
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modulation of charge transfer by N-alkylation to control photoluminescence energy and quantum yield†
Andrew T.
Turley
 a,
Andrew
Danos
b,
Antonio
Prlj
a,
Andrew P.
Monkman
b,
Basile F. E.
Curchod
a,
Paul R.
McGonigal
*a and
Marc K.
Etherington
*bc
a,
Andrew
Danos
b,
Antonio
Prlj
a,
Andrew P.
Monkman
b,
Basile F. E.
Curchod
a,
Paul R.
McGonigal
*a and
Marc K.
Etherington
*bc
aDepartment of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: paul.mcgonigal@durham.ac.uk
bDepartment of Physics, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: marc.k.etherington@northumbria.ac.uk
cDepartment of Mathematics, Physics and Electrical Engineering, Northumbria University, Ellison Place, NE1 8ST, UK
First published on 9th June 2020
Abstract
Charge transfer in organic fluorophores is a fundamental photophysical process that can be either beneficial, e.g., facilitating thermally activated delayed fluorescence, or detrimental, e.g., mediating emission quenching. N-Alkylation is shown to provide straightforward synthetic control of the charge transfer, emission energy and quantum yield of amine chromophores. We demonstrate this concept using quinine as a model. N-Alkylation causes changes in its emission that mirror those caused by changes in pH (i.e., protonation). Unlike protonation, however, alkylation of quinine's two N sites is performed in a stepwise manner to give kinetically stable species. This kinetic stability allows us to isolate and characterize an N-alkylated analogue of an ‘unnatural’ protonation state that is quaternized selectively at the less basic site, which is inaccessible using acid. These materials expose (i) the through-space charge-transfer excited state of quinine and (ii) the associated loss pathway, while (iii) developing a simple salt that outperforms quinine sulfate as a quantum yield standard. This N-alkylation approach can be applied broadly in the discovery of emissive materials by tuning charge-transfer states.
Introduction
‘Your “epipolic” dispersion has given me the clue to a most extensive field of research, which has occupied me during the last year when sunlight permitted’ wrote Sir George Stokes to Sir John Herschel on April 6, 1852.1 This prediction of an extensive field of research has held true through the continued use of spectroscopy to study epipolic dispersion that, also thanks to Stokes, we now know as fluorescence. In recent years, fluorescence spectroscopy has been used to characterize a wide range of different compounds and uncover new functional phenomena such as thermally activated delayed fluorescence (TADF),2–7 aggregation induced emission8–10 and room temperature phosphorescence.11–13TADF is of particular interest as it increases the efficiency of organic light-emitting diodes (OLEDs) used in displays and devices. The TADF process is contingent on the formation of charge-transfer (CT) states, i.e., the spatial redistribution of electron density in the excited state. Controlling and tuning CT states is crucial to the development of high efficiency TADF materials.14 This redistribution can result in reduced overlap between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO).4,15 CT states also have a significant impact on photochemistry and are commonplace in natural chromophores – for example, they have been invoked in photosynthesis16–18 and DNA repair19,20 mechanisms. CT states can contribute to enhancing light emission in organic compounds through mechanisms such as TADF, but their presence is not always desirable. Identification of their behaviour with respect to the electronic energy levels can guide material design.
Here, we utilize quinine (Qn) as a model system to demonstrate a strategy for modifying CT states of organic compounds. Recent literature has shown that some of the cinchona alkaloids display CT and proton transfer, which in turn dictate their photophysical properties.21,22 These studies have previously been performed using protonation equilibria in aqueous media. Here, we use N-alkylation as an alternative in order to permanently modify Qn, allowing investigation into the structure–property relationships between lone-pair availability, charge, emission energy and quantum yield. We demonstrate that N-alkylation is a versatile method for studying the effects of amine quaternization in aqueous and organic solvent media, not requiring acidic conditions or being subject to equilibria. The kinetic stabilities of the N-alkylated salts also make it possible to quaternize structures at positions other than their most basic N site. We identify one N site of Qn that can be manipulated to ‘turn off’ CT state formation and another whose modification tunes emission colour. The compound produced by double N-alkylation displays enhanced photoluminescence quantum yields (PLQYs) and solubilities across a range of solvents compared to acidified quinine sulfate (H2Qn·SO4), which is a common PLQY standard for characterizing blue emitters. Overall, this N-alkylation approach represents a simple, robust pathway for tuning the emission and functional properties of Qn and other tertiary amines23–26 to impart functional properties such as improved PLQYs and, potentially, TADF emission.
Results and discussion
Qn fluoresces blue with a high PLQY in acidified water (55% in 0.1 M aqueous H2SO4), but is only weakly emissive at UV wavelengths in basic solution.27 Recent work has shown that a similar pH-controlled increase in the PLQY of dehydroquinidine, an analogue of Qn, is linked to the availability of the quinuclidine lone pair electrons.21 It has been proposed that formation of a through-space CT state between the quinuclidine N and the quinoline chromophore leads to quenching of emission. This CT loss pathway is suppressed when the quinuclidine N is protonated, which increases the PLQY. We reasoned that N-alkylation of Qn could modulate its CT state in a similar manner and that investigation of N-alkylated derivatives would allow us to delineate the separate effects of quaternizing each of the two N sites (N1 and N2, Scheme 1) independently of one another, circumventing the intrinsic limitations of using protonation for this task.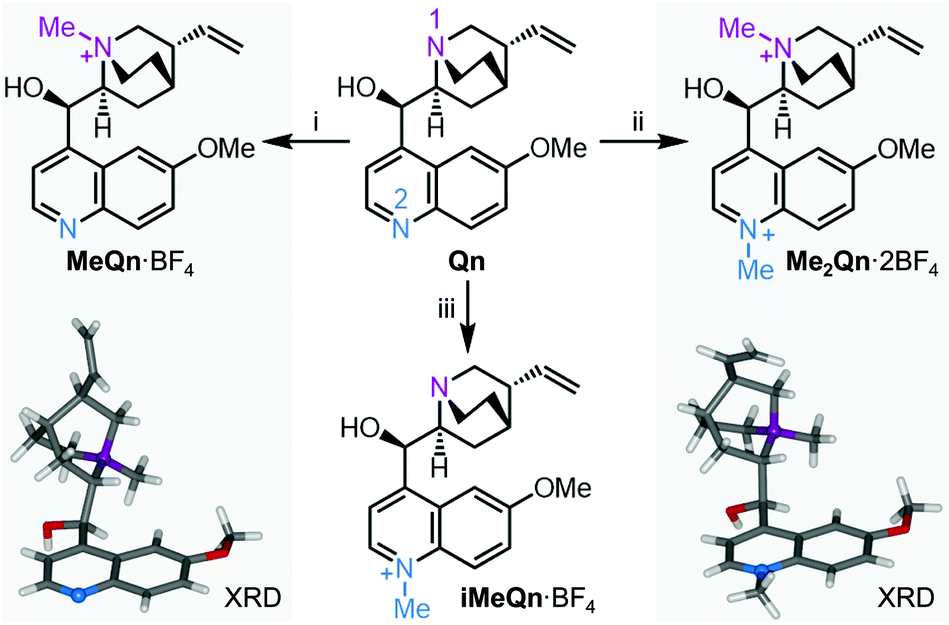 | ||
| Scheme 1 N-Alkylation of Qn to N-methylquinium tetrafluoroborate, MeQn·BF4, N,N′-dimethylquinium bis(tetrafluoroborate), Me2Qn·2BF4, and iso-N-methylquinium tetra-fluoroborate, iMeQn·BF4. XRD structures of MeQn·BF4 and Me2Qn·2BF4 are shown in stick representation with N atoms as balls. Further XRD structures can be found in Fig. S16, S17 and Table S1.† Reagents and conditions: (i) (a) MeI, rt, 3 d, (b) AgBF4, MeCN, 60 °C, 10 min, 74% over 2 steps; (ii) (a) MeI, MeCN, 100 °C, 4 h, (b) AgBF4, MeOH, rt, 10 min, 79% over 2 steps; (iii) (a) allyl bromide, CH2Cl2, rt, 16 h, (b) MeI, MeCN, 100 °C, 3 h, (c) barbituric acid, Pd(PPh3)4 (5 mol%), Me2SO, 40 °C, 16 h, (d) diisopropylaminomethyl polystyrene, MeOH, rt, 1 h, (e) AgBF4, MeOH, rt, 10 min, 79% over 5 steps. | ||
We synthesized three salts of methylated Qn (Scheme 1). Synthetic procedures and NMR spectroscopic characterisation can be found in Scheme S1 and Fig. S1–S13 in the ESI.† Selective N-methylation of the quinuclidine N (N1) is achieved by treating Qn with MeI at room temperature,28 giving MeQn+, whereas a reaction temperature of 100 °C leads to Me2Qn2+ by N-methylation of both N1 and the quinoline N (N2).29,30 This selectivity follows the known basicity trend of N1 and N2.21,31,32 Methylation at each site gives characteristic changes in 1H NMR chemical shifts (Fig. S14 and S15†) and is confirmed by X-ray diffraction (XRD) analysis of single crystals (Scheme 1).† By employing an allyl protecting group at N1 (Scheme S1†), however, it is also possible to prepare an isomeric form of MeQn+ that would be inaccessible using a thermodynamically controlled quaternization approach, such as reversible protonation under acidic conditions. After methylation at N2, the allyl protecting group at N1 is removed to give the kinetically stable ion iMeQn+. Each of the cations were isolated with halide counterions before exchanging to BF4 salts by metathesis with AgBF4 to avoid heavy-nucleus ions with large spin–orbit couplings that might complicate our photophysical investigations by quenching the singlet emission.33
The photophysical properties we have measured and modelled for Qn and its salts are summarized in Table 1.34 We first recorded (Fig. 1a) emission spectra of Qn dissolved in a series of solvents with a range of polarities. Although the absolute emission intensities from Qn in polar organic solvents are low on account of its near-zero PLQY, we could identify distinct emissions from a locally excited (LE) and a CT state.
| Compound | Φ /% | E em/eV | Calculated Eemd/eV | Presence of CTe | Red-shifted absorption and emissionf | |||
|---|---|---|---|---|---|---|---|---|
| MeCNb | H2O | MeCNb | H2O | LE | CT | |||
| a PLQYs (Φ) for Qn and MeQn·BF4 were measured with respect to a standard of 2-aminopyridine in 0.1 M aqueous H2SO4 (Φ = 60%)38 and those of Me2Qn·2BF4 and iMeQn·BF4 were measured with respect to a standard of H2Qn·SO4 in 0.1 M aqueous H2SO4 (Φ = 55%).27 b Anhydrous MeCN was used throughout the spectroscopic study. c Anhydrous MeCN solution with 10 mM Et3N used to suppress the formation of trace amounts of N1 protonated species resulting from adventitious water. d Excited-state energies in eV for protonated (rather than methylated) compounds calculated at the LR-TDDFT/ωB97X-D/6-31G* level of theory with state-specific implicit solvation (MeCN). e The presence or lack of an accessible CT state was determined by experimental observation (Fig. 1) and/or theoretical calculation (Tables S5 and S6). f Relative to the absorption and emission of Qn. | ||||||||
| Qn | 0 | 22 | 3.45 (2.30) | 3.20 | 3.75 | 1.41 | Yes | No |
| MeQn·BF4 | 5 | 32 | 3.40 | 3.24 | 3.69 | — | No | No |
| Me2Qn·2BF4 | 63 | 70 | 2.75 | 2.75 | 3.00 | — | No | Yes |
| iMeQn·BF4 | 0c | 60 | — | 2.75 | — | 1.01 | Yes | Yes |
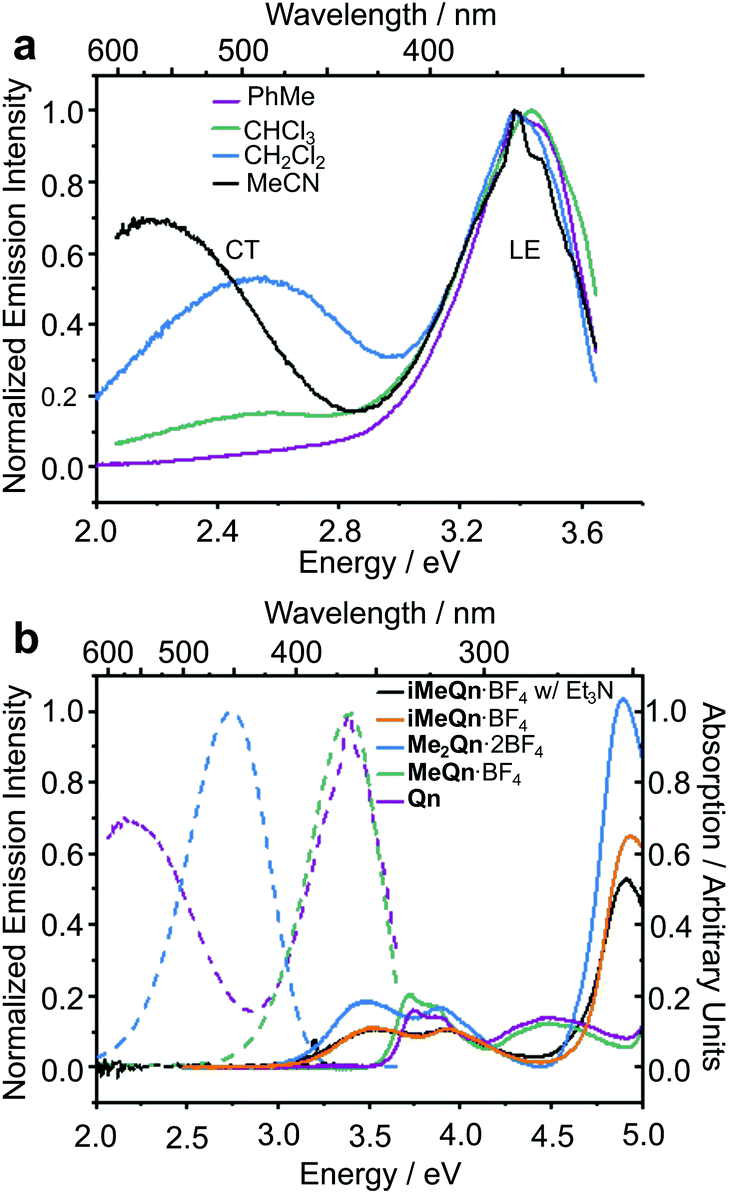 | ||
| Fig. 1 (a) Steady-state emission spectra of Qn dissolved in a series of solvents and excited at 3.75 eV, showing the LE and CT character of its emission. (b) The absorption (solid lines) and emission (dashed lines) spectra of Qn (purple), MeQn·BF4 (green), Me2Qn·2BF4 (blue), and iMeQn·BF4 (orange) in MeCN (20 μM). A significant shift in the absorption spectra is associated with the methylation of N2. The emission spectrum of iMeQn·BF4 in MeCN is omitted as partial protonation of N1 by adventitious water affects the spectrum. Instead, the absorption and emission spectra (black) of a MeCN solution of iMeQn·BF4 (20 μM) with Et3N (10 mM) are displayed to show there is no significant change in absorption with the addition of a base and that there is no emission in the visible region without protonation of N1. The excitation energies (Eex) used were 4.13 eV for Qn and MeQn·BF4 or 3.54 eV for Me2Qn·2BF4 and iMeQn·BF4 to allow comparison of PLQYs with known standards (Fig. S18–S26 and Table S2†). | ||
A peak in the UV region with emission energy, Eem, of 3.4 eV is present in low polarity solvent (PhMe), consistent with emission from an LE state. An additional peak is present in the visible region between 2.2 and 2.6 eV in more polar media. As the polarity is increased when moving from chlorinated solvents to MeCN, the increasing relative intensity of this second peak and its further bathochromic shift are indicative of emission from a CT state.35–37 The emission spectra are normalized relative to the peak of the LE (which has reduced intensity in polar solvents) in order to better demonstrate the solvatochromic shift. The intensities of the spectra alone do not illustrate the absolute populations of the states, but the changes in intensity can be rationalized. In apolar solvents there is a higher population of molecules emitting from the LE state, having a high oscillator strength. In polar solvents an important population of molecules is now emitting from the CT state, but the low oscillator strength results in a similar magnitude of emission relative to the LE state. Linear-response time-dependent density functional theory (LR-TDDFT) calculations performed at the ωB97X-D/6-31G* level of theory with state-specific implicit solvation (see ESI† for full all computational details and benchmarking) support the assignment of these LE and CT bands of Qn (Fig. 2). The CT state, in this case, is a result of a through-space charge transfer between the quinuclidine and quinoline system.
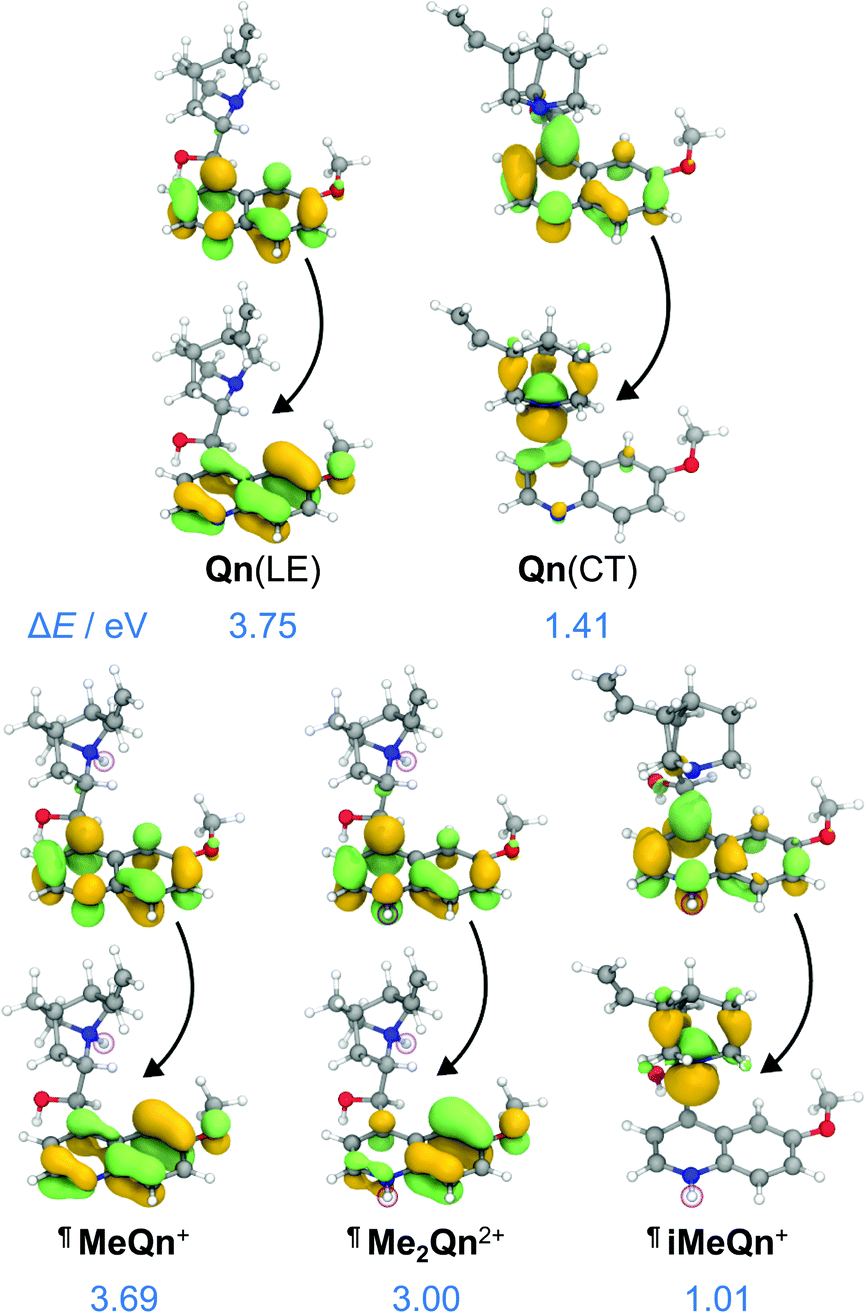 | ||
| Fig. 2 Natural transition orbitals (NTOs) and energies characterizing the singlet emission of Qn from its LE and CT states, as well as MeQn+, iMeQn+ and Me2Qn2+, calculated at the LR-TDDFT/ωB97X-D/6-31G* level of theory with state-specific implicit solvation. ¶ indicates that protonated structures were used as electronically similar (Table S7†) models for the methylated salts. | ||
The calculations predict (i) UV emission associated with an electronic transition localized on the quinoline ring system (π → π*) and (ii) a lower energy emission from a CT excited state, which is associated with an electronic transition from a nonbonding orbital located on N1 to an unoccupied π orbital on the quinoline ring (n → π*). We ascribe Qn's extremely low PLQY (Table 1) to the formation of this through-space CT state. Rapid nonradiative decay from the low-energy CT state, possibly through the low-lying LE triplet state (Fig. S27†), serves as a loss pathway that suppresses photoluminescence.39
To further confirm this hypothesis and to ‘switch on’ the UV emission of Qn, we investigated the effect of alkylating N1 selectively. Pleasingly, we observed (Fig. 1b) only a single peak in the emission spectrum of MeQn·BF4 at 3.4 eV in MeCN, indicative of emission from the π → π* LE state of the quinoline ring system. Compared to Qn, the peak wavelengths observed in the absorption and emission spectra of MeQn·BF4 are largely unchanged. However, MeQn·BF4 shows no evidence of the lower energy emission from a CT state found for Qn. Consequently, MeQn·BF4 emits (Table 1) with an increased PLQY of 5% in MeCN, compared to the near-zero PLQY of neutral Qn in MeCN. Quaternization at N1 prevents formation of a CT excited state, removing one of the pathways for rapid nonradiative decay and, as a result, enhancing the PLQY. Similarly, CT state formation is also suppressed for neutral H2O solutions of Qn, in which the major species at equilibrium is monoprotonated HQn+,31 giving a PLQY of 22%. The PLQY of MeQn·BF4 in H2O solution is 32%.
Methylation of N2 has a markedly different effect on the photoluminescence properties. We first investigated this effect by quaternizing both N1 and N2. The doubly methylated salt Me2Qn·2BF4 exhibits (Fig. 1b) significant bathochromic shifts in its absorption and emission energies relative to Qn and MeQn·BF4. These shifts are reproduced qualitatively by our LR-TDDFT (Table 1) and algebraic diagrammatic construction (ADC(2)) calculations (Tables S3 and S4†). Like MeQn·BF4, emission from Me2Qn·2BF4 (Fig. 2) comes exclusively from the π → π* LE state. However, the transition is brought into the blue region (2.8 eV) as a result of quaternizing N2.
Extrapolating from our observations based on MeQn·BF4 and Me2Qn·2BF4, we would expect that selective quaternization of only N2—the less basic N site—would give rise to an ion that retains the CT character of Qn on account of the available quinuclidine lone pair electrons (N1), but whose emission and absorption are red-shifted. The LR-TDDFT calculations fit with this hypothesis, predicting (Fig. 2) formation of a new low-energy CT state at lower electronic energy. Moreover, the LR-TDDFT indicates that, upon relaxation of the excited state, the CT state becomes the primary (singlet) ‘emissive’ state. The selectivity and kinetic stability of the N-alkylation approach allows us to test this hypothesis experimentally using iMeQn·BF4.
As predicted, the absorption spectrum of iMeQn·BF4 in MeCN matches closely (Fig. 1b) the spectrum of Me2Qn·2BF4. Photoluminescence measurements, which were carried out in MeCN with 10 mM triethylamine (Et3N) to prevent protonation of N1 by adventitious water, are also consistent. Unlike the other methylated derivatives, there is no detectable photoluminescence from iMeQn·BF4. Population of the extremely low-energy CT excited state predicted by calculation appears to lead solely to non-radiative decay. Overall, therefore, selective alkylation of different sites can be used to tune the photophysical behaviour of Qn, modulating independent properties in an orthogonal manner. It is a useful approach to control CT and emission energy. However, the kinetic stabilities and modified solubility profiles of the Qn salts also give them advantages over protonated analogues for use as functional materials. For example, while a doubly protonated Qn salt, H2Qn·SO4, is used routinely as a fluorescence standard for relative PLQY measurements in the blue region,10 its use is limited to acidic aqueous solutions – it is insoluble in common organic solvents and its photoluminescence is quenched in the aggregated state. The physical properties of Me2Qn·2BF4, on the other hand, make it appealing as a more versatile PLQY standard.
Our measurements show that it exhibits an enhanced PLQY of 70% in neutral H2O compared to the 55% PLQY of H2Qn·SO4 in 0.1 M aqueous H2SO4 (ref. 27) and, unlike H2Qn·SO4, it is soluble in organic solvents such as MeCN, EtOH, Me2CO, CH2Cl2, and EtOAc, giving rise to useful PLQYs (Fig. S18–S26 and Table S2†) of 63%, 43%, 54%, 48% and 13%, respectively. The variation in PLQY may be a result of modulating ion-pairing between the fluorescent cation and its counterion. Previous investigations of cationic fluorophores have also shown solvent-dependent PLQY and have suggested interactions with the counterion can provide radiationless decay pathways, e.g., through electron transfer and heavy atom effects.40,41 Overall, however, simple counterion exchange can be used to tune the solubility profile and emission properties. Work is ongoing in our laboratories to optimize these materials as PLQY standards and provide physical insight into the empirical changes in PLQY with solvent.
Conclusions
In summary, we have demonstrated an operationally simple method to tune and elucidate the CT excited states of emissive organic materials by N-alkylation. In the present investigation, this approach has allowed us to systematically toggle on and off the presence of a CT excited state, to change the emission colour, and to improve the PLQY of Qn – a compound whose photophysical properties were first studied by Herschel and Stokes over 150 years ago.42–44 This approach can be applied broadly to N-heterocycles, which are pervasive structural motifs in many organic chromophores. Resulting insights into their CT excited states will progress our understanding of natural photoactive systems16–18,45 and improve the performances of emissive2–7,39 and light-absorbing46–48 devices. The kinetic stabilities of the N-alkylated compounds open up the possibility of quaternizing the chromophores selectively at sites other than their most basic N site, achieving structures that are inaccessible by protonation, while the choice of counterion can serve as a handle to control physical properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Royal Society for access to their archives to retrieve the introductory quote, which was transcribed by Melody Bishop. We thank Dr Dmitry Yufit for solving single crystal XRD structures. A. T. T. acknowledges an EPSRC DTG. A. D., A. P. M. and M. K. E. acknowledge the HyperOLED Horizon 2020 project no. 732013. M. K. E. thanks the Hatfield Trust. This work made use of the facilities of the Hamilton HPC Service of Durham University.Notes and references
- G. G. Stokes, Sir George Gabriel Stokes to Sir John Frederick William Herschel, 6th April 1852, The Royal Society Archive, HS/17/27, 1852 Search PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef CAS PubMed.
- P. Stachelek, J. S. Ward, P. L. dos Santos, A. Danos, M. Colella, N. Haase, S. J. Raynes, A. S. Batsanov, M. R. Bryce and A. P. Monkman, ACS Appl. Mater. Interfaces, 2019, 11, 27125–27133 CrossRef CAS PubMed.
- D. G. Congrave, B. H. Drummond, P. J. Conaghan, H. Francis, S. T. E. Jones, C. P. Grey, N. C. Greenham, D. Credgington and H. Bronstein, J. Am. Chem. Soc., 2019, 141, 18390–18394 CrossRef CAS PubMed.
- S. Izumi, H. F. Higginbotham, A. Nyga, P. Stachelek, N. Tohnai, P. De Silva, P. Data, Y. Takeda and S. Minakata, J. Am. Chem. Soc., 2020, 142, 1482–1491 CrossRef CAS PubMed.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- J. Sturala, M. K. Etherington, A. N. Bismillah, H. F. Higginbotham, W. Trewby, J. A. Aguilar, E. H. C. Bromley, A.-J. Avestro, A. P. Monkman and P. R. McGonigal, J. Am. Chem. Soc., 2017, 139, 17882–17889 CrossRef CAS PubMed.
- H. Zhang, Z. Zhao, P. R. McGonigal, R. Ye, S. Liu, J. W. Y. Lam, R. T. K. Kwok, W. Z. Yuan, J. Xie, A. L. Rogach and B. Z. Tang, Mater. Today, 2020, 32, 275–292 CrossRef CAS.
- P. Pander, A. Swist, R. Turczyn, S. Pouget, D. Djurado, A. Lazauskas, R. Pashazadeh, J. V. Grazulevicius, R. Motyka, A. Klimash, P. J. Skabara, P. Data, J. Soloducho and F. B. Dias, J. Phys. Chem. C, 2018, 122, 24958–24966 CrossRef CAS.
- N. A. Kukhta, R. Huang, A. S. Batsanov, M. R. Bryce and F. B. Dias, J. Phys. Chem. C, 2019, 123, 26536–26546 CrossRef CAS.
- C. A. M. Salla, G. Farias, M. Rouzières, P. Dechambenoit, F. Durola, H. Bock, B. de Souza and I. H. Bechtold, Angew. Chem. Int. Ed., 2019, 58, 6982–6986 CrossRef CAS PubMed.
- R. Huang, N. A. Kukhta, J. S. Ward, A. Danos, A. S. Batsanov, M. R. Bryce and F. B. Dias, J. Mater. Chem. C, 2019, 7, 13224–13234 RSC.
- P. L. dos Santos, M. K. Etherington and A. P. Monkman, J. Mater. Chem. C, 2018, 6, 4842–4853 RSC.
- S. R. Meech, A. J. Hoff and D. A. Wiersma, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 9464–9468 CrossRef CAS PubMed.
- V. I. Novoderezhkin, J. P. Dekker and R. van Grondelle, Biophys. J., 2007, 93, 1293–1311 CrossRef CAS PubMed.
- T. K. Ahn, T. J. Avenson, M. Ballottari, Y.-C. Cheng, K. K. Niyogi, R. Bassi and G. R. Fleming, Science, 2008, 320, 794–797 CrossRef CAS PubMed.
- D. B. Bucher, C. L. Kufner, A. Schlueter, T. Carell and W. Zinth, J. Am. Chem. Soc., 2016, 138, 186–190 CrossRef CAS PubMed.
- L. Martinez-Fernandez, Y. Zhang, K. de La Harpe, A. A. Beckstead, B. Kohler and R. Improta, Phys. Chem. Chem. Phys., 2016, 18, 21241–21245 RSC.
- W. Qin, A. Vozza and A. M. Brouwer, J. Phys. Chem. C, 2009, 113, 11790–11795 CrossRef CAS.
- J. Qian and A. M. Brouwer, Phys. Chem. Chem. Phys., 2010, 12, 12562–12569 RSC.
- K.-L. Wang, W.-T. Liou, D.-J. Liaw and S.-T. Huang, Polymer, 2008, 49, 1538–1546 CrossRef CAS.
- J. Zhang, J. Chen, B. Xu, L. Wang, S. Ma, Y. Dong, B. Li, L. Ye and W. Tian, Chem. Commun., 2013, 49, 3878–3880 RSC.
- S. Achelle, J. Rodríguez-Lopez, C. Katan and F. Robin-Le Guen, J. Phys. Chem. C, 2016, 120, 26986–26995 CrossRef CAS.
- M. J. Matos, C. D. Navo, T. Hakala, X. Ferhati, A. Guerreiro, D. Hartmann, B. Bernardim, K. L. Saar, I. Compañón, F. Corzana, T. P. J. Knowles, G. Jiménez-Osés and G. J. L. Bernardes, Angew. Chem. Int. Ed., 2019, 58, 6640–6644 CrossRef CAS PubMed.
- W. H. Melhuish, J. Phys. Chem., 1961, 65, 229–235 CrossRef CAS.
- P. McNeice, F. M. F. Vallana, S. J. Coles, P. N. Horton, P. C. Marr, K. R. Seddon and A. C. Marr, J. Mol. Liq., 2020, 297, 111773 CrossRef CAS.
- O. Hesse, Justus Liebigs Ann. Chem., 1885, 230, 55–73 CrossRef.
- A. K. Yadav and A. Singh, Synlett, 2000, 8, 1199–1201 Search PubMed.
- S. G. Schulman, R. M. Threatte, A. C. Capomacchia and W. L. Paul, J. Pharm. Sci., 1974, 63, 876–880 CrossRef CAS PubMed.
- B. Xiang, K. M. Belyk, R. A. Reamer and N. Yasuda, Angew. Chem. Int. Ed., 2014, 53, 8375–8378 CrossRef CAS PubMed.
- J. H. Gutow, J. Chem. Educ., 2005, 82, 302–305 CrossRef CAS.
- Emission spectra are reported in eV and are Jacobian corrected to allow meaningful comparisons of Stokes shifts between compounds and emissive states, see: J. Mooney and P. Kambhampati, J. Phys. Chem. Lett., 2013, 4, 3316 CrossRef CAS.
- B. S. Brunschwig, S. Ehrenson and N. Sutin, J. Phys. Chem., 1987, 91, 4714–4723 CrossRef CAS.
- G. Verbeek, S. Depaemelaere, M. Van der Auweraer, F. C. De Schryver, A. Vaes, D. Terrell and S. De Meutter, Chem. Phys., 1993, 176, 195–213 CrossRef CAS.
- H. Laguitton-Pasquier, R. Pansu, J.-P. Chauvet, A. Collet, J. Faure and R. Lapouyade, Chem. Phys., 1996, 212, 437–455 CrossRef CAS.
- R. Rusakowicz and A. C. Testa, J. Phys. Chem., 1968, 72, 2680–2681 CrossRef CAS.
- M. K. Etherington, F. Franchello, J. Gibson, T. Northey, J. Santos, J. S. Ward, H. F. Higginbotham, P. Data, A. Kurowska, P. L. Dos Santos, D. R. Graves, A. S. Batsanov, F. B. Dias, M. R. Bryce, T. J. Penfold and A. P. Monkman, Nat. Commun., 2017, 8, 14987 CrossRef CAS PubMed.
- I. V. Avilov, A. Y. Panarin and V. S. Chirvony, Chem. Phys. Lett., 2004, 389, 352–358 CrossRef CAS.
- V. N. Knyukshto, K. N. Solovyov and G. D. Egorova, Biospectroscopy, 1998, 4, 121–133 CrossRef CAS PubMed.
- J. F. W. Herschel, Philos. Trans. R. Soc. London, 1845, 135, 143–145 CrossRef.
- G. G. Stokes, Philos. Trans. R. Soc. London, 1852, 142, 463–562 CrossRef.
- B. Valeur and M. N. Berberan-Santos, J. Chem. Educ., 2011, 88, 731–738 CrossRef CAS.
- S. Bai, V. Balevicius, E. Bittner, Y. C. Cheng, M. Chergui, J. Cina, N. Das Neves Rodrigues, A. Datta, J. Dawlaty, A. Dodin, B. Fingerhut, G. Fleming, N. Ginsberg, S. Hammes-Schiffer, V. Huxter, B. Kohler, Y. Lee, G. Leggett, A. Marcus, K. Morenz, J. Ogilvie, A. Olaya-Castro, T. A. A. Oliver, M. Son, Y. Song and V. Stavros, Faraday Discuss., 2019, 216, 133–161 RSC.
- A. Rao, P. C. Y. Chow, S. Gélinas, C. W. Schlenker, C. Li, H. Yip, A. K.-Y. Jen, D. S. Ginger and R. H. Friend, Nature, 2013, 500, 435–439 CrossRef CAS PubMed.
- M. K. Etherington, J. Wang, P. C. Y. Chow and N. C. Greenham, Appl. Phys. Lett., 2014, 104, 063304 CrossRef.
- W. Chang, D. N. Congreve, E. Hontz, M. E. Bahlke, D. P. McMahon, S. Reineke, T. C. Wu, V. Bulović, T. Van Voorhis and M. A. Baldo, Nat. Commun., 2015, 6, 6415 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1985987 (MeQn·BF4) and 1985988 (Me2Qn·2BF4). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02460k |
| This journal is © The Royal Society of Chemistry 2020 |