthesis of Functionalized Cyclopentanones Via a Multicatalytic Secondary Amine/N-Heterocyclic Carbene Catalyzed Cascade Sequence, J. Am. Chem. Soc., 2009, 131, 13628–13630 CrossRef CAS PubMed;
(c) N. A. White, D. A. DiRocco and T. Rovis, Asymmetric N-Heterocyclic Carbene Catalyzed Addition of Enals to Nitroalkenes: Controlling Stereochemistry Via the Homoenolate Reactivity Pathway to Access δ-Lactams, J. Am. Chem. Soc., 2013, 135, 8504–8507 CrossRef CAS PubMed;
(d) L. Wang, S. Li, M. Blümel, A. R. Philipps, A. Wang, R. Puttreddy, K. Rissanen and D. Enders, Asymmetric Synthesis of Spirobenzazepinones with Atroposelectivity and Spiro-1,2-Diazepinones by NHC-Catalyzed [3 + 4] Annulation Reactions, Angew. Chem., Int. Ed., 2016, 55, 11110–11114 CrossRef CAS PubMed;
(e) A. Chan and K. A. Scheidt, Direct Amination of Homoenolates Catalyzed by N-Heterocyclic Carbenes, J. Am. Chem. Soc., 2008, 130, 2740–2741 CrossRef CAS PubMed;
(f) A. Chan and K. A. Scheidt, Highly Stereoselective Formal [3 + 3] Cycloaddition of Enals and Azomethine Imines Catalyzed by N-Heterocyclic Carbenes, J. Am. Chem. Soc., 2007, 129, 5334–5335 CrossRef CAS PubMed;
(g) V. Nair, S. Vellalath, M. Poonoth and E. Suresh, N-Heterocyclic Carbene-Catalyzed Reaction of Chalcones and Enals Via Homoenolate: An Efficient Synthesis of 1,3,4-Trisubstituted Cyclopentenes, J. Am. Chem. Soc., 2006, 128, 8736–8737 CrossRef CAS PubMed;
(h) X. Fang, K. Jiang, C. Xing, L. Hao and Y. R. Chi, A Highly Regio- and Stereoselective Cascade Annulation of Enals and Benzodi(enone)s Catalyzed by N-Heterocyclic Carbenes, Angew. Chem., Int. Ed., 2011, 50, 1910–1913 CrossRef CAS PubMed;
(i) H. Lv, W.-Q. Jia, L.-H. Sun and S. Ye, N-Heterocyclic Carbene Catalyzed [4+3] Annulation of Enals and O-Quinone Methides: Highly Enantioselective Synthesis of Benzo-ε-Lactones, Angew. Chem., Int. Ed., 2013, 52, 8607–8610 CrossRef CAS PubMed;
(j) A. G. Kravina, J. Mahatthananchai and J. W. Bode, Enantioselective, NHC-Catalyzed Annulations of Trisubstituted Enals andCyclic N-Sulfonylimines Via α,β-Unsaturated Acyl Azoliums, Angew. Chem., Int. Ed., 2012, 51, 9433–9436 CrossRef CAS PubMed.
- (a) J. Cheng, Z. Huang and Y. R. Chi, NHC Organocatalytic Formal LUMO Activation of α,β-Unsaturated Esters for Reaction with Enamides, Angew. Chem., Int. Ed., 2013, 52, 8592–8596 CrossRef CAS PubMed; (b) Z. Fu, K. Jiang, T. Zhu, J. Torres and Y. R. Chi, Access to Oxoquinoline Heterocycles by N-Heterocyclic Carbene Catalyzed Ester Activation for Selective Reaction with an Enone, Angew. Chem., Int. Ed., 2014, 53, 6506–6510 CrossRef CAS PubMed.
- (a) X. Zhao, K. E. Ruhl and T. Rovis, N-Heterocyclic-Carbene-Catalyzed Asymmetric Oxidative Hetero-Diels–Alder Reactions with Simple Aliphatic Aldehydes, Angew. Chem., Int. Ed., 2012, 51, 12330–12333 CrossRef CAS PubMed; (b) X.-Y. Chen, Q. Liu, P. Chauhan, S. Li, A. Peuronen, K. Rissanen, E. Jafari and D. Enders, N-Heterocyclic Carbene Catalyzed [4+2] Annulation of Enals Via a Double Vinylogous Michael Addition: Asymmetric Synthesis of 3,5-Diaryl Cyclohexenones, Angew. Chem., Int. Ed., 2017, 56, 6241–6245 CrossRef CAS PubMed; (c) T. Zhu, C. Mou, B. Li, M. Smetankova, B.-A. Song and Y. R. Chi, N-Heterocyclic Carbene-Catalyzed δ-Carbon Lumo Activation of Unsaturated Aldehydes, J. Am. Chem. Soc., 2015, 137, 5658–5661 CrossRef CAS PubMed; (d) S. Bera, C. G. Daniliuc and A. Studer, Oxidative N-Heterocyclic Carbene Catalyzed Dearomatization of Indoles to Spirocyclic Indolenines with a Quaternary Carbon Stereocenter, Angew. Chem., Int. Ed., 2017, 56, 7402–7406 CrossRef CAS PubMed; (e) L. Lin, Y. Yang, M. Wang, L. Lai, Y. Guo and R. Wang, Oxidative N-Heterocyclic Carbene Catalyzed Stereoselective Annulation of Simple Aldehydes and 5-Alkenyl Thiazolones, Chem. Commun., 2015, 51, 8134–8137 RSC; (f) J. Mo, X. Chen and Y. R. Chi, Oxidative γ-Addition of Enals to Trifluoromethyl Ketones: Enantioselectivity Control Via Lewis Acid/N-Heterocyclic Carbene Cooperative Catalysis, J. Am. Chem. Soc., 2012, 134, 8810–8813 CrossRef CAS PubMed.
-
(a) B. Chan and L. Radom, Uncatalyzed Transfer Hydrogenation of Quinones and Related Systems: A Theoretical Mechanistic Study, J. Phys. Chem. A, 2007, 111, 6456–6467 CrossRef CAS PubMed;
(b) V. S. Batista, R. H. Crabtree, S. J. Konezny, O. R. Luca and J. M. Praetorius, Oxidative Functionalization of Benzylic C–H Bonds by DDQ, New J. Chem., 2012, 36, 1141–1144 RSC;
(c) S. Yamabe, S. Yamazaki and S. Sakaki, A DFT Study of Hydride Transfers to the Carbonyl Oxygen of DDQ, Int. J. Quantum Chem., 2015, 115, 1533–1542 CrossRef CAS;
(d) X. Guo, H. Zipse and H. Mayr, Mechanisms of Hydride Abstractions by Quinones, J. Am. Chem. Soc., 2014, 136, 13863–13873 CrossRef CAS PubMed.
- (a) J. Mo, L. Shen and Y. R. Chi, Direct β-Activation of Saturated Aldehydes to Formal Michael Acceptors through Oxidative NHC Catalysis, Angew. Chem., Int. Ed., 2013, 52, 8588–8591 CrossRef CAS PubMed; (b) O. R. Shehab and A. M. Mansour, Charge Transfer Complexes of 2-Arylaminomethyl-1H-Benzimidazole with 2,3-Dichloro-5,6-Dicyano-1,4-Benzoquinone: Experimental and DFT Studies, J. Mol. Struct., 2013, 1047, 121–135 CrossRef CAS; (c) A. K. Turek, D. J. Hardee, A. M. Ullman, D. G. Nocera and E. N. Jacobsen, Activation of Electron-Deficient Quinones through Hydrogen-Bond-Donor-Coupled Electron Transfer, Angew. Chem., Int. Ed., 2016, 55, 539–544 CrossRef CAS PubMed; (d) F. Liu, Z. Yang, Y. Yu, Y. Mei and K. N. Houk, Bimodal Evans–Polanyi Relationships in Dioxirane Oxidations of sp3 C–H: Non-Perfect Synchronization in Generation of Delocalized Radical Intermediates, J. Am. Chem. Soc., 2017, 139, 16650–16656 CrossRef CAS PubMed.
- (a) C. Höfler and C. Rüchardt, Bimolecular Formation of Radicals by Hydrogen Transfer, 10. On the Mechanism of Quinone Dehydrogenations, Liebigs Ann., 1996, 1996, 183–188 CrossRef; (b) C. Rüchardt, M. Gerst and J. Ebenhoch, Uncatalyzed Transfer Hydrogenation and Transfer Hydrogenolysis: Two Novel Types of Hydrogen-Transfer Reactions, Angew. Chem., Int. Ed., 1997, 36, 1406–1430 CrossRef; (c) D. A. Pratt, J. S. Wright and K. U. Ingold, Theoretical Study of Carbon–Halogen Bond Dissociation Enthalpies of Substituted Benzyl Halides. How Important Are Polar Effects?, J. Am. Chem. Soc., 1999, 121, 4877–4882 CrossRef CAS; (d) H. H. Jung and P. E. Floreancig, Mechanistic Analysis of Oxidative C–H Cleavages Using Inter- and Intramolecular Kinetic Isotope Effects, Tetrahedron, 2009, 65, 10830–10836 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- (a) Y. Zhao and D. G. Truhlar, Exploring the Limit of Accuracy of the Global Hybrid Meta Density Functional for Main-Group Thermochemistry, Kinetics, and Noncovalent Interactions, J. Chem. Theory Comput., 2008, 4, 1849–1868 CrossRef CAS PubMed; (b) Y. Zhao and D. G. Truhlar, Density Functionals with Broad Applicability in Chemistry, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed; (c) P. Zhang and C. Wolf, Catalytic Enantioselective Difluoroalkylation of Aldehydes, Angew. Chem., Int. Ed., 2013, 52, 7869–7873 CrossRef CAS PubMed.
- (a) R. B. Sunoj, Transition State Models for Understanding the Origin of Chiral Induction in Asymmetric Catalysis, Acc. Chem. Res., 2016, 49, 1019–1028 CrossRef CAS PubMed; (b) Y. Reddi, C.-C. Tsai, C. M. Avila, F. D. Toste and R. B. Sunoj, Harnessing Noncovalent Interactions in Dual-Catalytic Enantioselective Heck–Matsuda Arylation, J. Am. Chem. Soc., 2019, 141, 998–1009 CrossRef CAS PubMed; (c) M. Zhan, Z. Ding, S. Du, H. Chen, C. Feng, M. Xu, Z. Liu, M. Zhang, C. Wu, Y. Lan and P. Li, A Unified Approach for Divergent Synthesis of Contiguous Stereodiads Employing a Small Boronyl Group, Nat. Commun., 2020, 11, 792 CrossRef CAS PubMed.
- (a) J. Llaveria, Á. Beltrán, W. M. C. Sameera, A. Locati, M. M. Díaz-Requejo, M. I. Matheu, S. Castillón, F. Maseras and P. J. Pérez, Chemo-, Regio-, and Stereoselective Silver-Catalyzed Aziridination of Dienes: Scope, Mechanistic Studies, and Ring-Opening Reactions, J. Am. Chem. Soc., 2014, 136, 5342–5350 CrossRef CAS PubMed; (b) C. Hou, J. Jiang, Y. Li, C. Zhao and Z. Ke, When Bifunctional Catalyst Encounters Dual MLC Modes: DFT Study on the Mechanistic Preference in Ru-PNNH Pincer Complex Catalyzed Dehydrogenative Coupling Reaction, ACS Catal., 2017, 7, 786–795 CrossRef CAS; (c) J. Zhang, C. Shan, T. Zhang, J. Song, T. Liu and Y. Lan, Computational Advances Aiding Mechanistic Understanding of Silver-Catalyzed Carbene/Nitrene/Silylene Transfer Reactions, Coord. Chem. Rev., 2019, 382, 69–84 CrossRef CAS; (d) C. Shan, L. Zhu, L.-B. Qu, R. Bai and Y. Lan, Mechanistic View of Ru-Catalyzed C–H Bond Activation and Functionalization: Computational Advances, Chem. Soc. Rev., 2018, 47, 7552–7576 RSC; (e) Y. Li, Y. Luo, L. Peng, Y. Li, B. Zhao, W. Wang, H. Pang, Y. Deng, R. Bai, Y. Lan and G. Yin, Reaction Scope and Mechanistic Insights of Nickel-Catalyzed Migratory Suzuki–Miyaura Cross-Coupling, Nat. Commun., 2020, 11, 417 CrossRef CAS PubMed.
- (a) B. Mennucci and J. Tomasi, Continuum Solvation Models: A New Approach to the Problem of Solute's Charge Distribution and Cavity Boundaries, J. Chem. Phys., 1997, 106, 5151–5158 CrossRef CAS; (b) V. Barone and M. Cossi, Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- (a) L. R. Domingo, E. Chamorro and P. Pérez, An Analysis of the Regioselectivity of 1, 3-Dipolar Cycloaddition Reactions of Benzonitrile N-Oxides Based on Global and Local Electrophilicity and Nucleophilicity Indices, Eur. J. Org. Chem., 2009, 2009, 3036–3044 CrossRef; (b) L. R. Domingo, E. Chamorro and P. Perez, An Understanding of the Electrophilic/Nucleophilic Behavior of Electro-Deficient 2,3-Disubstituted 1,3-Butadienes in Polar Diels–Alder Reactions. A Density Functional Theory Study, J. Phys. Chem. A, 2008, 112, 4046–4053 CrossRef CAS PubMed; (c) L. R. Domingo and P. Perez, The Nucleophilicity N Index in Organic Chemistry, Org. Biomol. Chem., 2011, 9, 7168–7175 RSC.
- (a) R. G. Parr and R. G. Pearson, Absolute Hardness: Companion Parameter to Absolute Electronegativity, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS; (b) L. R. Domingo, M. T. Picher and J. A. Sáez, Toward an Understanding of the Unexpected Regioselective Hetero-Diels–Alder Reactions of Asymmetric Tetrazines with Electron-Rich Ethylenes: A DFT Study, J. Org. Chem., 2009, 74, 2726–2735 CrossRef CAS PubMed.
- (a) W. Kohn and L. J. Sham, Self-Consistent Equations Including Exchange and Correlation Effects, Phys. Rev., 1965, 140, A1133 CrossRef; (b) L. J. Sham and W. Kohn, One-Particle Properties of an Inhomogeneous Interacting Electron Gas, Phys. Rev., 1966, 145, 561 CrossRef CAS.
- L. R. Domingo, P. Pérez and J. A. Sáez, Understanding the Local Reactivity in Polar Organic Reactions through Electrophilic and Nucleophilic Parr Functions, RSC Adv., 2013, 3, 1486–1494 RSC.
- (a) A. D. Becke, Density-Functional Thermochemistry. III. The Role of Exact Exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, A New Hybrid Exchange–Correlation Functional Using the Coulomb-Attenuating Method (Cam-B3lyp), Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Long-Range Corrected Hybrid Density Functionals with Damped Atom-Atom Dispersion Corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- B. Liu, W. Wang, R. Huang, J. Yan, J. Wu, W. Xue, S. Yang, Z. Jin and Y. R. Chi, Direct Activation of β-sp3-Carbons of Saturated Carboxylic Esters as Electrophilic Carbons Via Oxidative Carbene Catalysis, Org. Lett., 2018, 20, 260–263 CrossRef CAS PubMed.
- (a) Y. Wang, Q.-Y. Wu, T.-H. Lai, K.-J. Zheng, L.-B. Qu and D. Wei, Prediction on the Origin of Selectivities of NHC-Catalyzed Asymmetric Dearomatization (CADA) Reactions, Catal. Sci. Technol., 2019, 9, 465–476 RSC; (b) Q.-C. Zhang, X. Li, X. Wang, S.-J. Li, L.-B. Qu, Y. Lan and D. Wei, Insights into Highly Selective Ring Expansion of Oxaziridines under Lewis Base Catalysis: A DFT Study, Org. Chem. Front., 2019, 6, 679–687 RSC; (c) S. Gehrke and O. Hollóczki, Are There Carbenes in N-Heterocyclic Carbene Organocatalysis?, Angew. Chem., Int. Ed., 2017, 56, 16395–16398 CrossRef CAS PubMed.
- (a) Y. Reddi and R. B. Sunoj, Mechanistic Studies on Stereoselective Organocatalytic Direct β-C–H Activation in an Aliphatic Chain by Chiral N-Heterocyclic Carbenes, ACS Catal., 2015, 5, 5794–5802 CrossRef CAS; (b) Y. Wang, D. Wei, Y. Wang, W. Zhang and M. Tang, N-Heterocyclic Carbene (NHC)-Catalyzed sp3 β-C–H Activation of Saturated Carbonyl Compounds: Mechanism, Role of Nhc, and Origin of Stereoselectivity, ACS Catal., 2016, 6, 279–289 CrossRef CAS.
- (a) X. Li, Y. Wang, Y. Wang, M. Tang, L.-B. Qu, Z. Li and D. Wei, Insights into the N-Heterocyclic Carbene (NHC)-Catalyzed Oxidative γ-C(sp3)–H Deprotonation of Alkylenals and Cascade [4 + 2] Cycloaddition with Alkenylisoxazoles, J. Org. Chem., 2018, 83, 8543–8555 CrossRef CAS PubMed; (b) X. Li, S.-J. Li, Y. Wang, Y. Wang, L.-B. Qu, Z. Li and D. Wei, Insights into NHC-Catalyzed Oxidative α-C(sp3)–H Activation of Aliphatic Aldehydes and Cascade [2 + 3] Cycloaddition with Azomethine Imines, Catal. Sci. Technol., 2019, 9, 2514–2522 RSC.
- R. E. Plata and D. A. Singleton, A Case Study of the Mechanism of Alcohol-Mediated Morita Baylis–Hillman Reactions. The Importance of Experimental Observations, J. Am. Chem. Soc., 2015, 137, 3811–3826 CrossRef CAS PubMed.
- A. Winter, Making a Bad Calculation, Nat. Chem., 2015, 7, 473–475 CrossRef CAS PubMed.
- (a) S. De Sarkar, S. Grimme and A. Studer, NHC Catalyzed Oxidations of Aldehydes to Esters: Chemoselective Acylation of Alcohols in Presence of Amines, J. Am. Chem. Soc., 2010, 132, 1190–1191 CrossRef CAS PubMed; (b) J. Xu, S. Yuan, J. Peng, M. Miao, Z. Chen and H. Ren, Enantioselective [2+2] Annulation of Simple Aldehydes with Isatin-Derived Ketimines Via Oxidative N-Heterocyclic Carbene Catalysis, Chem. Commun., 2017, 53, 3430–3433 RSC; (c) Z. Fu, J. Xu, T. Zhu, W. W. Y. Leong and Y. R. Chi, β-Carbon Activation of Saturated Carboxylic Esters through N-Heterocyclic Carbene Organocatalysis, Nat. Chem., 2013, 5, 835–839 CrossRef CAS PubMed; (d) J. Kaeobamrung and J. W. Bode, Stereodivergency of Triazolium and Imidazolium-Derived NHCs for Catalytic, Enantioselective Cyclopentane Synthesis, Org. Lett., 2009, 11, 677–680 CrossRef CAS PubMed; (e) J. Mo, R. Yang, X. Chen, B. Tiwari and Y. R. Chi, Direct α-Functionalization of Simple Aldehydes Via Oxidative N-Heterocyclic Carbene Catalysis, Org. Lett., 2013, 15, 50–53 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details, additional computational results, Cartesian coordinates and energy values of optimized structures. See DOI: 10.1039/d0sc01793k |
| This journal is © The Royal Society of Chemistry 2020 |