
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Intermolecular oxyarylation of olefins with aryl halides and TEMPOH catalyzed by the phenolate anion under visible light†
Kangjiang
Liang
,
Qian
Liu
,
Lei
Shen
,
Xipan
Li
,
Delian
Wei
,
Liyan
Zheng
 and
Chengfeng
Xia
*
and
Chengfeng
Xia
*
Key Laboratory of Medicinal Chemistry for Natural Resource (Ministry of Education and Yunnan Province), State Key Laboratory for Conservation and Utilization of Bio-Resources in Yunnan, School of Chemical Science and Technology, Yunnan University, 2 North Cuihu Road, Kunming 650091, China. E-mail: xiacf@ynu.edu.cn
First published on 4th June 2020
Abstract
The phenolate anion was discovered as a new photocatalyst with strong reduction potentials. Under visible light irradiation, the phenolate anion enabled the reduction of (hetero)aryl halides (including electron-rich aryl chlorides) to (hetero)aryl radicals through single electron transfer. Based on this new photocatalyst, a novel and efficient photocatalytic protocol for the intermolecular oxyarylation of olefins with aryl halides and TEMPOH was developed. The developed three-component coupling reaction proceeded under redox-neutral reaction conditions with stable and readily available synthons and exhibited broad substrate scope. The utility of this process was further highlighted by the diversified chemical manipulation of the resulting oxyarylation products and the late-stage modification of active pharmaceutical ingredients.
Introduction
Since the chemical reactivity of electronically excited molecules differs fundamentally from that of those in the ground state, the photochemistry induced by visible light provides fresh opportunities to expand the potential of organic chemistry. Various photocatalysts, including Ir or Ru complexes and organic dyes, have been developed and extensively explored.1 Phenolate anions are a useful model system for photoinduced electron ejection mechanism studies.2 Recent research showed that phenolate anions acted as photoreductants to activate perfluoroalkyl iodides in a perfluoroalkylation reaction.3 We also discovered that the visible light-excited vinylphenolate anions enabled the direct reduction of aryl halides to aryl radicals.4 However, attempts to use the vinylphenolate anion as a new photocatalyst failed and only afforded the Heck-type arylation products by coupling with aryl radicals. In this manuscript, we discovered that the tri-substituted phenolate anion possessed strong reduction potential and acted as a new photocatalyst with redox-neutral features. To demonstrate abilities of this new photocatalyst, we design an intermolecular oxyarylation of olefins with aryl halides under visible light irradiation.Intermolecular oxyarylation has attracted great attention since one oxygen substituent and one aryl group add across an olefin via a vicinal difunctionalization process. In the light of these benefits, transition-metal (Pd and Au) catalyzed oxidative oxyarylation has been extensively studied and several protocols for oxyarylation of olefins with arylboronic acids,5 arylstannanes,6 or arylsilanes7 were reported (Scheme 1a). In addition, Meerwein-type radical oxyarylation methods have been developed as an attractive alternative in recent years with aryl diazonium salts,8 diaryliodonium salts,8e,9 or aryl hydrazines10 as radical precursors (Scheme 1b). While numerous advances have been made in this area, the ability to access this molecular scaffold array with modular flexibility remains limited. Requirement of the use of toxic, unstable, or unavailable aryl precursors has impeded the further adaptation of these methods in the synthetic community. Furthermore, no general protocol for oxyarylation of olefins with aromatic heterocyclic motifs which are essential in a wide range of pharmaceuticals has yet been reported.
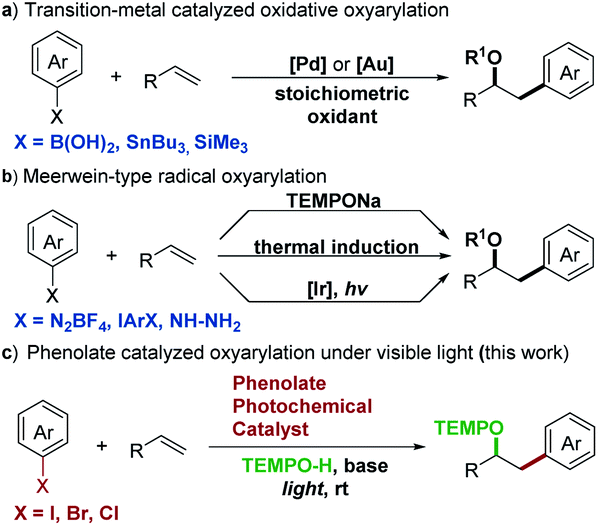 | ||
| Scheme 1 Strategies for intermolecular oxyarylation of olefins. (a) Transition-metal catalyzed oxidative oxyarylation. (b) Meerwein-type radical oxyarylation. (c) Phenolate catalyzed oxyarylation under visible light. | ||
In this context, a catalytic method for the direct oxyarylation of olefins with (hetero)aryl halides which are the cheapest and the most readily available arylation reagents would circumvent these issues and greatly expand the scope of aryl precursors. The single-electron reductive activation of aryl halides by photoredox catalysis has been a powerful strategy for aryl radical generation from stable starting materials.11 However, most of these methods require a large excess of sacrificial electron donors which result in a fast competing hydrogen atom abstraction by radical intermediates to deliver undesired side products and limit the choice of radical coupling partners. Additionally, aryl chlorides without electron withdrawing substituents are still a poorly accessible substrate for currently known visible light photocatalysts due to their large negative reduction potentials.12 Here, we demonstrated the application of the colored phenolate anion as a new visible light photocatalyst for the catalytic generation of (hetero)aryl radicals from non-activated (hetero)aryl halides and the successful development of an operationally simple and redox-neutral protocol for the intermolecular oxyarylation of a wide range of olefins (Scheme 1c).
Results and discussion
The details of our working hypothesis are described in Scheme 2. We envisioned a photocatalytic cycle wherein a suitable phenol catalyst A would first be deprotonated under basic conditions to generate the colored phenolate anion B, which could reach an electronically excited state (B*) under visible light irradiation. On the basis of our previous work,4 we proposed that a single electron transfer (SET) process between the excited phenolate anion B* and aryl halide D could occur to produce a phenoxyl radical C and an aryl radical E. The electrophilic aryl radical E would then undergo intermolecular addition to an olefin acceptor F to furnish a new C–C bond and an adjacent alkyl radical G. In the meantime, the phenoxyl radical C could abstract a hydrogen atom from TEMPO-H (the O–H BDFE in TEMPO-H ∼67 kcal mol−1)13 to recycle the phenol A (the O–H BDFE in phenol >75 kcal mol−1)13,14 and afford the persistent TEMPO radical. Finally, the highly selective radical–radical cross coupling of the transient alkyl radical G with TEMPO would be kinetically feasible to provide the desired oxyarylation product H based on the persistent radical effect.15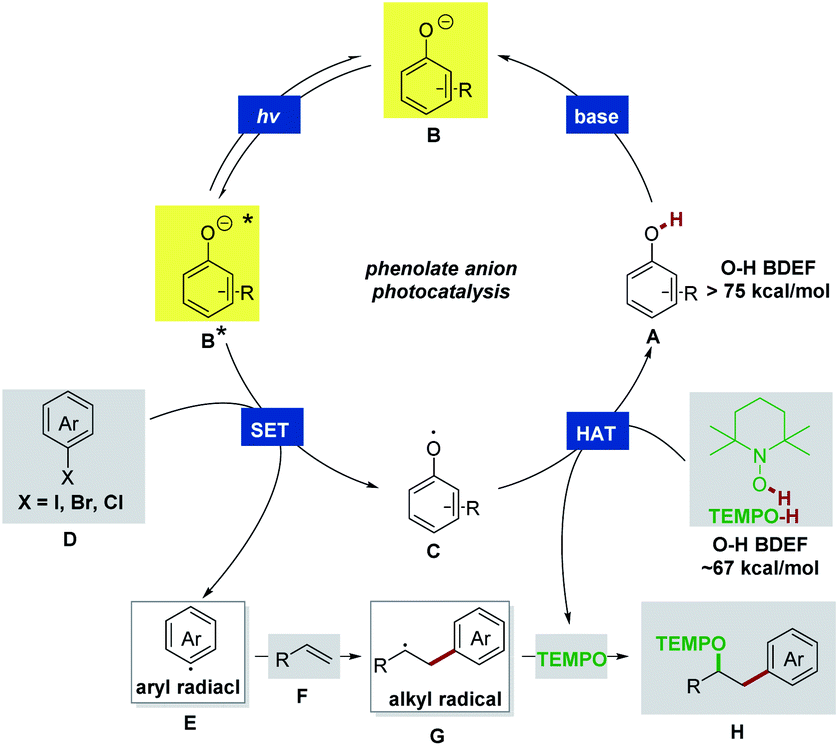 | ||
| Scheme 2 Proposed photocatalytic mechanism for intermolecular radical oxyarylation. | ||
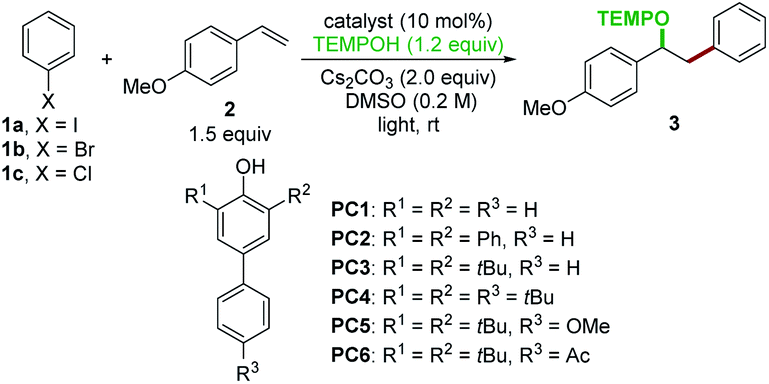
To evaluate these ideas, our initial exploration focused on the oxyarylation of 4-methoxystyrene 2 with iodobenzene 1a as a model reaction. The 4-phenylphenol derivatives were chosen as catalysts. After irradiation with two 18 W blue light emitting diode (LED) lamps for 16 h, a promising lead result was observed by using 4-phenylphenol (PC1) as the catalyst in the presence of TEMPOH and Cs2CO3 in DMSO, providing a 40% yield of the desired oxyarylation product 3 at room temperature (Table 1, entry 1). Further screening of catalysts revealed that 2,6-di-tert-butyl-4-phenylphenol (PC3) was the optimal catalyst for this protocol (entries 2–6), and product 3 was obtained in an 87% yield (entry 3). Interestingly, catalysts with a bis-tBu system (PC3–PC6) afforded higher yields compared to other tested catalysts (PC1 and PC2). The redox reversibility observed by electrochemical measurements (see ESI Fig. 5†) suggested that the introduction of bis-tBu remarkably prolongs the lifetime of the corresponding phenoxyl radicals. Therefore, we reasoned that the steric hindrance of the bis-tBu system might be good for the catalyst regeneration by improving the kinetic stability of the phenoxyl radicals, thus leading to high efficiency. In an attempt to seek better reaction conditions, a survey of other bases (KHCO3, Na2CO3, K2CO3, K3PO4, DBU, and TMG) and solvents (DMF, DMA, acetone, and CH3CN) indicated that they were not comparable to the combination of Cs2CO3 and DMSO (see ESI Table 1†). The control experiments revealed that the light, catalyst, and base were essential for this reaction (entries 7–9). Besides the bromobenzene 1b, the chlorobenzene 1c was also efficiently activated for the oxyarylation, albeit a moderate yield was achieved (entries 10 and 11). These results showed that the excited state phenolate anion was capable enough for reducing inert chlorobenzene.
|
|
||||
|---|---|---|---|---|
| Entrya | X | Catalyst | Light sources | Yieldb (%) |
| a General conditions: halobenzene 1 (0.30 mmol, 1.0 equiv.), 4-methoxystyrene 2 (0.45 mmol, 1.5 equiv.), TEMPOH (0.36 mmol, 1.2 equiv.), Cs2CO3 (0.60 mmol, 2.0 equiv.), catalyst (0.03 mmol, 10 mol%), and DMSO (1.5 mL, rigorously degassed by freezing/pumping/thawing). Halobenzene 1 and TEMPOH were dissolved in DMSO and added dropwise over 10 h. b Isolated yields by chromatography. c Reaction was run in the absence of Cs2CO3. | ||||
| 1 | I | PC1 | Blue LEDs | 40 |
| 2 | I | PC2 | Blue LEDs | 57 |
| 3 | I | PC3 | Blue LEDs | 87 |
| 4 | I | PC4 | Blue LEDs | 79 |
| 5 | I | PC5 | Blue LEDs | 73 |
| 6 | I | PC6 | Blue LEDs | 82 |
| 7 | I | PC3 | Dark | 0 |
| 8 | I | None | Blue LEDs | 0 |
| 9c | I | PC3 | Blue LEDs | 0 |
| 10 | Br | PC3 | Blue LEDs | 80 |
| 11 | Cl | PC3 | Blue LEDs | 42 |
With the optimized conditions in hand, we then explored the scope of the (hetero)aryl halides (Scheme 3, top). Iodides, bromides and chlorides (regardless of the electronic properties of the starting substrates) were successfully coupled to 4-methoxystyrene 2 with good to moderate yields (4–12). These results further emphasized the strong reducing properties of the excited phenolate anion of PC3. Moreover, the o- (4, 7, and 10), m- (5, 8, and 11) and p- (6, 9, and 12) substituted aryl halides readily participated in this transformation demonstrating that the site of the substitution on the aromatic ring had no detrimental effects on our method. It is worth highlighting that the mild conditions employed by this reaction were proved to be compatible with aryl halides containing a wide range of functional groups, including unprotected –CHO (13), –COCH3 (14), –COOH (15 and 16), –OH (17 and 18), and –NH2 (19), thereby eliminating the need for inefficient protection and deprotection procedures often required by traditional methodologies. Pleasingly, polycyclic aromatic hydrocarbon halides, such as naphthalene (20), anthracene (21), phenanthrene (22), and fluorene (23) can also be used as radical precursors under these conditions. Lastly, we investigated this photocatalytic transformation with a variety of privileged heterocyclic motifs, which were not involved in previously known methods. Remarkably, heterocycles such as pyridine (24–26), indole (27–29), quinoline (30), isoquinolone (31), pyrazine (32), thiophene (33) thianaphthene (34), benzofuran (35) and carbazole (36) were found to be efficient coupling partners in our reaction system.
 | ||
| Scheme 3 Reaction scope. General conditions: (hetero)aryl halide (0.30 mmol, 1.0 equiv.), olefin (0.45 mmol, 1.5 equiv.), TEMPOH (0.36 mmol, 1.2 equiv.), Cs2CO3 (0.60 mmol, 2.0 equiv.), PC3 (0.03 mmol, 10 mol%), and DMSO (1.5 mL, rigorously degassed by freezing/pumping/thawing). (Hetero)aryl halides and TEMPOH were dissolved in DMSO and added dropwise over 10 h. Isolated yields were reported. aWith 3 equiv. of Cs2CO3. bWith 3 equiv. of the olefin. | ||
We next surveyed the scope of olefin acceptors that could be used for the current transformation (Scheme 3, middle). Using iodobenzene 1a as a model aryl halide, various styrene derivatives were effectively converted to the corresponding oxyarylation products 37–42 in good to excellent yields under the standard conditions. Furthermore, this oxyarylation method was not only limited to aryl olefins; non-activated aliphatic olefins were also found to be compatible with this reaction system (43–52). We observed that a series of representative (hetero)aryl halides were successfully coupled to 1-hexene to give the desired products 43–47 with synthetically useful yields. To our delight, allylic sulfonamide (48) and alcohol derivatives (49) were also readily accommodated under these reaction conditions. In addition, the 1,1- or 1,2-disubstituted olefins were found to be competent coupling partners and provided the desired products with acceptable yields (50–52). Further studies revealed that enol ethers were also suitable substrates and yielded the oxyarylation products 53 and 54 in good yields. Subsequently, we found that intramolecular variants of this transformation were successful, and a variety of aryl iodides cyclized under our photocatalytic system to deliver a range of five- and six-membered heterocyclic products 55–60 (Scheme 3, bottom).
To expand the synthetic value of this method, we investigated the further chemical manipulation of these TEMPO-derived oxyarylation products (Scheme 4a). For instance, cleavage of the N–O bond in alkoxyamine 3 with Zn in acetic acid afforded the alcohol 61, oxidation of alkoxyamine 3 smoothly proceeded to provide ketone 62 with m-chloroperoxybenzoic acid (MCPBA) in DCM, and radical deoxygenation occurred upon heating alkoxyamine 3 in the presence of thiophenol to deliver compound 63.8a In addition, alkoxyamine 3 is also an ideal carbocation precursor in the Ir-catalyzed alkoxyamine radical cation mesolytic cleavage reaction, and the resulting carbocation intermediate can be trapped by different nucleophiles such as cyclohexanol, TMSN3, and indole to afford a series of useful molecular architectures 64–66.16 Finally, we evaluated our method for late-stage functionalization applications (Scheme 4b). Pharmaceutical ingredients (fenofibrate, furosemide, glibenclamide, and hydrochlorothiazide) that contained an aryl chloride were effectively converted into corresponding oxyarylation products 67–70 in 61–77% yield, further highlighting the utility of our method in a complex setting.
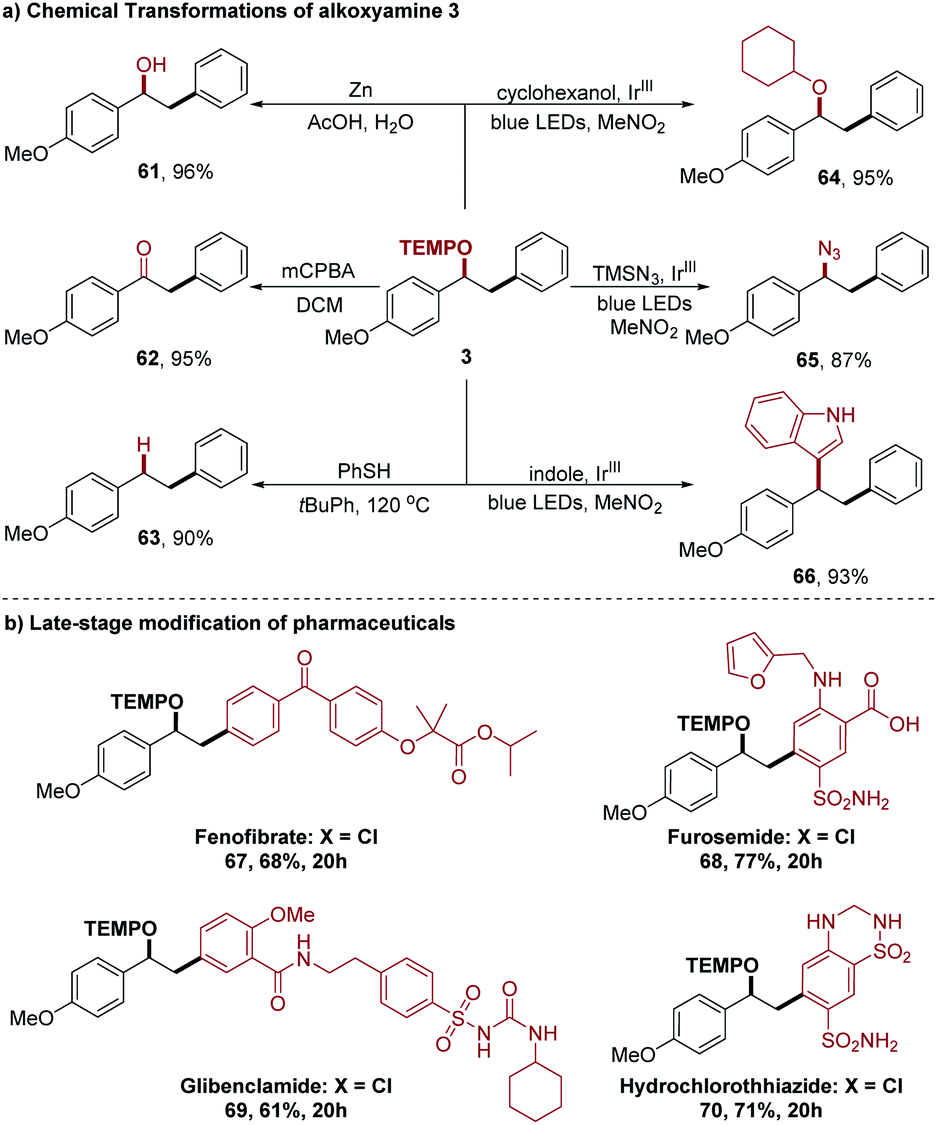 | ||
| Scheme 4 Synthetic applications of this photocatalytic oxyarylation reaction. (a) Chemical transformations of alkoxyamine 3. (b) Late-stage modification of pharmaceuticals. | ||
As a means of illuminating the mechanism of this proposed photocatalytic transformation, a number of UV-vis experiments were conducted (Fig. 1a). In analogy with our previous studies,4 we found that the colorless solution of PC3 (orange line in Fig. 1a) immediately turned primrose yellow upon addition of Cs2CO3 (blue line in Fig. 1a, the absorption of the phenolate anion of PC3) and did not observe any color change when iodobenzene 1a was added to the solution of the phenolate anion of PC3 (red line in Fig. 1a, perfectly overlapping with the absorption of the phenolate anion of PC3). These results excluded the formation of a ground-state EDA complex between the phenolate anion and aryl halide and indicated that the excited phenolate anion was responsible for triggering the formation of the aryl radical from its halide.3,17 Further support for this conclusion was obtained from Stern–Volmer quenching studies (Fig. 1b), in which the excited state of the phenolate anion of PC3 was effectively quenched by iodobenzene 1a. Moreover, a decline in the excited state lifetime also was observed in the time-resolved quenching studies, and the Stern–Volmer analysis revealed a linear correlation indicating a dynamic quenching of the excited phenolate anion of PC3 by 1a (see ESI note 5†).12b In addition, the thermodynamic feasibility of the photoinduced SET was analyzed from the oxidation-reduction potentials. The ground-state potential E1/2 (PC3˙/PC3−) was determined to be −0.10 V vs. SCE (Fig. 1c) and the excited-state energy E0–0 (PC3−*/PC3−) was read from the intersection of the normalized absorbance and emission spectra at 405 nm as 3.06 eV (Fig. 1d).18 Therefore, the redox potential of the excited phenolate anion of PC3 was calculated to be −3.16 V vs. SCE (E1/2 (PC3˙/PC3−*) = E1/2 ((PC3˙/PC3−) − E0–0 (PC3−*/PC3−)), which indicated that the reduction of iodobenzene 1a (−2.24 V vs. SCE), bromobenzene 1b (−2.44 V vs. SCE), and even chlorobenzene 1c (−2.78 V vs. SCE) by the excited phenolate anion of PC3 is thermodynamically feasible.19 To further probe whether the singlet excited state of the phenolate anion of PC3 could be responsible for the catalysis in our system, an oxygen tolerance experiment was performed open to air (Scheme 5a). Under these conditions, where triplet pathways should be inhibited by oxygen (a potent triplet quencher),20 a 72% yield was still observed after 16 h. These results indicated that the singlet state may be the primary mode of catalysis, but the combination catalysis of singlet and triplet states in our system cannot be completely ruled out at this time.20b
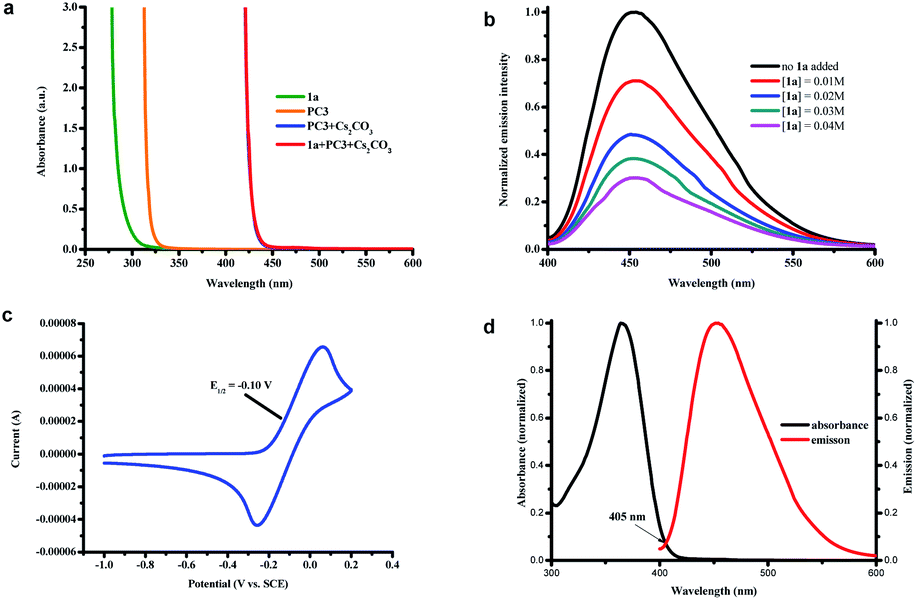 | ||
| Fig. 1 (a) UV/Vis absorption spectra of the DMSO solutions (0.10 M) of 1a, PC3, mixture of PC3 and Cs2CO3, and mixture of 1a, PC3 and Cs2CO3. (b) Quenching of the phenolate of PC3 emission (5 × 10−5 M in DMSO) in the presence of increasing amounts of 1a. (c) The cyclic voltammogram of the phenolate anion of PC3vs. SCE in DMSO at 0.1 V s−1. (d) Normalized absorption and emission spectra of the phenolate anion of PC3 in dry DMSO (5 × 10−5 M), and the intersection wavelength was considered to be 405 nm. | ||
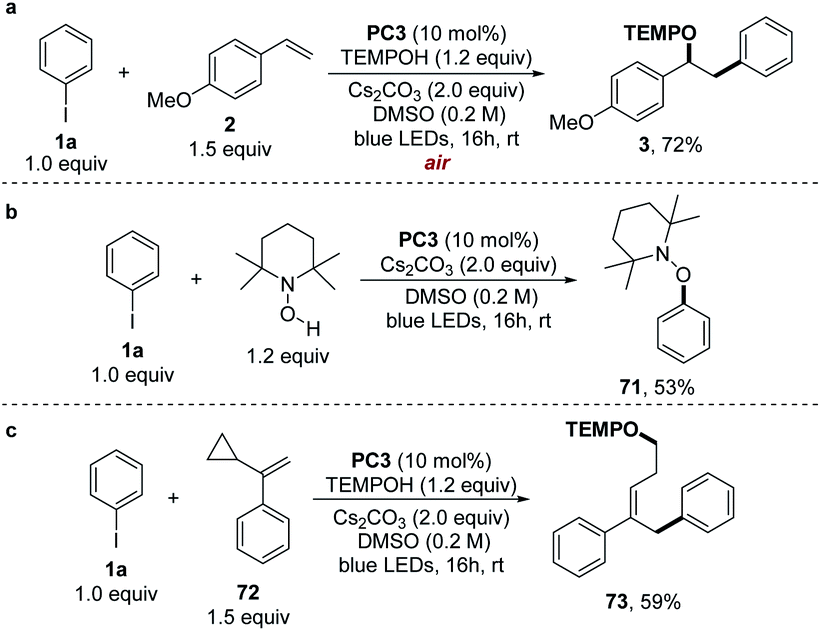 | ||
| Scheme 5 (a) Oxygen tolerance experiments. (b) Control experiments without 4-methoxystyrene. (c) Radical clock experiments. | ||
To support the radical mechanism suggested in Scheme 2, we performed several mechanism experiments. In the absence of 4-methoxystyrene, the reaction provided TEMPO-Ph (71) in a 53% yield (Scheme 5b), proving that a phenyl radical along with a TEMPO was formed under reaction conditions. We then carried out the radical clock experiments with α-cyclopropylstyrene (72) and found that the ring-opened adduct 73 was obtained in a 59% yield (Scheme 5c).These results strongly suggested the formation of a benzyl radical as the intermediate.21
Besides the proposed pathway of coupling between the alkyl radical and TEMPO as shown in Scheme 2, it is also possible that the alkyl radical is first oxidized by the phenoxyl radical in the SET pathway to give the carbocation and then is trapped by the TEMPOH alcohol to afford the oxyarylation product. In order to exclude the possible SET pathway between the benzyl radical and the phenoxy radical, 2-iodophenylmethanol 74 was chosen as a substrate to trap the carbocation intermediate by the intramolecular alcohol (Scheme 6). If the carbocation 76 was generated by the SET process with the phenoxyl radical (path b), an intermolecular cyclization will follow to give product 77. However, only the intermolecular oxyarylation product 78 was obtained in a 85% yield without any formation of intramolecular cyclization product 77. Instead, the alkoxyamine 78 was efficiently cyclized to provide 77 in a 92% yield through a carbocation intermediate in the Ir-catalyzed alkoxyamine radical cation mesolytic cleavage reaction,16 confirming that the cyclization could easily take place if the carbocation 76 was generated. These results implied that the radical–radical cross coupling of the benzyl radical with the HAT product TEMPO to provide the desired oxyarylation product would be more plausible.
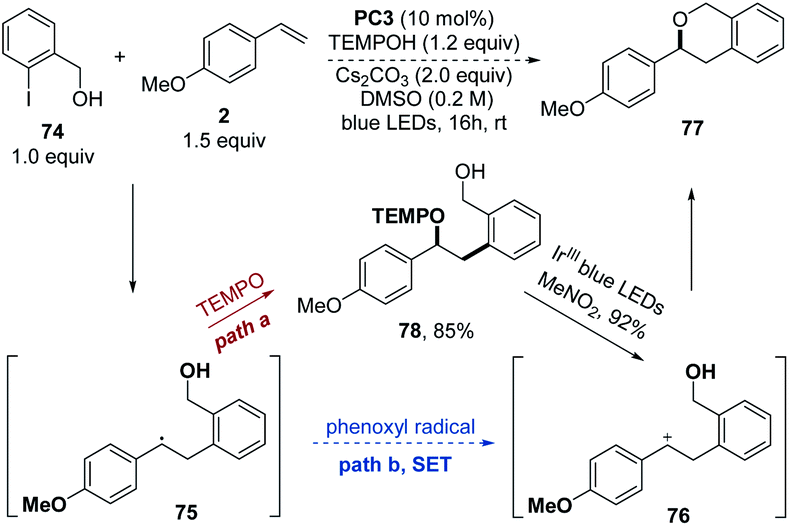 | ||
| Scheme 6 Intramolecular carbocation trapping experiments. | ||
Conclusions
In conclusion, the phenolate anion was discovered as a new photocatalyst under visible light and enabled the intermolecular oxyarylation of olefins with aryl and heteroaryl halides. This process utilized stable and readily available halogenated arenes as radical precursors, avoided the use of sacrificial reductants, and exhibited broad functional group tolerance. Moreover, the developed three-component coupling reaction was further applied to the late-stage modification of active pharmaceutical ingredients, and the resulting TEMPO-derived oxyarylation products are ideal precursors for diversified chemical manipulations to afford a series of useful molecular architectures. The highly reducing character of this photocatalytic system together with the mild reaction conditions will open new avenues for the development of aryl radical-based transformations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Natural Science Foundation of Yunnan Province (2018FY001015), the China Postdoctoral Science Foundation (2018M643540), the National Natural Science Foundation of China (21871228), and the Program for Changjiang Scholars and Innovative Research Team in University (IRT_17R94).Notes and references
- For recent reviews, see: (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (b) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (c) M. D. Kärkäs, J. A. Porco and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683–9747 CrossRef PubMed; (d) M. Silvi and P. Melchiorre, Nature, 2018, 554, 41 CrossRef CAS PubMed; (e) J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Acc. Chem. Res., 2016, 49, 1911–1923 CrossRef CAS PubMed; (f) M. N. Hopkinson, A. Tlahuext-Aca and F. Glorius, Acc. Chem. Res., 2016, 49, 2261–2272 CrossRef CAS PubMed; (g) D. Staveness, I. Bosque and C. R. J. Stephenson, Acc. Chem. Res., 2016, 49, 2295–2306 CrossRef CAS PubMed; (h) X. Huang and E. Meggers, Acc. Chem. Res., 2019, 52, 833–847 CrossRef CAS PubMed; (i) X. Lang, J. Zhao and X. Chen, Chem. Soc. Rev., 2016, 45, 3026–3038 RSC; (j) S. Protti, S. Garbarino, D. Ravelli and A. Basso, Angew. Chem., Int. Ed., 2016, 55, 15476–15484 CrossRef CAS PubMed; (k) J. Xie, H. Jin and A. S. K. Hashmi, Chem. Soc. Rev., 2017, 46, 5193–5203 RSC.
- (a) X. Chen, D. S. Larsen, S. E. Bradforth and I. H. M. van Stokkum, J. Phys. Chem. A, 2011, 115, 3807–3819 CrossRef CAS PubMed; (b) T. Ichino and R. W. Fessenden, J. Phys. Chem. A, 2003, 107, 9257–9268 CrossRef CAS; (c) R. W. Fessenden and N. C. Verma, J. Am. Chem. Soc., 1976, 98, 243–244 CrossRef CAS; (d) G. N. Lewis and D. Lipkin, J. Am. Chem. Soc., 1942, 64, 2801–2808 CrossRef CAS.
- G. Filippini, M. Nappi and P. Melchiorre, Tetrahedron, 2015, 71, 4535–4542 CrossRef CAS.
- K. Liang, T. Li, N. Li, Y. Zhang, L. Shen, Z. Ma and C. Xia, Chem. Sci., 2020, 11, 2130–2135 RSC.
- (a) S. Kirchberg, R. Fröhlich and A. Studer, Angew. Chem., Int. Ed., 2010, 49, 6877–6880 CrossRef CAS PubMed; (b) A. D. Melhado, W. E. Brenzovich, A. D. Lackner and F. D. Toste, J. Am. Chem. Soc., 2010, 132, 8885–8887 CrossRef CAS PubMed.
- A. D. Satterfield, A. Kubota and M. S. Sanford, Org. Lett., 2011, 13, 1076–1079 CrossRef CAS PubMed.
- (a) L. T. Ball, M. Green, G. C. Lloyd-Jones and C. A. Russell, Org. Lett., 2010, 12, 4724–4727 CrossRef CAS PubMed; (b) W. E. Brenzovich, J.-F. Brazeau and F. D. Toste, Org. Lett., 2010, 12, 4728–4731 CrossRef CAS PubMed; (c) L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, Chem.–Eur. J., 2012, 18, 2931–2937 CrossRef CAS PubMed.
- (a) M. Hartmann, Y. Li and A. Studer, J. Am. Chem. Soc., 2012, 134, 16516–16519 CrossRef CAS PubMed; (b) C.-J. Yao, Q. Sun, N. Rastogi and B. König, ACS Catal., 2015, 5, 2935–2938 CrossRef CAS; (c) S. Kindt, K. Wicht and M. R. Heinrich, Angew. Chem., Int. Ed., 2016, 55, 8744–8747 CrossRef CAS PubMed; (d) M. R. Heinrich, A. Wetzel and M. Kirschstein, Org. Lett., 2007, 9, 3833–3835 CrossRef CAS PubMed; (e) M. N. Hopkinson, B. Sahoo and F. Glorius, Adv. Synth. Catal., 2014, 356, 2794–2800 CrossRef CAS; (f) M. Bu, T. F. Niu and C. Cai, Catal. Sci. Technol., 2015, 5, 830–834 RSC; (g) R. Govindarajan, J. Ahmed, A. K. Swain and S. K. Mandal, J. Org. Chem., 2019, 84, 13490–13502 CrossRef CAS PubMed.
- (a) G. Fumagalli, S. Boyd and M. F. Greaney, Org. Lett., 2013, 15, 4398–4401 CrossRef CAS PubMed; (b) M. Hartmann, Y. Li, C. Mück-Lichtenfeld and A. Studer, Chem.–Eur. J., 2016, 22, 3485–3490 CrossRef CAS PubMed.
- (a) Y. Su, X. Sun, G. Wu and N. Jiao, Angew. Chem., Int. Ed., 2013, 52, 9808–9812 CrossRef CAS PubMed; (b) T. Taniguchi, H. Zaimoku and H. Ishibashi, Chem.–Eur. J., 2011, 17, 4307–4312 CrossRef CAS PubMed; (c) S. Kindt, H. Jasch and M. R. Heinrich, Chem.–Eur. J., 2014, 20, 6251–6255 CrossRef CAS PubMed; (d) Y.-H. Chen, M. Lee, Y.-Z. Lin and D. Leow, Chem.–Asian J., 2015, 10, 1618–1621 CrossRef CAS PubMed.
- For selected reviews and examples, see: (a) I. Ghosh, L. Marzo, A. Das, R. Shaikh and B. König, Acc. Chem. Res., 2016, 49, 1566–1577 CrossRef CAS PubMed; (b) G. Qiu, Y. Li and J. Wu, Org. Chem. Front., 2016, 3, 1011–1027 RSC; (c) R. Lekkala, R. Lekkala, B. Moku, K. P. Rakesh and H.-L. Qin, Eur. J. Org. Chem., 2019, 2019, 2769–2806 CrossRef CAS; (d) J. D. Nguyen, E. M. D'Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854–859 CrossRef CAS PubMed; (e) I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS PubMed; (f) A. J. Boyington, C. P. Seath, A. M. Zearfoss, Z. Xu and N. T. Jui, J. Am. Chem. Soc., 2019, 141, 4147–4153 CrossRef CAS PubMed.
- (a) A. U. Meyer, T. Slanina, A. Heckel and B. König, Chem.–Eur. J., 2017, 23, 7900–7904 CrossRef CAS PubMed; (b) M. Schmalzbauer, I. Ghosh and B. König, Faraday Discuss., 2019, 215, 364–378 RSC; (c) H. Kim, H. Kim, T. H. Lambert and S. Lin, J. Am. Chem. Soc., 2020, 142, 2087–2092 CrossRef CAS PubMed; (d) I. A. MacKenzie, L. Wang, N. P. R. Onuska, O. F. Williams, K. Begam, A. M. Moran, B. D. Dunietz and D. A. Nicewicz, Nature, 2020, 580, 76–80 CrossRef CAS PubMed; (e) N. G. W. Cowper, C. P. Chernowsky, O. P. Williams and Z. K. Wickens, J. Am. Chem. Soc., 2020, 142, 2093–2099 CrossRef CAS PubMed.
- J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS PubMed.
- G. Brigati, M. Lucarini, V. Mugnaini and G. F. Pedulli, J. Org. Chem., 2002, 67, 4828–4832 CrossRef CAS PubMed.
- (a) H. Fischer, Chem. Rev., 2001, 101, 3581–3610 CrossRef CAS PubMed; (b) A. Studer, Chem.–Eur. J., 2001, 7, 1159–1164 CrossRef CAS PubMed; (c) A. Studer, Chem. Soc. Rev., 2004, 033, 267–273 RSC; (d) D. Leifert and A. Studer, Angew. Chem., Int. Ed., 2020, 59, 74–108 CrossRef CAS PubMed.
- Q. Zhu, E. C. Gentry and R. R. Knowles, Angew. Chem., Int. Ed., 2016, 55, 9969–9973 CrossRef CAS PubMed.
- (a) M. Silvi, E. Arceo, I. D. Jurberg, C. Cassani and P. Melchiorre, J. Am. Chem. Soc., 2015, 137, 6120–6123 CrossRef CAS PubMed; (b) A. Bahamonde and P. Melchiorre, J. Am. Chem. Soc., 2016, 138, 8019–8030 CrossRef CAS PubMed.
- M. Liang, Z.-Y. Wang, L. Zhang, H.-Y. Han, Z. Sun and S. Xue, Renewable Energy, 2011, 36, 2711–2716 CrossRef CAS.
- L. Pause, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 1999, 121, 7158–7159 CrossRef CAS.
- (a) C. D. McTiernan, S. P. Pitre and J. C. Scaiano, ACS Catal., 2014, 4, 4034–4039 CrossRef CAS; (b) E. H. Discekici, N. J. Treat, S. O. Poelma, K. M. Mattson, Z. M. Hudson, Y. Luo, C. J. Hawker and J. R. de Alaniz, Chem. Commun., 2015, 51, 11705–11708 RSC.
- (a) H. Zhang, W. Pu, T. Xiong, Y. Li, X. Zhou, K. Sun, Q. Liu and Q. Zhang, Angew. Chem., Int. Ed., 2013, 52, 2529–2533 CrossRef CAS PubMed; (b) L. Guo, H.-Y. Tu, S. Zhu and L. Chu, Org. Lett., 2019, 21, 4771–4776 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc02160a |
| This journal is © The Royal Society of Chemistry 2020 |