
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA polycyclic aromatic hydrocarbon diradical with pH-responsive magnetic properties†
Xiangyu
Fu‡
,
Han
Han‡
,
Di
Zhang
,
Han
Yu
,
Qilin
He
and
Dahui
Zhao
 *
*
Beijing National Laboratory for Molecular Sciences, Centre for the Soft Matter Science and Engineering, Key Lab of Polymer Chemistry & Physics of the Ministry of Education, College of Chemistry, Peking University, Beijing, 100871, China. E-mail: dhzhao@pku.edu.cn
First published on 11th May 2020
Abstract
By integrating azulene with a quinoid moiety, a novel non-alternant polycyclic aromatic hydrocarbon molecule BCHF1 exhibiting manifold zwitterionic, quinoidal and diradical behaviors is designed and synthesized. Its zwitterionic feature is evidenced by the changes shown by the 1H-NMR and absorption spectra when the molecule undergoes reversible protonation and deprotonation reactions at varied pH. The diradical facet, manifesting a small singlet–triplet energy gap (ΔES–T), is characterized with a paramagnetic resonance signal detected by the EPR spectroscopy at room temperature. As the diradical properties are not observed in the protonated form, BCHF1+H+, a pH-controlled reversible magnetic switching behavior is illustrated by monitoring the on and off cycles of EPR signals upon successively adding bases and acids to a solution or exposing a thin film of BCHF1+H+ to base vapor followed by acid vapor.
Introduction
Owing to the unique properties conferred by the magnetic spin and open-shell electronic feature, organic radicals have attracted growing research interest as emerging functional materials with potential applications in diverse fields, including molecular magnetism,1,2 magneto- and opto-electronics,3–7etc. Among the various capacities, molecules exhibiting different spin states and thus switchable magnetic properties responsive to external stimuli, such as light,8–10 temperature,3,11–14 pH15,16 and redox conditions,17 are of special value.18–23 The major challenge in developing functional materials lies in the evasive nature and low stability of organic (poly)radicals, due to their open-shell electronic characteristics. Compared to the light hetero-atom (N or O) analogues, polycyclic aromatic hydrocarbon (PAH) polyradicals are even more difficult to stabilize. Nonetheless, owing to their unique optical, electronic and magnetic properties which are valuable to versatile applications,24,25 continual research endeavours are made for designing and synthesizing new persistent PAH radicals, and tremendous progress has been witnessed in past decades.26–30 In particular, devising suitably extended polycyclic quinoidal scaffolds has been demonstrated as an effective strategy to induce the open-shell diradical state, by gaining stabilizing energy from aromatizing multiple quinoid rings.Among the different stimulating mechanisms, we are interested in developing a pH-sensitive molecular magnetic switch by harnessing the diradical–quinoidal bistable states. This necessarily entails integrating a pH-responsive unit with a hydrocarbon moiety capable of swapping between the open-shell and closed-shell electronic structures. To this end, azulene31–36 attracted our attention for possessing a unique bicyclic zwitterionic scaffold (Fig. 1a) comprising an electron-rich five-membered ring which is capable of reversible protonation under suitable pH conditions.37–39 We thus propose that, by integrating the azulene skeleton with a quinoid moiety, a diradical molecule equipped with a zwitterionic feature may be devised, and hence a pH-responsive magnetic capacity is attained. Based on these considerations, we design BCHF1, which consists of a polycyclic benzo[5,6]cyclohepta[1,2-b]fluorene framework. That is, a quinoid moiety is fused in-between the 5- and 7-membered rings of azulene and thereby a novel PAH manifesting three different resonance forms of diradical, quinoidal and zwitterionic features is created. Among the three states, the quinoidal form exhibits only one Clar sextet. Yet, the diradical possesses three of such sextets, and the zwitterion gains further stabilizing energy from the pseudo-aromatization of the five- and seven-membered-ring moieties, besides three benzene sextets. Hence, the molecule is expected to manifest more pronounced diradical and zwitterionic properties, bestowing the designed pH-responsive magnetic-switching capability (Fig. 1c).
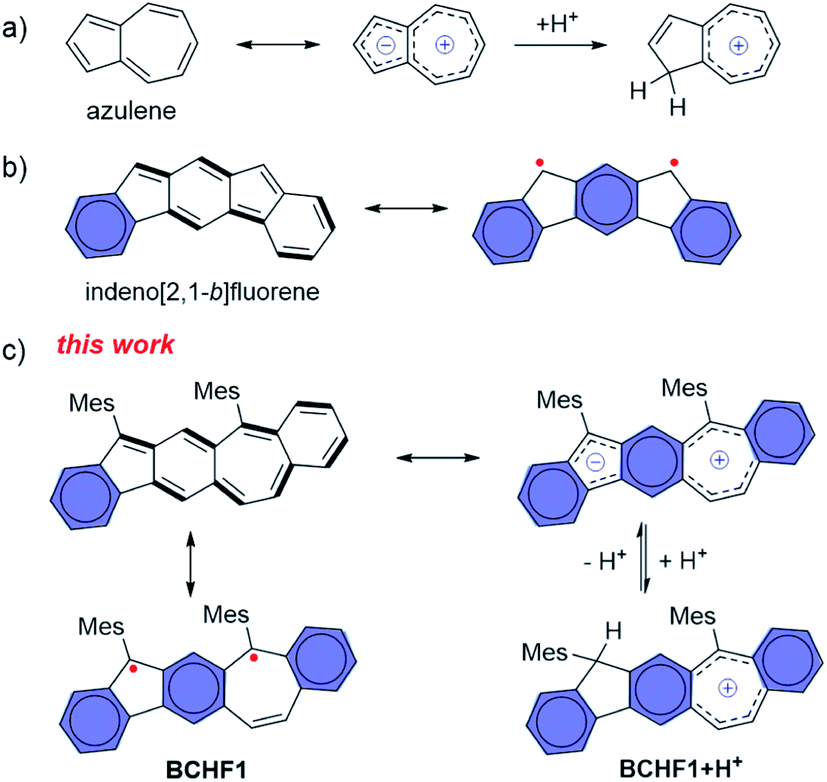 | ||
| Fig. 1 (a) Azulene and its protonated form; (b) indeno[2,1-b]fluorene with a distinct diradical feature; (c) quinoidal, zwitterionic, diradical and protonated forms of BCHF1. | ||
Interestingly, in comparison to BCHF1, its regio-isomeric molecule benzo[4,5]cyclohepta[1,2-b]fluorene (BCHF2, Scheme 1), which was previously synthesized and studied by Chi and Miao et al.,40 was shown to be a stable compound with no evident diradical properties. Distinct diradical properties are anticipated in BCHF1 and BCHF2, by drawing analogy with a pair of indenofluorene (IF) isomers,27,41–45 indeno[1,2-b]fluorene and indeno[2,1-b]fluorene. According to previous studies,43 indeno[2,1-b]fluorene exhibits pronounced open-shell characteristics (Fig. 1b), by displaying a large diradical component (y) of 0.65 with a small singlet–triplet energy gap (ΔES–T), whereas indeno[1,2-b]fluorene is shown to be a closed-shell molecule (y = 0.26) with a dominant quinoid structure.44 Such disparate behaviours are attributed to the extra Clar sextet gained by indeno[2,1-b]fluorene compared to indeno[1,2-b]fluorene, upon transforming from a quinoidal to a diradical form. This additional aromatizing energy helps stabilize the diradical state of indeno[2,1-b]fluorene. On the basis of these findings with IF isomers, we speculate that such contrasting diradical behaviours should similarly be manifested by the benzocycloheptafluorene isomers. That is, as an analogue of indeno[2,1-b]fluorene, BCHF1 would possess more pronounced diradical character with a smaller ΔES–T than BCHF2, which is an analogue of indeno[1,2-b]fluorene.
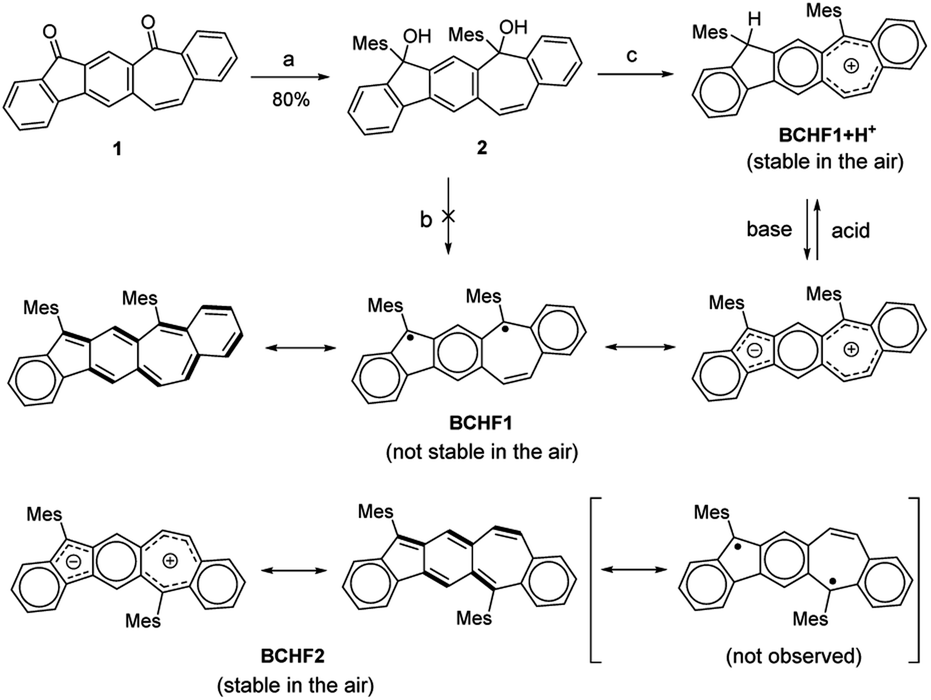 | ||
| Scheme 1 Chemical structures and synthetic routes. Reactions and conditions: (a) MesLi, THF; (b) SnCl2, ethyl acetate; (c) BF3·Et2O, Et3SiH, CH2Cl2. | ||
Moreover, one extra Clar sextet is also acquired by the zwitterionic form of BCHF1 compared to BCHF2, relative to their respective quinoidal states. Therefore, a more pronounced charge separation feature is also expectable from BCHF1 than BCHF2, which should enable more facile protonation of the zwitterionic form of BCHF1, resembling the properties of azulene. As the paramagnetic state of BCHF1 is switched “off” upon protonation, for eliminating the diradical resonance (Fig. 1c), superior pH sensitivity and magnetic-switching capability can thus be achieved.
Results and discussion
DFT calculations
Computational simulations were conducted to analyze and compare the properties of BCHF1 and BCHF2. The calculations were conducted at the level of (U)B3LYP/6-311G(d). The calculation results very well supported our hypothesis that BCHF1 should possess significant open-shell characters, and a small singlet–triplet energy gap (ΔES–T = ES − ET, where ES and ET represent the energy of the open-shell singlet and triplet, respectively) of merely −1.84 kcal mol−1 (Table S1†) was predicted, which was sufficiently small for the existence triplet species around room temperature. In comparison, a closed-shell structure with a much larger ΔES–T of −18.49 kcal mol−1 was obtained with BCHF2. Moreover, evident differences were also noted with the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO). While both HOMO and LUMO of BCHF2 were much delocalized over the entire polycyclic framework, the HOMO of BCHF1 was fairly localized to the fluorene moiety and the LUMO was mainly distributed among the seven-membered ring and two fused benzene units (Fig. S10†). Such more localized charge distribution predicted for BCHF1 was verified by a considerably larger dipole moment (4.75 D) calculated for BCHF1 than BCHF2 (2.76 D). Such properties evidently demonstrated the more pronounced charge separation feature of BCHF1. Therefore, all these calculation results basically proved that, because of the extra Clar sextet gained by the zwitterionic and diradical states of BCHF1 than BCHF2, compared to their respective quinoidal forms, the diradical and zwitterionic forms are more important than the quinoidal form in BCHF1, whereas the quinoidal form is comparatively more significant in BCHF2, which was confirmed by its crystallographic data.40Synthesis and structural characterization
Based on the above molecular design and calculation results, we set out to synthesize and investigate the properties of BCHF1, with BCHF2 examined for comparison. The preparation of BCHF1 was first attempted by adapting a similar synthetic route previously developed for BCHF2.40 Specifically, dione 1 (Scheme 1) was obtained from 6-phenethyl-4-phenyl substituted m-phthalate via dual intramolecular Friedel–Crafts acylation, followed by bromination and elimination to convert the ethylene linker to a conjugated vinylene bridge (see the ESI†). Then, by carrying out a double nucleophilic addition to dione 1 with mesityllithium, diol 2 was obtained as a direct precursor to BCHF1. However, when a commonly applied reductive reagent tin(II) chloride was used to reduce diol 2, we were not able to isolate BCHF1. In order to confirm that the synthetic procedures were suitably performed, we independently prepared the regio-isomer BCHF2 and found that under the same conditions BCHF2 was successfully obtained as a green solid from analogous substrates. The structure of BCHF2 was verified by comparing the characterization data to those reported in the literature.40 Then, the molecule was confirmed to be diamagnetic by its silent electron-paramagnetic resonance (EPR) spectrum and the quite normal NMR signals. The molecule was also observed to display adequate stability under ambient conditions.As the reducing protocol using SnCl2 did not provide the desired molecule BCHF1, alternative reducing conditions were employed. After diol 2 was subjected to reaction with Et3SiH in the presence of Lewis acid BF3·Et2O and maintained at 0 °C for about an hour, a substantial amount of red precipitation was observed upon solvent evaporation. Besides the disparate colour, the 1H NMR spectrum of this red product was evidently dissimilar to that of BCHF2. While all 16 aromatic and vinylic protons were detected in a relatively narrow chemical shift range of 6.4 to 7.6 ppm with BCHF2 (see the ESI†), a set of resonance signals shown by this red compound were found to range widely from 5.6 to 9.6 ppm (Fig. 2a). Although the appearance of both up- and down-field shifted signals was initially suspected to arise from the zwitterionic feature of BCHF1, inconsistency emerged unambiguously when 17 rather than 16 proton resonances were identified between 5 and 10 ppm. While this observation ruled out the possibility that the red product was the simple form of BCHF1, subsequently performed high-resolution mass spectrometry confirms the presence of an ion peak corresponding to the molecular mass of BCHF1+H+. Combined with the 1H NMR data, we concluded that the red compound was a protonated salt of BCHF1 (i.e., BCHF1+H+).
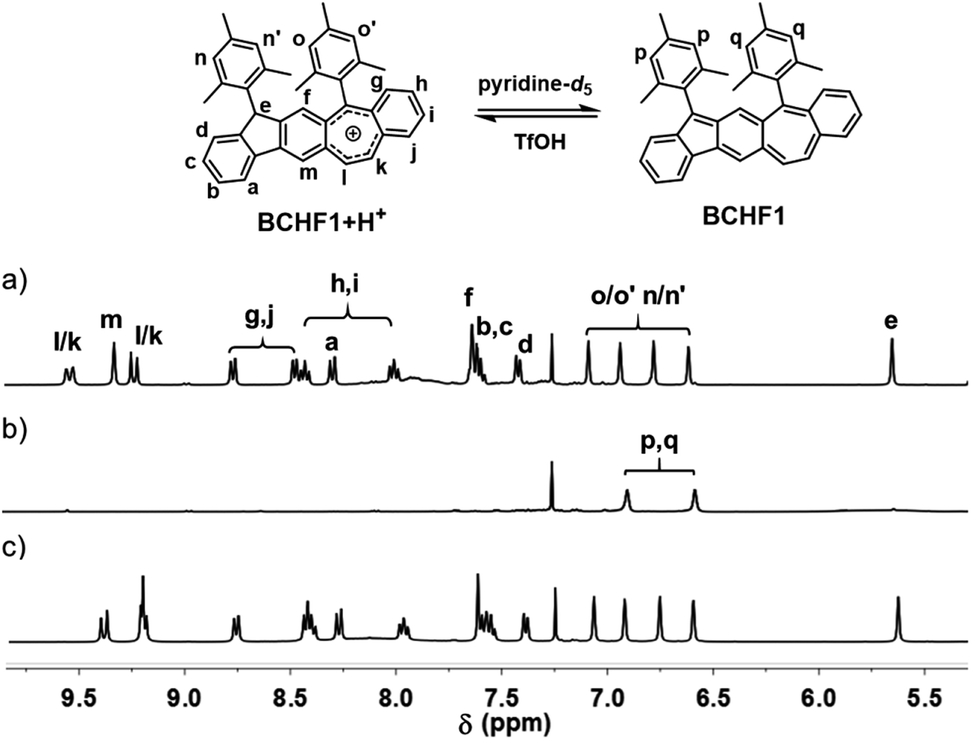 | ||
| Fig. 2 1H NMR spectra of BCHF1+H+ in CDCl3 (a) and upon the addition of pyridine-d5 (b) followed by excess TfOH acid (c). | ||
The formation of such a protonated structure was plausible considering the reduction mechanism of triethylsilane, which offered a hydride upon the Lewis acid-facilitated removal of both hydroxide groups from diol 2. The reduction by a second hydride did not occur likely because of the high stability of the cationic aromatic species BCHF1+H+, which boasted a seven-membered ring hosting 6 electrons. Moreover, reasonable assignments were made for all 17 proton signals detected in the aromatic regime of the 1H NMR spectrum (Fig. 2a). The furthest up-field shifted singlet at about 5.6 ppm was ascribed to the tertiary H in the 9-position of the fluorene moiety, and the substantially down-field shifted signals appearing beyond 8 ppm were mostly assignable to protons around the polycyclic moiety bearing the positive charge.
Finally, the unambiguous structure of BCHF1+H+, including the identity of the counter ion, was revealed by X-ray crystallography performed with a single crystal of the red compound (Fig. 3 and S13†). The molecule was shown to have a planar polycyclic skeleton composed of linearly fused 6-5-6-7-6 membered rings, to which two mesityl groups were attached almost perpendicularly via the five- and seven-membered rings. Nonetheless, from the bond angles it was perceptible that the 9-position of the fluorene moiety was protonated, assuming the sp3 hybridization state. Moreover, the counter ion of this protonated structure was identified to be tetrafluoroborate (BF4−), which was positioned closer to the centre of the seven-membered ring bearing a positive charge. The average bond length of the seven-membered ring was 1.418 Å, slightly shorter than that found with a dibenzotropylium cation derivative (1.425 Å) reported in the literature.46 Thus, the calculated harmonic oscillator model of aromaticity (HOMA) index47 of the seven-membered ring in BCHF1+H+ (0.52) was also slightly higher than that of the dibenzotropylium cation (0.48),46 suggesting a comparable aromatic character.
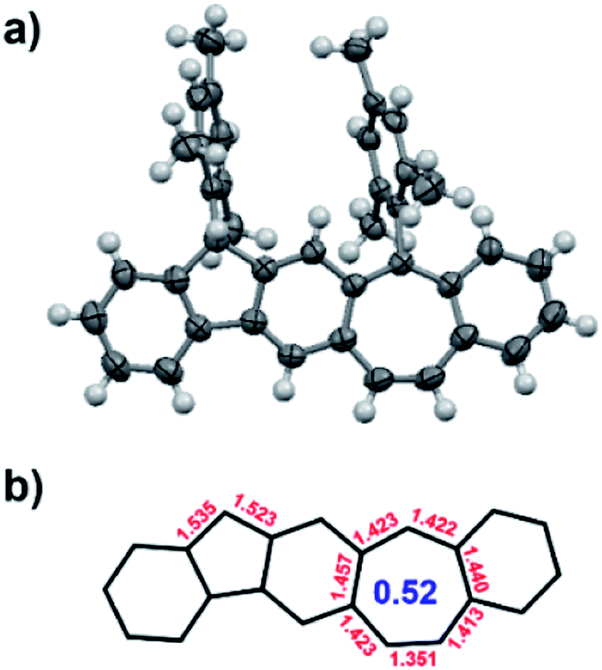 | ||
| Fig. 3 (a) X-ray crystallographic structure of BCHF1+H+ (counter ion BF4− omitted for clarity); (b) selected bond lengths (Å) and the calculated HOMA index of the seven-membered ring. | ||
Acid/base-responsive properties
Subsequently, acid–base reaction experiments were conducted to examine the properties of neutral BCHF1. The reversible deprotonation of BCHF1+H+ and re-protonation of BCHF1 were first examined by monitoring the 1H NMR spectral changes in response to consecutive additions of a base and an acid to a solution of BCHF1+H+. As the neutral form of BCHF1 was found to be unstable under ambient conditions, a small amount of deuterated pyridine (pyridine-d5) was added to the solution of BCHF1+H+ in CDCl3 under a nitrogen atmosphere. Such sensitivity toward oxygen also corroborated the diradical propensity of BCHF1. Upon addition of the base, almost all resonances belonging to BCHF1+H+ vanished from the 1H NMR spectrum (Fig. 2b), suggesting the formation of a paramagnetic species. Only two aromatic signals remained at about 6.6 and 6.9 ppm, which were assigned to the aromatic protons on two mesityl groups positioned relatively far away from the spins distributed over the polycyclic framework. The fact that four different mesityl protons observed with BCHF1+H+ merged into two sets in BCHF1 further corroborated the assumption that deprotonation took place with the sp3-hybridized carbon-9 of the fluorene unit and resulted in a more symmetrized, planar polycyclic skeleton. Next, an excess amount of trifluoromethanesulfonic acid (TfOH) was added to this solution containing in situ generated BCHF1, followed by some deuterated water to extract the excess acid and pyridinium triflate salt. After these procedures, all proton resonances characteristic of BCHF1+H+ were recovered (Fig. 2c), with minor chemical shift variations likely due to the solvent polarity and ionic strength changes. These results clearly verified the occurrence of reversible deprotonation and protonation processes occurring to BCHF1+H+ and BCHF1, and that the latter displayed paramagnetic properties around room temperature, in agreement with the small ΔES–T predicted by the theoretical calculations.Next, the absorption spectral features of BCHF1+H+ and BCHF1 were examined by varying the pH conditions and compared to those of BCHF2. A solution of BCHF1+H+ in CH2Cl2 depicted a series of absorption bands in the UV-visible range from ca. 300 to 630 nm (Fig. S1†), which accounted for the red colour of the compound. Upon the addition of pyridine (ca. 1000 equiv.), the red colour of the solution turned lighter and the absorption intensity was found to decrease only by about 20%, while the overall spectral band shape remained nearly unchanged (Fig. S2†). Such a phenomenon suggested that the basicity of pyridine was not strong enough to completely deprotonate BCHF1+H+. Although in the earlier experiments, most of the 1H NMR signals of BCHF1+H+ were observed to disappear completely upon treatment with pyridine, it was likely due to the rapid exchange processes between the deprotonated paramagnetic and protonated diamagnetic species. Thus, in order to completely deprotonate BCHF1+H+, a stronger base was necessarily applied. We then found that some strongly reductive bases, such as triethylamine (Et3N), caused noticeable decomposition of BCHF1, as evidenced by the partial recovery of the absorption spectra in repeated acid–base titrations (Fig. S2†).
Upon screening a set of different bases, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) appeared to offer suitable basicity while entailing minimum decomposition of BCHF1. Upon adding ca. 2 equiv. DBU to a solution of BCHF1+H+ in CH2Cl2, a drastic colour change from red to green was observed. Meanwhile, the absorption spectrum showed evident changes, with the original band replaced by a completely new set of absorption peaks (Fig. 4). Particularly notable was the emergence of some pronounced long-wavelength absorption activities between 520 and 700 nm, which evidenced the formation of a more delocalized framework featuring a quinoidal/diradical structure, attributable to BCHF1. More importantly, the absorption spectrum of BCHF1+H+ was completely restored when the solution of BCHF1 was treated with TFA, evidencing the conversion back to BCHF1+H+. When a larger amount of DBU was then added to the same solution such as to re-generate BCHF1, slightly attenuated absorptivity was observed, indicative of minor decomposition. Overall, BCHF1 produced by treating BCHF1+H+ with DBU exhibited decent stability. By continuously monitoring the absorption spectrum, its half-life time in CH2Cl2 under a nitrogen atmosphere, in the presence of a minimal amount of DBU, was estimated to be about 35 hours (Fig. S6†).
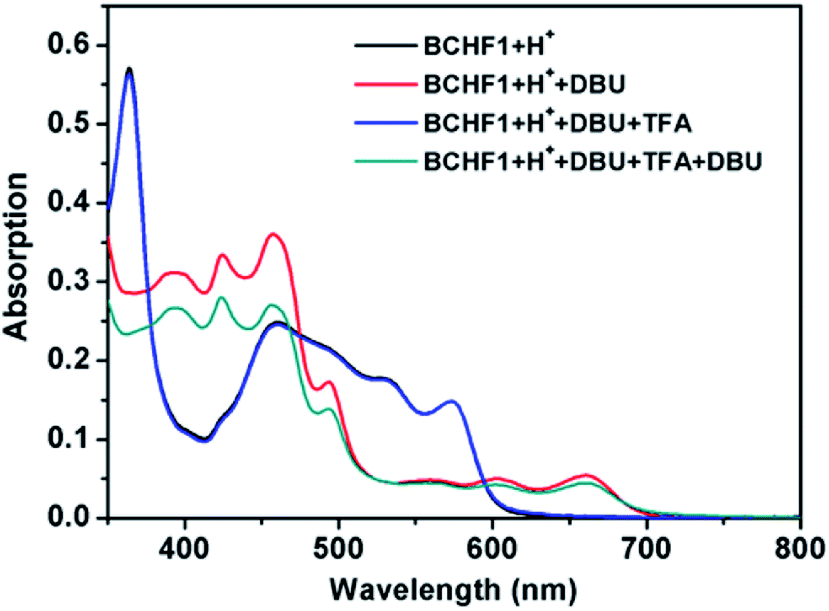 | ||
| Fig. 4 Absorption spectra of BCHF1+H+ in CH2Cl2 upon sequential additions of DBU, TFA, and DBU again (the black and blue lines overlap almost completely; spectra were collected from the same sample subjected to repeated acid/base additions). | ||
Analogous to the reversible protonation–deprotonation behaviours demonstrated with BCHF1, similar processes were also exhibited by BCHF2. The latter displayed completely reversible absorption changes upon reacting with acids and bases (Fig. S4†). Compared to the low-energy band shown by BCHF1 curtailed at about 700 nm, the long-wavelength absorption of BCHF2 was extended to over 900 nm (Fig. S1†). Such spectral differences were explained by TD-DFT calculations (Tables S2 and S4†), which indicated that the low-energy transition from the HOMO to the LUMO was allowed for BCHF2, but forbidden in BCHF1. This might as well be related to the more pronounced diradical nature and considerably smaller ΔES–T of BCHF1. In spite of the similar acid-based responsive spectral behaviours, much differed air stability was observed with BCHF2. All the above acid–base reactions were conducted under ambient conditions and no evidence of decomposition was detected even when the solution of BCHF2 was exposed to a large excess of Et3N. Such optimal stability was also a good indication that BCHF2 did not possess observable diradical properties, which was confirmed by its silence in the EPR spectroscopy.
Besides the absorption spectral properties, both molecules also manifested distinctive fluorescence emission responses to pH changes. The protonated form of BCHF1+H+ was found to be highly emissive in solution by showing an intense fluorescence band around 600 nm. The fluorescence was found to be completely quenched upon de-protonation with DBU (Fig. S5†). The non-emissive properties were consistent with the diradical characteristic of BCHF1. Upon adding TFA to the solution, the emission band was restored. Similarly, the neutral form of BCHF2 was non-emissive in solution, but an intense emission band was observed around 640 nm upon treatment with TFA, which could be quenched again with excessive Et3N (Fig. S4†).
Next, the pH-sensitive magnetic-switching behaviours were investigated with EPR spectroscopy. While BCHF2 was completely EPR silent, the conjugated acid–base pair of BCHF1+H+/BCHF1 demonstrated clearly pH-responsive EPR intensity changes (Fig. 5). As a diamagnetic molecule, BCHF1+H+ unsurprisingly showed no EPR signal in CH2Cl2 solution. When 2 equiv. DBU was added to this solution, a conspicuous paramagnetic resonance signal immediately appeared at g = 2.0032, which was assignable to in situ generated BCHF1. Then, upon the addition of 20 equiv. TFA to the same solution the EPR signal vanished, again. Subsequently, by adding 20 more equivalents of DBU the EPR signal clearly regenerated, with its intensity very close to that of the first time (Fig. S7†). This entire process was in complete consistency with the absorption spectral changes observed earlier (Fig. 4). The slight EPR intensity drop, indicative of minor BCHF1 decomposition, was mainly due to the relatively large amount of DBU necessarily applied to neutralize excess TFA. Hence, we turned to a different acid, p-toluenesulfonic acid (TsOH) with a pKa value better matching the pKb of DBU, such that minimized amounts of acids and bases could be used to achieve better reproducibility in the repeated interconversion between BCHF1 and BCHF1+H+, for inducing minimal decomposition. Next, by successively adding a finite amount of DBU (2 equiv.) followed by TsOH (2 equiv.), many more cycles of switching between the “on” and “off” states of the EPR signal were realized (Fig. 5 and S3†). These experiments very well demonstrated that BCHF1 could be swapped reversibly between the deprotonated paramagnetic and protonated diamagnetic forms via the dynamic acid/base reactions in response to pH changes.
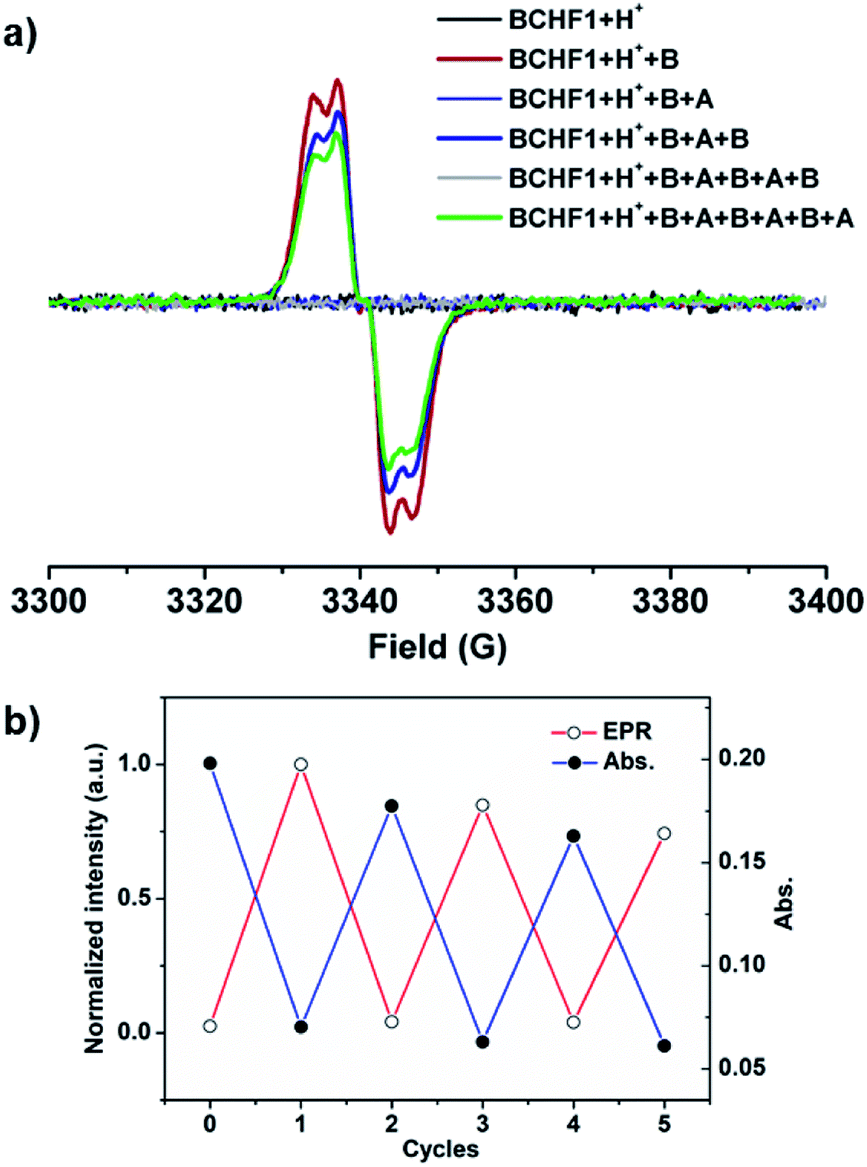 | ||
| Fig. 5 (a) EPR spectra of BCHF1+H+ in CH2Cl2 upon three cycles of the addition of 2 equiv. DBU (B) followed by 2 equiv. TsOH (A); (b) normalized EPR (@3336 G) and absorption (@570 nm) intensity changes in each cycle. | ||
Next, the intrinsic magnetic properties of BCHF1 were investigated. A featureless EPR signal with a g factor of 2.0036 was produced by BCHF1 in the solid state (Fig. 6a). The intensity of this resonance peak was observed to decrease at lowered temperatures, indicating the existence of a singlet ground state with a thermally accessed triplet state. By fitting the values of IT as a function of T using the modified Bleaney–Bowers equation (Fig. 6b),48 a ΔES–T value of −0.95 kcal mol−1 was estimated for BCHF1, which was in a reasonable agreement with the calculated value (−1.84 kcal mol−1). Also, such a singlet–triplet energy gap was much smaller compared to those reported for various studied indeno-fluorene isomers,28,43 suggesting that a more pronounced diradical propensity was manifested by BCHF1.
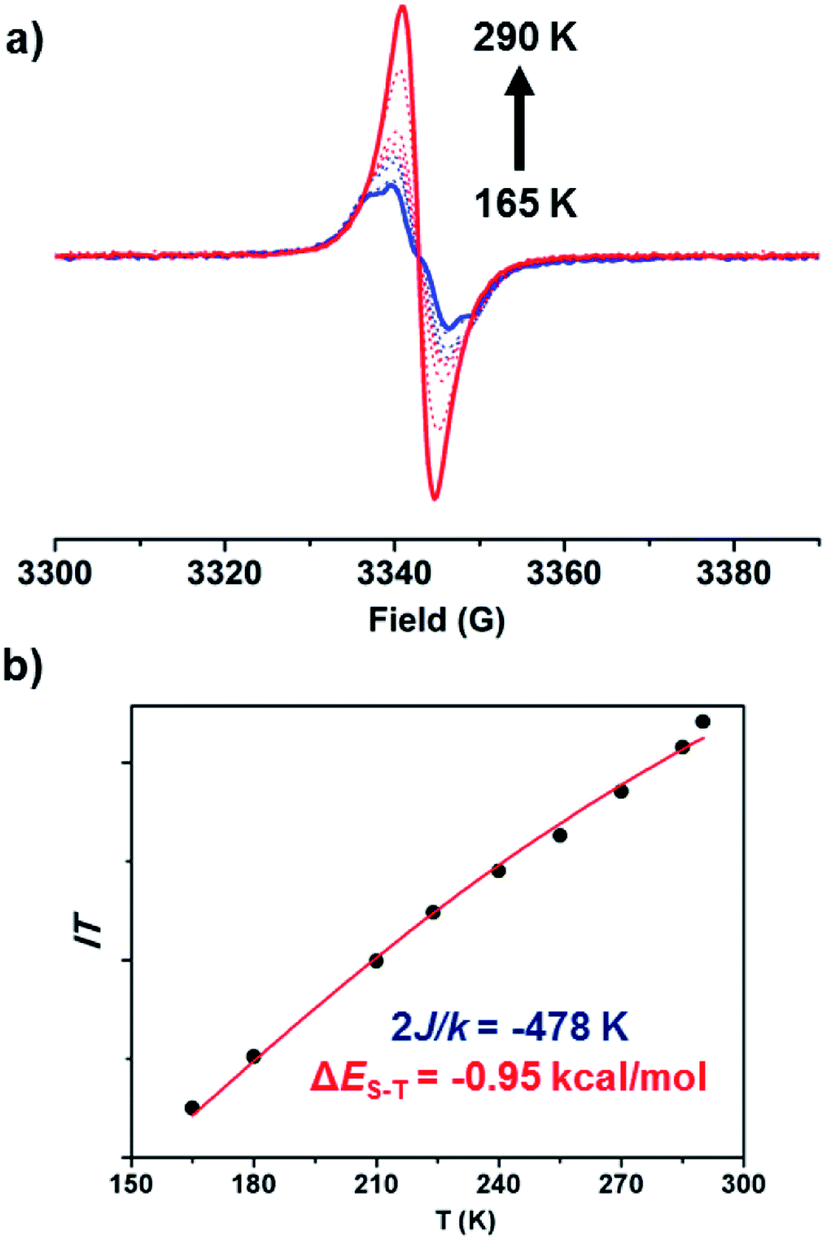 | ||
| Fig. 6 (a) Varied-temperature EPR spectra of BCHF1 in the solid state (obtained by treating BCHF1+H+ in CH2Cl2 with 2 equiv. DBU followed by evaporation under an inert atmosphere); (b) plot of IT vs. T (I, double integral of the EPR signal; the fitting curve shown as a red line). | ||
In light of its impressive pH-sensitive magnetic switching properties illustrated in solution, the capability of BCHF1+H+ for responding to gaseous acids and bases in the solid state was also tested. First, a thin film of BCHF1+H+ was prepared by evaporating its CH2Cl2 solution under a nitrogen atmosphere. Then, the vapour of DBU was introduced to induce a reaction with the film. The dark red film was observed to turn green gradually over 5 minutes. The absorption spectrum change, particularly the emergence of the peak around 670 nm, clearly indicated the formation of BCHF1 in the thin film (Fig. S9†). Subsequently, the film was exposed to the vapour of TFA, and its green colour quickly turned back to dark red in merely one minute, with the absorption spectrum restored to the protonated state as well. The faster kinetics observed with re-protonation was assumed to be related to the higher vapour pressure of TFA. These deprotonation and re-protonation processes were also monitored with EPR spectroscopy. A clear rise in the EPR signal was detected when the thin film of BCHF1+H+ was exposed to DBU vapour, and a complete silence of the resonance signal was also recorded when the film was then exposed to the TFA vapour (Fig. S9b†).
Conclusions
In summary, by integrating a quinoid moiety with azulene, a novel non-alternant hydrocarbon BCHF1 featuring multi-facet zwitterionic and diradical properties was designed. By virtue of the zwitterionic feature conferred by the azulene-like structure, BCHF1 easily adopted a proton and formed a stable compound in its conjugated acid salt state (i.e., BCHF1+H+). This salt was successfully isolated and characterized by 1H NMR spectroscopy and single-crystal X-ray crystallography. It could be converted to its neutral form of BCHF1 by reacting with organic bases, such as DBU. The diradical properties of BCHF1, featuring a small ΔES–T, were clearly evidenced by the conspicuous EPR signal detectable around room temperature, since the closed-shell quinoidal and zwitterionic states should both be EPR silent. The varied-temperature EPR experiments further revealed an impressively small ΔES–T of ca. −0.95 kcal mol−1 for BCHF1, indicative of a singlet ground state with a thermally accessed triplet state. By monitoring the recurrent disappearance and re-appearance of 1H NMR and EPR signals in response to repeated additions of acids and bases to the solutions of BCHF1+H+, the molecule was clearly demonstrated to undergo reversible deprotonation and protonation reactions, thereby switching between the paramagnetic and diamagnetic forms. Additionally, the thin film of BCHF1+H+/BCHF1 was shown to be responsive to the vapour of DBU and TFA by manifesting dramatic absorption changes and reversibly turning on and off its EPR signal. Basically, by harnessing a dynamic protonation process of a unique zwitterionic/diradical molecule, a pH-controlled switching mechanism between the paramagnetic and diamagnetic states was realized. To the best of our knowledge, this is the first example of a PAH showing acid/base-responsive magnetic-switching properties, which is envisioned to open up new opportunities for designing stimuli-responsive organic magnetic materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work is financially supported by the National Natural Science Foundation of China (Nos 21925501, 21790363 and 21674001) and the Beijing National Laboratory for Molecular Sciences (BNLMS-CXXM-201902). The support from the High-performance Computing Platform of Peking University for the computational resources and the Steady High Magnetic Field Facilities, High Magnetic Field Laboratory, CAS is acknowledged.Notes and references
- A. Rajca, Chem. Rev., 1994, 94, 871–893 CrossRef CAS.
- A. Rajca, J. Wongsriratanakul and S. Rajca, Science, 2001, 294, 1503–1505 CrossRef CAS PubMed.
- M. E. Itkis, X. Chi, A. W. Cordes and R. C. Haddon, Science, 2002, 296, 1443–1445 CrossRef CAS PubMed.
- X. Yu, A. Mailman, K. Lekin, A. Assoud, C. M. Robertson, B. C. Noll, C. F. Campana, J. A. K. Howard, P. A. Dube and R. T. Oakley, J. Am. Chem. Soc., 2012, 134, 2264–2275 CrossRef CAS PubMed.
- A. Coskun, A. J. M. Spruell, G. Barin, W. R. Dichtel, A. H. Flood, Y. Y. Botros and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 4827–4859 RSC.
- Q. Peng, A. Obolda, M. Zhang and F. Li, Angew. Chem., Int. Ed., 2015, 54, 7091–7095 CrossRef CAS PubMed.
- I. Ratera and J. Veciana, Chem. Soc. Rev., 2012, 41, 303–349 RSC.
- N. Tanifuji, M. Irie and K. Matsuda, J. Am. Chem. Soc., 2005, 127, 13344–13353 CrossRef CAS PubMed.
- J. Wang, L. Hou, W. R. Browne and B. L. Feringa, J. Am. Chem. Soc., 2011, 133, 8162–8164 CrossRef CAS PubMed.
- K. Lekin, H. Phan, S. M. Winter, J. W. L. Wong, A. A. Leitch, D. Laniel, W. Yong, R. A. Secco, J. S. Tse, S. Desgreniers, P. A. Dube, M. Shatruk and R. T. Oakley, J. Am. Chem. Soc., 2014, 136, 8050–8062 CrossRef CAS PubMed.
- W. Fujita and K. Awaga, Science, 1999, 286, 261–262 CrossRef CAS PubMed.
- D. A. Shultz, R. M. Fico, P. D. Boyle and J. W. Kampf, J. Am. Chem. Soc., 2001, 123, 10403–10404 CrossRef CAS PubMed.
- J. L. Brusso, O. P. Clements, R. C. Haddon, M. E. Itkis, A. A. Leitch, R. T. Oakley, R. W. Reed and J. F. Richardson, J. Am. Chem. Soc., 2004, 126, 8256–8265 CrossRef CAS PubMed.
- M. B. Mills, T. Wohlhauser, B. Stein, W. R. Verduyn, E. Song, P. Dechambenoit, M. Rouzieres, R. Clerac and K. E. Preuss, J. Am. Chem. Soc., 2018, 140, 16904–16908 CrossRef CAS PubMed.
- G. Gryn’ova, D. L. Marshall, S. J. Blanksby and M. L. Coote, Nat. Chem., 2013, 5, 474–481 CrossRef PubMed.
- T. Krchova, V. Herynek, A. Galisova, J. Blahut, P. Hermann and J. Kotek, Inorg. Chem., 2017, 56, 2078–2091 CrossRef CAS PubMed.
- Y. Li, X. Lei, S. Jockusch, J. Y.-C. Chen, M. Frunzi, J. A. Johnson, R. G. Lawler, Y. Murata, M. Murata, K. Komatsu and N. J. Turro, J. Am. Chem. Soc., 2010, 132, 4042–4043 CrossRef CAS PubMed.
- D. Bardelang, G. Casano, F. Poulhes, H. Karoui, J. Filippini, A. Rockenbauer, R. Rosas, V. Monnier, D. Siri, A. Gaudel-Siri, O. Ouari and P. Tordo, J. Am. Chem. Soc., 2014, 136, 17570–17577 CrossRef CAS PubMed.
- A. C. Fahrenbach, Z. Zhu, D. Cao, W. Liu, H. Li, S. K. Dey, S. Basu, A. Trabolsi, Y. Y. Botros, W. A. Goddard and J. F. Stoddart, J. Am. Chem. Soc., 2012, 134, 16275–16288 CrossRef CAS PubMed.
- S. Venkataramani, U. Jana, M. Dommaschk, F. D. Sönnichsen, F. Tuczek and R. Herges, Science, 2011, 331, 445–448 CrossRef CAS PubMed.
- F. Touti, P. Maurin and J. Hasserodt, Angew. Chem., Int. Ed., 2013, 52, 4654–4658 CrossRef CAS PubMed.
- B. P. Timko, T. Dvir and D. S. Kohane, Adv. Mater., 2010, 22, 4925–4943 CrossRef CAS PubMed.
- H. Lee, T. Shin, J. Cheon and R. Weissleder, Chem. Rev., 2015, 115, 10690–10724 CrossRef CAS PubMed.
- M. Nakano and B. Champagne, J. Phys. Chem. Lett., 2015, 6, 3236–3256 CrossRef CAS.
- Y. Morita, S. Suzuki, K. Sato and T. Takui, Nat. Chem., 2011, 3, 197–204 CrossRef CAS PubMed.
- Z. Sun, Q. Ye, C. Chi and J. Wu, Chem. Soc. Rev., 2012, 41, 7857–7889 RSC.
- Z. Zeng, X. Shi, C. Chi, J. T. L. Navarrete, J. Casado and J. Wu, Chem. Soc. Rev., 2015, 44, 6578–6596 RSC.
- G. E. Rudebusch, J. L. Zafra, K. Jorner, K. Fukuda, J. L. Marshall, I. Arrechea-Marcos, G. L. Espejo, R. P. Ortiz, C. J. Gómez-García, L. N. Zakharov, M. Nakano, H. Ottosson, J. Casado and M. M. Haley, Nat. Chem., 2016, 8, 753–759 CrossRef CAS PubMed.
- A. Konishi, Y. Okada, M. Nakano, K. Sugisaki, K. Sato, T. Takui and M. Yasuda, J. Am. Chem. Soc., 2017, 139, 15284–15287 CrossRef CAS PubMed.
- M. R. Ajayakumar, Y. Fu, J. Ma, F. Hennersdorf, H. Komber, J. J. Weigand, A. Alfonsov, A. A. Popov, R. Berger, J. Liu, K. Müllen and X. Feng, J. Am. Chem. Soc., 2018, 140, 6240–6244 CrossRef CAS PubMed.
- Z. Zhang, G. M. Ferrence and T. D. Lash, Org. Lett., 2009, 11, 101–104 CrossRef CAS PubMed.
- Y. Yamaguchi, M. Takubo, K. Ogawa, K. Nakayama, T. Koganezawa and H. Katagiri, J. Am. Chem. Soc., 2016, 138, 11335–11343 CrossRef CAS PubMed.
- E. Puodziukynaite, H. W. Wang, J. Lawrence, A. J. Wise, T. P. Russell, M. D. Barnes and T. Emrick, J. Am. Chem. Soc., 2014, 136, 11043–11049 CrossRef CAS PubMed.
- Y. Sasaki, M. Takase, T. Okujima, S. Mori and H. Uno, Org. Lett., 2019, 21, 1900–1903 CrossRef CAS PubMed.
- M. Murai, S. Iba, H. Ota and K. Takai, Org. Lett., 2017, 19, 5585–5588 CrossRef CAS PubMed.
- H. Xin and X. Gao, ChemPlusChem, 2017, 82, 945–956 CrossRef CAS PubMed.
- K. H. Grellmann, E. Heilbronner, P. Seiler and A. Weller, J. Am. Chem. Soc., 1968, 90, 4238–4242 CrossRef CAS.
- X. B. Wang, J. K.-P. Ng, P. Jia, T. Lin, C. M. Cho, J. Xu, X. Lu and C. He, Macromolecules, 2009, 42, 5534–5544 CrossRef CAS.
- E. Amir, R. J. Amir, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2011, 133, 10046–10049 CrossRef CAS PubMed.
- X. Yang, X. Shi, N. Aratani, T. P. Goncalves, K.-W. Huang, H. Yamada, C. Chi and Q. Miao, Chem. Sci., 2016, 7, 6176–6181 RSC.
- M. Abe, Chem. Rev., 2013, 113, 7011–7088 CrossRef CAS PubMed.
- A. Shimizu and Y. Tobe, Angew. Chem., Int. Ed., 2011, 50, 6906–6910 CrossRef CAS PubMed.
- A. Shimizu, R. Kishi, M. Nakano, D. Shiomi, K. Sato, T. Takui, I. Hisaki, M. Miyata and Y. Tobe, Angew. Chem., Int. Ed., 2013, 52, 6076–6079 CrossRef CAS PubMed.
- J. J. Dressler, Z. Zhou, J. L. Marshall, R. Kishi, S. Takamuku, Z. Wei, S. N. Spisak, M. Nakano, M. A. Petrukhina and M. M. Haley, Angew. Chem., Int. Ed., 2017, 56, 15363–15367 CrossRef CAS PubMed.
- D. T. Chase, A. G. Fix, B. D. Rose, C. D. Weber, S. Nobusue, C. E. Stockwell, L. N. Zakharov, M. C. Lonergan and M. M. Haley, Angew. Chem., Int. Ed., 2011, 50, 11103–11106 CrossRef CAS PubMed.
- A. Konishi, K. Horii, D. Shiomi, K. Sato, T. Takui and M. Yasuda, J. Am. Chem. Soc., 2019, 141, 10165–10170 CrossRef CAS PubMed.
- T. M. Krygowski and M. K. Cyranski, Chem. Rev., 2001, 101, 1385–1419 CrossRef CAS PubMed.
- Y. Matsuta, D. Sakamaki, R. Kurata, A. Ito and S. Seki, Chem.–Asian J., 2017, 12, 1889–1894 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis details, additional spectroscopic data, and DFT calculations. CCDC 1982861. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc00770f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |