
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing catalytic alkane hydroxylation by tuning the outer coordination sphere in a heme-containing metal–organic framework†
David Z.
Zee
 a and
T. David
Harris
*ab
a and
T. David
Harris
*ab
aDepartment of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, Illinois 60208, USA
bDepartment of Chemistry, University of California, Berkeley, Berkeley, California 94720, USA. E-mail: dharris@berkeley.edu
First published on 7th May 2020
Abstract
Catalytic heme active sites of enzymes are sequestered by the protein superstructure and are regulated by precisely defined outer coordination spheres. Here, we emulate these protective functions in the porphyrinic metal–organic framework PCN-224 by post-synthetic acetylation and subsequent hydroxylation of the Zr6 nodes. A suite of physical methods demonstrates that both transformations preserve framework structure, crystallinity, and porosity without modifying the inner coordination spheres of the iron sites. Single-crystal X-ray analyses establish that acetylation replaces the mixture of formate, benzoate, aqua, and terminal hydroxo ligands at the Zr6 nodes with acetate ligands, and hydroxylation affords nodes with seven-coordinate, hydroxo-terminated Zr4+ ions. The chemical influence of these reactions is probed with heme-catalyzed cyclohexane hydroxylation as a model reaction. By virtue of passivated reactive sites at the Zr6 nodes, the acetylated framework oxidizes cyclohexane with a yield of 68(8)%, 2.6-fold higher than in the hydroxylated framework, and an alcohol/ketone ratio of 5.6(3).
Introduction
Nature widely employs the iron porphyrin, or heme, prosthetic group to catalyze a remarkably diverse range of challenging oxidative transformations, including C–H bond functionalization.1 These reactions require the heme active site to form highly reactive intermediates,2 which must be enveloped by the local protein environment in order to function.3 Without sequestration by the protein superstructure, molecular heme complexes readily condense into oxo-bridged species.4 In addition to immobilization provided by the protein, its folding pattern precisely regulates the chemical environment of the heme. For example, structural and molecular dynamics analyses of cytochrome P450, catalase, and peroxidase enzymes suggest excess water is expelled from catalytic sites to control hydrogen-bonding interactions, manage proton delivery, and tune heme redox potentials.5 Together, these design elements have inspired synthetic chemists to pursue steric protection,6 second coordination sphere hydrogen-bonding,7 macromolecular encapsulation,8 and immobilization9 strategies in developing structural and functional models of heme enzymes.
species.4 In addition to immobilization provided by the protein, its folding pattern precisely regulates the chemical environment of the heme. For example, structural and molecular dynamics analyses of cytochrome P450, catalase, and peroxidase enzymes suggest excess water is expelled from catalytic sites to control hydrogen-bonding interactions, manage proton delivery, and tune heme redox potentials.5 Together, these design elements have inspired synthetic chemists to pursue steric protection,6 second coordination sphere hydrogen-bonding,7 macromolecular encapsulation,8 and immobilization9 strategies in developing structural and functional models of heme enzymes.
Metal–organic frameworks possess a number of traits well-suited to the study of biomimetic heme chemistry. In particular, porphyrinic frameworks rigidly separate metalloporphyrin units with atomic-level precision,10–15 thus replicating the sequestering role of proteins. Moreover, through judicious selection of pore size, shape, and environment, solution-phase substrates and reagents can readily diffuse into microporous catalysts.16 Crucially, metal–organic frameworks can often be prepared in single-crystalline form, which enables structural characterization of the first coordination sphere at reactive metal centers with atomic resolution. Indeed, researchers have harnessed this feature to study unusual metal–ligand binding modes17 and to structurally elucidate intermediates in catalytic,18 photoinduced,19 and cooperative20 reactions. Along these lines, we have recently shown that four-coordinate metalloporphyrins within the Zr-based framework PCN-224 (ref. 13b) can support reactive dioxygen14a,b,d and carbonyl14c complexes that otherwise elude structural characterization.
In the context of framework-based catalysis, precise synthetic control of pore environment is vital because the pores constitute the outer coordination spheres of catalytic metal active sites. For instance, one challenge here is addressing non-periodic defects, such as the replacement of a multitopic structural linker with monotopic ligands that introduce anomalously reactive sites.21 This linker replacement phenomenon is especially prominent in PCN-224 and related Zr-based frameworks,13,22 where as few as half of the coordination sites at the Zr6 nodes bind structural linkers. Depending on the synthesis conditions, non-structural ligands in Zr frameworks have been variously identified as Brønsted-acidic23 pairs of aqua and hydroxo ligands,13,22 nucleophilic hydroxide counteranions,24 or formate.25 Herein, we report the selective post-synthetic acetylation and subsequent hydroxylation of the Zr6 nodes in free-base and heme-containing PCN-224. Using cyclohexane hydroxylation as a model reaction, we find that these treatments afford pore environments with contrasting influences on the catalytic activity of the heme centers.
Results and discussion
In the absence of post-synthetic treatments, preparations of PCN-224 (ref. 13b,14,15) result in a material best formulated as (H2TCPP)3{Zr6O4(OH)4.7(HCO2)2.3(C6H5CO2)3(H2O)x}2 (H2TCPP4− = tetraanion of 5,10,15,20-tetrakis(4-carboxyphenyl)porphyrin, see Fig. S1 and Table S1†), which we refer to as “as-synthesized PCN-224.” Here, the incorporation of benzoate and formate ligands stems from the use of benzoic acid as the modulator and decomposition of the solvent dimethylformamide, respectively. Inspired by the wealth of post-synthetic modifications that preserve crystallinity of metal–organic frameworks,26 in addition to the incorporation of monocarboxylates into the Zr6 nodes of NU-1000 (ref. 22b and d) and UiO-66,25,27 we initially treated as-synthesized PCN-224 with a variety of carboxylic acids in an attempt to passivate the Zr6 clusters. However, we observed incomplete incorporation of acetate ligands, and both acetic and trifluoroacetic acid led to noticeable dissolution of the framework to give green solutions of H8TCPP2+. We therefore reasoned that PCN-224 is better suited to electrophilic, non-acidic reagents. In support of this hypothesis, treatment of as-synthesized PCN-224 with acetic anhydride afforded the material (H2TCPP)3{Zr6O4(OH)4(CH3CO2)6}2, PCN-224′ (1, see Fig. 1 and ESI† for synthetic details). Importantly, the 1H NMR spectrum of 1 digested in D2SO4/dimethylsulfoxide-d6 shows no detectable traces of formate or benzoate, suggesting that acetic anhydride exchanges formate and benzoate ligands for acetate (see Fig. S1†). Moreover, the mole ratio of H2TCPP4− to acetate is near the ideal stoichiometry of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12, suggesting the terminal aqua and hydroxo ligands are also replaced (see Table S1†). The acetate ligands in 1 carry out the esterification of methanol to form methyl acetate (see Fig. S2†). Thus, treatment of 1 with methanol at 60 °C, followed by soaking in wet acetone, resulted in removal of acetate ligands to afford (H2TCPP)3{Zr6O4(μ-OH)4(OH)6}2 (2, see Fig. 1). The 1H NMR spectrum of digested 2 indicates that ∼90% of the acetate ligands are removed (see Fig. S1†) and that methanol is not readily incorporated (see Table S1†). Finally, heating 1 with FeCl3 and 2,6-lutidine in dimethylformamide, followed by treatment with acetic anhydride,‡ affords 1FeCl (see Fig. 2 and ESI† for experimental details), which in turn can be hydroxylated to afford 2FeCl.
:12, suggesting the terminal aqua and hydroxo ligands are also replaced (see Table S1†). The acetate ligands in 1 carry out the esterification of methanol to form methyl acetate (see Fig. S2†). Thus, treatment of 1 with methanol at 60 °C, followed by soaking in wet acetone, resulted in removal of acetate ligands to afford (H2TCPP)3{Zr6O4(μ-OH)4(OH)6}2 (2, see Fig. 1). The 1H NMR spectrum of digested 2 indicates that ∼90% of the acetate ligands are removed (see Fig. S1†) and that methanol is not readily incorporated (see Table S1†). Finally, heating 1 with FeCl3 and 2,6-lutidine in dimethylformamide, followed by treatment with acetic anhydride,‡ affords 1FeCl (see Fig. 2 and ESI† for experimental details), which in turn can be hydroxylated to afford 2FeCl.
 | ||
| Fig. 1 Treatment of as-synthesized PCN-224, which contains a mixture of formate, benzoate, hydroxo, and aqua ligands, with acetic anhydride affords acetylated six-connected Zr6 nodes (1), as revealed by single-crystal X-ray crystallography. Subsequent treatment of 1 with methanol and water removes the acetate ligands to give Zr6 nodes with terminal hydroxo ligands (2). Green octahedra represent Zr6 clusters; red, blue, gray, and light blue spheres represent O, N, C, and H atoms, respectively; H atoms, except those of terminal hydroxo ligands, are omitted for clarity. The node structure of as-synthesized PCN-224 is reproduced from ref. 14a; the formate and benzoate ligands could not be crystallographically located. | ||
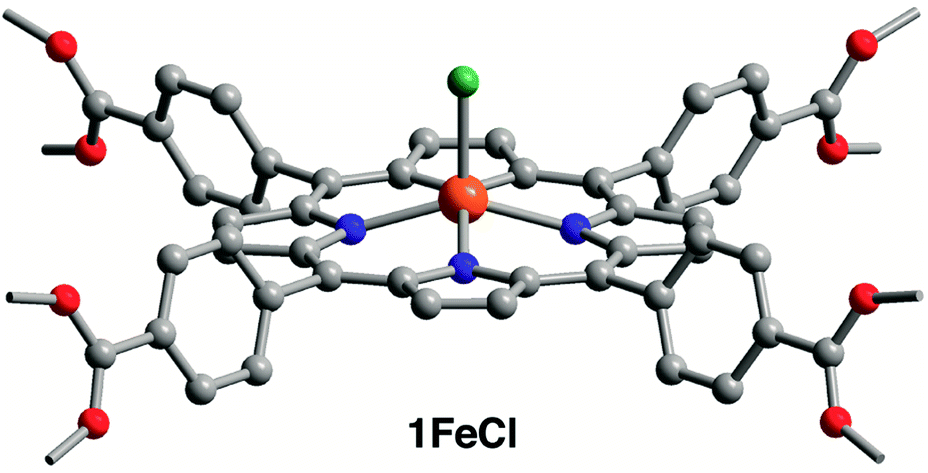 | ||
| Fig. 2 Crystal structure of the metalloporphyrin unit in 1FeCl. Orange, teal, red, blue, and gray spheres represent Fe, Cl, O, N, and C atoms, respectively; H atoms are omitted for clarity. Selected interatomic distances (Å): Fe–Cl 2.23(2), Fe–N 2.084(6), Fe⋯N4 plane 0.575(8). | ||
A suite of physical and spectroscopic methods demonstrates that acetylation and hydroxylation at the Zr6 nodes are chemically orthogonal to the first coordination sphere of the heme. The powder X-ray diffractograms show that bulk crystallinity is retained by ligand substitution at the Zr6 nodes (see Fig. S3†). UV-vis spectroscopy of 1 and 2 (see Fig. S4†) versus1FeCl and 2FeCl (see Fig. S5†), in addition to trace metals analysis, indicate that the metalloporphyrins are not demetalated and the chloride ion remains bound to the heme. The zero-field 57Fe Mössbauer spectra for 1FeCl and 2FeCl display broad, asymmetric quadrupole doublets characteristic of high-spin, chloro-ligated ferric hemes28 and are identical to the spectrum for (TPP)FeCl (H2TPP = 5,10,15,20-tetraphenylporphyrin, see Fig. S6 and Table S2†).
The acetylation and hydroxylation of the Zr6 nodes were confirmed with single-crystal X-ray analysis. Structures of 1 and 1FeCl were both solved in the space group Im![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, with similar unit cell parameters, as previously reported structures of PCN-224 (ref. 13b,14,15) (see Table S3† for crystallographic details and Fig. S7, S8† for ellipsoid plots). At the equatorial face of each Zr6 octahedron, the O atoms refine to two positions of near 50:50 occupancy, reflecting the stoichiometry of bridging oxo versus hydroxo ligands (see Fig. S10†). In addition, each Zr6 node coordinates six acetates (see Fig. 1), each either bridging neighboring Zr4+ ions or chelating a single Zr4+ ion in a κ2 mode (see Fig. S11†). The X-ray structure of 2 reveals each Zr6 node to coordinate only six terminal hydroxo ligands, with no additional aqua ligands (see Fig. 1 and S9†), thus establishing seven-coordinate Zr centers in 2. At 1.910(6) Å, the short Zr–O distance is consistent with Zr–OH bonds (see Table S4†). To our knowledge, this result provides the first structurally characterized example of seven-coordinate Zr within a metal–organic framework, which supports the spectral and density functional theory investigations that suggest rigorous activation of Zr-based frameworks can expose undercoordinated Zr atoms.22f Finally, the X-ray structure of 1FeCl shows the expected square pyramidal Fe and confirms that the chloro ligand is not lost upon acetylation (see Fig. 2).
m, with similar unit cell parameters, as previously reported structures of PCN-224 (ref. 13b,14,15) (see Table S3† for crystallographic details and Fig. S7, S8† for ellipsoid plots). At the equatorial face of each Zr6 octahedron, the O atoms refine to two positions of near 50:50 occupancy, reflecting the stoichiometry of bridging oxo versus hydroxo ligands (see Fig. S10†). In addition, each Zr6 node coordinates six acetates (see Fig. 1), each either bridging neighboring Zr4+ ions or chelating a single Zr4+ ion in a κ2 mode (see Fig. S11†). The X-ray structure of 2 reveals each Zr6 node to coordinate only six terminal hydroxo ligands, with no additional aqua ligands (see Fig. 1 and S9†), thus establishing seven-coordinate Zr centers in 2. At 1.910(6) Å, the short Zr–O distance is consistent with Zr–OH bonds (see Table S4†). To our knowledge, this result provides the first structurally characterized example of seven-coordinate Zr within a metal–organic framework, which supports the spectral and density functional theory investigations that suggest rigorous activation of Zr-based frameworks can expose undercoordinated Zr atoms.22f Finally, the X-ray structure of 1FeCl shows the expected square pyramidal Fe and confirms that the chloro ligand is not lost upon acetylation (see Fig. 2).
Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) supports the assignment of terminal hydroxo ligands in 2 and 2FeCl. The DRIFTS spectra for 1 and 1FeCl show the expected μO–H stretches at 3620–3700 cm−1 (see Fig. 3; see Fig. S12† for full spectrum), while the DRIFTS spectra for 2 and 2FeCl show higher-energy peaks between 3720–3800 cm−1. These energies are in good agreement with the bridging and terminal hydroxides of zirconia.29
 | ||
| Fig. 3 DRIFTS spectra highlighting the ν(O–H), ν(μO–H), and ν(N–H) modes in 1 (black), 2 (navy), 1FeCl (orange), and 2FeCl (red). The absence of porphyrin N–H stretches indicates quantitative iron insertion into 1FeCl and 2FeCl. The features between 3720–3800 cm−1 are assigned to ν(O–H) modes of terminal hydroxo ligands at the Zr6 clusters of 2 and 2FeCl. | ||
Surface area measurements show that porosity is maintained in these frameworks upon acetylation and subsequent hydroxylation. The N2 adsorption isotherms for desolvated 1, 2, 1FeCl, and 2FeCl display uptakes characteristic of microporous adsorbents (see Fig. S12†). Fitting the isotherms to the Brunauer–Emmett–Teller (BET) equation afforded surface areas of 3586(15), 3638(16), 3011(9), and 3204(9) m2 g−1 for 1, 2, 1FeCl and 2FeCl, respectively (see Fig. S13–S16 and Table S5†). These experimental BET surface areas are in excellent agreement with the N2-accessible surface areas calculated30 from their corresponding crystal structures (see Table S6†). Notably, the BET surface areas measured here are higher than the 2400–3000 m2 g−1 of reported PCN-224 frameworks.13b,14–15 The higher gravimetric surface areas may be attributed to the removal of benzoate ligands, thereby decreasing crystal densities of the frameworks, to our solvent exchange and activation procedure (see ESI† for details), or to the improved phase purity afforded by our PCN-224 preparations.
With the Zr6 coordination environments well-established, we subsequently probed the chemical ramifications of acetylation and hydroxylation using catalytic cyclohexane oxidation as a model reaction. While a number of metalloporphyrin-based frameworks have successfully used olefin epoxidation to demonstrate the accessibility of catalytic metal sites,31 we eschewed this strategy largely because molecular hemes are already proficient in epoxidation catalysis.6b,32 Furthermore, mounting evidence implicates both metalloporphyrin-oxidant adducts and metalloporphyrin oxo complexes as active intermediates capable of epoxidizing olefins,33 which could potentially complicate the interpretation of catalytic results. The molecular complex (TPP)FeCl reacts with iodosylbenzene and cyclohexane in a solution of CH2Cl2 to produce a mixture of cyclohexanol, cyclohexanone, and chlorocyclohexane in a combined yield of 9(5)%, consistent with previous reports (see Fig. 4 and Table S7†).6b–d In contrast, oxidation yields for 1FeCl and 2FeCl are 68(8)% and 26(5)%, respectively. Assuming every Fe atom in the frameworks participates in catalysis, these yields correspond to turnover numbers of 14(2) and 5(1) for 1FeCl and 2FeCl, respectively (see Table S7†). The improvement in oxidation yields with 1FeCl and 2FeClversus (TPP)FeCl arises from the ability of PCN-224 to afford isolated, catalytically active heme sites, and agrees well with the greater alkane hydroxylation yields observed for sterically encumbered molecular heme complexes.6c,d,f On the basis of the oxidation yield of 68(8)%, acetate exchange between 1FeCl and iodosylbenzene to form (diacetoxyiodo)benzene34 is likely slow.
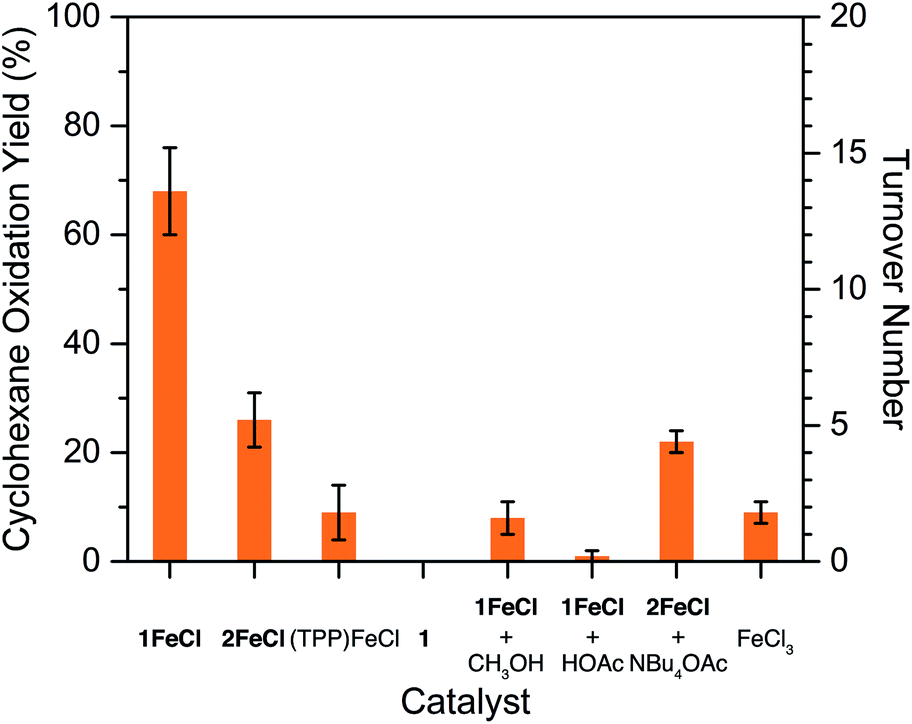 | ||
| Fig. 4 Cyclohexane oxidation yields with iodosylbenzene. With the acetylated framework 1FeCl as the catalyst, yields are 2.6-times larger than with the hydroxylated framework 2FeCl. Error bars reflect one standard deviation (n = 3); OAc− = CH3COO−. | ||
While a number of framework-based manganese porphyrins have been shown to oxidize unactivated C–H bonds in alkanes,10a,31b,35 to our knowledge the present results represent only the third example of such reactions with heme sites.16c,36 Previously, heme frameworks have been shown to oxidize cyclohexane with tert-butylhydroperoxide and cis-decalin with 2-tert-butylsulfonyliodosylbenzene (tBuSO2PhIO). Among all reported Fe and Mn porphyrin frameworks capable of oxidizing cyclohexane, 1FeCl ranks fifth-highest based on overall yield, and second-highest among frameworks that use iodosylbenzene as the terminal oxidant (see Table S8†). Notably, the more active Fe and Mn frameworks all favor the formation of cyclohexanone, with alcohol/ketone (A/K) ratios ≤ 0.81. An A/K ratio of one or less implies a radical chain autoxidation mechanism, in which the role of the metal is to generate free radicals.37 In contrast, 1FeCl favors the formation of cyclohexanol, with an A/K ratio of 5.6(3) that is consistent with metal-based oxidation.37 Of note, the selectivity of 2FeCl for cyclohexanol is even greater (see Table S7†). The lack of over-oxidized cyclohexanone product may be due to the decreased activity of the hydroxylated framework (see below).
Crucially, the cyclohexane oxidation yields of 1FeClversus2FeCl demonstrate that acetylation of the Zr6 nodes improves the catalytic activity by 2.6-fold (see Fig. 4). Several control experiments suggest that acidic protons within the framework pores impair the oxidative reactivity at the heme centers. For instance, addition of methanol or acetic acid to catalytic reactions with 1FeCl results in a 9- to 70-fold decrease in cyclohexane oxidation yields (see Fig. 4). Addition of tetrabutylammonium acetate to reactions with 2FeCl has no significant impact on yields, suggesting that catalysis is enhanced only if reactive proton sources are removed from the Zr6 nodes, and not improved by acetate ions alone. Control experiments with the free-base framework 1 and FeCl3 confirm that the heme unit is responsible for catalysis (see Fig. 4).
A similar reactivity trend arises when tBuSO2PhIO,38 a more soluble iodosylarene reagent, is instead used as the terminal oxidant. Again, oxidation yields are highest with the acetylated framework 1FeCl, a 30-fold increase relative to the hydroxylated framework 2FeCl, and a 70-fold increase relative to (TPP)FeCl (see Table S9†). Notably, the yields with tBuSO2PhIO are significantly lower than with iodosylbenzene. Since tBuSO2PhIO is more soluble in CH2Cl2, it affords higher concentrations of iodosylarene, and thus faster rates of metal-catalyzed disproportionation.6d,38 Also, tBuSO2PhIO may be inactivated by the Zr6 nodes in PCN-224, as the reagent was recently demonstrated to ligate molecular Zr6 clusters.34
Conclusions
The foregoing results demonstrate the facile and quantitative acetylation and hydroxylation of the Zr6 nodes in free-base and heme PCN-224, with the former reaction giving significant enhancement of C–H bond activation chemistry by virtue of removing labile acidic protons within framework pores. These results outline a path toward isolating and interrogating catalytically competent models of fleeting intermediates in heme enzymes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U. S. Army Research Office (W911NF-14-1-0168/P00005) and made use of the IMSERC at Northwestern University (NU), which received support from NSF ECCS-1542205, the State of Illinois, and the International Institute for Nanotechnology. The catalysis studies made use of a gas chromatograph purchased by Prof. SonBinh T. Nguyen. Surface area and DRIFTS measurements were performed at the NU REACT Core (DE-FG02-03ER15457). Metals analysis was performed at the NU Quantitative Bio-element Imaging Center. We thank Mr Youwei Shu for experimental assistance.Notes and references
- (a) M. Sono, M. P. Roach, E. D. Coulter and J. H. Dawson, Chem. Rev., 1996, 96, 2841–2888 CrossRef CAS PubMed; (b) Cytochrome P450: Structure, Mechanism, and Biochemistry, ed. P. R. Ortiz de Montellano, Springer US, Boston, MA, 3rd edn, 2005 Search PubMed; (c) T. L. Poulos, Chem. Rev., 2014, 114, 3919–3962 CrossRef CAS PubMed.
- (a) J. T. Groves, J. Chem. Educ., 1985, 62, 928–931 CrossRef CAS; (b) J. Rittle and M. T. Green, Science, 2010, 330, 933–937 CrossRef CAS PubMed; (c) C. M. Krest, E. L. Onderko, T. H. Yosca, J. C. Calixto, R. F. Karp, J. Livada, J. Rittle and M. T. Green, J. Biol. Chem., 2013, 288, 17074–17081 CrossRef CAS PubMed; (d) X. Huang and J. T. Groves, Chem. Rev., 2018, 118, 2491–2553 CrossRef CAS PubMed; (e) M. Guo, T. Corona, K. Ray and W. Nam, ACS Cent. Sci., 2019, 5, 13–28 CrossRef CAS PubMed.
- (a) J. P. Klinman, Acc. Chem. Res., 2007, 40, 325–333 CrossRef CAS PubMed; (b) R. L. Shook and A. S. Borovik, Inorg. Chem., 2010, 49, 3646–3660 CrossRef CAS PubMed; (c) K. M. Lancaster, in Molecular Electronic Structures of Transition Metal Complexes I, ed. D. M. P. Mingos, P. Day and J. P. Dahl, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, vol. 142, pp. 119–153 Search PubMed; (d) S. P. de Visser, Chem.–Eur. J., 2020, 26, 5308–5327 CrossRef CAS PubMed.
- (a) A. B. Hoffman, D. M. Collins, V. W. Day, E. B. Fleischer, T. S. Srivastava and J. L. Hoard, J. Am. Chem. Soc., 1972, 94, 3620–3626 CrossRef CAS PubMed; (b) D.-H. Chin, G. N. La Mar and A. L. Balch, J. Am. Chem. Soc., 1980, 102, 4344–4350 CrossRef CAS.
- (a) T. I. Oprea, G. Hummer and A. E. García, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2133–2138 CrossRef CAS PubMed; (b) P. Jones, J. Biol. Chem., 2001, 276, 13791–13796 CrossRef CAS PubMed; (c) K. D. Dubey and S. Shaik, Acc. Chem. Res., 2019, 52, 389–399 CrossRef PubMed.
- (a) J. P. Collman, Acc. Chem. Res., 1977, 10, 265–272 CrossRef CAS; (b) J. T. Groves, R. C. Haushalter, M. Nakamura, T. E. Nemo and B. J. Evans, J. Am. Chem. Soc., 1981, 103, 2884–2886 CrossRef CAS; (c) J. T. Groves and T. E. Nemo, J. Am. Chem. Soc., 1983, 105, 6243–6248 CrossRef CAS; (d) M. J. Nappa and C. A. Tolman, Inorg. Chem., 1985, 24, 4711–4719 CrossRef CAS; (e) B. R. Cook, T. J. Reinert and K. S. Suslick, J. Am. Chem. Soc., 1986, 108, 7281–7286 CrossRef CAS; (f) D. Dolphin, T. G. Traylor and L. Y. Xie, Acc. Chem. Res., 1997, 30, 251–259 CrossRef CAS; (g) W. Liu and J. T. Groves, Acc. Chem. Res., 2015, 48, 1727–1735 CrossRef CAS PubMed.
- (a) G. E. Wuenschell, C. Tetreau, D. Lavalette and C. A. Reed, J. Am. Chem. Soc., 1992, 114, 3346–3355 CrossRef CAS; (b) C. K. Chang, Y. Liang, G. Aviles and S.-M. Peng, J. Am. Chem. Soc., 1995, 117, 4191–4192 CrossRef CAS; (c) C.-Y. Yeh, C. J. Chang and D. G. Nocera, J. Am. Chem. Soc., 2001, 123, 1513–1514 CrossRef CAS PubMed; (d) S. A. Cook and A. S. Borovik, Acc. Chem. Res., 2015, 48, 2407–2414 CrossRef CAS PubMed.
- (a) J. T. Groves and R. Neumann, J. Am. Chem. Soc., 1989, 111, 2900–2909 CrossRef CAS; (b) R. Breslow, Acc. Chem. Res., 1995, 28, 146–153 CrossRef CAS; (c) I. C. Reynhout, J. J. L. M. Cornelissen and R. J. M. Nolte, Acc. Chem. Res., 2009, 42, 681–692 CrossRef CAS PubMed.
- (a) J. H. Wang, J. Am. Chem. Soc., 1958, 80, 3168–3169 CrossRef CAS; (b) P. Battioni, E. Cardin, M. Louloudi, B. Schöllhorn, G. A. Spyroulias, D. Mansuy and T. G. Traylor, Chem. Commun., 1996, 2037–2038 RSC; (c) L. L. Welbes and A. S. Borovik, Acc. Chem. Res., 2005, 38, 765–774 CrossRef CAS PubMed.
- Selected reviews of porphyrinic metal–organic frameworks: (a) K. S. Suslick, P. Bhyrappa, J. H. Chou, M. E. Kosal, S. Nakagaki, D. W. Smithenry and S. R. Wilson, Acc. Chem. Res., 2005, 38, 283–291 CrossRef CAS PubMed; (b) M. Zhao, S. Ou and C.-D. Wu, Acc. Chem. Res., 2014, 47, 1199–1207 CrossRef CAS PubMed; (c) W.-Y. Gao, M. Chrzanowski and S. Ma, Chem. Soc. Rev., 2014, 43, 5841–5866 RSC; (d) Z. Guo and B. Chen, Dalton Trans., 2015, 44, 14574–14583 RSC.
- (a) B. F. Abrahams, B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1991, 113, 3606–3607 CrossRef CAS; (b) B. F. Abrahams, B. F. Hoskins, D. M. Michail and R. Robson, Nature, 1994, 369, 727–729 CrossRef CAS.
- A. M. Shultz, O. K. Farha, J. T. Hupp and S. T. Nguyen, J. Am. Chem. Soc., 2009, 131, 4204–4205 CrossRef CAS PubMed.
- (a) D. Feng, Z.-Y. Gu, J.-R. Li, H.-L. Jiang, Z. Wei and H.-C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 10307–10310 CrossRef CAS PubMed; (b) D. Feng, W.-C. Chung, Z. Wei, Z.-Y. Gu, H.-L. Jiang, Y.-P. Chen, D. J. Darensbourg and H.-C. Zhou, J. Am. Chem. Soc., 2013, 135, 17105–17110 CrossRef CAS PubMed; (c) N. Huang, S. Yuan, H. Drake, X. Yang, J. Pang, J. Qin, J. Li, Y. Zhang, Q. Wang, D. Jiang and H.-C. Zhou, J. Am. Chem. Soc., 2017, 139, 18590–18597 CrossRef CAS PubMed.
- (a) J. S. Anderson, A. T. Gallagher, J. A. Mason and T. D. Harris, J. Am. Chem. Soc., 2014, 136, 16489–16492 CrossRef CAS PubMed; (b) A. T. Gallagher, M. L. Kelty, J. G. Park, J. S. Anderson, J. A. Mason, J. P. S. Walsh, S. L. Collins and T. D. Harris, Inorg. Chem. Front., 2016, 3, 536–540 RSC; (c) A. T. Gallagher, C. D. Malliakas and T. D. Harris, Inorg. Chem., 2017, 56, 4654–4661 CrossRef PubMed; (d) A. T. Gallagher, J. Y. Lee, V. Kathiresan, J. S. Anderson, B. M. Hoffman and T. D. Harris, Chem. Sci., 2018, 9, 1596–1603 RSC.
- (a) J. M. Zadrozny, A. T. Gallagher, T. D. Harris and D. E. Freedman, J. Am. Chem. Soc., 2017, 139, 7089–7094 CrossRef CAS PubMed; (b) C.-J. Yu, M. D. Krzyaniak, M. S. Fataftah, M. R. Wasielewski and D. E. Freedman, Chem. Sci., 2019, 10, 1702–1708 RSC.
- (a) F. Song, C. Wang, J. M. Falkowski, L. Ma and W. Lin, J. Am. Chem. Soc., 2010, 132, 15390–15398 CrossRef CAS PubMed; (b) D. J. Xiao, J. Oktawiec, P. J. Milner and J. R. Long, J. Am. Chem. Soc., 2016, 138, 14371–14379 CrossRef CAS PubMed; (c) A. D. Cardenal, H. J. Park, C. J. Chalker, K. G. Ortiz and D. C. Powers, Chem. Commun., 2017, 53, 7377–7380 RSC; (d) C.-H. Wang, A. Das, W.-Y. Gao and D. C. Powers, Angew. Chem., Int. Ed., 2018, 57, 3676–3681 CrossRef CAS PubMed; (e) A. W. Stubbs, L. Braglia, E. Borfecchia, R. J. Meyer, Y. Román- Leshkov, C. Lamberti and M. Dincă, ACS Catal., 2018, 8, 596–601 CrossRef CAS; (f) W.-Y. Gao, A. D. Cardenal, C.-H. Wang and D. C. Powers, Chem.–Eur. J., 2019, 25, 3465–3476 CrossRef CAS PubMed.
- (a) T. Sawano, Z. Lin, D. Boures, B. An, C. Wang and W. Lin, J. Am. Chem. Soc., 2016, 138, 9783–9786 CrossRef CAS PubMed; (b) M. I. Gonzalez, J. A. Mason, E. D. Bloch, S. J. Teat, K. J. Gagnon, G. Y. Morrison, W. L. Queen and J. R. Long, Chem. Sci., 2017, 8, 4387–4398 RSC; (c) Z. Niu, X. Cui, T. Pham, P. C. Lan, H. Xing, K. A. Forrest, L. Wojtas, B. Space and S. Ma, Angew. Chem., Int. Ed., 2019, 58, 10138–10141 CrossRef CAS PubMed.
- (a) K. Ikemoto, Y. Inokuma, K. Rissanen and M. Fujita, J. Am. Chem. Soc., 2014, 136, 6892–6895 CrossRef CAS PubMed; (b) W. M. Bloch, A. Burgun, C. J. Coghlan, R. Lee, M. L. Coote, C. J. Doonan and C. J. Sumby, Nat. Chem., 2014, 6, 906–912 CrossRef CAS PubMed; (c) A. Burgun, C. J. Coghlan, D. M. Huang, W. Chen, S. Horike, S. Kitagawa, J. F. Alvino, G. F. Metha, C. J. Sumby and C. J. Doonan, Angew. Chem., Int. Ed., 2017, 56, 8412–8416 CrossRef CAS PubMed; (d) M. T. Huxley, A. Burgun, H. Ghodrati, C. J. Coghlan, A. Lemieux, N. R. Champness, D. M. Huang, C. J. Doonan and C. J. Sumby, J. Am. Chem. Soc., 2018, 140, 6416–6425 CrossRef CAS PubMed; (e) R. J. Young, M. T. Huxley, E. Pardo, N. R. Champness, C. J. Sumby and C. J. Doonan, Chem. Sci., 2020, 11, 4031–4050 RSC.
- A. J. Blake, N. R. Champness, T. L. Easun, D. R. Allan, H. Nowell, M. W. George, J. Jia and X.-Z. Sun, Nat. Chem., 2010, 2, 688–694 CrossRef CAS PubMed.
- (a) R. L. Siegelman, T. M. McDonald, M. I. Gonzalez, J. D. Martell, P. J. Milner, J. A. Mason, A. H. Berger, A. S. Bhown and J. R. Long, J. Am. Chem. Soc., 2017, 139, 10526–10538 CrossRef CAS PubMed; (b) P. J. Milner, R. L. Siegelman, A. C. Forse, M. I. Gonzalez, T. Runčevski, J. D. Martell, J. A. Reimer and J. R. Long, J. Am. Chem. Soc., 2017, 139, 13541–13553 CrossRef CAS PubMed; (c) C. M. McGuirk, R. L. Siegelman, W. S. Drisdell, T. Runčevski, P. J. Milner, J. Oktawiec, L. F. Wan, G. M. Su, H. Z. H. Jiang, D. A. Reed, M. I. Gonzalez, D. Prendergast and J. R. Long, Nat. Commun., 2018, 9, 5133 CrossRef PubMed.
- (a) M. Taddei, Coord. Chem. Rev., 2017, 343, 1–24 CrossRef CAS; (b) S. Dissegna, K. Epp, W. R. Heinz, G. Kieslich and R. A. Fischer, Adv. Mater., 2018, 30, 1704501 CrossRef PubMed; (c) D. Yang and B. C. Gates, ACS Catal., 2019, 9, 1779–1798 CrossRef CAS.
- (a) J. E. Mondloch, W. Bury, D. Fairen-Jimenez, S. Kwon, E. J. DeMarco, M. H. Weston, A. A. Sarjeant, S. T. Nguyen, P. C. Stair, R. Q. Snurr, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 10294–10297 CrossRef CAS PubMed; (b) P. Deria, J. E. Mondloch, E. Tylianakis, P. Ghosh, W. Bury, R. Q. Snurr, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2013, 135, 16801–16804 CrossRef CAS PubMed; (c) N. Planas, J. E. Mondloch, S. Tussupbayev, J. Borycz, L. Gagliardi, J. T. Hupp, O. K. Farha and C. J. Cramer, J. Phys. Chem. Lett., 2014, 5, 3716–3723 CrossRef CAS PubMed; (d) P. Deria, W. Bury, J. T. Hupp and O. K. Farha, Chem. Commun., 2014, 50, 1965–1968 RSC; (e) S. Yuan, Y.-P. Chen, J. Qin, W. Lu, X. Wang, Q. Zhang, M. Bosch, T.-F. Liu, X. Lian and H.-C. Zhou, Angew. Chem., Int. Ed., 2015, 54, 14696–14700 CrossRef CAS PubMed; (f) D. Yang, V. Bernales, T. Islamoglu, O. K. Farha, J. T. Hupp, C. J. Cramer, L. Gagliardi and B. C. Gates, J. Am. Chem. Soc., 2016, 138, 15189–15196 CrossRef CAS PubMed.
- (a) J. Jiang and O. M. Yaghi, Chem. Rev., 2015, 115, 6966–6997 CrossRef CAS PubMed; (b) R. C. Klet, Y. Liu, T. C. Wang, J. T. Hupp and O. K. Farha, J. Mater. Chem. A, 2016, 4, 1479–1485 RSC; (c) S. Ling and B. Slater, Chem. Sci., 2016, 7, 4706–4712 RSC.
- C. A. Trickett, K. J. Gagnon, S. Lee, F. Gándara, H.-B. Bürgi and O. M. Yaghi, Angew. Chem., Int. Ed., 2015, 54, 11162–11167 CrossRef CAS PubMed.
- D. Yang, M. A. Ortuño, V. Bernales, C. J. Cramer, L. Gagliardi and B. C. Gates, J. Am. Chem. Soc., 2018, 140, 3751–3759 CrossRef CAS PubMed.
- (a) K. K. Tanabe and S. M. Cohen, Chem. Soc. Rev., 2011, 40, 498–519 RSC; (b) S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS PubMed; (c) J. D. Evans, C. J. Sumby and C. J. Doonan, Chem. Soc. Rev., 2014, 43, 5933–5951 RSC; (d) S. M. Cohen, J. Am. Chem. Soc., 2017, 139, 2855–2863 CrossRef CAS PubMed.
- R. Wei, C. A. Gaggioli, G. Li, T. Islamoglu, Z. Zhang, P. Yu, O. K. Farha, C. J. Cramer, L. Gagliardi, D. Yang and B. C. Gates, Chem. Mater., 2019, 31, 1655–1663 CrossRef CAS.
- J. R. Sams and T. B. Tsin, in The Porphyrins, ed. D. Dolphin, Academic Press, New York, NY, 1st edn, 1979, ch. 9, vol. 4, pp. 425–478 Search PubMed.
- (a) F. Ouyang, J. N. Kondo, K.-i. Maruya and K. Domen, J. Phys. Chem. B, 1997, 101, 4867–4869 CrossRef CAS; (b) K. T. Jung and A. T. Bell, J. Catal., 2001, 204, 339–347 CrossRef CAS.
- D. Ongari, P. G. Boyd, S. Barthel, M. Witman, M. Haranczyk and B. Smit, Langmuir, 2017, 33, 14529–14538 CrossRef CAS PubMed.
- Examples of olefin epoxidation by Fe porphyrin metal–organic frameworks: (a) Z. Zhang, L. Zhang, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, J. Am. Chem. Soc., 2012, 134, 928–933 CrossRef CAS PubMed; (b) C. Zou, T. Zhang, M.-H. Xie, L. Yan, G.-Q. Kong, X.-L. Yang, A. Ma and C.-D. Wu, Inorg. Chem., 2013, 52, 3620–3626 CrossRef CAS PubMed.
- J. T. Groves and T. E. Nemo, J. Am. Chem. Soc., 1983, 105, 5786–5791 CrossRef CAS.
- (a) W. Nam, M. H. Lim, H. J. Lee and C. Kim, J. Am. Chem. Soc., 2000, 122, 6641–6647 CrossRef CAS; (b) W. J. Song, Y. J. Sun, S. K. Choi and W. Nam, Chem.–Eur. J., 2006, 12, 130–137 CrossRef CAS PubMed; (c) M. Guo, H. Dong, J. Li, B. Cheng, Y.-q. Huang, Y.-q. Feng and A. Lei, Nat. Commun., 2012, 3, 1190 CrossRef PubMed; (d) M. Guo, Y.-M. Lee, M. S. Seo, Y.-J. Kwon, X.-X. Li, T. Ohta, W.-S. Kim, R. Sarangi, S. Fukuzumi and W. Nam, Inorg. Chem., 2018, 57, 10232–10240 CrossRef CAS PubMed.
- A. D. Cardenal, A. Maity, W.-Y. Gao, R. Ashirov, S.-M. Hyun and D. C. Powers, Inorg. Chem., 2019, 58, 10543–10553 CrossRef CAS PubMed.
- (a) M. H. Alkordi, Y. Liu, R. W. Larsen, J. F. Eubank and M. Eddaoudi, J. Am. Chem. Soc., 2008, 130, 12639–12641 CrossRef CAS PubMed; (b) O. K. Farha, A. M. Shultz, A. A. Sarjeant, S. T. Nguyen and J. T. Hupp, J. Am. Chem. Soc., 2011, 133, 5652–5655 CrossRef CAS PubMed; (c) M.-H. Xie, X.-L. Yang, Y. He, J. Zhang, B. Chen and C.-D. Wu, Chem.–Eur. J., 2013, 19, 14316–14321 CrossRef CAS PubMed; (d) W. Zhang, P. Jiang, Y. Wang, J. Zhang, J. Zheng and P. Zhang, Chem. Eng. J., 2014, 257, 28–35 CrossRef CAS.
- D. Feng, H.-L. Jiang, Y.-P. Chen, Z.-Y. Gu, Z. Wei and H.-C. Zhou, Inorg. Chem., 2013, 52, 12661–12667 CrossRef CAS PubMed.
- (a) M. W. Grinstaff, M. G. Hill, J. A. Labinger and H. B. Gray, Science, 1994, 264, 1311–1313 CrossRef CAS PubMed; (b) J. Kim, R. G. Harrison, C. Kim and L. Que, J. Am. Chem. Soc., 1996, 118, 4373–4379 CrossRef CAS; (c) A. Gunay and K. H. Theopold, Chem. Rev., 2010, 110, 1060–1081 CrossRef CAS PubMed.
- D. Macikenas, E. Skrzypczak-Jankun and J. D. Protasiewicz, J. Am. Chem. Soc., 1999, 121, 7164–7165 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and characterization data for all new compounds; crystallographic data for 1, 1FeCl, and 2; detailed results of catalytic cyclohexane oxidation reactions. CCDC 1992907–1992909. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc01796e |
| ‡ Additional treatment with acetic anhydride after metalation in dimethylformamide is required to ensure the Zr6 nodes remain quantitatively acetylated. We found that soaking 1 in neat dimethylformamide at ambient temperature for 12 h replaces ∼5% of the acetate ligands with formate. At 100 °C, ∼50% of the acetates are replaced. |
| This journal is © The Royal Society of Chemistry 2020 |