
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePrecipitation-free high-affinity multivalent binding by inline lectin ligands†
Philipp
Rohse
,
Sabrina
Weickert
,
Malte
Drescher
 and
Valentin
Wittmann
*
and
Valentin
Wittmann
*
University of Konstanz, Department of Chemistry, Konstanz Research School Chemical Biology (KoRS-CB), Universitätsstraße 10, 78457 Konstanz, Germany. E-mail: mail@valentin-wittmann.de
First published on 27th April 2020
Abstract
Multivalent ligand–protein interactions are a key concept in biology mediating, for example, signalling and adhesion. Multivalent ligands often have tremendously increased binding affinities. However, they also can cause crosslinking of receptor molecules leading to precipitation of ligand–receptor complexes. Plaque formation due to precipitation is a known characteristic of numerous fatal diseases limiting a potential medical application of multivalent ligands with a precipitating binding mode. Here, we present a new design of high-potency multivalent ligands featuring an inline arrangement of ligand epitopes with exceptionally high binding affinities in the low nanomolar range. At the same time, we show with a multi-methodological approach that precipitation of the receptor is prevented. We distinguish distinct binding modes of the ligands, in particular we elucidate a unique chelating binding mode, where four receptor binding sites are simultaneously bridged by one multivalent ligand molecule. The new design concept of inline multivalent ligands, which we established for the well-investigated model lectin wheat germ agglutinin, has great potential for the development of high-potency multivalent inhibitors as future therapeutics.
Introduction
Multivalency can drastically enhance the binding affinity between interacting species and it is a common concept used in nature.1 Accordingly, numerous artificial multivalent ligands have been designed to interfere with natural systems. Many of these ligands have tremendously high binding affinities, but multivalent binding often results in crosslinking of the receptors. This becomes critical when receptor–ligand complexes precipitate. Plaque formation due to precipitation of proteins is known to result in numerous fatal diseases such as amyloidosis. Here, we present a solution to the problem of receptor precipitation in multivalent systems that is based on a conceptionally new design of the multivalent ligand.A prime example for low affinity interactions that are enhanced by multivalent presentation to become physiologically relevant is the recognition of carbohydrates by lectins. Lectins participate in many important processes,1,2 such as cell–cell, cell–pathogen, and cell–toxin interactions, for example during bacterial or viral infection, making lectins a potential target for future therapeutics.3
In the past, many different multivalent lectin ligands have been developed.1,4 Typically, a scaffold is used to which mono- or oligosaccharides are attached via a suitable linker. Fig. 1 schematically shows different architectures that have been applied. Examples are: (A) ligands that have a central small core structure,5 (B) ligands based on cyclic scaffolds such as cyclic peptides,6 calixarenes7 or cyclodextrins,8 (C) dendrimers,9 and (D) glycopolymers.10 Ligands of type (E) are based on DNA or PNA11 which allows to create rigid scaffolds with defined distances between the carbohydrate residues. (F) is a specific example where the lectin subunit of cholera toxin is modified with carbohydrates and acts as an inhibitor of native cholera toxin.12 Example (G) illustrates ligands based on nanomaterials such as nanoparticles,13 quantum dots,14 carbon nanotubes,15 or fullerenes.16 For some multivalent ligands structural information on the binding mode has been obtained by X-ray crystallography.5c,17 In many cases tremendously increased binding affinities compared to the monovalent systems have been achieved.1,4a–c,e,18
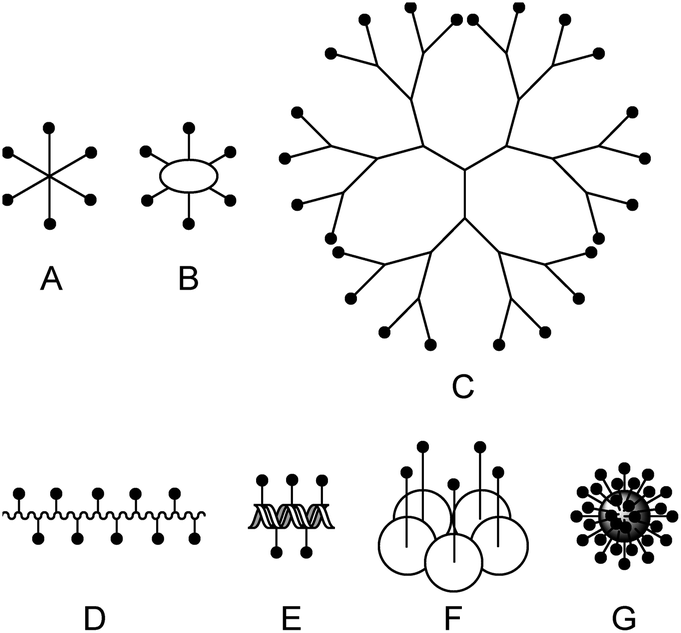 | ||
| Fig. 1 Scaffolds for multivalent lectin ligands: (A) central core, (B) cyclic core, (C) dendrimer, (D) polymer, (E) DNA based, (F) protein scaffold, (G) nanomaterial. | ||
High binding affinities, however, are often associated with crosslinking leading to precipitation of receptor–ligand complexes17b,c,19 thus limiting a potential medical application of a multivalent ligand. Precipitation of proteins is a hallmark of severe diseases such as Alzheimer's, Parkinson's and sickle cell disease to mention a few.20 Hence, there is an urgent need for multivalent ligands, designed to bind with high affinity but without precipitation. Here, we present such multivalent ligands that bind to the model lectin wheat germ agglutinin (WGA) with exceptionally high binding affinities without any concomitant precipitation. By combining several analytical techniques including distance measurements by electron paramagnetic resonance (EPR) spectroscopy, we were able to obtain detailed information on the ligand binding mode.
WGA is a plant lectin isolated from Triticum vulgaris that has been studied extensively in the past.21 The protein forms a stable homodimer at neutral pH exhibiting eight binding sites at the interface of the two monomers each of which is specific to N-acetylglucosamine (GlcNAc) and its oligomers.17d,22 They are classified into primary binding sites with higher affinity and secondary binding sites that could be identified by X-ray crystallography but are reported to bind only weakly in solution.23 Many multivalent ligands for WGA based on different scaffolds have been developed in the past including polymers,24 dendrimers,9b,25 cyclodextrins,26 calixarenes,7a small glycoclusters,6b,27 octasilsesquioxanes,28 and quantum dots.14 The ligands with the highest affinities reported so far are cyclic peptides presented by Fiore et al.6b and our group,19d octasilsesquioxanes by Lo Conte et al.,28 and glycodendrimers by Ghirardello et al.25c In these examples precipitation has been observed6b,19d or is expected to occur.25c,28
Here, we present an approach, that enables high-affinity binding to WGA without precipitation. The ligand design is based on a recently solved crystal structure of WGA in complex with a divalent ligand,17d which showed a chelating binding mode of the ligand bridging pairs of adjacent primary WGA binding sites (Fig. 2A). From this structure it became obvious that the 6-hydroxy groups of the GlcNAc residues point away from the protein, thus offering the opportunity to link two divalent ligands in a linear fashion (Fig. 2B). In earlier studies we already used this position to link a nitroxide spin label to GlcNAc without loss of affinity.27c The carbohydrates are now arranged inline with the inner two GlcNAc residues being integrated into the ligand backbone, an architecture which we term inline lectin ligands (iLecs). The design is especially efficient in terms of molecular size because the carbohydrates are directly attached to each other without the need for a central scaffold. With optimized linkers, the central carbohydrate moieties are held in the binding site by two linkers leaving them less mobility to dissociate once the ligand has bound to the protein. This ligand design leads to high affinity and a defined binding mode without precipitating the protein.
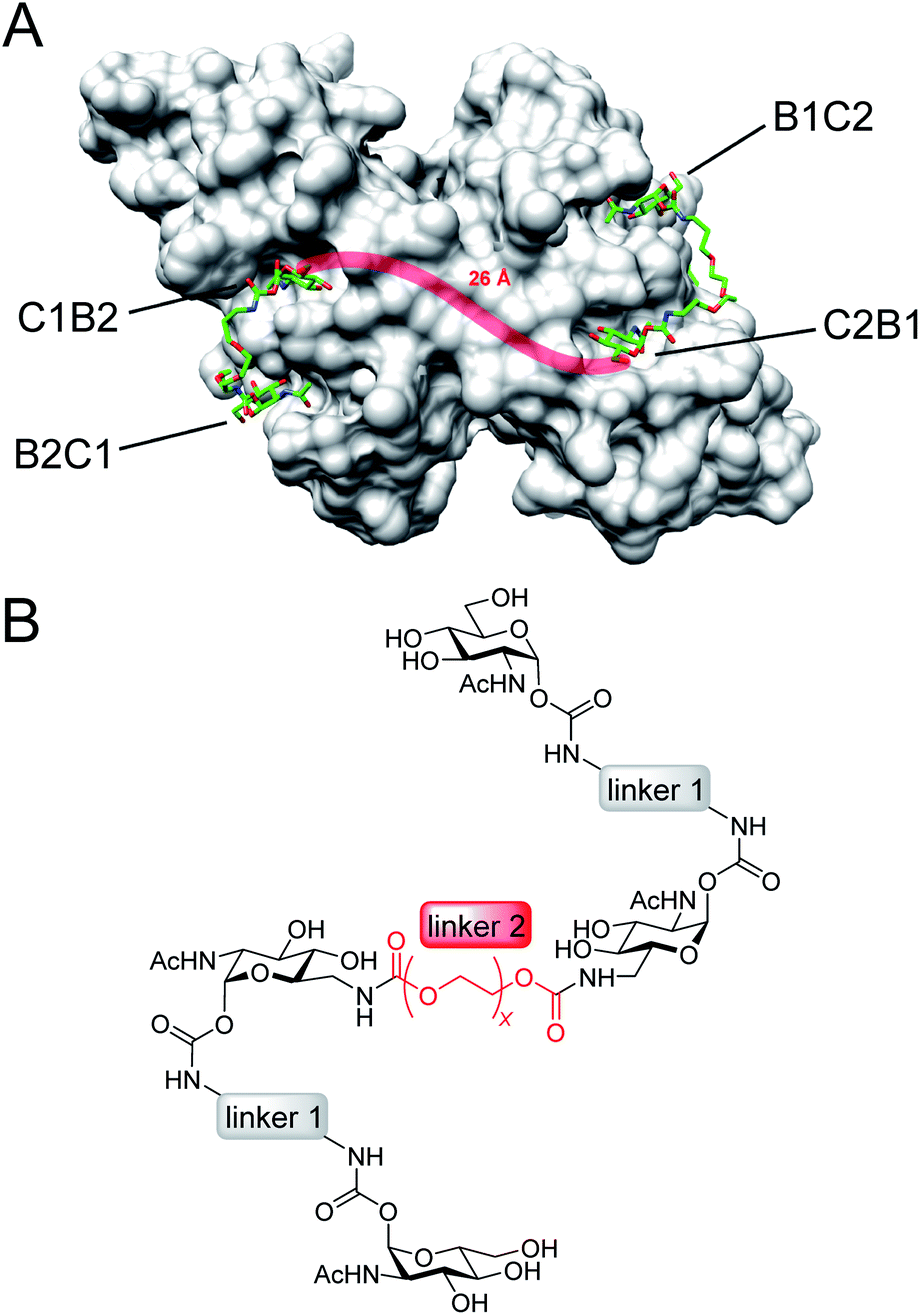 | ||
| Fig. 2 (A) Crystal structure of the WGA dimer (grey, surface representation) in complex with divalent ligands (green) bridging pairs of primary binding sites named B1C2, C2B1, C1B2, and B2C1. A linker connecting two divalent ligands is sketched as red line. (B) Inline lectin ligand design. | ||
Results and discussion
Optimization of linker 1
The design of the iLecs was carried out in two steps. Firstly, we optimized the length of linker 1 by investigating various divalent ligands. Secondly, the connection of two divalent ligands (linker 2) was addressed. To find the best linker 1 to bridge adjacent binding sites we synthesized the series of divalent ligands 1–5 (Scheme 1A) differing in the length of linker 1. Known carbonate 6 (ref. 17d and 29) was coupled to different diamines (Scheme 1B) that were derived from the corresponding diols. The resulting compounds were then O-deacetylated yielding divalent ligands 1–5 in up to 84% yield over two steps. Compounds 2 and 3 have already been described before.17d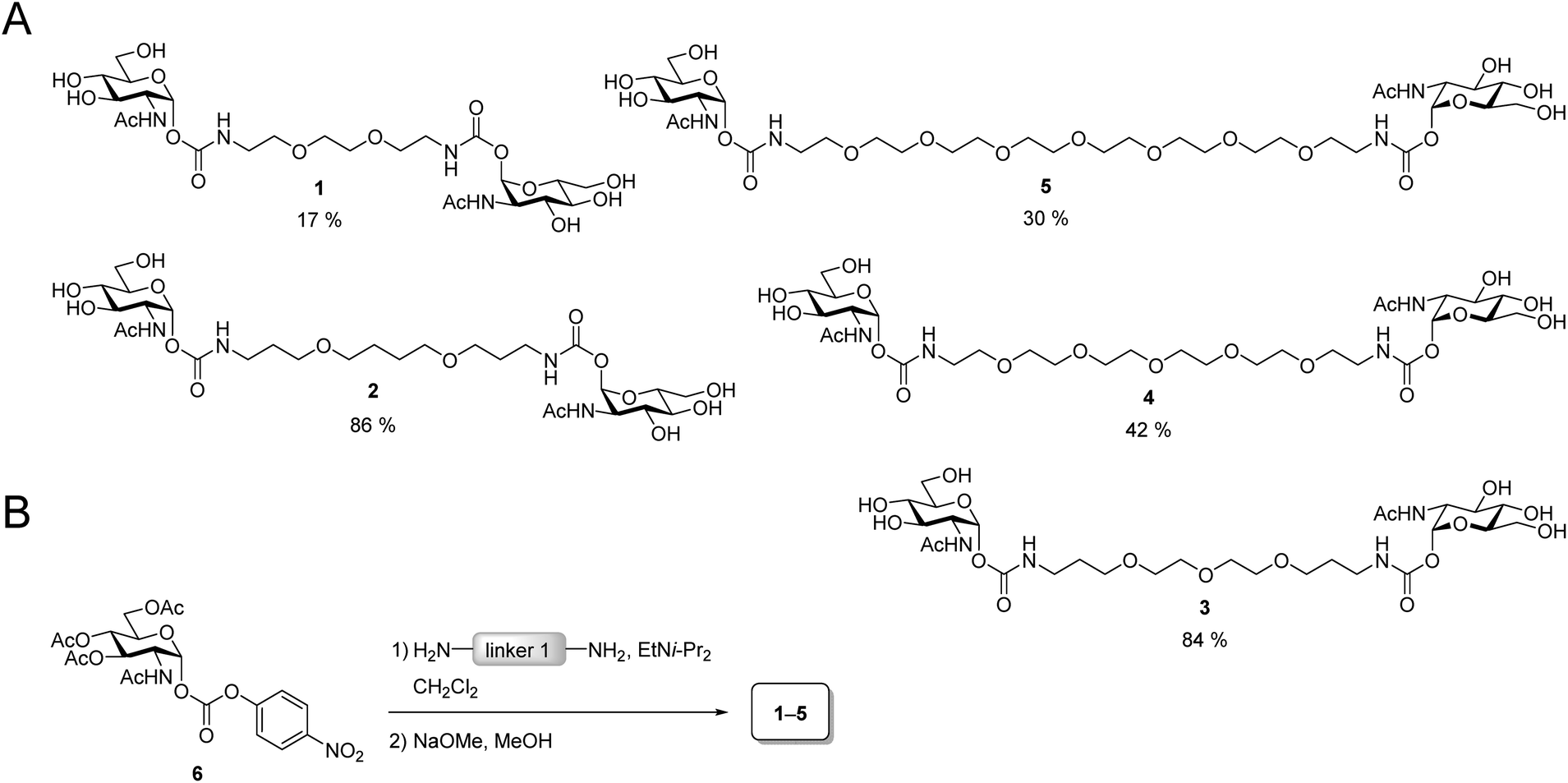 | ||
| Scheme 1 (A) Divalent ligands 1–5. (B) Synthesis of 1–5. | ||
Binding of the divalent ligands to WGA was investigated by isothermal titration calorimetry (ITC). ITC is a widely used method that provides full thermodynamic characterization and the stoichiometry of a binding event. It has been used extensively for studying carbohydrate–lectin interactions30 and has also been used to analyze interactions of WGA with multivalent ligands.6b,19d The results from the ITC measurements are summarized in Table S1 (see ESI†).
The linker length has a strong impact on the binding affinity. Compound 3 has the highest binding affinity with a Kd value of 102 nM corresponding to a relative binding affinity βKd of 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 940 relative to GlcNAc. Both with increasing as well as decreasing length of linker 1, the binding affinity drops to a Kd of 730 nM for ligand 5 with the longest linker 1 and to 1.92 μM for ligand 1 which has the shortest linker 1 in the series. The stoichiometry for all ligands is approximately two ligands binding to one WGA dimer or somewhat lower. This suggests a chelating binding mode with only the primary binding sites being occupied which is in line with the results obtained previously for divalent peptidic ligands.19d During the ITC experiments, no precipitation was observed. However, to test, whether the divalent ligands form crosslinks leading to small aggregates, we performed dynamic light scattering (DLS) experiments.
940 relative to GlcNAc. Both with increasing as well as decreasing length of linker 1, the binding affinity drops to a Kd of 730 nM for ligand 5 with the longest linker 1 and to 1.92 μM for ligand 1 which has the shortest linker 1 in the series. The stoichiometry for all ligands is approximately two ligands binding to one WGA dimer or somewhat lower. This suggests a chelating binding mode with only the primary binding sites being occupied which is in line with the results obtained previously for divalent peptidic ligands.19d During the ITC experiments, no precipitation was observed. However, to test, whether the divalent ligands form crosslinks leading to small aggregates, we performed dynamic light scattering (DLS) experiments.
DLS is a method that allows the determination of hydrodynamic radii of particles or macromolecules in solution and it was previously applied to investigate binding of multivalent ligands to WGA.19d For DLS experiments, WGA was incubated with the ligands in the stoichiometries determined by ITC. The resulting solutions were filtered through a 100 nm cutoff filter to remove dust and insoluble material before the measurements. Indeed, for ligands 1–5 no increase in the hydrodynamic radii compared to the protein alone was observed which confirms the absence of crosslinks between protein receptors in presence of the divalent ligands (see ESI†). In summary, ligand 3 which had also been crystallized with WGA before17d (Fig. 2A), is the optimal candidate for the development of high-affinity tetravalent ligands, since it features the highest binding affinity of all tested divalent ligands.
Synthesis of tetravalent iLecs
After the optimal length of linker 1 had been determined, we optimized the central linker (linker 2). We chose oligo(ethylene glycol) (OEG) as material as it is water-soluble, flexible and can be obtained in defined lengths. Although a rigid, perfectly fitting linker is expected to result in a maximal increase in binding affinity,1,31 flexible linkers allow a perfect adaption of the attached carbohydrates to the binding sites, maximizing the binding enthalpy. In order to connect two divalent ligands generating the anticipated iLecs, linker 2 has to be long enough to bridge the 26 Å distance shown in Fig. 2A. OEG crystallizes in a 2/7 helix with an average monomer length of 2.78 Å.32 The helical structure is also conserved in aqueous solution.33 Taking this into consideration, 9.3 ethylene glycol monomers would be needed to span the distance. Assuming a completely stretched conformation (antiperiplanar conformation of all C–C and C–O bonds) the number of necessary monomers would be 7.2. In polymer science, the average end-to-end distance of a given polymer can be calculated as the Flory radius RF = bN3/5 with Kuhn length b and the number of monomers N.34 Using a Kuhn length b of 7 Å,35 the number of monomers N needed for an end-to-end distance of 26 Å is 8.9. Therefore, we decided to use hexa- to dodeca(ethylene glycol) as linker 2. These linkers include examples, which are too short to bridge the necessary distance even in a completely stretched conformation as well as linkers that are exceeding the necessary number of monomers.The synthesis of the series of iLecs started with the reaction of OEGs 7–13 with p-nitrophenyl chloroformiate 14 to form dicarbonates 15–21 in yields up to 68% (Scheme 2). The carbonates were then coupled to divalent compound 22 (ref. 27c) in which one of the 6-hydroxy groups is replaced with an amino group allowing the formation of carbamates. iLecs 23–29 were obtained in yields up to 80% after RP-HPLC purification.
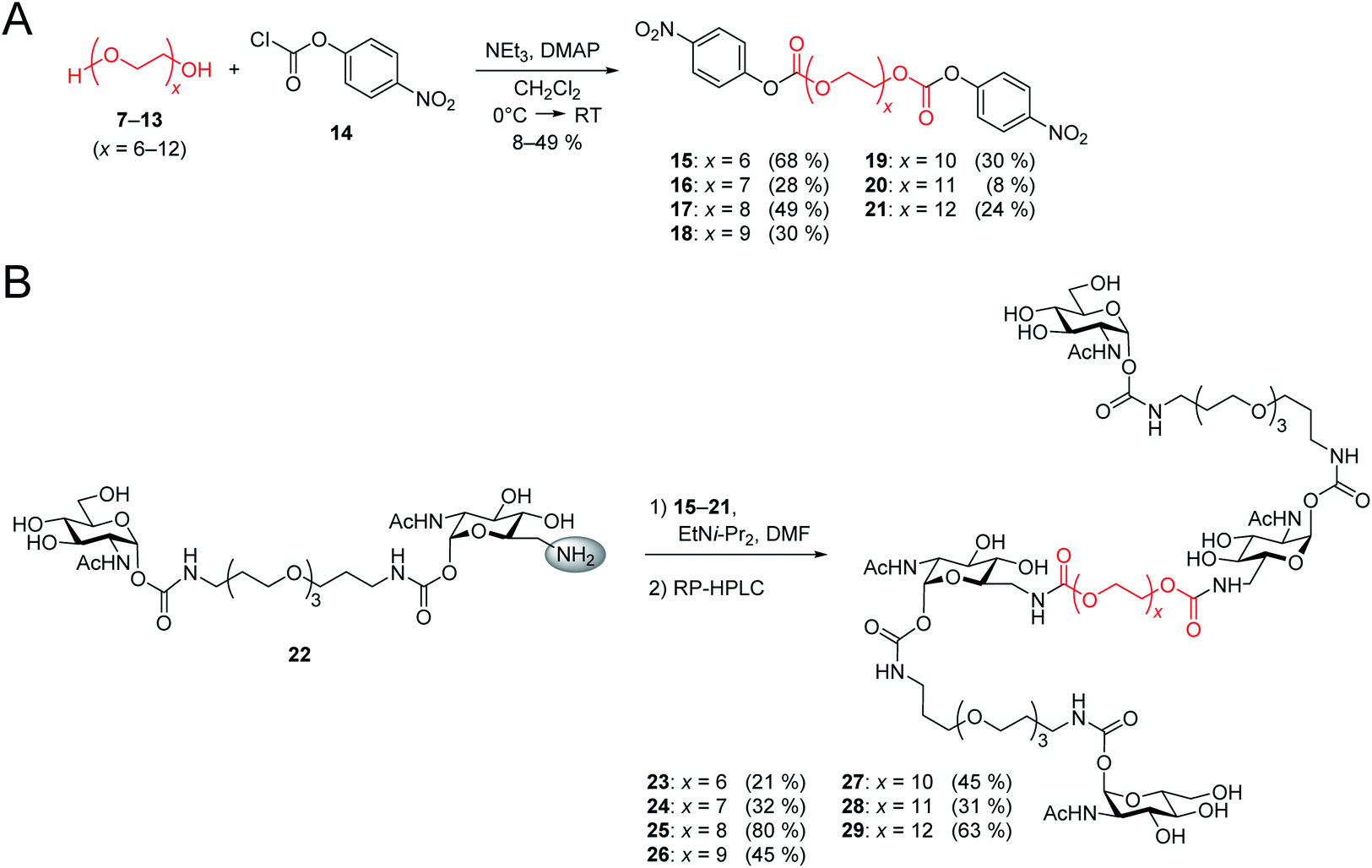 | ||
| Scheme 2 (A) Synthesis of oligo(ethylene glycol) carbonates 15–21. (B) Synthesis of tetravalent iLecs 23–29. | ||
Binding assays
The series of iLecs was investigated by ITC (Table 1). The highest binding affinity was found for ligand 29 with a Kd of 1.6 nM. To the best of our knowledge, this is the lowest Kd value reported for oligovalent WGA ligands so far. Also the other ligands have remarkably high affinities in the low nanomolar range. It is worth mentioning that ligands 23–26 with shorter central linkers (linker 2) have somewhat lower affinities than ligands 27–29 with longer central linkers. The binding enthalpies of the iLecs are between −31.1 kcal mol−1 and −36.8 kcal mol−1, which is about twice as high as the binding enthalpy of ligand 3 confirming that all four GlcNAc residues bind to the protein. The stoichiometry for all ligands is close to one ligand binding per protein dimer.| Compound | K d (nM) |
n
L:P |
ΔH (kcal mol−1) | −TΔS (kcal mol−1) | ΔG (kcal mol−1) | β K d |
|---|---|---|---|---|---|---|
| a Binding stoichiometry, L = ligand, P = protein (dimeric WGA). b Fixed during fit.36 c Relative binding affinity. | ||||||
| GlcNAc | (1.83 ± 0.081) × 106 | 4b | −7.06 ± 0.57 | 3.33 ± 0.61 | −3.73 ± 0.03 | 1 |
| 23 (x = 6) | 3.8 ± 1.8 | 0.98 ± 0.02 | −33.9 ± 1.5 | 22.2 ± 1.9 | −11.6 ± 0.3 | 482000 |
| 24 (x = 7) | 4.5 ± 0.8 | 0.95 ± 0.01 | −33.0 ± 0.1 | 21.5 ± 0.1 | −11.4 ± 0.1 | 407000 |
| 25 (x = 8) | 4.3 ± 0.3 | 1.00 ± 0.02 | −32.0 ± 0.4 | 20.6 ± 0.4 | −11.5 ± 0.1 | 426000 |
| 26 (x = 9) | 5.4 ± 0.8 | 0.96 ± 0.05 | −33.5 ± 2.8 | 22.6 ± 2.3 | −10.9 ± 0.5 | 389000 |
| 27 (x = 10) | 2.2 ± 0.4 | 1.03 ± 0.03 | −31.1 ± 0.8 | 19.3 ± 0.9 | −11.8 ± 0.1 | 832000 |
| 28 (x = 11) | 3.3 ± 0.2 | 1.01 ± 0.02 | −36.8 ± 0.7 | 25.2 ± 0.8 | −11.6 ± 0.1 | 555000 |
| 29 (x = 12) | 1.6 ± 0.5 | 0.90 ± 0.01 | −34.4 ± 0.7 | 22.3 ± 0.9 | −12.1 ± 0.2 | 1144000 |
We also investigated the iLecs using an enzyme-linked lectin assay (ELLA). Here, the ability of a ligand to inhibit the binding of the lectin to a surface coated with a reference ligand is tested. The assay results in IC50 values which are the concentrations at which 50% of the binding is inhibited.27a The resulting dose–response curves and IC50 values are reported in Fig. S1 (see ESI†) and Table 2, respectively. The highest inhibition potency was found for ligand 28 with an IC50 value of 6.6 nM, closely followed by ligands 27 and 29 with IC50 values of 6.9 and 7.4 nM, respectively. Once again, the group of ligands 23–26 with the shorter central linkers have lower affinities ranging from 15 nM for ligand 24 to 56 nM for ligand 23.
| Compound | IC50 (nM) | β IC50 |
|---|---|---|
| a Relative inhibitory potency. | ||
| GlcNAc | 15 × 106 | 1 |
| 23 | 56 | 268000 |
| 24 | 15 | 1000000 |
| 25 | 37 | 405000 |
| 26 | 35 | 429000 |
| 27 | 6.9 | 2174000 |
| 28 | 6.6 | 2273000 |
| 29 | 7.4 | 2027000 |
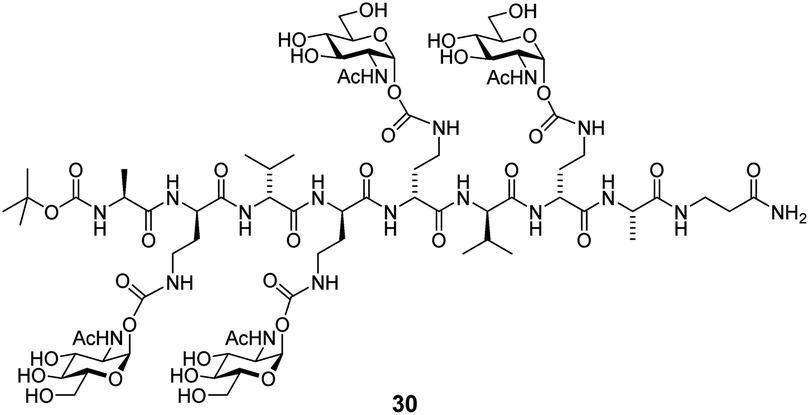
The lowest Kd values accessible by ITC are in the low nanomolar range (10−9–10−8 M).37 This is due to the fact that high affinity requires a low protein concentration in the experiment, which leads to a low signal to noise ratio. To determine Kd values of high-affinity interactions, competitive binding experiments have been reported.38 The protein is pre-incubated with a known ligand of lower affinity and then the high-affinity ligand is titrated into the solution. The affinity of the strong ligand is lowered by the competing ligand, allowing the investigation of ligands with a Kd value below the detection limit. Here, we performed competitive ITC experiments using GlcNAc as a competing ligand (Table 3). We employed iLecs 23 and 29, which have the shortest and the longest central linker. In addition, we investigated tetravalent glycopeptide 30, one of the best WGA ligands (Kd = 7 nM) previously published.19d The obtained Kd values are reported as apparent Kd,app values because there is no appropriate evaluation model available for competitive ITC experiments that accounts for multivalency effects. However, since the experimental conditions for all three ligands were the same, the Kd,app values can be compared qualitatively. Ligand 29 has the highest affinity with Kd,app = 13 nM. This affinity is 9.5-fold higher than that of iLec 23 and even 46-fold higher than that of peptide 30. These numbers illustrate the advantage of the new iLec design over conventional multivalent WGA ligands, especially under competitive binding conditions.
|
|
||
|---|---|---|
| Compound | K d,app [nM] | β K d,app |
| a Relative binding affinity. | ||
| 30 | 602 | 1 |
| 23 | 124 | 4.8 |
| 29 | 13 | 46 |
Investigation of ligand-induced protein aggregation
The aim of the new ligand design was to create a ligand that binds with high affinity without precipitating the protein. To quantify potential protein precipitation, we incubated WGA solutions (17 μM) with buffer containing 0–3 equivalents of a multivalent ligand. The solutions were then centrifuged and the protein concentration of the supernatant was determined photometrically at 280 nm. The absorption A0 of the sample containing no ligand served as reference for 0% precipitation. With the absorption A of the samples containing ligand the precipitation was calculated according to the following formula: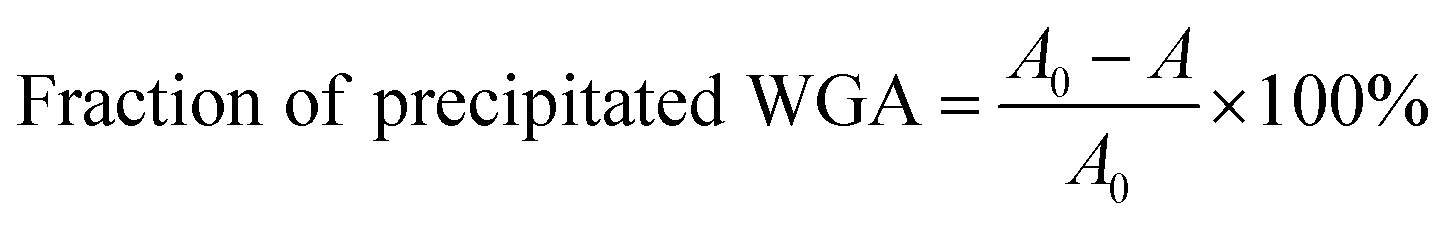
As in the case of the competitive ITC measurements, iLec 23 with the shortest and 29 with the longest central linker (linker 2) were used. Remarkably, both ligands did not result in protein precipitation (Fig. 3A and B). In contrast, addition of tetravalent glycopeptide 30 (ref. 19d) induced up to 20% precipitation of WGA at ligand/WGA ratios of 1.5 and higher.
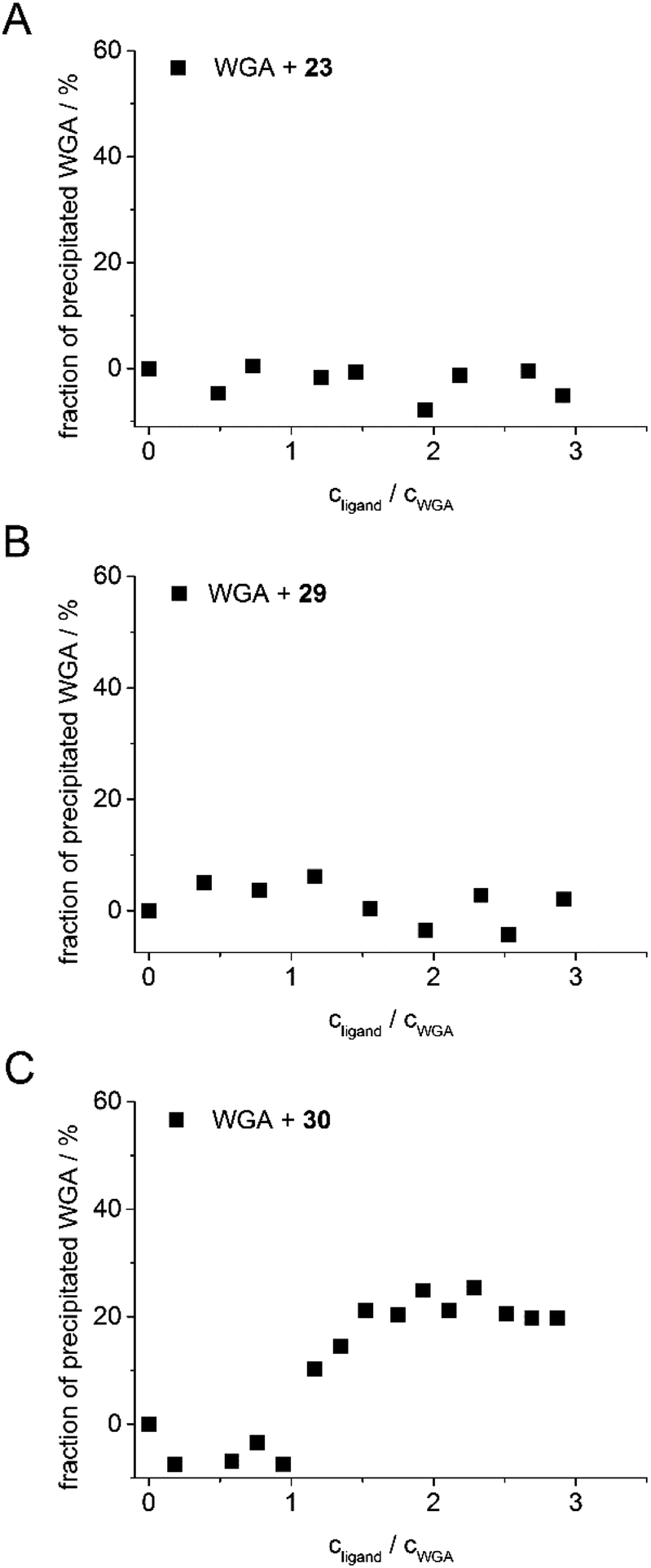 | ||
| Fig. 3 Precipitation profiles of WGA incubated with ligand 23 (A), with ligand 29 (B), and with ligand 30 (ref. 19d) (C). WGA solutions were incubated with varying amounts of the corresponding ligand (0–3 equiv.) for 1 h, centrifuged to remove precipitated material and the remaining protein content of the supernatant was determined photometrically at 280 nm. The fraction of precipitated WGA is plotted against the ratio of ligand to WGA. | ||
Since we did not observe any precipitation upon addition of iLecs 23 or 29 to WGA, we investigated the potential formation of soluble aggregates using DLS. In Fig. 4A the intensity distributions of the hydrodynamic radius of WGA alone and of mixtures of WGA and ligand 23 or ligand 29 are shown.
 | ||
| Fig. 4 Results of DLS experiments. (A) Exemplary intensity distributions of hydrodynamic radii r of mixtures of iLec 23 and WGA dimer at a ratio of 1:1 (red curve) and iLec 29 and WGA dimer at a ratio of 1:1 (green curve) in comparison to pure WGA (blue curve) determined by DLS after filtration through a 100 nm cutoff filter. (B) Mean hydrodynamic radii r (derived from intensity distributions) of species present in solutions of WGA alone (blue) and after addition of iLecs 23–26 (red) and 27–29 (green) determined by DLS after filtration through a 100 nm cutoff filter. (C) Molecular masses M of the species calculated from the hydrodynamic radii shown in (B) by OmniSIZE 3.0 using the built-in protein model. | ||
While addition of ligand 23 resulted in a significant shift of the maximum of the intensity distribution to higher radii, the maximum did not change upon addition of 29 (Fig. 4A). Fig. 4B shows the mean hydrodynamic radii of mixtures of all synthesized iLecs (23–29) with WGA and Fig. 4C the corresponding calculated molecular masses. The ligands separate in two groups. Mixing WGA with ligands 23–26 that have shorter central linkers resulted in complexes with increased radii that correspond to approximately twice the molecular weight (red bars) of WGA (blue bar). In contrast, complexes formed with ligands 27–29 (green bars) do not have a significantly increased radius compared to WGA alone. In order to get further insight into the formed complexes, we performed electron paramagnetic resonance (EPR) spectroscopy measurements of selected spin-labeled iLec derivatives.
Synthesis of spin-labeled iLec derivatives
EPR spectroscopy allows the determination of distance distributions between two spin labels using the double electron–electron resonance (DEER) technique, also called PELDOR (pulsed electron–electron double resonance).39 The method has been proven powerful for the investigation of aggregation-prone proteins relevant in Alzheimer's and Parkinson's diseases40 as well as in the context of multivalency. In recent publications,27c,41 we showed that DEER spectroscopy can be employed in the targeted design of multivalent ligands and that DEER distance measurements with spin-labeled divalent ligands and WGA allow to draw conclusions on the binding mode of the ligand, including the discrimination between chelating and monovalent binding. In order to apply this technique to iLecs, we attached nitroxide spin labels to the 6-positions of the terminal carbohydrates of the ligands. From EPR distance measurements between these spin labels in iLecs bound to WGA we expected to obtain valuable information on the iLec binding mode. Exemplarily, we used ligands 23 and 29 having the shortest and longest central linker 2, respectively. The short linker 2 of ligand 23 does not allow a simultaneous binding of the ligand to four binding sites of one WGA dimer. On the other hand, ligand 29 enables such a binding mode. This should lead to distinct distance distributions for both ligands in complex with WGA.As shown in Scheme 3, the synthesis started from mono Boc-protected diamine 31 (ref. 42) which was coupled to 6-azido GlcNAc carbonate 32 (ref. 27c) in a yield of 80%. O-deacetylation of compound 33 lead to compound 34 which was hydrogenated to give 35. At this point, the differentiation between the two different lengths of linker 2 was made. Amine 35 was coupled to either hexa- or dodeca(ethylene glycol) carbonate 15 or 21 followed by acetylation to give 36 or 37 in 78% yield in both cases. Removal of the Boc-protecting group and coupling of the free amine to carbonate 32 gave 38 and 39 in 83% and 90% yield, respectively. Removal of the acetyl groups led to 40 and 41 in excellent yields. Hydrogenation of the azide and subsequent coupling of the spin label using active ester 42 resulted in the spin-labeled iLecs 43 and 44 in 14% and 39% yield, respectively, after RP-HPLC purification.
 | ||
| Scheme 3 Synthesis of spin-labeled iLecs 43 and 44. | ||
EPR measurements
Spin-labeled iLecs 43 and 44 were employed in EPR distance measurements with a four-pulse DEER experiment at Q-band frequency (∼34 GHz). The samples contained ligands 43 or 44 in 150 μM concentration and were prepared either in the absence or presence of 200 μM WGA dimer in aqueous solutions containing 20% (v/v) glycerol-d8 as a cryoprotectant. The DEER experiments were performed at a temperature of 50 K after shock-freezing the samples in liquid nitrogen to trap the room temperature conformational ensemble. Distance distributions were extracted from background-corrected raw data by a model-free analysis using DEERAnalysis 2016.43The distance distributions obtained with the doubly spin labeled iLecs 43 and 44 in the absence of WGA are shown in Fig. 5 (grey lines). Both ligands 43 and 44 result in rather broad distance distributions with considerable contributions ranging from short distances of below 2 nm and up to 4 nm. The distance distributions are mainly unstructured, which suggests that the iLecs adopt a variety of conformations in solution when not bound to a protein. Upon addition of 200 μM WGA, drastic changes occur in the distance distributions of iLecs 43 (red line) and 44 (green line). As the corresponding dissociation constants are in the low nanomolar range (Table 1), both iLecs 43 and 44 bind quantitatively to WGA under the applied conditions.
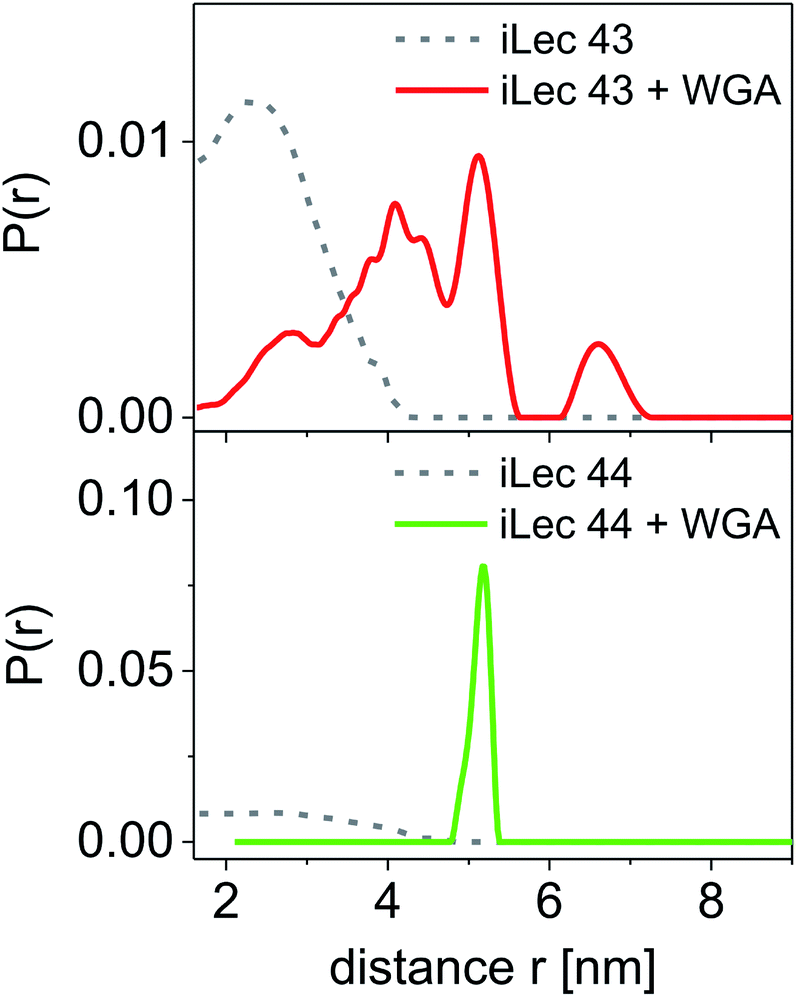 | ||
| Fig. 5 Distance distributions for spin labeled ligand 43 and 44 (ciLec = 150 μM). Distance distributions are normalized to the same area. Results for iLecs 43 and 44 with pure ligand are displayed as gray dotted lines and results obtained with 200 μM WGA are displayed as compact red and green lines for iLec 43 and 44, respectively. All experiments were performed at Q-band frequency (∼34 GHz), T = 50 K, and 20% glycerol-d8. | ||
For iLec 43, that is too short to simultaneously attach to four binding sites on one WGA dimer, the distance distribution (red line) is broad, but structured and shows contributions between 2 and up to approximately 7 nm (Fig. 5). This indicates several different conformations of bound iLec 43 on WGA, which produce a variety of interspin distances. Furthermore, the largest experimentally found distances exceed the maximal possible distance between two binding sites within one WGA dimer.17d This suggests that spin labels located on different WGA dimers contribute to this distance distribution implying that iLec 43 crosslinks WGA dimers.
For iLec 44 the distance distribution significantly simplifies to a single narrow peak when WGA dimer is added (green line). The maximum at a distance r = 5.1 nm suggests that iLec 44 binds to the four primary binding sites of one WGA dimer leading to the observed end-to-end distance of the bound ligand. Comparison with the X-ray structural model confirms that the distance is in agreement with spin labeled GlcNAc moieties bound to binding sites B2C1 and B1C2 (Fig. 2A). As this distance exceeds the distances found in the absence of WGA, the protein obviously forces the ligand into a stretched conformation upon binding.
A modulation depth analysis of the background-corrected DEER raw data (Fig. 6) was used to further clarify the binding mode of iLecs 43 and 44. The modulation depth of a DEER time trace can be exploited for counting the number of interacting spins per nanoobject.44 In order to apply this method, a calibration of the modulation depth is required. Here, we used the doubly spin labeled iLecs 43 and 44 in the absence of WGA to calibrate the modulation depth to a value of 2 spin labels per nanoobject (Fig. 6, gray lines). The modulation depth of iLec 43 in the presence of WGA corresponds to 3.9 spin-labeled GlcNAc moieties per nanoobject (WGA-ligand complex). Since iLec 43 carries two spin labeled GlcNAc moieties, this is consistent with two iLec 43 molecules per WGA-ligand complex. For iLec 44, the modulation depth translates to 2.1 spin-labeled GlcNAc moieties, which corresponds to one molecule of 44 per WGA-ligand complex.
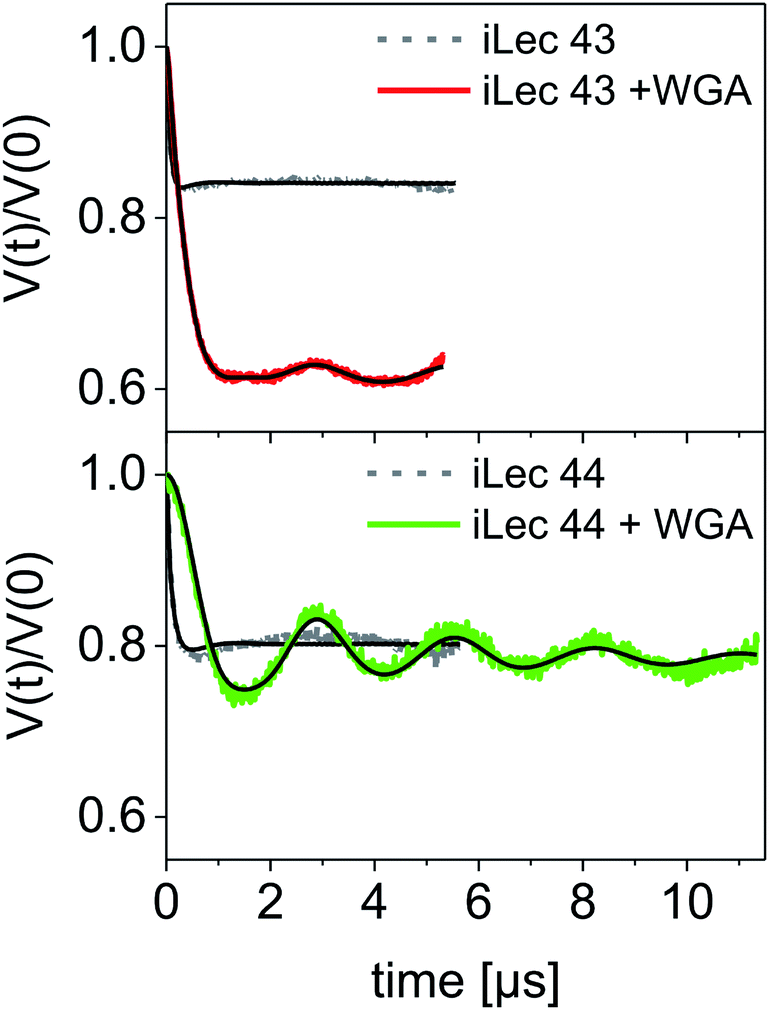 | ||
| Fig. 6 DEER form factors with Tikhonov fits for ligands 43 (top) and 44 (bottom) in the absence (grey lines with black fits) and presence of WGA (red and green lines with black fits, respectively). The modulation depth of the iLec 43 experiment clearly increases upon addition of WGA to the ligand. | ||
Discussion of binding modes
The combined evidence from ITC, precipitation assays, DLS, and EPR experiments allows a precise description of the binding mode of the iLecs. The binding enthalpies derived from ITC measurements were approximately four times the value of the monosaccharide GlcNAc which indicates that all four carbohydrate residues of a ligand bind to protein. The stoichiometry of binding was approximately 1 for all iLecs meaning that complexes of ligand and WGA dimer with ratios of 1:1, 2:2, etc. are present. The existence of big, insoluble complexes could be excluded by precipitation assays. The species that remained in solution were characterized by DLS in terms of their hydrodynamic radii. Complexes of iLecs 23–26 with shorter central linkers and WGA showed increased hydrodynamic radii of about 4 nm with a calculated molecular weight that corresponds to twice the weight of the protein alone. Complexes of iLecs 27–29 and WGA did not show an increased hydrodynamic radius. DEER experiments with spin-labeled iLec 23 (43) confirm the complex formation of 2 ligands with 2 WGA dimers, leading to several distances in the distance distribution and 4 spin labels per nanoobject. Spin-labeled iLec 29 (44) produces a single sharp distance when bound to WGA. These results allow a description of the binding mode (Fig. 7). iLecs 23–26 form complexes of two iLecs crosslinking two WGA dimers in a 2:2 ratio (Fig. 7A). It needs to be mentioned that several orientations of the two ligands bound to the WGA dimers are possible as it is indicated in Fig. 7A explaining the variety of interspin distances observed in the EPR experiments. iLecs 27–29 form complexes where one iLec is binding in a chelating manner, simultaneously bridging four binding sites on one WGA dimer (Fig. 7B). The distance between the terminal GlcNAc moieties is well-defined, allowing the conclusion that there is exactly one conformation the ligand adopts on the receptor surface as visualized in Fig. 7B.
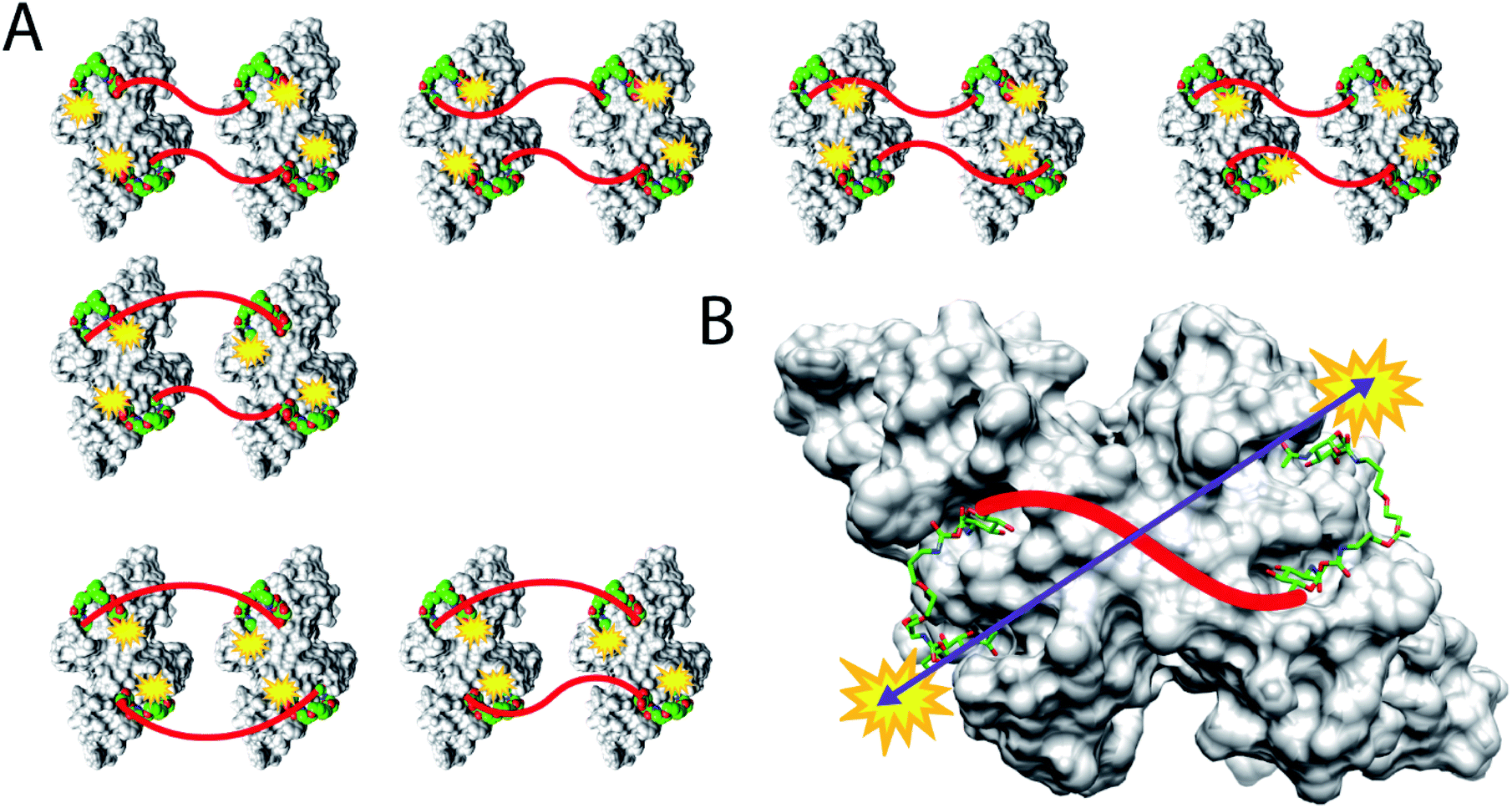 | ||
| Fig. 7 Schematic representation of the binding modes of iLecs 43 and 44. (A) Crosslinking binding of ligand 43 and the class of iLecs 23–26. (B) Chelating binding mode for ligand 44 and the class of iLecs 27–29 resulting in a single distance between the spin labels. For clarity, linker 2 is depicted as red line. | ||
Conclusion
Multivalent binding of oligo- and multivalent lectin ligands is not only a means to achieve strong binding affinities but often also results in crosslinking and precipitation of the receptor. We altered the common design strategy for multivalent lectin ligands of attaching glycans to a central scaffold and instead developed lectin ligands that integrate the carbohydrates into the ligand backbone structure making them function as scaffold and as binding partner for the lectin. The inner carbohydrates are fixed in the ligand structure by two linkers, hampering dissociation, which is reflected by the exceptionally high binding affinities. Using ITC, precipitation assays, DLS, and EPR experiments, we characterized the binding mechanism of the ligands. With optimized linkers, multivalent ligands with exceptionally high binding affinity and a distinct binding mode are achievable that bind in a chelating manner bridging four binding sites of WGA without precipitation of the protein. We showed the importance of using a combination of different analytical techniques for the elucidation of multivalent binding to lectins. Especially EPR distance measurements proved to be a powerful tool for the analysis of binding modes. Our new concept of inline lectin ligands is a promising approach for the future development of high-affinity ligands of medically relevant lectins that bind their targets without precipitation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This article is dedicated to Professor Richard R. Schmidt on the occasion of his 85th birthday. This work was supported by the German Excellence Initiative thorough the Konstanz Research School Chemical Biology (KoRS-CB) and COST Action GLYCONanoProbes (CA18132). P. R. and S. W. acknowledge stipends from the KoRS-CB.Notes and references
- M. Mammen, S.-K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2755–2794 CrossRef CAS
.
- H. Lis and N. Sharon, Chem. Rev., 1998, 98, 637–674 CrossRef CAS PubMed
-
(a) A. Bernardi, J. Jimenez-Barbero, A. Casnati, C. De Castro, T. Darbre, F. Fieschi, J. Finne, H. Funken, K.-E. Jaeger, M. Lahmann, T. K. Lindhorst, M. Marradi, P. Messner, A. Molinaro, P. V. Murphy, C. Nativi, S. Oscarson, S. Penades, F. Peri, R. J. Pieters, O. Renaudet, J.-L. Reymond, B. Richichi, J. Rojo, F. Sansone, C. Schaffer, W. B. Turnbull, T. Velasco-Torrijos, S. Vidal, S. Vincent, T. Wennekes, H. Zuilhof and A. Imberty, Chem. Soc. Rev., 2013, 42, 4709–4727 RSC
-
(a) J. J. Lundquist and E. J. Toone, Chem. Rev., 2002, 102, 555–578 CrossRef CAS PubMed
-
(a) P. Langer, S. J. Ince and S. V. Ley, J. Chem. Soc., Perkin Trans. 1, 1998, 3913–3916 RSC
-
(a) V. Wittmann and S. Seeberger, Angew. Chem., Int. Ed., 2004, 43, 900–903 CrossRef CAS PubMed
-
(a) G. M. L. Consoli, F. Cunsolo, C. Geraci and V. Sgarlata, Org. Lett., 2004, 6, 4163–4166 CrossRef CAS PubMed
-
(a) I. Baussanne, J. M. Benito, C. O. Mellet, J. M. Garcia Fernandez, H. Law and J. Defaye, Chem. Commun., 2000, 1489–1490 RSC
-
(a) R. Roy, D. Zanini, S. J. Meunier and A. Romanowska, J. Chem. Soc., Chem. Commun., 1993, 1869–1872 RSC
-
(a) A. Spaltenstein and G. M. Whitesides, J. Am. Chem. Soc., 1991, 113, 686–687 CrossRef CAS
-
(a) K. Gorska, K.-T. Huang, O. Chaloin and N. Winssinger, Angew. Chem., Int. Ed., 2009, 48, 7695–7700 CrossRef CAS PubMed
- T. R. Branson, T. E. McAllister, J. Garcia-Hartjes, M. A. Fascione, J. F. Ross, S. L. Warriner, T. Wennekes, H. Zuilhof and W. B. Turnbull, Angew. Chem., Int. Ed., 2014, 53, 8323–8327 CrossRef CAS PubMed
-
(a) J. M. de la Fuente, A. G. Barrientos, T. C. Rojas, J. Rojo, J. Cañada, A. Fernández and S. Penadés, Angew. Chem., Int. Ed., 2001, 40, 2257–2261 CrossRef
- A. Robinson, J.-M. Fang, P.-T. Chou, K.-W. Liao, R.-M. Chu and S.-J. Lee, ChemBioChem, 2005, 6, 1899–1905 CrossRef CAS PubMed
- M. E. Ragoussi, S. Casado, R. Ribeiro-Viana, G. de la Torre, J. Rojo and T. Torres, Chem. Sci., 2013, 4, 4035–4041 RSC
- H. Kato, A. Yashiro, A. Mizuno, Y. Nishida, K. Kobayashi and H. Shinohara, Bioorg. Med. Chem. Lett., 2001, 11, 2935–2939 CrossRef CAS PubMed
-
(a) Y. Bourne, B. Bolgiano, D.-I. Liao, G. Strecker, P. Cantau, O. Herzberg, T. Feizi and C. Cambillau, Nat. Struct. Biol., 1994, 1, 863–870 CrossRef CAS PubMed
- H. Zuilhof, Acc. Chem. Res., 2016, 49, 274–285 CrossRef CAS PubMed
-
(a) T. K. Dam, R. Roy, S. K. Das, S. Oscarson and C. F. Brewer, J. Biol. Chem., 2000, 275, 14223–14230 CrossRef CAS PubMed
-
(a) S. S. Wang, J. W. Wu, S. Yamamoto and H. S. Liu, Biotechnol. J., 2008, 3, 165–192 CrossRef CAS PubMed
-
(a)
J. P. Grivet, P. Midoux, P. Gatellier, F. Delmotte and M. Monsigny, in Structure, Dynamics, Interactions and Evolution of Biological Macromolecules, ed. C. Hélène, Springer, Dordrecht, 1983, pp. 329–349 Search PubMed
-
(a) C. S. Wright, J. Mol. Biol., 1980, 141, 267–291 CrossRef CAS PubMed
- C. S. Wright and G. E. Kellogg, Protein Sci., 1996, 5, 1466–1476 CrossRef CAS PubMed
-
(a) S.-I. Nishimura, T. Furuike, K. Matsuoka, K. Maruyama, K. Nagata, K. Kurita, N. Nishi and S. Tokura, Macromolecules, 1994, 27, 4876–4880 CrossRef CAS
-
(a) D. Zanini and R. Roy, Bioconjugate Chem., 1997, 8, 187–192 CrossRef CAS PubMed
- T. Furuike, S. Aiba and S.-I. Nishimura, Tetrahedron, 2000, 56, 9909–9915 CrossRef CAS
-
(a) C. Maierhofer, K. Rohmer and V. Wittmann, Bioorg. Med. Chem., 2007, 15, 7661–7676 CrossRef CAS PubMed
- M. Lo Conte, S. Staderini, A. Chambery, N. Berthet, P. Dumy, O. Renaudet, A. Marra and A. Dondoni, Org. Biomol. Chem., 2012, 10, 3269–3277 RSC
-
T. Seitz, C. Maierhofer, D. Matzner and V. Wittmann, in Carbohydrate Chemistry: Proven Synthetic Methods, ed. R. Roy and S. Vidal, CRC Press, Boca Raton, 2015, vol. 3, pp. 215–218 Search PubMed
-
(a)
T. K. Dam and C. F. Brewer, in Lectins, ed. L. N. Carol, Elsevier Science B.V., Amsterdam, 2007, pp. 75–101 Search PubMed
-
(a) F. Pertici, N. de Mol, J. Kemmink and R. J. Pieters, Chem.–Eur. J., 2013, 19, 16923–16927 CrossRef CAS PubMed
-
(a) H. Tadokoro, Y. Chatani, T. Yoshihara, S. Tahara and S. Murahashi, Makromol. Chem., 1964, 73, 109–127 CrossRef CAS
-
(a) R. Begum and H. Matsuura, J. Chem. Soc., Faraday Trans., 1997, 93, 3839–3848 RSC
-
M. Rubinstein and R. H. Colby, Polymer Physics, Oxford University Press, Oxford, 2003 Search PubMed
- F. Kienberger, V. P. Pastushenko, G. Kada, H. J. Gruber, C. Riener, H. Schindler and P. Hinterdorfer, Single Mol., 2000, 1, 123–128 CrossRef CAS
- W. B. Turnbull and A. H. Daranas, J. Am. Chem. Soc., 2003, 125, 14859–14866 CrossRef CAS PubMed
- E. Freire, O. L. Mayorga and M. Straume, Anal. Chem., 1990, 62, 950A–959A CrossRef CAS
- B. W. Sigurskjold, Anal. Biochem., 2000, 277, 260–266 CrossRef CAS PubMed
-
(a) A. D. Milov, A. B. Ponomarev and Y. D. Tsvetkov, Chem. Phys. Lett., 1984, 110, 67–72 CrossRef CAS
- S. Weickert, J. Cattani and M. Drescher, Electron Paramagn. Reson., 2019, 26, 1–37 CAS
- S. Weickert, T. Seitz, W. K. Myers, C. R. Timmel, M. Drescher and V. Wittmann, J. Phys. Chem. Lett., 2018, 9, 6131–6135 CrossRef CAS PubMed
- T. T. Hai, D. E. Pereira, D. J. Nelson, J. Catarello and A. Srnak, Bioconjugate Chem., 2000, 11, 705–713 CrossRef CAS PubMed
- G. Jeschke, V. Chechik, P. Ionita, A. Godt, H. Zimmermann, J. Banham, C. R. Timmel, D. Hilger and H. Jung, Appl. Magn. Reson., 2006, 30, 473–498 CrossRef CAS
-
(a) B. E. Bode, D. Margraf, J. Plackmeyer, G. Dürner, T. F. Prisner and O. Schiemann, J. Am. Chem. Soc., 2007, 129, 6736–6745 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc01744b |
| This journal is © The Royal Society of Chemistry 2020 |