
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFocusing, sorting, and separating microplastics by serial faradaic ion concentration polarization†
Collin D.
Davies
and
Richard M.
Crooks
 *
*
Department of Chemistry and Texas Materials Institute, The University of Texas at Austin, 105 E. 24th St., Stop A5300, Austin, Texas, 78712-1224, USA. E-mail: crooks@cm.utexas.edu; Tel: +1-512-475-8674
First published on 20th May 2020
Abstract
In this article, we report continuous sorting of two microplastics in a trifurcated microfluidic channel using a new method called serial faradaic ion concentration polarization (fICP). fICP is an electrochemical method for forming ion depletion zones and their corresponding locally elevated electric fields in microchannels. By tuning the interplay between the forces of electromigration and convection during a fICP experiment, it is possible to control the flow of charged objects in microfluidic channels. The key findings of this report are threefold. First, fICP at two bipolar electrodes, configured in series and operated with a single power supply, yields two electric field gradients within a single microfluidic channel (i.e., serial fICP). Second, complex flow variations that adversely impact separations during fICP can be mitigated by minimizing convection by electroosmotic flow in favor of pressure-driven flow. Finally, serial fICP within a trifurcated microchannel is able to continuously and quantitatively focus, sort, and separate microplastics. These findings demonstrate that multiple local electric field gradients can be generated within a single microfluidic channel by simply placing metal wires at strategic locations. This approach opens a vast range of new possibilities for implementing membrane-free separations.
Introduction
In this paper, we report focusing, sorting, and separating of charged microplastics using electrokinetic phenomena at bipolar electrodes (BPEs) operated in a series configuration. The results demonstrate that serial bipolar electrochemistry provides an experimentally simple approach to form and maintain multiple electric field gradients (EFGs) within a single microfluidic channel. Moreover, by tuning the orientation and dimensions of the BPEs, it is possible to simultaneously manipulate the flow of charged microplastics at multiple positions along the channel length. The results reported here illustrate the utility of serial bipolar electrochemistry for continuous separations generally and of microplastics specifically.One of the most interesting approaches for controlling the motion of charged species within microchannels involves the use of ion depletion zones (IDZs) and their associated electric fields.1–4 An IDZ refers to a region of solution containing fewer charge carriers, and thus exhibiting higher solution resistance, than the bulk. Consequently, when a voltage is applied across a microchannel containing an IDZ, a disproportionate amount of the voltage drops within the IDZ thereby yielding an EFG. Charged species experience enhanced electromigration along the EFG and, by tuning the rate of solution convection, their motion can be controlled within the microchannel.
EFG focusing is a relatively simple method for controlling the motion of charged species.5–7 For example, consider the case of the negatively charged object shown in Scheme 1a. In this microfluidic experiment, the motion of the object is controlled by the interplay of two forces: convection and electromigration. When the object is at position A, the magnitude of the electric field is small, convection dominates electromigration, and the analyte moves from right to left. As it moves to the left, however, it encounters the IDZ and its associated locally enhanced electric field. Accordingly, at axial position B, the electric field strength is such that electromigration and convection are equal in magnitude but opposite in direction and therefore the net velocity of the analyte is zero. If the analyte were to diffuse to position C, the higher electric field would restore the object to position B. The net effect is that the object is focused at axial position B. In this way, EFGs have been used to enrich,8–13 deplete,14,15 and separate charged species.16–19
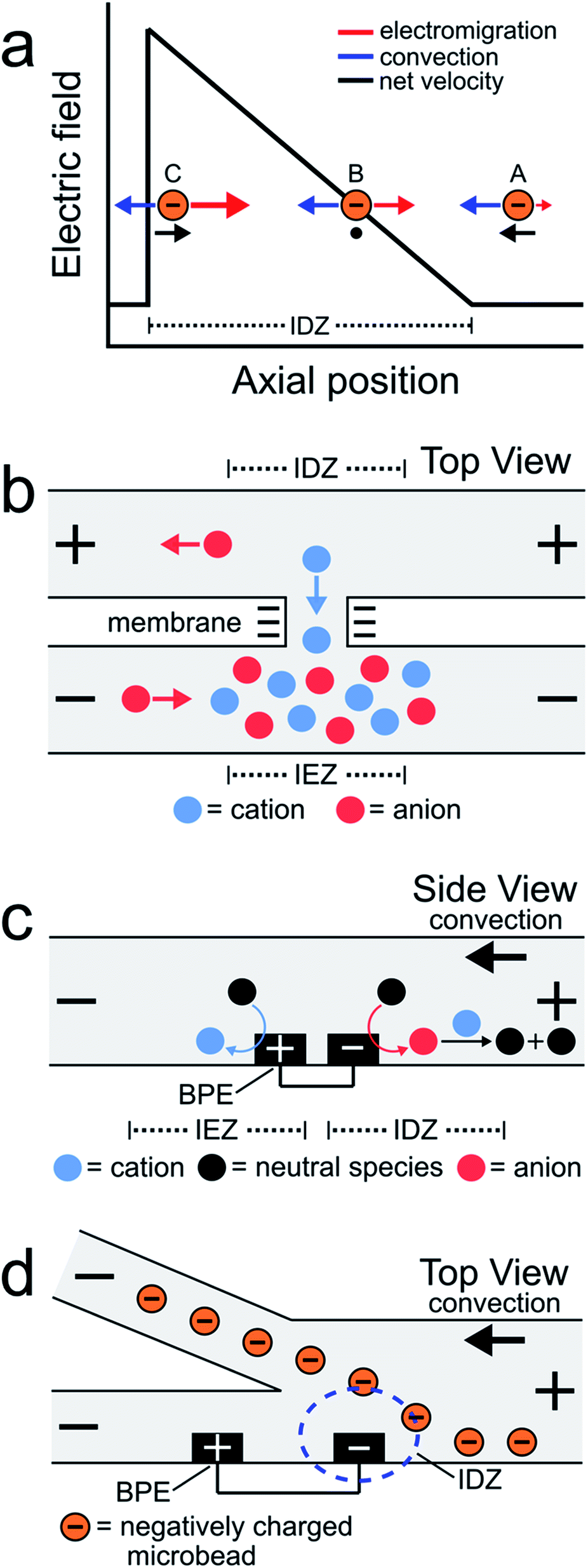 | ||
| Scheme 1 | ||
Some of the earliest reports describing EFG focusing in microfluidic systems relied on IDZs formed by ion concentration polarization (ICP).1,3,8,9,20 ICP usually refers to the formation of concentration gradients near an ion-selective nanopore or membrane.21Scheme 1b depicts a cation-selective membrane separating two microfluidic channels. When a voltage of the indicated polarity is applied across the microfluidic channel network, the negatively charged membrane passes cations from the top channel to the bottom channel but rejects the passage of anions. To maintain electroneutrality throughout the system, anions migrate as indicated in the illustration. Accordingly, an IDZ forms near the membrane in the top channel and a region of enhanced ion concentration, termed an ion enrichment zone (IEZ), forms near the membrane in the bottom channel.
Several years ago we reported an electrochemical variant of ICP.22,23 In this case, the IDZ forms via faradaic reactions proceeding at a BPE embedded within a microchannel containing a Tris buffer solution. This process, called faradaic ion concentration polarization (fICP),24 is illustrated in Scheme 1c. In this configuration, a BPE is patterned along the floor of a microfluidic channel. When a sufficient voltage is applied across the channel, faradaic water splitting proceeds at the ends of the BPE. Specifically, water reduction at the cathodic edge produces OH− (red circle), which reacts with TrisH+ (blue circle) present in solution (eqn (1) and (2)), to produce neutral Tris. This neutralization reaction reduces the number of charge carriers in the vicinity of the cathodic pole of the BPE, thereby yielding an IDZ. To conserve charge, H+ generation at the anodic edge of the BPE (eqn (3)) leads to an IEZ.22
Cathode:
| 2H2O + 2e− → 2OH− + H2 | (1) |
| TrisH+ + OH− → Tris + H2O | (2) |
Anode:
| 2H2O → 4H+ + O2 + 4e− | (3) |
In 2018 we used fICP to continuously redirect and separate plastic microbeads within a bifurcated microchannel.25 There were two main outcomes of this work. First, positioning the BPE, and thus the IDZ and EFG, across a portion of the channel width in the vicinity of the bifurcation enabled control over the direction of microbead flow (Scheme 1d). Second, the IDZ and IEZ led to local variations in the rate of electroosmotic flow (EOF) throughout the microchannel, and this also affected the trajectory of the microbeads.
In the present study, we used fICP to control the flow of two microplastics having different electrophoretic mobilities within a trifurcated microchannel. This was accomplished by configuring two BPEs in series and actuating them with a single power supply.26–29 This approach makes it possible to continuously focus, sort, and separate microplastics, and it represents a significant advance over our prior report because: (1) there are interesting, new challenges associated with implementing the complete and continuous separation of multiple objects (compared to steering one type of object in a particular direction) and (2) generating multiple local electric fields within a single channel by simply placing metal wires at strategic locations opens up a vast range of new possibilities for membrane-free separations.30,31
Experimental section
Chemicals
Polystyrene microbeads functionalized with surface carboxyl groups (diameter = 0.99 μm (μP1), 1.04 μm (μP2), and 0.20 μm (μP3)) were obtained from Bangs Laboratories, Inc. (Fishers, IN); FC04N, PC04N, and PC02N, respectively. BODIPY disulfonate fluorophore (BODIPY2−) was obtained from Molecular Probes (Eugene, OR). Pluronic F108 was purchased from BASF (Florham Park, NJ). Tris–HCl buffer solution was prepared by dissolving reagent grade Trizma base (Sigma-Aldrich, St. Louis, MO) in deionized water (18.0 MΩ cm, Milli-Q Gradient System, MilliporeSigma, Burlington, MA) and then adjusting the solution pH to 8.1 using 1.0 M HCl (Fisher Scientific, Hampton, NH). Microfluidic channels were fabricated using poly(dimethylsiloxane) (PDMS) prepared from a silicone elastomer kit (Sylgard 184, Dow, Midland, MI).Microfluidic device fabrication
Hybrid glass/PDMS microfluidic devices were fabricated using a previously reported procedure.32 First, Pt microbands (10 nm Ti adhesion layer + 100 nm Pt, Kurt J. Lesker Company, Jefferson Hills, PA) were fabricated atop a glass slide using standard lift-off photo-patterning procedures. Second, 3 mm reservoirs were punched at the ends of a PDMS microfluidic channel (5 mm length, 200 μm width, 11.5 μm height). Third, the glass and PDMS were exposed to an O2 plasma for 45 s (medium power, 60 W, PDC-32G, Harrick Scientific Products Inc., Ossining, NY) and then joined together. Fourth, the assembled device was heated in an oven at 65 °C for 5 min to ensure irreversible bonding. Trifurcated microfluidic channels were used in this study, and to ensure a uniform flow rate the width of each of the smaller channels was ∼1/3 that of the main channel. The microfluidic channel was positioned so that the Pt microbands spanned 20–25% of the main channel width.Sorting experiments
Sorting experiments were performed as follows. First, equal heights of 10.0 mM Tris buffer solution (pH 8.1), containing either 1.0 μM BODIPY2− and 1.5 pM μP1 or 150 fM μP2 and 190 pM μP3, were placed into the inlet and outlet reservoirs. To ensure that there was no pressure-driven flow (PDF) in the absence of a driving voltage, the motion of the plastic microbeads was monitored.33 Second, a driving voltage was applied across the channel length using a power supply (PWS 4721, Tektronix, Beaverton, OR) connected to Pt electrodes located on the floor of each reservoir. Third, a BPE of the desired length was formed by connecting two Pt microbands by an external jumper wire. Finally, after sorting, the experiment was terminated by disconnecting the jumper wire from the Pt microbands and turning off the driving voltage. An electrometer (6517b, Keithley Instruments, Cleveland, OH), connected in series with the power supply, and a hand-held multimeter (AM-1118, Aktakom, Russia), connected in series with the BPE, were used to measure the current passed through the driving electrodes (itot) and the BPE (iBPE), respectively.Optical and fluorescence microscopy
The motion of microplastic particles and the BODIPY2− fluorophore during sorting experiments was visualized using an inverted fluorescence microscope (Eclipse TE 2000-U, Nikon, Japan) equipped with a CCD camera (Cascade 512, Photometrics, Tucson, AZ) controlled by V++ software (DigitalOptics Corporation, San Jose, CA). Image analysis was performed using ImageJ (National Institutes of Health, Bethesda, MD). Fluorescence measurements were performed with the fluorescence lamp (X-Cite 120Q, Excelitas Technologies Corp., Waltham, MA) on, while optical observations were performed with the lamp off.Solution conductivity measurements
Solution conductivity measurements were obtained using a previously reported, home-built conductivity instrument.34,35 Measurements were performed as follows. A function generator (Model 182A, Wavetek, San Diego, CA) was used to apply an ac sine wave (±0.30 V amplitude, 5 kHz frequency) to one Pt microband. The resulting current passed through solution to a detection microband patterned 30 μm (center-to-center) downstream, where it was amplified and converted back to a voltage by a transimpedance amplifier. The attenuation of the current passing through solution to the detection microband was related to solution conductivity using a calibration curve (ESI†). Conductivity measurements were carried out at 20 ± 2 °C.Numerical simulations
Finite element simulations were carried out using the COMSOL Multiphysics version 5.4 software package (COMSOL Inc., Burlington, MA). Simulations were performed on a workstation (Precision T7500, Dell, Round Rock, TX) outfitted with 108 GB of RAM and dual Intel Xeon processors (2.40 GHz). All simulations were performed at steady state. A complete discussion of the theoretical background and details of the numerical simulations is provided in the ESI.†Results and discussion
Sorting by electrophoretic mobility
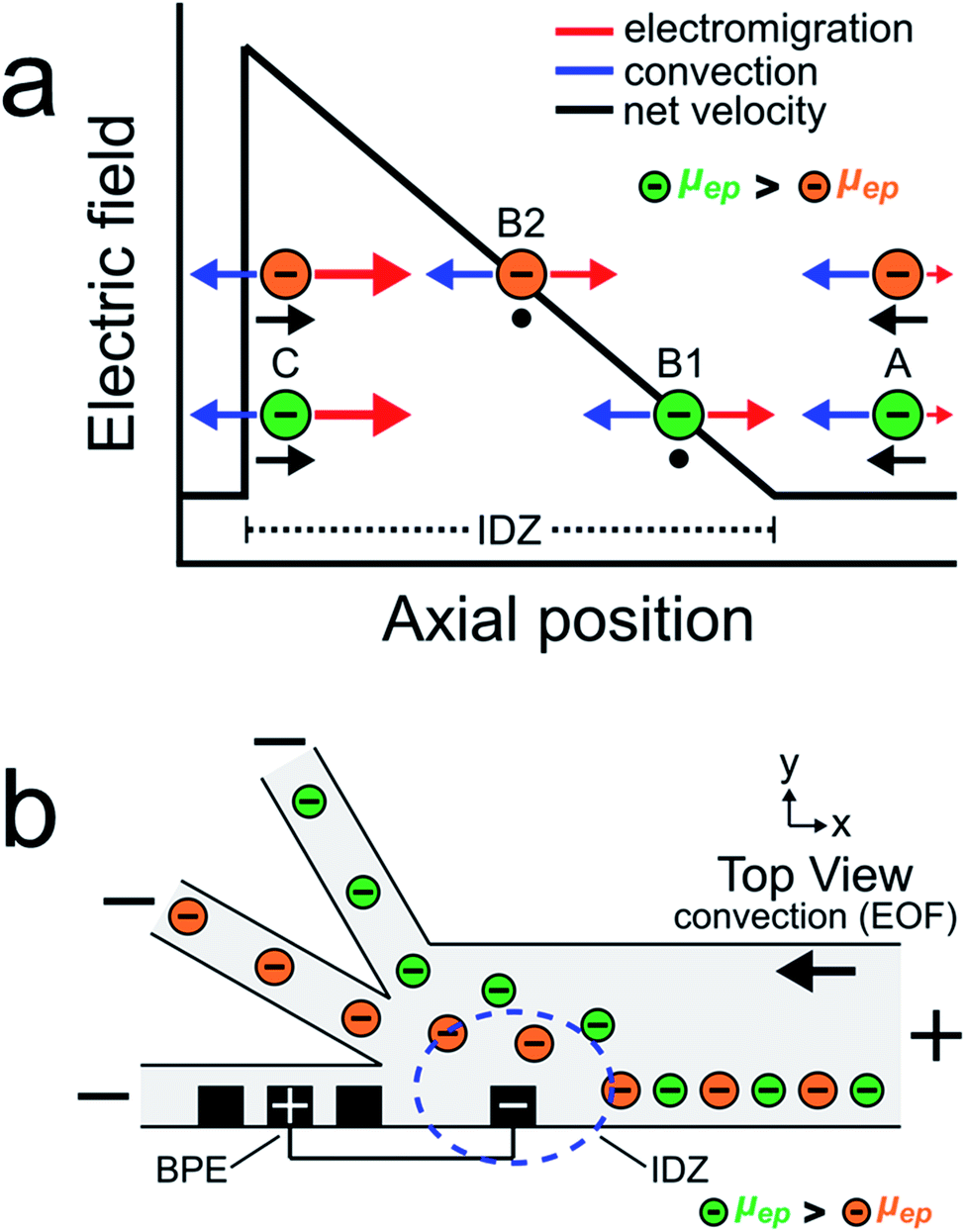
The goal of this study is to continuously sort and separate two microplastics having different electrophoretic mobilities using fICP within a trifurcated microchannel. We begin, however, by controlling the motion of the BODIPY2− fluorophore and a negatively charged microplastic (μP1) to develop a better understanding of processes fundamental to continuous sorting by fICP.Charged objects having different electrophoretic mobilities interact with EFGs in different ways.5,6,19,36–39 Consider, for example, EFG focusing of the two charged objects shown in Scheme 2a.16,17,40 The interpretation of the motion of the charged objects in this illustration is essentially the same as that shown in Scheme 1a, except now there are two particles having different properties. The key point is that the object with the larger electrophoretic mobility (μep, green) is focused at a position with a relatively low electric field (position B1), while the object with the smaller electrophoretic mobility (μep, orange) is focused at a position having a relatively high electric field (position B2). Extrapolating from the concept embodied by this scheme, we thought it would be possible to continuously separate two charged objects having different electrophoretic mobilities.
 | ||
| Scheme 2 | ||
Scheme 2b illustrates the experimental system used to continuously sort two charged objects having different electrophoretic mobilities. It shows a BPE, formed from two microband electrodes,13,16 positioned across a portion of the width of a trifurcated microchannel. Two additional microband electrodes are shown on either side of the anodic pole of the BPE. These make it possible to change the length of the BPE in other experiments that will be described later. When a sufficient driving voltage is applied across the channel length, an IDZ forms near the cathodic end of the BPE (blue dashed circle) due to fICP (eqn (1) and (2)). The magnitude of the corresponding EFG is largest near the cathodic end of the BPE and decreases as a function of position across the channel width. The non-uniform EFG directs the trajectory of charged objects away from the BPE cathode and toward the upper sidewall of the microfluidic channel. Here, the electric field is weak, convection dominates electromigration, and the objects flow downstream.25
If two charged objects interact with the EFG near the leading edge of the BPE in a similar way, their motion is controlled in a manner proportional to the magnitude of their electrophoretic mobility. Specifically, the object having the larger electrophoretic mobility (green) is repelled further away from the BPE cathode than the object having the smaller electrophoretic mobility (orange). Accordingly, by tuning the magnitude of electromigration and convection (by EOF) in the trifurcated channel, the flow of the green and orange objects can be directed into the top and middle outlet channels, respectively. Thus, the two objects are continuously sorted by their electrophoretic mobility and separated from the plastic-free solution flowing into the bottom outlet channel.
Actual sorting experiments were performed in a glass/PDMS microelectrochemical device having a trifurcated microchannel. The microchannel was positioned so that an array of microbands extended across 20–25% of the width of the main 200 μm-wide channel (Scheme 2b). A 750 μm-long BPE was formed near the trifurcation by connecting the two microbands shown in Scheme 2b with a jumper wire. The magnitude of the potential difference between the BPE ends and the solution (ΔEelec) is approximately proportional to the length of the BPE and the magnitude of the driving voltage.41–45 As mentioned earlier, the rate of PDF was set to zero prior to the experiment, and therefore solution convection was controlled only by EOF. Because the channel surfaces are negatively charged in the presence of Tris buffer (pH 8.1), application of a driving voltage having the polarity shown in Scheme 2b results in solution flow from right to left (in opposition to electromigration).46,47
The sorting experiments were carried out as follows. First, equal heights of 10.0 mM Tris buffer solution (pH 8.1) containing 1.0 μM BODIPY2− (electrophoretic mobility = −3.4 μm cm V−1 s−1)22 and 1.5 pM μP1 (electrophoretic mobility = −2.0 ± 0.5 μm cm V−1 s−1, see ESI† for electrophoretic mobility measurement details) were placed into the inlet and outlet reservoirs. Second, a driving voltage of 25.0 V was applied across the channel length, resulting in EOF toward the negative driving electrodes (Scheme 2b). Third, the two microbands indicated in Scheme 2b were connected with a jumper wire to form a 750 μm-long BPE (ΔEelec = ∼3.8 V).
Fig. 1 is a pair of micrographs captured at steady state that display the location of μP1 and BODIPY2− in the trifurcated channel during a typical sorting experiment. Fig. 1a is an optical micrograph showing that μP1 is directed away from the bottom outlet channel and into the middle and top channels. Fig. 1b is a fluorescence micrograph showing that BODIPY2− is directed away from the bottom and middle outlet channels and into the top channel. A movie of the complete experiment is provided in the ESI (Movie ESI-1†).
 | ||
| Fig. 1 Sorting BODIPY2− and μP1 in a trifurcated microchannel. (a) Optical micrograph showing the location of μP1 during a sorting experiment. (b) Fluorescence micrograph showing the location of BODIPY2− during the same sorting experiment. The dashed white line in (b) indicates the location of the cathodic pole of the BPE. The BPE is formed by connecting the two microwires illustrated in Scheme 2b. The solution contained 1.0 μM BODIPY2−, 1.5 pM μP1, and 10.0 mM Tris buffer (pH 8.1). The driving voltage was 25.0 V and the BPE was 750 μm long. EOF was from right to left. Both micrographs were captured at steady state. The scale bar shown in (a) also applies to (b). | ||
The important point is that these results demonstrate that two charged objects can be continuously redirected and sorted along an EFG as illustrated in Scheme 2b. In this case, BODIPY2− has the larger electrophoretic mobility and is repelled further from the BPE than μP1 during fICP. Fig. 1 also shows that sorting is imperfect. Specifically, all of the BODIPY2− flowing past the cathodic pole of the BPE is directed into the top outlet channel, but μP1 is directed into both the middle and top outlet channel. The principal reason for some of μP1 being present in the upper outlet channel is that it does not encounter the strongest region of the EFG. That is, the magnitude of the EFG decreases as the distance from the BPE increases along the width of the microchannel. To address this issue, and thereby improve sorting efficiency, we thought it would be possible to focus BODIPY2− and μP1 toward the lower sidewall of the main channel prior to their encounter with the strongest part of the EFG. This hypothesis is represented schematically in Fig. 2a, discussed next.
 | ||
| Fig. 2 (a) Schematic illustration of the microfluidic configuration used for serial fICP experiments. For frames (b–e), only BPE1 was active. For frames (f) and (g), both BPE1 and BPE2 were active. (b–g) Series of optical and fluorescence micrographs showing the location of μP1 and BODIPY2− during serial fICP. With reference to the three dotted lines at the bottom of (a), the micrographs were captured along the portion of the channel length indicated by (b and c) the dotted black line; (d) the dotted green line; and (e–g) the dotted red line. The curved black arrow in (b) indicates the location and rotation direction (counterclockwise) of the vortex downstream of the cathodic pole of BPE1 (the vortex can be more easily visualized in Movie ESI-2 in the ESI†). The dashed white lines in (c) and (g) indicate the locations of the cathodic poles of BPE1 and BPE2, respectively. The solution contained 1.0 μM BODIPY2−, 1.5 pM μP1, and 10.0 mM Tris buffer (pH 8.1). The driving voltage was 25.0 V. BPE1 was 750 μm long and BPE2 was 1000 μm long. EOF was from right to left. All micrographs were captured at steady state. The scale bar shown in (b) also applies to (c–g). | ||
Focusing and sorting by serial fICP
So far we have shown that BODIPY2− and μP1 can be continuously redirected and sorted at a single BPE embedded within a trifurcated microchannel. However, only a fraction of the objects in solution interact with the highest EFG (in the vicinity of the BPE) in this configuration and, therefore, sorting is imperfect. Accordingly, to improve sorting efficiency, we designed a more sophisticated microelectrochemical device comprising two BPEs.The diagram in Fig. 2a is a top view of two BPEs (BPE1 and BPE2) positioned across a fraction of the width of a trifurcated channel. BPE1 is positioned near the upper sidewall of the channel upstream from the trifurcation, while BPE2 is positioned along the lower sidewall of the channel near the trifurcation. Two additional microband electrodes are shown near the anodic pole of both BPEs. These make it possible to change the length of the BPEs. When a sufficient driving voltage is applied across the channel length, fICP (eqn (1) and (2)) results in an IDZ near the cathodic pole of both BPE1 and BPE2 (blue dashed circles). Importantly, fICP at BPE1 continuously directs the flow of BODIPY2− and μP1 toward the lower sidewall of the channel. Accordingly, both objects encounter the strongest part of the EFG at BPE2 and both are more effectively redirected compared to when only a single BPE is present near the trifurcation.
The two-BPE focusing and sorting experiments were carried out as follows. First, equal heights of 10.0 mM Tris buffer solution (pH 8.1) containing 1.0 μM BODIPY2− and 1.5 pM μP1 were placed into the inlet and outlet reservoirs to ensure zero PDF. Second, a driving voltage of 25.0 V was applied across the channel length, resulting in EOF toward the negative driving electrodes. Third, two pairs of microband electrodes were connected with jumper wires to yield the two BPEs shown in Fig. 2a. These BPEs extend across 20–25% of the width of the main 200 μm-wide channel, and the distance from the upstream edge of BPE1 to the upstream edge of BPE2 is 1460 μm. BPE1 is 750 μm long (ΔEelec = ∼3.8 V) and BPE2 is 1000 μm long (ΔEelec = ∼5.0 V).
The remaining frames in Fig. 2 are a series of micrographs, captured at steady state, that reveal the location of BODIPY2− and μP1 along the channel length during a focusing and sorting experiment. Fig. 2b is an optical micrograph captured along the portion of the channel length indicated by the dotted black line at the bottom of Fig. 2a. It shows that the presence of the cathodic pole of BPE1, and the associated fICP, changes the distribution of μP1 in the channel. Although it is difficult to see in this still image, there is also a single vortex (indicated by the curved black arrow in Fig. 2b) that forms just downstream from the cathodic pole of BPE1. Both of these observations are more clearly visible in Movie ESI-2.†Fig. 2c is a fluorescence micrograph captured at the same axial position as Fig. 2b, and it shows that BODIPY2− is directed away from the cathodic pole of BPE1 during fICP. Fig. 2d is an optical micrograph captured along the portion of the channel length indicated by the dotted green line in Fig. 2a. It shows that μP1 is directed away from the anodic pole of BPE1 and toward the lower sidewall of the channel during fICP.
Taken together, Fig. 2b–d show that fICP at BPE1 significantly impacts the trajectory of BODIPY2− and μP1 as they traverse the channel length. While redirection near the cathodic edge of BPE1 results from the locally enhanced electric field in solution, the vortex shown in Fig. 2b and the redirection in the vicinity of the BPE anode shown in Fig. 2d are more complicated to explain. These latter two phenomena are related to formation of ionic concentration gradients in solution and will be discussed in more detail later.25 For now, however, the key point is that BPE1 focuses BODIPY2− and μP1 toward the lower sidewall of the channel upstream from BPE2 (Fig. 2c and d).
Fig. 2e is an optical micrograph captured along the portion of the channel length indicated by the dotted red line in Fig. 2a. This micrograph was captured prior to connecting the microbands comprising BPE2 but with BPE1 active. It shows that μP1 is still focused in the lower half of the channel when it reaches the cathodic edge of BPE2.
Fig. 2f is an optical micrograph captured at the same axial position as in Fig. 2e, but with fICP proceeding at both BPE1 and BPE2 (i.e., serial fICP). It shows that the majority of μP1 is directed away from the cathodic pole of BPE2 and into the middle outlet channel. However, a portion of μP1 passes through the EFG formed near the cathodic pole of BPE2 and enters the bottom outlet channel. Fig. 2g is a fluorescence micrograph captured at the same axial position as Fig. 2e and f, and it shows that BODIPY2− is directed away from the cathodic pole of BPE2 and into the middle and top outlet channels.
Fig. 2f and g confirm that the EFG near the cathodic pole of BPE2 sorts μP1 and BODIPY2− according to their electrophoretic mobility. Unfortunately, despite both species interacting with the strongest part of the EFG at BPE2, sorting is still imperfect. The results shown in Fig. 2b–g demonstrate, however, that two BPEs, operated in series with a single power supply, can be used to form two IDZs and that the corresponding EFGs control the motion of charged species in a microchannel. This was a surprising finding because we thought that processes occurring at and near BPE1 might inhibit formation of a significant IDZ and EFG at BPE2. To better understand the distribution of solution species during serial fICP, as well as why sorting is incomplete, we performed in situ solution conductivity measurements.
Solution conductivity measurements
Fig. 3a is a simplified schematic illustration of the experimental configuration shown in Fig. 2a. A table summarizing both experimental and simulated (discussed later) results from the solution conductivity measurements is also shown. The experimental measurements were carried out as described for Fig. 2, except conductivity microbands (shown in red), positioned at least 1000 μm downstream from the trifurcation intersection, were used to measure the solution conductivity at steady state in each outlet channel. Conductivity measurements performed in each outlet channel with only the microbands comprising BPE1 connected (i.e., BPE1 “on”) were assumed to represent, approximately, the conductivity of the solution between the downstream edge of BPE1 and the upstream edge of BPE2 during focusing and sorting experiments.48–52 Conductivity measurements performed with the microbands comprising both BPE1 and BPE2 connected (i.e., BPE1 and BPE2 “on”) are characteristic of the conductivity of solution in each outlet channel during focusing and sorting experiments. The results in the table are normalized to the conductivity of the bulk solution (bulk = 1.00) and represent the average of three measurements.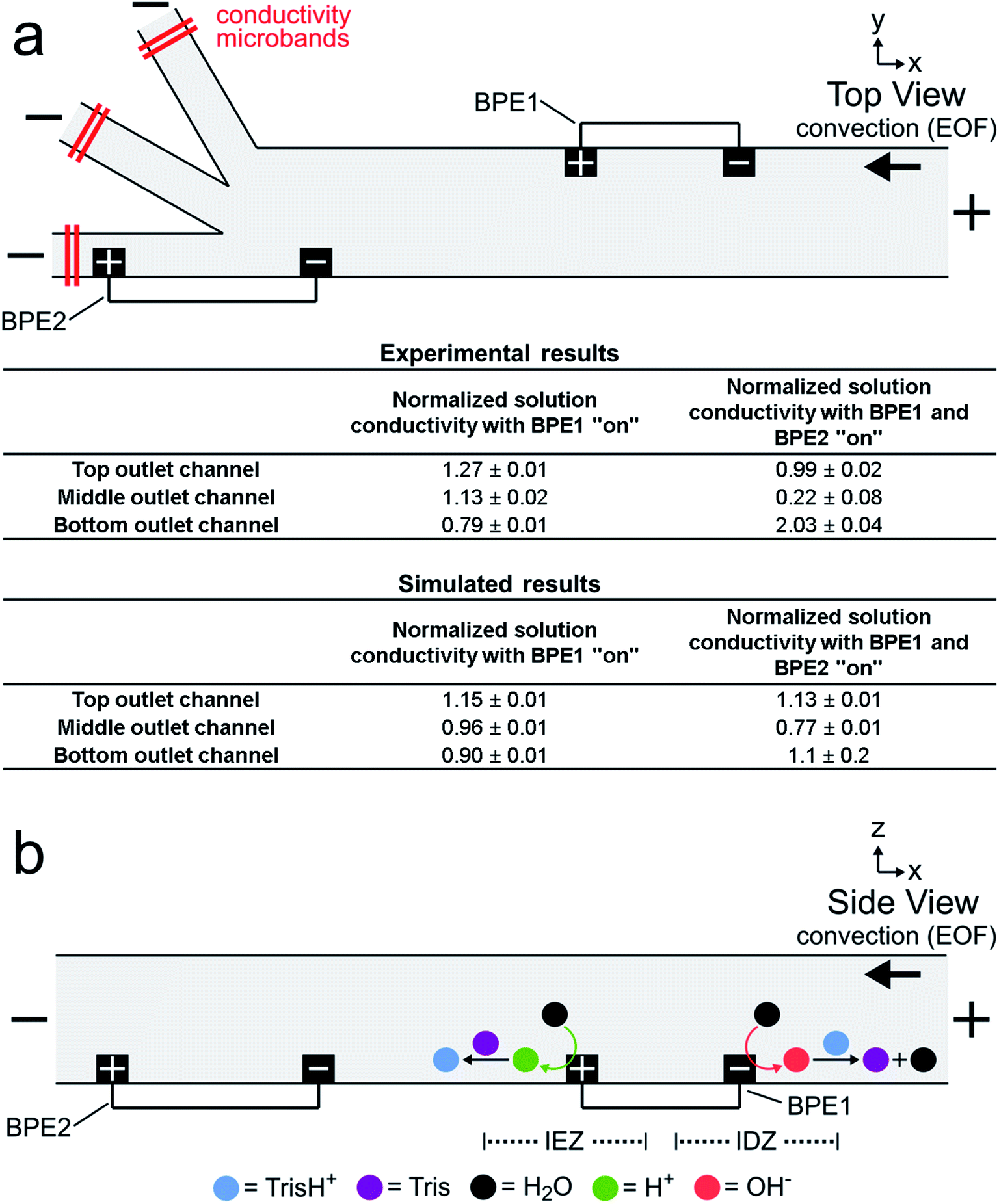 | ||
| Fig. 3 (a) Schematic illustration of the microfluidic configuration used to measure solution conductivity, and a table summarizing the results from direct, in situ solution conductivity measurements (n = 3) and analogous numerical simulations. The conductivity measurements are normalized to the conductivity of the bulk solution. The solution contained 1.0 μM BODIPY2−, 1.5 pM μP1, and 10.0 mM Tris buffer (pH 8.1). The driving voltage was 25.0 V. Both BPE1 (750 μm long) and BPE2 (1000 μm long) were connected for this experiment. EOF was from right to left. All measurements were performed at steady state. (b) Schematic illustration of the Tris buffer chemistry at BPE1 during serial fICP. | ||
The tabulated experimental data in Fig. 3a reveal two key findings. First, the three values shown in the first column of the table are fairly close to the normalized bulk conductivity of 1.00. This means that forming an IDZ and IEZ near the anodic and cathodic poles, respectively, of BPE1 does not greatly change the conductivity of solution downstream from BPE1. We believe this is related to Tris buffer chemistry. Specifically, just as OH− produced at the cathodic end of a BPE neutralizes TrisH+ (eqn (1) and (2)), H+ produced at the anodic pole converts Tris to TrisH+ (eqn (3) and (4)).53 In this way, TrisH+ molecules neutralized near the cathodic pole of BPE1 are reionized in the vicinity of the anodic pole of the same BPE, as illustrated in Fig. 3b. Regeneration of TrisH+ acts to conserve the conductivity (and presumably the composition) of the solution upstream from BPE1 and BPE2 and is critical to the success of serial fICP.
| Tris + H+ → TrisH+ | (4) |
Second, the data presented in the second column show that solution conductivity is significantly different in each outlet channel during serial fICP experiments. Specifically, solution conductivity is increased in the bottom outlet channel, decreased in the middle outlet channel, and nearly unchanged in the top outlet channel vs. bulk solution. This is important because conductivity variations of the magnitudes measured with BPE1 and BPE2 “on” are known to yield non-uniform EOF within microfluidic channels.2,25,54–60 For the experiments discussed so far, where solution convection is controlled by EOF, local variations in the rate of EOF can impact the performance of focusing and sorting by serial fICP.
To summarize, solution conductivity measurements performed with BPE1 “on”, as well as with BPE1 and BPE2 “on”, improve our understanding of the mechanism of serial fICP and reveal significant variation between the conductivity of solution in each outlet channel. To better understand how these local variations in solution conductivity impact the rate of solution convection throughout the microfluidic channel, we performed numerical simulations.
Numerical simulations
We used the finite element method to solve numerical simulations and gain additional insight into the experimental results of focusing and sorting by serial fICP. Steady-state simulations were performed using a two-dimensional (2D) model based on the xy-plane of the microelectrochemical device illustrated in Fig. 2a.A complete description of the theoretical background and simulation methods is provided in the ESI.† Briefly, however, solution convection was calculated using the Navier–Stokes equation. The electric double layer is considerably smaller than the channel dimensions (thin double layer approximation), and thus EOF was modeled as a slip condition imposed at the channel walls and formulated according to the Helmholtz–Smoluchowski equation.23,61 Mass transport was resolved by the Nernst–Planck equation and the electroneutrality condition. The electric field in solution was determined from the steady-state currents passed through the driving electrodes (itot) and the BPEs (iBPE1 and iBPE2) as well as solution conductivity. The electrochemical reactions (eqn (1) and (3)) at the BPEs were modeled as fluxes, and the Tris buffer chemistry (eqn (2) and (4)) was modeled using reaction rate constants.23
Fig. 4a is a plot of the simulation domain. This asymmetric experimental system was approximated in 2D by positioning the BPEs along the channel sidewalls. The location of the poles of BPE1 and BPE2 are indicated by the small red and blue rectangles, respectively. The x- and y-axes refer to the channel length and width, respectively.
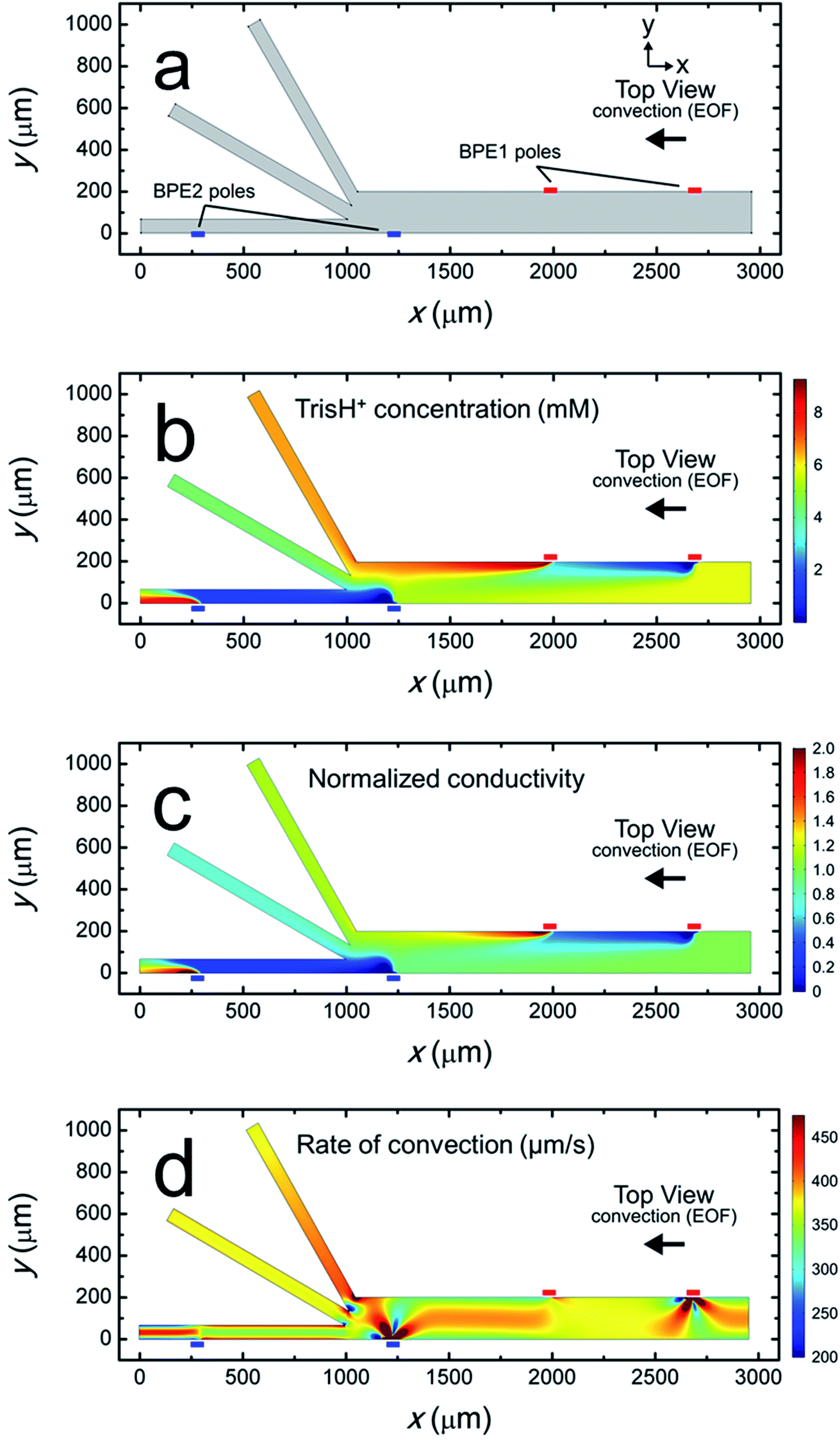 | ||
| Fig. 4 Steady-state simulation results for a solution containing 10.0 mM Tris buffer (pH 8.1), and with itot = 1.32 μA, iBPE1 = 0.60 μA, and iBPE2 = 0.74 μA (i.e., BPE1 and BPE2 “on”). (a) Plot of the 2D model domain. The red and blue rectangles indicate the positions of the poles of BPE1 and BPE2, respectively. (b) Distribution of TrisH+ along the channel length. (c) Plot of solution conductivity throughout the channel. Conductivity values are normalized to the conductivity of the bulk solution. (d) Plot of the rate of convection along the channel length during serial fICP. | ||
Fig. 4b is a plot of the distribution of TrisH+ during a focusing and sorting experiment (i.e., both BPEs “on”). It shows that the concentration of TrisH+ decreases in the vicinity of the cathodic poles of each BPE but increases near their anodic poles. This finding is in accord with our understanding of Tris buffer chemistry (eqn (1)–(4)). Furthermore, Fig. 4b shows that the concentration of TrisH+ near the leading edge of BPE2 (∼5.3 mM at x = 1300 μm, y = 0–100 μm) is similar to that of the bulk solution upstream from BPE1 (5.8 mM at x = 2900 μm). This finding confirms that fICP at BPE1 does not significantly impact the concentration of TrisH+ near the leading edge of BPE2.
Fig. 4c is a plot of the solution conductivity normalized to the conductivity of bulk solution. The trends in this plot are nearly identical to those shown in Fig. 4b for the concentration of TrisH+. Specifically, the prominent features of this plot are IDZs and IEZs in the vicinity of the cathodic and anodic poles, respectively, of the BPEs. Additionally, the normalized solution conductivity just upstream from BPE2 (x = 1300 μm) is ∼0.9 at the lower sidewall of the channel (y = 0 μm) and ∼1.2 at the upper sidewall (y = 200 μm). Finally, the solution conductivity 1000 μm downstream from the trifurcation is increased in the bottom outlet channel, decreased in the middle outlet channel, and nearly unchanged in the top outlet channel vs. bulk solution. Importantly, the conclusions relating to Fig. 4c, and those derived from the experimental conductivity measurements (Fig. 3a), are nearly the same.
To better compare the results of the numerical simulations to the solution conductivity measurements in Fig. 3a, we also performed simulations with only BPE1 “on” (ESI†). The conductivity of the simulated solution at the left end of each outlet channel (1000 μm downstream from the trifurcation) in this case is shown in the first column of the lower part of the table in Fig. 3a. Each value represents the average conductivity of solution across the width of the indicated channel and is normalized to the conductivity of bulk solution. These results are in reasonable agreement with the experimental results in the top part of the table. We conclude that the electrochemical (eqn (1) and (3)) and chemical (eqn (2) and (4)) reactions included in the numerical model appropriately describe fICP at a BPE.
Simulation results analogous to those discussed in the previous paragraph and related to those in Fig. 4c (both BPEs “on”) are shown in the second column of the lower part of the table in Fig. 3a. The significant differences between the simulated and experimentally measured conductivities in this case are likely related to geometric effects. Specifically, in the experimental device, the BPE poles extend across 20–25% of the width of the 200 μm-wide main channel (as shown in Fig. 2a), whereas in the simulations the BPEs are flush with the sidewalls. Therefore, in the experiments (but not in the simulations), a significant portion of the TrisH+ neutralized near the cathodic pole of BPE2 flows downstream and enters the middle outlet channel. Thus, flow displacement of the IDZ leads to the measurement of a significant decrease in solution conductivity in the middle outlet channel.25 Likewise, the amount of H+ generated at the anodic pole of BPE2 exceeds the amount of neutralized TrisH+ in the bottom outlet channel, and thus a significant increase in solution conductivity is measured in the bottom outlet channel. Additional details are provided in the context of Fig. ESI-3 in the ESI.†
The rate of convection along the channel length is plotted in Fig. 4d. It shows that the rate of convection varies significantly near the poles of the BPEs and the channel trifurcation. The main finding extracted from Fig. 4d is that serial fICP produces significant and complex flow variations within the microelectrochemical device when convection is controlled by EOF. Flow variations of the type shown in Fig. 4d account for the experimentally observed microplastic vortexing near the cathodic pole of BPE1 (shown in Fig. 2b) and microplastic redirection near the anodic pole of BPE1 (Fig. 2d). Furthermore, it is likely that complex flow near the cathodic pole of BPE2 and the channel trifurcation limits the performance of sorting and focusing by serial fICP. More detailed information relating to Fig. 4d is provided in the ESI.† To address the complexity of the flow, however, we developed a method for minimizing these effects. This is discussed next.
Focusing, sorting, and separating by serial fICP
The effectiveness of serial fICP depends on convection via EOF. As we demonstrated in the previous section (Fig. 4d), however, EOF is complex and varies along the channel length. To address this issue, we now turn our attention to suppressing EOF and controlling convection by PDF.60,62Serial fICP experiments were performed in a microelectrochemical device identical to the one previously described in the context of Fig. 2a. Following fabrication, however, the surfaces of the microchannel were modified by flowing 10.0 mM Tris–HCl solution (pH 8.1) containing 5.0 μM of the non-ionic surfactant Pluronic F108 through the channel for 21 h by PDF.40,63,64 Pluronic treatment suppressed the rate of EOF within the microchannel by 49 ± 8% (n = 3) for at least 2 h following treatment.65
Following treatment of the channel surfaces with Pluronic, sorting experiments were carried out as follows. First, different heights of 10.0 mM Tris buffer solution (pH 8.1) containing 150 fM μP2 (electrophoretic mobility = −2.4 ± 0.5 μm cm V−1 s−1) and 190 pM μP3 (electrophoretic mobility = −4 ± 1 μm cm V−1 s−1, see ESI† for electrophoretic mobility measurement details) were placed into the inlet and outlet reservoirs resulting in PDF (32 ± 3 nL min−1, n = 7) toward the trifurcation. Second, a driving voltage of 25.0 V was applied across the channel length resulting in EOF toward the negative driving electrodes. Therefore, in this experiment, convection is by both EOF and PDF. Third, two pairs of microband electrodes were connected with jumper wires to yield a pair of BPEs configured as in Fig. 2a. In this case, BPE1 and BPE2 are both 1000 μm long (ΔEelec = ∼5.0 V).
Fig. 5 is a series of optical micrographs captured at steady state during serial fICP. Fig. 5a was captured along the portion of the channel length indicated by the dotted black line at the bottom of Fig. 2a. It shows that the EFG in the vicinity of the cathodic pole of BPE1 directs μP2 and μP3 toward the lower sidewall of the channel. Note that μP3 is too small (0.20 μm) to be visualized as discrete particles, and therefore it appears as a slightly darker gray haze below the white line that has been added to this image. The red arrow in Fig. 5a also indicates that some microplastics stack just upstream from the BPE1 cathode. At this location, the forces of electromigration and convection are equal in magnitude but opposite in direction, and thus the microplastics are focused here.
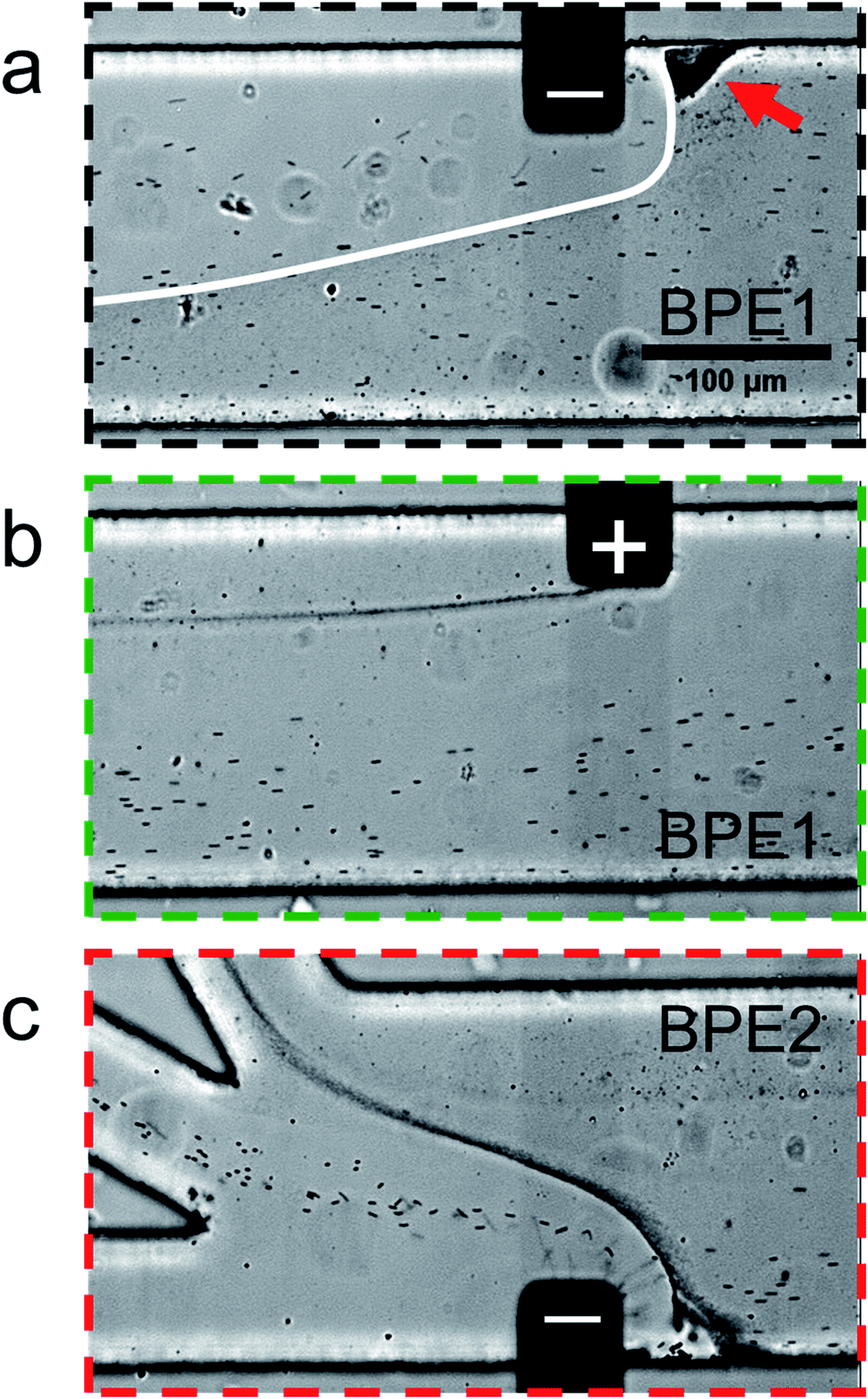 | ||
| Fig. 5 Series of micrographs showing the location of μP2 and μP3 during serial fICP (both BPE1 and BPE2 active and 1000 μm in length). Optical micrographs captured along the portion of the channel length indicated by (a) the dotted black line, (b) the dotted green line, and (c) the dotted red line at the bottom of Fig. 2a. The white line in (a) indicates the approximate boundary between solution containing μP3 (below) and μP3-free solution (above). The red arrow in (a) indicates a region of the channel in which microplastics are retained. The solution contained 150 fM μP2, 190 pM μP3, and 10.0 mM Tris buffer (pH 8.1). The driving voltage was 25.0 V. PDF (32 ± 3 nL min−1, n = 7) and EOF were from right to left. All micrographs were captured at steady state. The scale bar shown in (a) also applies to (b) and (c). | ||
Fig. 5b was captured along the portion of the channel length indicated by the dotted green line in Fig. 2a. It shows that processes at the anodic pole of BPE1 have little to no impact on the flow of the majority of the microplastics. However, this micrograph shows a stream of μP3 downstream of the edge of the anodic pole. This is likely due to electrostatic interactions between the negatively charged microplastics and the positively charged anodic pole of BPE1.
Fig. 5c is a micrograph captured along the portion of the channel length indicated by the dotted red line in Fig. 2a. It shows that the IDZ and corresponding EFG near the cathodic pole of BPE2 direct μP2 and μP3 into the middle and top outlet channels, respectively. Accordingly, the two microplastics are nearly quantitatively sorted according to their electrophoretic mobility, and they are completely separated from the solution flowing into the bottom outlet channel. A movie of the complete experiment is included in the ESI (Movie ESI-3†). The main point relating to Fig. 5 is that by suppressing EOF and controlling convection primarily by PDF, the types of flow variations apparent in Fig. 4d are minimized. This results in greatly enhanced separation efficiency of the two types of microplastics.
Summary and conclusions
In this article, we have reported three key findings. First, serial bipolar electrochemistry enables simultaneous formation of two IDZs, and their corresponding EFGs, within a single microfluidic channel and using just a single power supply. Second, complex flow variations that adversely impact separations during fICP can be mitigated by minimizing EOF in favor of PDF. Finally, serial fICP within a trifurcated microchannel results in continuous and quantitative focusing, sorting, and separating of microplastics. These results represent a significant advancement of our understanding of processes fundamental to fICP and their application to continuous and precise control of the flow of multiple charged objects. In the future, we plan to develop alternative approaches to fICP that do not rely on buffer chemistry. Success in this endeavor will increase the types of solutions in which the motion of ions and charged nano- and micro-scale objects can be controlled by fICP. The results of these experiments will be reported in due course.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge support from the Chemical Sciences, Geosciences, and Biosciences Division, Office of Basic Energy Sciences, Office of Science, U.S. Department of Energy (Grant: DE-FG02-06ER15758). We thank the Robert A. Welch Foundation (Grant F-0032) for sustained support of our research program. We gratefully acknowledge the Center for Electrochemistry at The University of Texas at Austin for access to modeling software and computational resources.References
- M. Li and R. K. Anand, Analyst, 2016, 141, 3496–3510 RSC.
- S. J. Kim, Y.-A. Song and J. Han, Chem. Soc. Rev., 2010, 39, 912–922 RSC.
- T. A. Zangle, A. Mani and J. G. Santiago, Chem. Soc. Rev., 2010, 39, 1014–1035 RSC.
- S. Kim, B. Ganapathysubramanian and R. K. Anand, J. Am. Chem. Soc., 2020, 142, 3196–3204 CrossRef CAS PubMed.
- W. S. Koegler and C. F. Ivory, J. Chromatogr. A, 1996, 726, 229–236 CrossRef CAS.
- W. S. Koegler and C. F. Ivory, Biotechnol. Prog., 1996, 12, 822–836 CrossRef CAS.
- R. T. Kelly and A. T. Woolley, J. Sep. Sci., 2005, 28, 1985–1993 CrossRef CAS PubMed.
- Y.-C. Wang, A. L. Stevens and J. Han, Anal. Chem., 2005, 77, 4293–4299 CrossRef CAS PubMed.
- J. Dai, T. Ito, L. Sun and R. M. Crooks, J. Am. Chem. Soc., 2003, 125, 13026–13027 CrossRef CAS PubMed.
- D.-T. Phan, S. A. M. Shaegh, C. Yang and N.-T. Nguyen, Sens. Actuators, B, 2016, 222, 735–740 CrossRef CAS.
- J. Choi, K. Huh, D. J. Moon, H. Lee, S. Y. Son, K. Kim, H. C. Kim, J.-H. Chae, G. Y. Sung, H.-Y. Kim, J. W. Hong and S. J. Kim, RSC Adv., 2015, 5, 66178–66184 RSC.
- S. I. Han, K. S. Hwang, R. Kwak and J. H. Lee, Lab Chip, 2016, 16, 2219–2227 RSC.
- R. K. Perdue, D. R. Laws, D. Hlushkou, U. Tallarek and R. M. Crooks, Anal. Chem., 2009, 81, 10149–10155 CrossRef CAS PubMed.
- E. Sheridan, K. N. Knust and R. M. Crooks, Analyst, 2011, 136, 4134–4137 RSC.
- S. J. Kim, S. H. Ko, K. H. Kang and J. Han, Nat. Nanotechnol., 2010, 5, 297–301 CrossRef CAS PubMed.
- K. N. Knust, E. Sheridan, R. K. Anand and R. M. Crooks, Lab Chip, 2012, 12, 4107–4114 RSC.
- K. Scida, E. Sheridan and R. M. Crooks, Lab Chip, 2013, 13, 2292–2299 RSC.
- L. Liu, M.-R. Xie, Y.-Z. Chen and Z.-Y. Wu, Lab Chip, 2019, 19, 845–850 RSC.
- H. Jeon, H. Lee, K. H. Kang and G. Lim, Sci. Rep., 2013, 3, 3483 CrossRef PubMed.
- R. Dhopeshwarkar, R. M. Crooks, D. Hlushkou and U. Tallarek, Anal. Chem., 2008, 80, 1039–1048 CrossRef CAS PubMed.
- Q. Pu, J. Yun, H. Temkin and S. Liu, Nano Lett., 2004, 4, 1099–1103 CrossRef CAS.
- R. Dhopeshwarkar, D. Hlushkou, M. Nguyen, U. Tallarek and R. M. Crooks, J. Am. Chem. Soc., 2008, 130, 10480–10481 CrossRef CAS PubMed.
- D. Hlushkou, R. K. Perdue, R. Dhopeshwarkar, R. M. Crooks and U. Tallarek, Lab Chip, 2009, 9, 1903–1913 RSC.
- R. K. Anand, E. Sheridan, K. N. Knust and R. M. Crooks, Anal. Chem., 2011, 83, 2351–2358 CrossRef CAS PubMed.
- C. D. Davies, E. Yoon and R. M. Crooks, ChemElectroChem, 2018, 5, 877–884 CrossRef CAS.
- J.-C. Bradley, H.-M. Chen, J. Crawford, J. Eckert, K. Ernazarova, T. Kurzeja, M. Lin, M. McGee, W. Nadler and S. G. Stephens, Nature, 1997, 389, 268–271 CrossRef CAS.
- K.-F. Chow, F. Mavré, J. A. Crooks, B.-Y. Chang and R. M. Crooks, J. Am. Chem. Soc., 2009, 131, 8364–8365 CrossRef CAS PubMed.
- Y. Koizumi, N. Shida, M. Ohira, H. Nishiyama, I. Tomita and S. Inagi, Nat. Commun., 2016, 7, 10404 CrossRef CAS PubMed.
- S. Hu and J. Gao, Adv. Funct. Mater., 2019 DOI:10.1002/adfm.201907003.
- R. K. Anand, E. S. Johnson and D. T. Chiu, J. Am. Chem. Soc., 2015, 137, 776–783 CrossRef CAS PubMed.
- M. Li and R. K. Anand, J. Am. Chem. Soc., 2017, 139, 8950–8959 CrossRef CAS PubMed.
- J. C. McDonald, D. C. Duffy, J. R. Anderson, D. T. Chiu, H. Wu, O. J. A. Schueller and G. M. Whitesides, Electrophoresis, 2000, 21, 27–40 CrossRef CAS PubMed.
- C. D. Davies, S. E. Johnson and R. M. Crooks, ChemElectroChem, 2019, 6, 4867–4876 CrossRef CAS.
- M. Galloway, W. Stryjewski, A. Henry, S. M. Ford, S. Llopis, R. L. McCarley and S. A. Soper, Anal. Chem., 2002, 74, 2407–2415 CrossRef CAS PubMed.
- E. Yoon, C. D. Davies, T. A. Hooper and R. M. Crooks, Lab Chip, 2017, 17, 2491–2499 RSC.
- J. M. Burke, Z. Huang and C. F. Ivory, Anal. Chem., 2009, 81, 8236–8243 CrossRef CAS PubMed.
- M. M. Meighan, S. J. R. Staton and M. A. Hayes, Electrophoresis, 2009, 30, 852–865 CrossRef CAS PubMed.
- J. G. Shackman and D. Ross, Electrophoresis, 2007, 28, 556–571 CrossRef CAS PubMed.
- M. Courtney and C. L. Ren, Electrophoresis, 2019, 40, 643–658 CAS.
- D. R. Laws, D. Hlushkou, R. K. Perdue, U. Tallarek and R. M. Crooks, Anal. Chem., 2009, 81, 8923–8929 CrossRef CAS PubMed.
- R. M. Crooks, ChemElectroChem, 2016, 3, 357–359 CrossRef CAS.
- S. E. Fosdick, K. N. Knust, K. Scida and R. M. Crooks, Angew. Chem., Int. Ed., 2013, 52, 10438–10456 CrossRef CAS PubMed.
- L. Koefoed, S. U. Pedersen and K. Daasbjerg, Curr. Opin. Electrochem., 2017, 2, 13–17 CrossRef CAS.
- G. Loget, D. Zigah, L. Bouffier, N. Sojic and A. Kuhn, Acc. Chem. Res., 2013, 46, 2513–2523 CrossRef CAS PubMed.
- F. Mavré, R. K. Anand, D. R. Laws, K.-F. Chow, B.-Y. Chang, J. A. Crooks and R. M. Crooks, Anal. Chem., 2010, 82, 8766–8774 CrossRef PubMed.
- D. C. Duffy, J. C. McDonald, O. J. A. Schueller and G. M. Whitesides, Anal. Chem., 1998, 70, 4974–4984 CrossRef CAS PubMed.
- V. Tandon, S. K. Bhagavatula, W. C. Nelson and B. J. Kirby, Electrophoresis, 2008, 29, 1092–1101 CrossRef CAS PubMed.
- S. Takayama, J. C. McDonald, E. Ostuni, M. N. Liang, P. J. A. Kenis, R. F. Ismagilov and G. M. Whitesides, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 5545–5548 CrossRef CAS PubMed.
- S. Takayama, E. Ostuni, P. LeDuc, K. Naruse, D. E. Ingber and G. M. Whitesides, Nature, 2001, 411, 1016 CrossRef CAS PubMed.
- M. A. Holden, S. Kumar, E. T. Castellana, A. Beskok and P. S. Cremer, Sens. Actuators, B, 2003, 92, 199–207 CrossRef CAS.
- G. H. Seong and R. M. Crooks, J. Am. Chem. Soc., 2002, 124, 13360–13361 CrossRef CAS PubMed.
- S. K. Sia and G. M. Whitesides, Electrophoresis, 2003, 24, 3563–3576 CrossRef CAS PubMed.
- R. W. Ramette, C. H. Culberson and R. G. Bates, Anal. Chem., 1977, 49, 867–870 CrossRef CAS.
- S. J. Kim, Y.-C. Wang, J. H. Lee, H. Jang and J. Han, Phys. Rev. Lett., 2007, 99, 044501 CrossRef.
- S. J. Kim, L. D. Li and J. Han, Langmuir, 2009, 25, 7759–7765 CrossRef CAS PubMed.
- K.-D. Huang and R.-J. Yang, Microfluid. Nanofluid., 2008, 5, 631–638 CrossRef CAS.
- V. S. Pham, Z. Li, K. M. Lim, J. K. White and J. Han, Phys. Rev. E, 2012, 86, 046310 CrossRef PubMed.
- N. A. Mishchuk and P. V. Takhistov, Colloids Surf., A, 1995, 95, 119–131 CrossRef CAS.
- I. Cho, G. Y. Sung and S. J. Kim, Nanoscale, 2014, 6, 4620–4626 RSC.
- B. Qiu, L. Gong, Z. Li and J. Han, Theor. Appl. Mech. Lett., 2019, 9, 36–42 CrossRef.
- Z. Li, W. Liu, L. Gong, Y. Zhu, Y. Gu and J. Han, Int. J. Appl. Mech. Eng., 2017, 09, 1750107 CrossRef.
- S. Ehlert, D. Hlushkou and U. Tallarek, Microfluid. Nanofluid., 2008, 4, 471–487 CrossRef CAS.
- R. K. Anand, E. Sheridan, D. Hlushkou, U. Tallarek and R. M. Crooks, Lab Chip, 2011, 11, 518–527 RSC.
- E. Sheridan, D. Hlushkou, R. K. Anand, D. R. Laws, U. Tallarek and R. M. Crooks, Anal. Chem., 2011, 83, 6746–6753 CrossRef CAS PubMed.
- X. Huang, M. J. Gordon and R. N. Zare, Anal. Chem., 1988, 60, 1837–1838 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Solution conductivity calibration curve; electrophoretic mobility measurements; numerical simulations; normalized conductivity with BPE1 “on”; Tris concentration with BPE1 and BPE2 “on”; simulated rate of convection; movies showing sorting by fICP; focusing and sorting by serial fICP; focusing, sorting, and separating by serial fICP. See DOI: 10.1039/d0sc01931c |
| This journal is © The Royal Society of Chemistry 2020 |