
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRing expansion of alumoles with organic azides: selective formation of six-membered aluminum–nitrogen heterocycles†
Regina
Drescher‡
a,
Shujuan
Lin‡
b,
Alexander
Hofmann
a,
Carsten
Lenczyk
a,
Stephanie
Kachel
a,
Ivo
Krummenacher
a,
Zhenyang
Lin
 *b and
Holger
Braunschweig
*a
*b and
Holger
Braunschweig
*a
aInstitute for Inorganic Chemistry, Institute for Sustainable Chemistry & Catalysis with Boron, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: h.braunschweig@uni-wuerzburg.de
bDepartment of Chemistry, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China. E-mail: chzlin@ust.hk
First published on 14th May 2020
Abstract
Aside from simple Lewis acid–base chemistry, the reaction chemistry of aluminacyclopentadienes, which are commonly referred to as aluminoles or simply alumoles, remains relatively underdeveloped. To date, few attempts to extend their inherent insertion and cycloaddition reactivity to the construction of stable aluminum-containing heterocycles have been reported. Herein, we demonstrate the selective ring expansion of a cyclopentadienyl-substituted alumole with a series of organic azides to form unsaturated six-membered AlN heterocycles. Depending on the substituent on the azide, the reaction proceeds either with or without loss of dinitrogen, leading to incorporation of only the “NR” unit of the azide or the entire azo substituent into the periphery of the heterocycle. A deeper understanding of these ring expansion reactions is reached through computational studies, illustrating deviations in the mechanism as a function of the organic azide employed.
Introduction
Fully unsaturated, five-membered ring systems containing a main-group or transition metal atom and a dianionic C4R4 backbone, a class of compounds known as metallacyclopentadienes, are highly versatile intermediates for synthesis, participating in numerous valuable transformations such as C–C and C-heteroatom coupling reactions.1 This is probably best illustrated by the facile conversions of zirconacyclopentadienes into a plethora of conjugated cyclic and acyclic compounds2 and their ability to transfer the diene unit to main group elements and transition metals, thereby generating a broad spectrum of unsaturated heterocycles in a single transmetalation step.3The aluminum-containing analogues of this system, commonly referred to as alumoles, have equally been shown to engage in coupling reactions where carbon–carbon and carbon–heteroatom bonds are formed, although reported examples are few.4–8 Moreover, with the exception of Tokitoh's work on alkyne insertions (vide infra),8 these studies fail to provide direct evidence for the proposed Al-heterocycles. Their instability and high reactivity has in general left the chemistry of alumoles relatively underdeveloped,9–19 despite their first synthesis dating back to 1962.20 While the mono- and bicyclic products formed in organic coupling reactions imply both insertion and cycloaddition reactivity of alumoles, the latter being unique to this family of organoaluminum compounds, intermediate adducts of the alumoles with the substrates appear to be reactive and have so far eluded isolation.4–8 In fact, isolable aluminacycles derived from either cycloaddition or ring expansion of alumoles are exceptionally rare.8 This reactivity contrasts with that of their lighter congeners, the boroles, which are well-known to insert a variety of substrates into the five-membered ring, thereby providing access to a diverse array of stable boracycles.21,22 For instance, while reactions with aldehydes,23 elemental sulfur24 or alkynes25 lead to expansion of the ring system in the case of boroles, in the case of alumoles they preferentially proceed with extrusion of the aluminum atom from the ring system and formation of aromatic coupling products such as cyclopentadienes,5 thiophenes7 and benzenes.4,7,8
In 2015, the group of Tokitoh reported the first and thus far only example of an alumole ring expansion by isolation of a nine-membered aluminacycle through reaction of a bromoalumole with an excess of 3-hexyne (see Scheme 1).8 Using only two equivalents of alkyne resulted in exclusive formation of the C–C coupling product, namely hexaethylbenzene. The same reaction pathway, yielding a naphthalene derivative, was predominant when Eisch studied the reaction of a benzannulated alumole with diphenylacetylene.4 Although these reactions are thought to proceed through a seven-membered alumepin (or aluminepin) intermediate, which is initially formed by alkyne insertion into the endocyclic Al–C bond of the alumole, it could neither be isolated nor spectroscopically detected (Scheme 1).26
 | ||
| Scheme 1 Previous report of an alumole ring expansion reaction by Tokitoh, with competing C–C coupling pathway.8 | ||
Rather than using alumoles in organic coupling reactions, we sought to exploit their inherent insertion reactivity as a route to larger aluminacycles. Herein, we demonstrate that alumoles undergo selective ring expansion reactions with organic azides to give stable aluminum–nitrogen heterocycles, which are structural analogs of 1,2-azaborinines, an emerging class of compounds with potential utility as therapeutic agents, catalysts and components in functional materials.27 Despite being known for more than half a century by the pioneering work of Eisch, synthetic access to this class of compounds remains very limited and restricted to benzo-fused derivatives.28,29 Reminiscent of the reactivity of boroles, ring expansion reactions of a new cyclopentadienyl-substituted tetraethylalumole with organic azides take different courses depending on the substituents of the azide. With the help of density functional theory (DFT) calculations we provide insight into the substituent effects on the ring expansion pathways.
Results and discussion
Two new alumole derivatives, 1 and 2, were synthesized by salt metathesis of the dilithium salt of 1,2,3,4-tetraethyl-1,3-butadiene with tert-butyl30 aluminum dichloride and 1,2,4-tris(tert-butyl)cyclopentadienyl (Cp3t) aluminum dibromide, respectively (Fig. 1).31 The isolated crystalline yields of 1 (6%) and 2 (30%) are rather low, presumably due to the oily nature and the facile hydrolysis of the products, although poor selectivity in these cyclometalation reactions cannot be entirely ruled out. In both cases, the 1H and 13C NMR spectra as well as the elemental analysis results are consistent with the proposed molecular connectivity. Their structures in the solid state, as determined by single-crystal X-ray analysis, are shown in Fig. 1. Alumole 1, bearing a tert-butyl group on the aluminum atom, favors a dimeric structure in the solid state, stabilized by interactions between an endocyclic carbon atom of one alumole unit to the Lewis acidic aluminum center of another. The bonding situation in the resulting Al2C2 ring (Al⋯Al* 2.638(1) Å; Al1–C1–Al1* 76.27(6)°) is comparable to those previously described for the 1-bromo16,18 and 1-chloro derivatives,18 and 1-ethyl-2,5-dimethyl-3,4-bis(trimethylsilyl)alumole.7,32 Likewise, the dimer-induced structural changes in the alumole ring are in good agreement with the reported data. Compound 2 remains monomeric in the solid state. The five-membered alumole ring in 2 is not completely planar and adopts a slightly twisted conformation. The C–C bond distances in the aluminacycle alternate between single (1.518(4) Å) and double bonds (1.345(3) and 1.357(4) Å) and resemble those of the closely related 1-(2,4,6-tri-tert-butylphenyl)-substituted derivative reported by Tokitoh, in which the alumole unit is perfectly planar.15 By comparison, the endocyclic Al–C bonds are slightly elongated owing to the higher coordination number of the aluminum atom in 2. Although the Cp3t ligand is somewhat unsymmetrically coordinated to Al with the C(Cp3t)–Al bond distances ranging from 2.253(3) to 2.314(3) Å, its hapticity is best described as η5.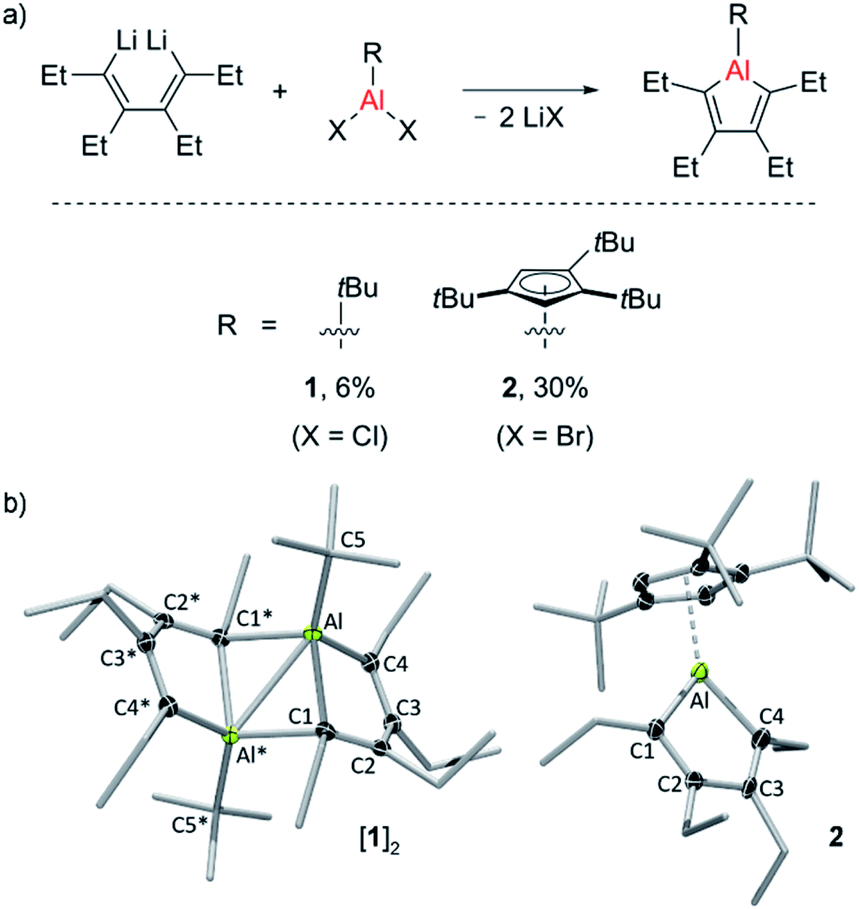 | ||
| Fig. 1 (a) Alumole formation via salt metathesis. (b) Molecular structures of dimeric 1 and monomeric 2 with displacement ellipsoids set at the 50% probability level, and H atoms removed for clarity. Selected bond distances (Å) and angles (°): 1: Al–C1 2.117(2), Al–C1* 2.155(2), Al⋯Al* 2.638(1); Al–C1–Al* 76.27(6); 2: Al–C1 1.977(3), Al–C4 1.966(3); C1–Al–C4 91.59(1). | ||
According to a series of temperature-dependent 1H NMR spectra between −50 and 90 °C, alumole 1 shows a fluxional behavior in toluene solution, likely resulting from a temperature-dependent monomer–dimer equilibrium. While at room temperature and above the 1H NMR spectra suggest a C2 symmetric structure consistent with the monomeric form, upon cooling to −50 °C, characteristic signals for the dimer are revealed (see ESI† for more details). The dimer-to-monomer dissociation in toluene solution is also corroborated by a 1H DOSY (diffusion-ordered spectroscopy) experiment at room temperature: the hydrodynamic radius of rH = 4.0 Å estimated from the diffusion coefficient (D = 8.74 × 1010 m2 s−1) is much smaller than that expected for a dinuclear species (r = 5.7 Å, rotational radius derived from X-ray crystallographic data). Thus, the data suggests that the monomeric form of 1 prevails at higher temperatures, and the dimeric form at lower temperatures. In addition, we note a peculiar broadening of the methylene proton resonances upon warming above room temperature, which could be indicative of conformational changes in the arrangement of the ethyl groups. By contrast, alumole 2 retains its monomeric structure in toluene solution over the same temperature range, as confirmed by variable-temperature proton NMR spectroscopy.
While the alumoles 1 and 2 do not form adducts with tetrahydrofuran (donor ligand) at room temperature, a stable adduct forms between alumole 2 and 4-dimethylaminopyridine (DMAP, Scheme 2). Adduct 2–DMAP was characterized by 1H and 13C NMR spectroscopy and X-ray diffraction analysis, confirming the expected Lewis acid–base complex formation via coordination of the pyridine nitrogen atom to the aluminum atom (for spectroscopic and crystal data, see ESI†). As revealed by the molecular structure of 2–DMAP, the coordination of the nitrogen base induces a change in the coordination mode of the Cp3t ligand from η5 to η1, leaving a single Al–C interaction of 2.081(2) Å. While the length of the newly formed Al–N bond (1.967(1) Å) is within the range of those found in other DMAP adducts of organoaluminum compounds,33 the carbon–carbon distances in the five-membered AlC4 ring are relatively unchanged by base coordination.
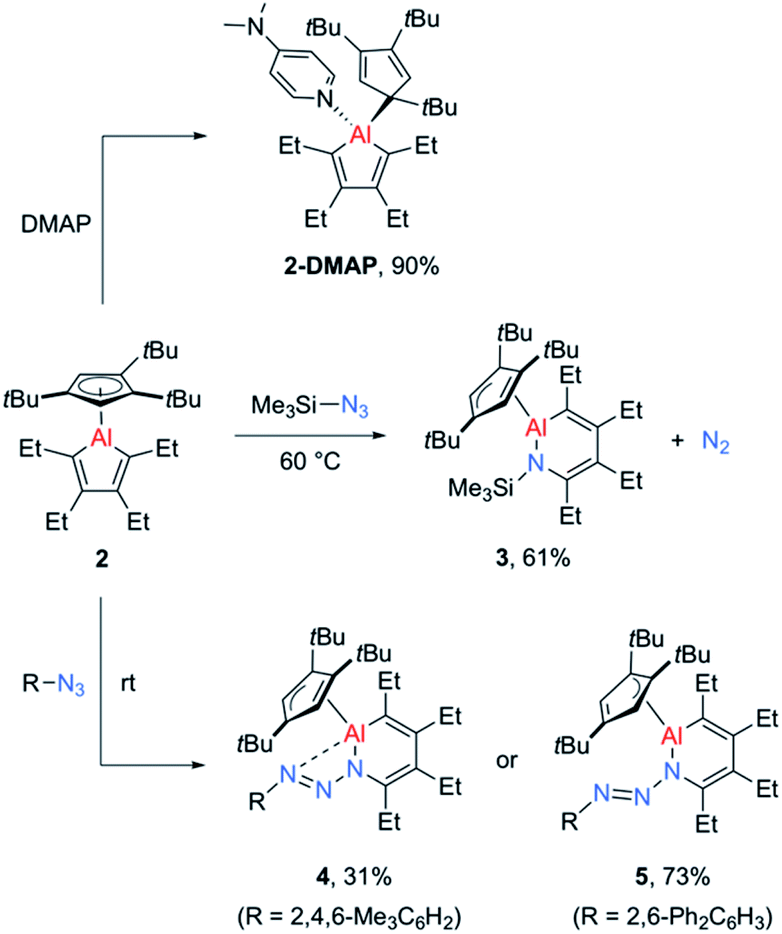 | ||
| Scheme 2 Reactivity of alumole 2 toward 4-dimethylaminopyridine (DMAP) and organic azides. | ||
Given the low yield and difficulties in the isolation of 1, we started exploring the reactivity of alumole 2 with a series of organic azides (Scheme 2). Treatment of equimolar amounts of alumole 2 and trimethylsilyl azide in benzene at 60 °C resulted in clean formation of product 3, which after recrystallization in pentane was obtained in 61% yield as colorless crystals. While the 1H NMR spectrum of 3 indicated the nonequivalence of the four methyl groups of the ethyl substituents and the incorporation of the trimethylsilyl group, its complete molecular constitution was revealed by X-ray structural analysis. The solid-state structure, as depicted in Fig. 2, shows the expected product of formal trimethylsilyl nitrene insertion into the endocyclic Al–C bond. The resulting six-membered aluminacycle adopts a distorted boat conformation with alternating C–C single (1.501(2) Å) and double bonds (1.358(2) and 1.359(2) Å). The Al–N bond length of 1.821(1) Å is at the shorter range of those seen in monomeric aluminium amide complexes of the form tBu2AlNR2 (ref. 34) and slightly shorter than those in two related aluminacycles reported by Roesky and co-workers (1.832(2)–1.857(2) Å) where the aluminum atom is intramolecularly stabilized by an amino group.29 Since the Al–N bond in 3 is highly twisted (∠C4–N1–Al1–C1 = 60.2°) and the nitrogen atom is considerably pyramidalized (Σ°(N) = 353.8), any Al–N π-bonding can be safely neglected. The Cp3t ligand is coordinated in an η2-fashion to Al, as indicated by two short (2.135(1) and 2.143(1) Å) and three long aluminum-ring carbon atom distances (2.594(1), 2.604(1), and 2.808(1) Å).
 | ||
| Fig. 2 Molecular structures of 3, 4 and 5 as determined by single-crystal X-ray diffraction. Pertinent bond distances (Å) and angles (°): 3: Al–N 1.828(1), Al–C1 1.940(1); N–Al–C1 102.75(5); 4: Al–N 1.921(1), Al–C1 1.995(1), Al–N3 2.080(1), N1–N2 1.320(2), N2–N3 1.308(2); N1–Al–C1 91.26(5); 5: Al–N1 1.884(3), Al–C1 1.940(4), N1–N2 1.342(4), N2–N3 1.277(4); N1–Al–C1 97.5(2). | ||
In contrast, the reaction of alumole 2 with mesityl (Mes = 2,4,6-Me3C6H2) or 2,6-diphenylphenyl azide led to a color change from colorless to orange and the formation of 4 or 5, respectively (Scheme 2). Mass spectrometry and X-ray diffraction analysis confirmed the products as being the arylazo-substituted derivatives in which the N3 linkage of the azide remained intact (see Fig. 2). Due to the presence of an intramolecular nitrogen–aluminum interaction (N3–Al1 2.080(1) Å), enabled by the sterically less hindered mesityl group, the azo N–N bond length in 4 (1.308(2) Å) is notably longer than that in 5 (1.277(4) Å). The intramolecular electron donation by the nitrogen atom leads to distinct structural changes around the aluminum atom. While the coordination mode of the Cp3t ligand in 4 is closer to η2, it is essentially η3 in 5. The additional nitrogen coordination to the aluminum atom in 4 is reflected in the elongation of the endocyclic Al–N (1.921(1) vs. 1.884(3) Å) and Al–C bonds (1.995(1) vs. 1.940(4) Å). The intraring C–N bonds in 4 (1.427(2) Å) and 5 (1.427(5) Å) are slightly contracted compared to that in 3 (1.436(2) Å), but well within the normal range for a C–N single bond. Lastly, while the endocyclic nitrogen atom in 5 is perfectly trigonal planar (Σ°(N) = 360.0), the nitrogen atom in 4 is strongly pyramidalized (Σ°(N) = 340.5) and thus not expected to be involved in π-bonding with the aluminum atom. Furthermore, the sterically-induced nonplanar conformation across the Al–N bond in 5, with a twist angle of 50.0°, inhibits overlap of the respective p-orbitals. As a result of the distinct structure and bonding of the triazene unit, the UV-vis absorption maxima differ between 4 (λmax = 338 nm, ε = 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 285 M−1 cm−1) and 5 (λmax = 370 nm, ε = 7923 M−1 cm−1).
285 M−1 cm−1) and 5 (λmax = 370 nm, ε = 7923 M−1 cm−1).
Furthermore, the differences in the N3 linkage are also expected to be revealed by their N–N stretching frequencies in the IR vibrational spectra. Although experimental assessment of the N–N stretching frequencies was complicated by the presence of multiple bands in the 1300–1500 cm−1 region, the calculated IR data (see ESI† for details of the DFT calculations) clearly mirror the differences in the azo bond lengths observed in the solid state, with lower frequencies for 4 (1343, 1347 cm−1) and higher for 5 (1357 cm−1).
In view of the reaction conditions required for the synthesis of 3 and its azo-substituted derivatives 4 and 5, it becomes apparent that the divergent ring expansion reactions involve very different energy barriers. While the reaction of alumole 2 with trimethylsilyl azide shows no conversion at room temperature and necessitates elevated temperatures, the aryl azides react readily with 2 at room temperature. Both reactions proceed without detectable intermediates as determined by 1H NMR spectroscopy, prompting us to pursue mechanistic studies by DFT calculations. For this analysis, we considered the reactions of trimethylsilyl and mesityl azides with the Cp3t-substituted alumole, whose structure was slightly simplified by substituting the ethyl for methyl groups in the periphery of the alumole and the tert-butyl for methyl groups in the Cp ring. Since the reactivity of alumole 2 towards organic azides resembles those of related boroles, we considered similar mechanistic pathways previously established for the latter, including initial attack of the terminal (γ) and internal (α) nitrogen atoms of the azide on the aluminum atom and various 1,3-dipolar cycloadditions between the azide and the alumole.21,24b,35 Overall, six different mechanisms were explored, which are illustrated in Scheme S2 in the ESI.†
For the modeled reaction of alumole 2′ with trimethylsilyl azide, the kinetically most favored pathway involves initial 1,3-dipolar addition of the azide across the endocyclic Al–C bond, resulting in the formation of the eight-membered ring system P3-IN1 in which the trimethylsilyl group is adjacent to the aluminum atom (see Fig. 3).35b,e Ring isomerization of the Cp ligand from η1 to η3, followed by stepwise migration of the silyl group to the nitrogen atom furthest from the Al atom yields the aluminacyclobutene P3-IN4 after loss of dinitrogen. Intramolecular Al–N coordination with concomitant cleavage of the endocyclic Al–C bond finally yields the observed ring-expanded product 3′. Of these steps, the elimination of dinitrogen has the highest energy barrier (28.7 kcal mol−1). This energy barrier more than doubles when mesityl instead of trimethylsilyl azide is used in the same reaction sequence (see Fig. S35 in the ESI†), reflecting the greater ease of 1,2-migration of the silyl group. An alternative pathway shown in red in Fig. 3 involving the cycloaddition transition states P3-TS1-5 and P3-TS5-6 is kinetically much less favorable. A similar high overall energy barrier is found for the denitrogenative pathway initiated by coordination of the α-nitrogen atom of the azide to the aluminum center (see Fig. S28 in the ESI†).
 | ||
| Fig. 3 Energy profile for the transformation of alumole 2′ into the trimethylsilyl-substituted AlN heterocycle 3′. The relative free energy and electronic energies (in brackets) are given in kcal mol−1. | ||
In contrast, for the formation of the arylazo-substituted azaaluminabenzene 4′, direct insertion of the terminal nitrogen atom of the azide into the Al–C bond of the alumole is favored. The energy profile, which is illustrated in Fig. 4, shows that the reaction proceeds through a single transition state (P2′-TS) to form the ring-expanded intermediate P2′-IN1, which furnishes the final product 4′ after intramolecular Al–N coordination. The overall free energy barrier for this process is about 7 kcal mol−1 lower than for the denitrogenative ring expansion and is in line with the experimental fact that the reaction of alumole 2 with mesityl azide takes place rapidly at room temperature. Although it is the kinetic product, azo derivative 4 (also 5) cannot be converted to the corresponding arylated AlN heterocycle by thermal (60–80 °C) or photolytic extrusion of nitrogen. The computational studies indicate that, in order for the nitrogen elimination to occur, a relatively high barrier has to be overcome, and this is facilitated by 1,2-silyl-group shifts in the silyl-containing compounds. For this reason, only the kinetic product is observed in the case of the aryl azides, the aryl group of which is less prone to migration.
 | ||
| Fig. 4 Energy profile for the transformation of alumole 2′ into the arylazo-substituted AlN heterocycle 4′. The relative free energy and electronic energies (in brackets) are given in kcal mol−1. | ||
Conclusions
In summary, two new alumole derivatives with a common tetraethyl-substituted backbone have been prepared and extensively characterized in solution and the solid state. The cyclopentadienyl-substituted alumole, which is monomeric in solution and the solid state, was found to undergo ring expansion reactions with organic azides to afford six-membered aluminum–nitrogen heterocycles, the first non-annulated 1,2-azaaluminabenzenes. The ring expansion products adopt non-planar structures due to the absence of π-bonding between aluminum and nitrogen. A considerable sensitivity toward the nature of the nitrogen substituent was observed in the ring expansion reactions. While trimethylsilyl azide reacts with loss of nitrogen, the N–N bonds of aryl azides are preserved in the products, leading to aluminum–nitrogen heterocycles bearing azo units. Mechanistic DFT studies suggest that the differences in the reactivity of the organic azides arise from the relative ease with which the silyl group can undergo 1,2-shifts, which in turn lower the activation barrier for nitrogen elimination.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Julius-Maximilians-Universität Würzburg and the Research Grants Council of Hong Kong (HKUST16304017 and 16302418) for financial support of this work.Notes and references
- (a) W. Ma, C. Yu, T. Chen, L. Xu, W.-X. Zhang and Z. Xi, Chem. Soc. Rev., 2017, 46, 1160–1192 RSC; (b) W. Ma, C. Yu, T. Chen, L. Xu, W.-X. Zhang and Z. Xi, Chem. Soc. Rev., 2018, 47, 2217 RSC.
- T. Takahashi and Y. Li, in Titanium and Zirconium in Organic Synthesis, ed. I. Marek, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2002, pp. 50–85 Search PubMed.
- (a) X. Yan and C. Xi, Acc. Chem. Res., 2015, 48, 935–946 CrossRef CAS PubMed; (b) P. J. Fagan, W. A. Nugent and J. C. Calabrese, J. Am. Chem. Soc., 1994, 116, 1880–1889 CrossRef CAS; (c) P. J. Fagan and W. A. Nugent, J. Am. Chem. Soc., 1988, 110, 2310–2312 CrossRef CAS.
- J. J. Eisch and R. L. Harrell Jr, J. Organomet. Chem., 1969, 20, 257–260 CrossRef CAS.
- (a) Z. Xi and P. Li, Angew. Chem., Int. Ed., 2000, 39, 2950–2952 CrossRef CAS; (b) C. Zhao, P. Li, X. Cao and Z. Xi, Chem.–Eur. J., 2002, 8, 4292–4298 CrossRef CAS PubMed.
- L. O. Khafizova, L. R. Yakupova, R. A. Tuktarova, V. A. D’yakonov, L. M. Khalilov, A. G. Ibragimov and U. M. Dzhemilev, Russ. J. Org. Chem., 2008, 44, 1311–1315 CrossRef CAS.
- Y. Zhang, J. Wei, W.-X. Zhang and Z. Xi, Inorg. Chem., 2015, 54, 10695–10700 CrossRef CAS PubMed.
- T. Agou, T. Wasano, T. Sasamori, J.-D. Guo, S. Nagase and N. Tokitoh, Angew. Chem., Int. Ed., 2015, 54, 9568–9571 CrossRef CAS PubMed.
- J. J. Eisch and W. C. Kaska, J. Am. Chem. Soc., 1966, 88, 2976–2983 CrossRef CAS.
- H. Hoberg, R. Krause-Göing, C. Krüger and J. C. Sekutowski, Angew. Chem., Int. Ed. Engl., 1977, 16, 183–184 CrossRef.
- H. Hoberg and R. Krause-Göing, J. Organomet. Chem., 1977, 127, C29–C31 CrossRef CAS.
- C. Krüger, J. C. Sekutowski, H. Hoberg and R. Krause-Göing, J. Organomet. Chem., 1977, 141, 141–148 CrossRef.
- H. Hoberg and W. Richter, J. Organomet. Chem., 1980, 195, 347–353 CrossRef CAS.
- H.-Y. Fang and Z.-F. Xi, Progr. Chem., 2005, 17, 1041–1047 CAS.
- T. Agou, T. Wasano, P. Jin, S. Nagase and N. Tokitoh, Angew. Chem., Int. Ed., 2013, 52, 10031–10034 CrossRef CAS PubMed.
- T. Wasano, T. Agou, T. Sasamori and N. Tokitoh, Chem. Commun., 2014, 50, 8148–8150 RSC.
- T. Agou, T. Wasano, T. Sasamori and N. Tokitoh, Organometallics, 2014, 33, 6963–6966 CrossRef CAS.
- N. Tokitoh, T. Agou, T. Wasano and T. Sasamori, Phosphorus, Sulfur Silicon Relat. Elem., 2016, 191, 584–587 CrossRef CAS.
- J. Li, P. Wu, W. Jiang, B. Wang, H. Zhu and H. Roesky, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202000899.
- J. J. Eisch and W. C. Kaska, J. Am. Chem. Soc., 1962, 84, 1501–1502 CrossRef CAS.
- J. H. Barnard, S. Yruegas, K. Huang and C. D. Martin, Chem. Commun., 2016, 52, 9985–9991 RSC.
- For general reviews on boroles, see: (a) J. Eisch, Adv. Organomet. Chem., 1996, 39, 355–391 CrossRef CAS; (b) A. Steffen, R. M. Ward, W. D. Jones and T. B. Marder, Coord. Chem. Rev., 2010, 254, 1950–1976 CrossRef CAS; (c) H. Braunschweig and T. Kupfer, Chem. Commun., 2011, 47, 10903–10914 RSC; (d) H. Braunschweig, I. Krummenacher and J. Wahler, Adv. Organomet. Chem., 2013, 61, 1–53 CrossRef CAS; (e) H. Braunschweig and I. Krummenacher, in Organic Redox Systems: Synthesis, Properties, and Applications, ed. T. Nishinaga, John Wiley & Sons, New Jersey, 2016, pp. 503–522 Search PubMed; (f) B. Su and R. Kinjo, Synthesis, 2017, 49, 2985–3034 CrossRef CAS; (g) Y. Su and R. Kinjo, Chem. Soc. Rev., 2019, 48, 3613–3659 RSC.
- K. Huang and C. D. Martin, Inorg. Chem., 2015, 54, 1869–1875 CrossRef CAS PubMed.
- (a) S. Yruegas and C. D. Martin, Chem.–Eur. J., 2016, 22, 18358–18361 CrossRef CAS PubMed; (b) X. Su, J. J. Baker and C. D. Martin, Chem. Sci., 2020, 11, 126–131 RSC.
- (a) J. J. Eisch and J. E. Galle, J. Am. Chem. Soc., 1975, 97, 4436–4437 CrossRef CAS; (b) J. J. Eisch, J. E. Galle, B. Shafii and A. L. Rheingold, Organometallics, 1990, 9, 2342–2349 CrossRef CAS; (c) C. Fang, W. E. Piers, M. Parvez and R. McDonald, Organometallics, 2010, 29, 5132–5139 CrossRef; (d) Y. Shoji, N. Tanaka, S. Muranaka, N. Shigeno, H. Sugiyama, K. Takenouchi, F. Hajjaj and T. Fukushima, Nat. Commun., 2016, 7, 12704 CrossRef PubMed.
- A stable aluminepin derivative is known, see: K. Yoshida, T. Furuyama, C. Wang, A. Muranaka, D. Hashizume, S. Yasuike and M. Uchiyama, J. Org. Chem., 2012, 77, 729–732 CrossRef CAS PubMed.
- For recent reviews, see: (a) Z. X. Giustra and S.-Y. Liu, J. Am. Chem. Soc., 2018, 140, 1184–1194 CrossRef CAS PubMed; (b) G. Bélanger-Chabot, H. Braunschweig and D. K. Roy, Eur. J. Inorg. Chem., 2017, 4353–4368 CrossRef; (c) P. G. Campbell, A. J. V. Marwitz and S.-Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 6074–6092 CrossRef CAS PubMed.
- J. J. Eisch, J. Am. Chem. Soc., 1964, 86, 4221–4222 CrossRef CAS.
- S. Chen, B. Li, X. Wang, Y. Huang, J. Li, H. Zhu, L. Zhao, G. Frenking and H. W. Roesky, Chem.–Eur. J., 2017, 23, 13633–13637 CrossRef CAS PubMed.
- A. Hofmann, T. Tröster, T. Kupfer and H. Braunschweig, Chem. Sci., 2019, 10, 3421–3428 RSC.
- H. Lehmkuhl, O. Olbrysch and H. Nehl, Liebigs Ann. Chem., 1973, 708–714 CrossRef CAS.
- See, also: W. Uhl, A. Hepp, H. Westenberg, S. Zemke, E.-U. Würthwein and J. Hellmann, Organometallics, 2010, 29, 1406–1412 CrossRef CAS.
- (a) F. Thomas, T. Bauer, S. Schulz and M. Nieger, Z. Anorg. Allg. Chem., 2003, 629, 2018–2027 CrossRef CAS; (b) K. Leszczyńska, I. Madura, A. R. Kunicki and J. Zachara, J. Organomet. Chem., 2006, 691, 5970–5979 CrossRef; (c) J. J. Weigand, N. Burford, A. Decken and A. Schulz, Eur. J. Inorg. Chem., 2007, 4868–4872 CrossRef CAS; (d) T. Nakamura, K. Suzuki and M. Yamashita, J. Am. Chem. Soc., 2014, 136, 9276–9279 CrossRef CAS PubMed; (e) S. J. Urwin, D. M. Rogers, G. S. Nichol and M. J. Cowley, Dalton Trans., 2016, 45, 13695–13699 RSC.
- M. A. Petrie, K. Ruhlandt-Senge and P. P. Power, Inorg. Chem., 1993, 32, 1135–1141 CrossRef CAS.
- (a) H. Braunschweig, C. Hörl, L. Mailänder, K. Radacki and J. Wahler, Chem.–Eur. J., 2014, 20, 9858–9861 CrossRef CAS PubMed; (b) S. A. Couchman, T. K. Thompson, D. J. D. Wilson, J. L. Dutton and C. D. Martin, Chem. Commun., 2014, 50, 11724–11726 RSC; (c) H. Braunschweig, M. A. Celik, F. Hupp, I. Krummenacher and L. Mailänder, Angew. Chem., Int. Ed., 2015, 54, 6347–6351 CrossRef CAS PubMed; (d) H. Braunschweig, F. Hupp, I. Krummenacher, L. Mailänder and F. Rauch, Chem.–Eur. J., 2015, 21, 17844–17849 CrossRef CAS PubMed; (e) H. Braunschweig, M. A. Celik, T. Dellermann, G. Frenking, K. Hammond, F. Hupp, H. Kelch, I. Krummenacher, F. Lindl, L. Mailänder, J. H. Müssig and A. Ruppert, Chem.–Eur. J., 2017, 23, 8006–8013 CrossRef CAS PubMed; (f) S. Yruegas, J. J. Martinez and C. D. Martin, Chem. Commun., 2018, 54, 6808–6811 RSC; (g) W. Zhang, G. Li, L. Xu, W. Wan, N. Yan and G. He, Chem. Sci., 2018, 9, 4444–4450 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1987117–1987122. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02032j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |