
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mixing and matching genes of marine and terrestrial origin in the biosynthesis of the mupirocin antibiotics†
Luoyi
Wang
 a,
Zhongshu
Song
a,
Paul R.
Race
b,
James
Spencer
c,
Thomas J.
Simpson
a,
Matthew P.
Crump
*a and
Christine L.
Willis
*a
a,
Zhongshu
Song
a,
Paul R.
Race
b,
James
Spencer
c,
Thomas J.
Simpson
a,
Matthew P.
Crump
*a and
Christine L.
Willis
*a
aSchool of Chemistry, University of Bristol, Cantock's Close, BS8 1TS, Bristol, UK. E-mail: chris.willis@bristol.ac.uk; matt.crump@bristol.ac.uk
bSchool of Biochemistry, University of Bristol, University Walk, BS8 1TD, Bristol, UK
cSchool of Cellular and Molecular Medicine, University of Bristol, BS8 1TD, Bristol, UK
First published on 9th May 2020
Abstract
With growing understanding of the underlying pathways of polyketide biosynthesis, along with the continual expansion of the synthetic biology toolkit, it is becoming possible to rationally engineer and fine-tune the polyketide biosynthetic machinery for production of new compounds with improved properties such as stability and/or bioactivity. However, engineering the pathway to the thiomarinol antibiotics has proved challenging. Here we report that genes from a marine Pseudoalternomonas sp. producing thiomarinol can be expressed in functional form in the biosynthesis of the clinically important antibiotic mupirocin from the soil bacterium Pseudomonas fluorescens. It is revealed that both pathways employ the same unusual mechanism of tetrahydropyran (THP) ring formation and the enzymes are cross compatible. Furthermore, the efficiency of downstream processing of 10,11-epoxy versus 10,11-alkenic metabolites are comparable. Optimisation of the fermentation conditions in an engineered strain in which production of pseudomonic acid A (with the 10,11-epoxide) is replaced by substantial titres of the more stable pseudomonic acid C (with a 10,11-alkene) pave the way for its development as a more stable antibiotic with wider applications than mupirocin.
Introduction
Rapid acceleration in the emergence of antibiotic resistance in pathogenic bacteria is severely eroding our ability to treat bacterial infections.1 Central to an effective response to this problem is the development of novel antibiotics with activity against bacteria resistant to existing agents. A majority of antibiotics in clinical use are natural products or their derivatives, possessing chemotypes and properties often lacking in “drug-like” chemical libraries. Thus, rational engineering of natural product biosynthetic pathways for production of new compounds with improved stability and/or bioactivity is an attractive route to overcoming resistance.2 However, the success of such approaches requires detailed understanding of the biosynthetic process.3The antibiotic mupirocin produced by Pseudomonas fluorescens NCIMB 10586 is among the most effective topical treatments for Gram-positive bacterial infections including methicillin resistant Staphylococcus aureus (MRSA).4 It was identified as one of the first examples of the now extensive family of antibiotics produced by the “trans-AT” class of modular polyketides synthases (PKSs).5 Mupirocin is a mixture of pseudomonic acids of which the major component (>90%, Fig. 1) is pseudomonic acid A (1, PA-A) assembled on a C17-polyketide derived moiety (monic acid) esterified by 9-hydroxynonanoic acid (9-HN).6 Further minor components of mupirocin include ca. 5% PA-B (2) containing an additional 8-hydroxyl group and a minor product, <1%, PA-C (3), with a 10,11-alkene rather than the 10,11-epoxide found in PA-A and PA-B. Studies on mupirocin biosynthesis including analyses of wild-type and mutant strains of P. fluorescens and chemical complementation studies have revealed that PA-B is formed first and contains the tetrahydropyran (THP) ring necessary for activity as shown in Fig. 1A.7 The 8-OH is subsequently lost through a series of transformations which begins with oxidation at C-6 to form mupirocin P (4), requiring the enzymes MupU (CoA ligase), MupO (P450), MacpE (acyl carrier protein, ACP) and MupV (putative didomain oxidoreductase/thioesterase). Dehydration (C7–C8) of 4 catalysed by MupP (dehydratase) followed by two consecutive reductions by the oxidoreductases MupC (C6) and MupF (C7) gives PA-A (1).8 PA-C (3) is the product of a minor parallel pathway that branches from the main pathway (to PA-A).9
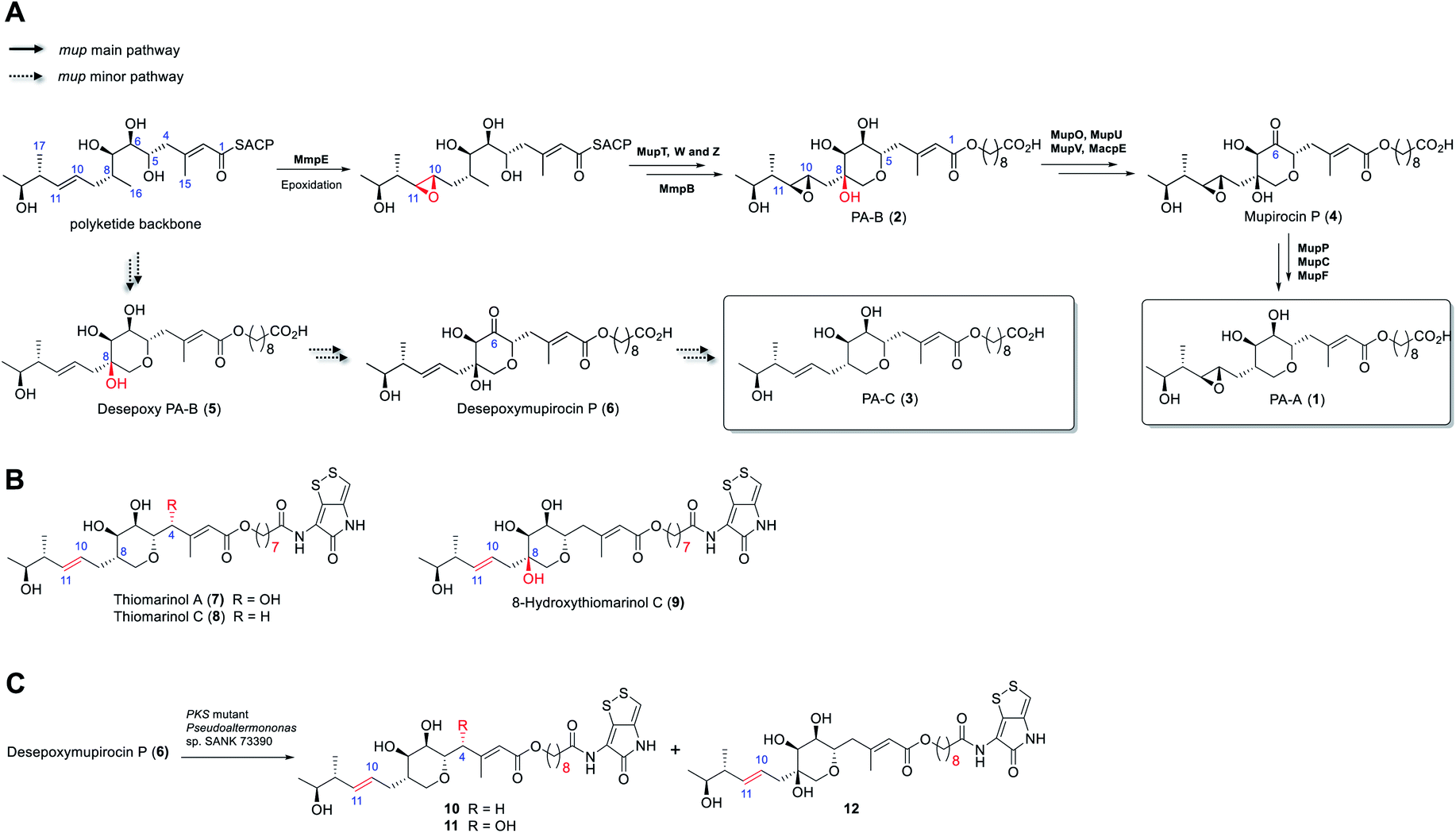 | ||
| Fig. 1 (A) Summary of the later stages of mupirocin biosynthesis showing the parallel pathways. (B) Structures of selected thiomarinols. (C) Transformations of desepoxymupirocin P by the PKS mutant strain of Pseudoalteromonas SANK 73390. | ||
A practical limitation of the major bioactive component, PA-A, is that the 10,11-epoxide moiety renders the molecule unstable outside a narrow pH range, due to formation of inactive bicyclic products arising from attack of the 7-OH onto the epoxide.4 In contrast, the minor metabolite PA-C, containing the 10,11-alkene, is much more stable. It is intriguing that in wild-type P. fluorescens 97% of total PAs and related metabolites contain or are derived from a 10,11-epoxide whereas the parallel minor biosynthetic pathway produces <3% of compounds with the 10,11-alkene (e.g. PA-C). In contrast, the closely related thiomarinols (e.g.7 and 8) all possess a 10,11-alkene (Fig. 1B).
The thiomarinols are a family of marine natural products isolated from Pseudoalternomonas sp. SANK 73390 which also exhibit potent antibiotic activity against MRSA.10 They are assembled on a similar C17-polyketide moiety to the pseudomonic acids but differ in their esterification with an 8-hydroxyoctanoic acid (rather than 9-HN) side chain further linked as an amide to a pyrrothine subunit of the holomycin class of antibiotics. The major metabolite thiomarinol A (7) is also hydroxylated at C-4. Labelling studies are in accord with a PKS/NRPS origin of the thiomarinols.11 The mupirocin and thiomarinol gene clusters show significant homology but differences are evident.12 For example, homologues of the mupirocin standalone ACP MacpE along with MupU and MupV, necessary for conversion of PA-B to PA-A, are absent from SANK 73390. TmpB however contains additional ketosynthase (KS) and ACP domains as compared to MmpB, which are proposed to replace the apparent missing activities.12
Compared with the mupirocin pathway which has been readily modified,9 elucidating and engineering of the thiomarinol biosynthetic pathway in the marine organism has proved remarkably challenging. So far, only the ΔtmlF, ΔtmlU, and the mutant strains of Pseudoaltermonas sp. SANK 73390 in which the PKS and NRPS parts of the gene cluster have been deactivated have been successfully generated,8 all other gene knock-out studies (e.g., ΔtmlW) have failed to produce any isolable metabolites (unpublished results). Characterisation of the family of metabolites, including 8-hydroxythiomarinol C (9) (Fig. 1B) isolated from the ΔtmlF mutant of Pseudoaltermonas sp. SANK 73390, combined with the processing of the THP ring observed on feeding desepoxymupirocin P (6) to the PKS mutant strain (Fig. 1C),8 indicates that it is likely that the late stage biosynthetic machinery generating both thiomarinols and PAs are closely related. However, details of the THP ring formation in thiomarinol biosynthesis remains to be proven experimentally. We now report in vitro functional characterisation of key enzymes from the thiomarinol pathway and results from engineering genes from the thiomarinol pathway into the mupirocin system as well as a comparison of the processing efficiency of 10,11-epoxy versus 10,11-alkenic metabolites at later stages of the mupirocin biosynthetic pathway. Furthermore, by optimisation of fermentation conditions, a significant increase in titres in cultures of mutants of P. fluorescens producing PA-C as the major metabolite has been achieved.
Results
To investigate the apparent differences in metabolite flux involving intermediates containing the 10,11-epoxide versus 10,11-alkenes, we focussed our investigations on two key parts of the mupirocin biosynthetic pathway involving mid and late stage transformations. The latter involves loss of the 8-OH going from PA-B (2) to PA-A (1) whereas the mid-stage steps occur post PKS-mediated backbone assembly and encompass 10,11-epoxidation, THP ring formation and fatty acid chain extension (Fig. 1A).First to late stage processing: the conversion of desepoxy-PA-B (5) to PA-C (3) (i.e. loss of the 8-hydroxyl group in metabolites with the 10,11-alkene) was compared with the major pathway involving PA-B with the analogous 10,11-epoxide. PA-B (2) was isolated from cultures of the ΔmupU mutant of P. fluorescens as previously described.9 However, a new mutant was required to access the corresponding 10,11-alkene, desepoxy-PA-B (5). MmpEOR has been identified as the epoxidase9 and thus the double mutant mmpEΔOR/ΔmupU was generated and 5 was isolated from fermentations of this strain (see ESI, Fig. S1†) in similar titres to that obtained for PA-B from the ΔmupU mutant.
Two mutant strains, ΔmupA and ΔmupH, of P. fluorescens NCIMB 10586 were selected for the late stage mutasynthesis studies as neither produce any metabolites assembled on 6,7-dihydroxy-tetrahydropyrans.9 The same quantity of PA-B (2) and desepoxy-PA-B (5) was fed to cultures of each of the mutants and after 2 days' growth, products were analysed by LC-MS by comparison with standards for starting materials and products (PA-A 1 and PA-C 3). Both substrates 2 (35% conversion) and 5 (75% conversion) were efficiently metabolised with overall loss of the 8-OH to PA-A and PA-C respectively (Fig. 2). Thus the steps involved in the removal of the 8-OH are not rate limiting in PA-C production and so do not account for the significantly different titres of PAs and related metabolites containing a 10,11-epoxide (97% of the total) compared with compounds with a 10,11-alkene including PA-C (<3%) in wild-type P. fluorescens.
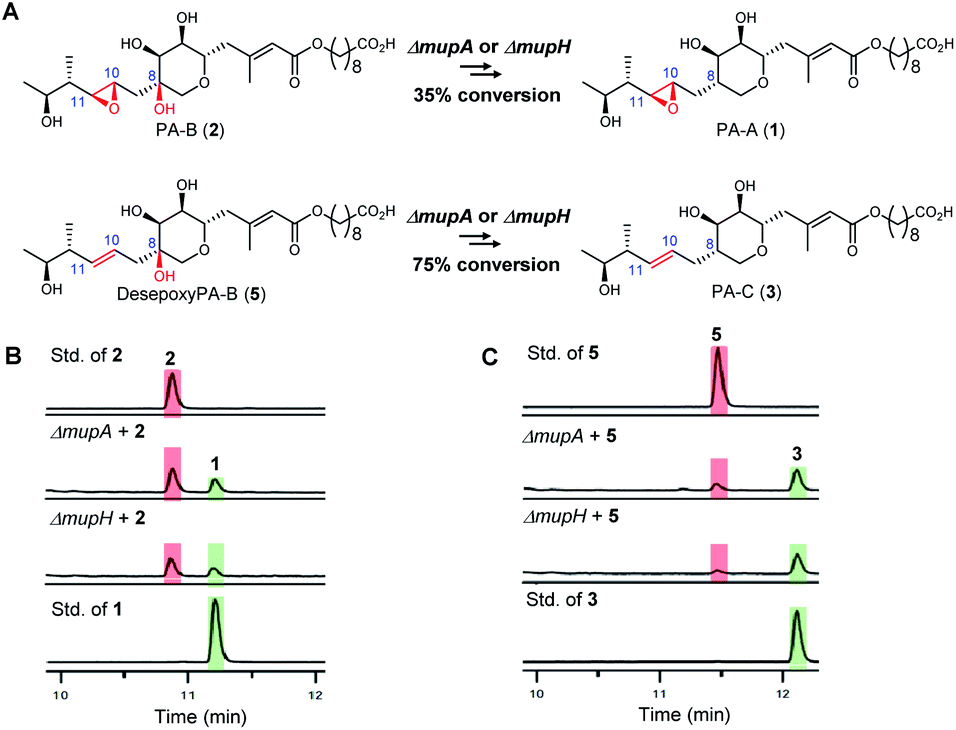 | ||
| Fig. 2 (A) Comparison of the processing of the 8-OH-10,11-epoxy PA (PA-B) with the analogous 10,11-alkene, desepoxy-PA-B. (B) HPLC traces: Conversion of PA-B (2) to PA-A (1) by the ΔmupA and ΔmupH mutants. (C) HPLC traces: Conversion of desepoxy-PA-B (5) to PA-C (3) by the ΔmupA and ΔmupH mutants. | ||
Next, we turned to the mid-stage of mupirocin assembly and the key step involving tetrahydropyran (THP) ring formation. It has been shown previously that deletion of the oxygenase MupW (or its redox partner MupT) abolished production of the pseudomonic acids and led to accumulation of mupirocin W1 (14a) and W2 (14b) albeit in low titres.9,13 These metabolites lack the THP ring but contain a new tetrahydrofuran ring and are likely shunt products formed from the linear compounds 13a and 13b which undergo spontaneous intra-molecular attack by the 7-OH onto the 10,11-epoxide (Fig. 3A).13 Thus studies on THP formation on substrates possessing the 10,11-epoxide cannot be undertaken with linear substrates 13a/b. However, using the corresponding 10,11-alkenes 15a/b isolated from the mmpEΔOR/ΔmupW strain we have recently shown that THP formation occurs via an oxidative enzyme-catalysed cascade by the dual action of the Rieske non-haem oxygenase MupW and the epoxide hydrolase MupZ.14 Selective oxidation of the C8–C16 single bond in the acyclic precursors mupirocin W4 and W5 (15a/b) is proposed to give the corresponding 8,16-epoxide 16 (Fig. 3). In the absence of MupZ, the five-membered tetrahydrofuran ring products mupirocin Z1 and Z2 (17a/b) were isolated via spontaneous cyclisation (Fig. 3B).14
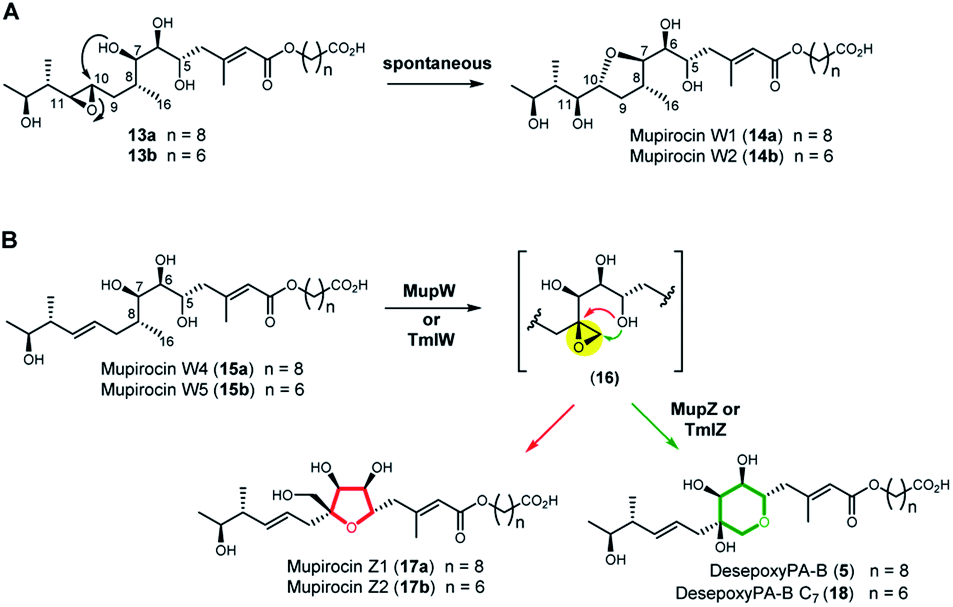 | ||
| Fig. 3 (A) Structures of mupirocin W1 (14a) and W2 (14b) isolated from the ΔmupW mutant of P. fluorescens. (B) THP ring formation in mupirocin biosynthesis catalysed by the dual action of the Rieske non-haem oxygenase MupW and the epoxide hydrolase MupZ.14 The functions of their equivalents TmlW/TmlZ in thiomarinol biosynthesis were confirmed in the present study. | ||
TmlW and TmlZ from the thiomarinol gene cluster, show 55.8% and 56.3% identity, respectively, to MupW and MupZ. To investigate if the same mechanism of tetrahydropyran formation operates in both thiomarinol and mupirocin biosynthesis, TmlW was heterologously overexpressed in E. coli which is able to provide the additional redox partners and cofactors necessary to constitute an active cascade.14 Linear substrate mupirocin W5 (15b) was added to a whole-cell system using resting TmlW overexpressing E. coli cells supplemented with glucose to enable NAD(P)H regeneration (ESI†). LC-MS indicated full conversion to a new product had occurred which on isolation and analysis by NMR spectroscopy was confirmed to be tetrahydrofuran mupirocin Z2 (17b), identical to that previously obtained from the whole live cells of the MupW overexpressing E. coli strain (Fig. S2†). Furthermore, incubation of mupirocin W4 (15a) with E. coli cells overexpressing both MupW and TmlZ afforded the THP ring product desepoxy-PA-B (5) (Fig. S2†). These results not only confirm that thiomarinol biosynthesis employs the same THP ring forming mechanism as that of mupirocin, but also demonstrate that these enzymes are cross compatible.
Unlike the major metabolites from wild-type P. fluorescens which contain the 10,11-epoxide, all thiomarinols isolated from Pseudoalteromonas sp. SANK 73390 have a 10,11-alkene. Having demonstrated MupW/TmlW and MupZ/TmlZ are functionally interchangeable we therefore investigated if replacing mupW with tmlW in the mupirocin gene cluster (Fig. 4A) would lead to more efficient processing of 10,11-alkenes versus 10,11-epoxides and hence alteration of the PA-C (3) to PA-A (1) ratio.
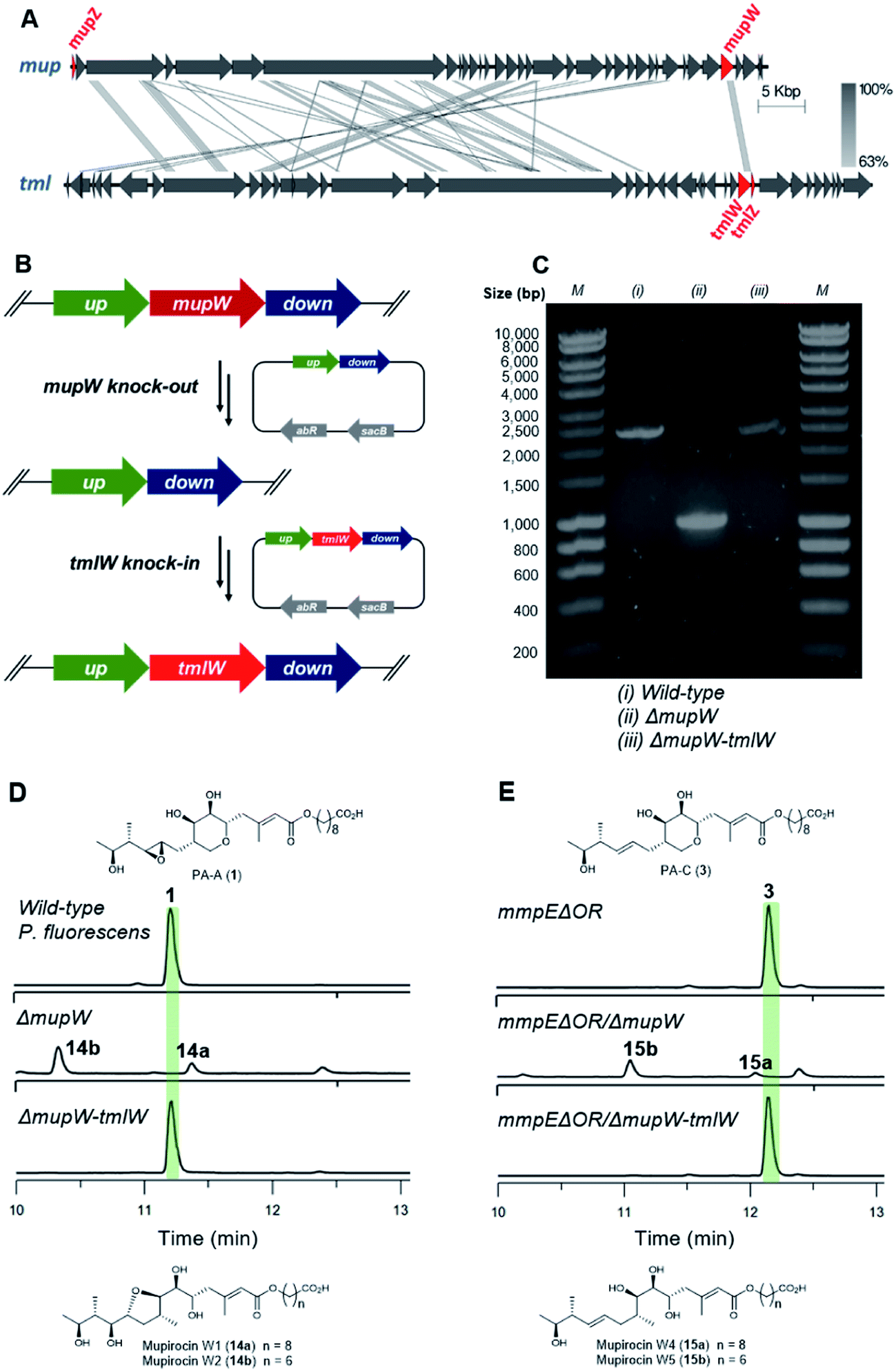 | ||
| Fig. 4 (A) Comparison of the mupirocin (mup) and thiomarinol (tml) biosynthetic gene cluster, key genes involved in THP ring formation are highlighted in red, mupW is replaced by tmlW to form the chimeric pathway in this study. (B) Gene knock-out and knock-in by the two-step allelic exchange method. (C) PCR identification of the knock-out and knock-in mutants. (D) HPLC traces of the wild-type, ΔmupW and ΔmupW-tmlW mutants of P. fluorescens. (E) HPLC traces of the mmpEΔOR, mmpEΔOR/ΔmupW and mmpEΔOR/ΔmupW-tmlW mutants of P. fluorescens. | ||
The entire mupW gene was firstly knocked out from the wild-type strain of P. fluorescens using the two-step allelic exchange method (Fig. 4B).14,15 Desired mutants were screened by PCR and confirmed by Sanger sequencing (Fig. 4C). The resulting ΔmupW mutant produced only the THF-containing shunt products mupirocin W1 and W2 (14a/b) (Fig. 4D). TmlW was then introduced into the ΔmupW strain at the same locus from which mupW was removed, using the same genetic manipulation approach. This new hybrid mutant, P. fluorescens ΔmupW-tmlW, was cultured in parallel with the original wild-type P. fluorescens under the same fermentation conditions. This restored PA-A production and the ratio of PA-C (trace) to PA-A (major) in cultures of the mutant strain was equivalent to that obtained for wild-type P. fluorescens (Fig. 4D).
Next, the same knockout–knockin approach was applied to the mmpEΔOR mutant of P. fluorescens which only produces PA-C. The mmpEΔOR/ΔmupW mutant abolished PA-C production and gave only the acyclic metabolites mupirocin W4 and W5 (15a/b) (Fig. 4E). The new hybrid mutant mmpEΔOR/ΔmupW-tmlW restored PA-C production, and titres were similar as compared to mmpEΔOR (Fig. 4E).
During our investigations we found that the titres of mupirocin metabolites from both wild-type P. fluorescens and mutants were variable and hence we undertook a systematic investigation to improve both the reproducibility and importantly the levels of metabolite production. Fermentation media, incubation time and temperature, shaking speed etc. were optimised for PA-C production. The key observation was that when the mmpEΔOR mutant was grown for 2 days at 22 °C in LB-broth supplemented with 4% glucose, the titres of PA-C (3) reached a reproducible level of ca. 40 mg L−1 in baffled flasks compared to ca. 2 mg L−1 in unbaffled flasks (Fig. 5 and S3†). Minor metabolites isolated under these conditions include a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of desepoxy-PA-B (5) with mupirocin Z1 (17a) (Fig. 5 and S4†), and the newly identified desepoxy-PA-D (19) (Fig. 5, Table S1 and Fig. S5–S10†). The new hybrid mutant mmpEΔOR/ΔmupW-tmlW showed a similar result, again indicating their equivalent effectiveness in initiating THP ring formation. Interestingly, increased aeration during fermentation of cultures of mutants acting early in the pathway led to no detectible differences in titres of accumulated acyclic intermediates such as 15a and 15b.
:1 mixture of desepoxy-PA-B (5) with mupirocin Z1 (17a) (Fig. 5 and S4†), and the newly identified desepoxy-PA-D (19) (Fig. 5, Table S1 and Fig. S5–S10†). The new hybrid mutant mmpEΔOR/ΔmupW-tmlW showed a similar result, again indicating their equivalent effectiveness in initiating THP ring formation. Interestingly, increased aeration during fermentation of cultures of mutants acting early in the pathway led to no detectible differences in titres of accumulated acyclic intermediates such as 15a and 15b.
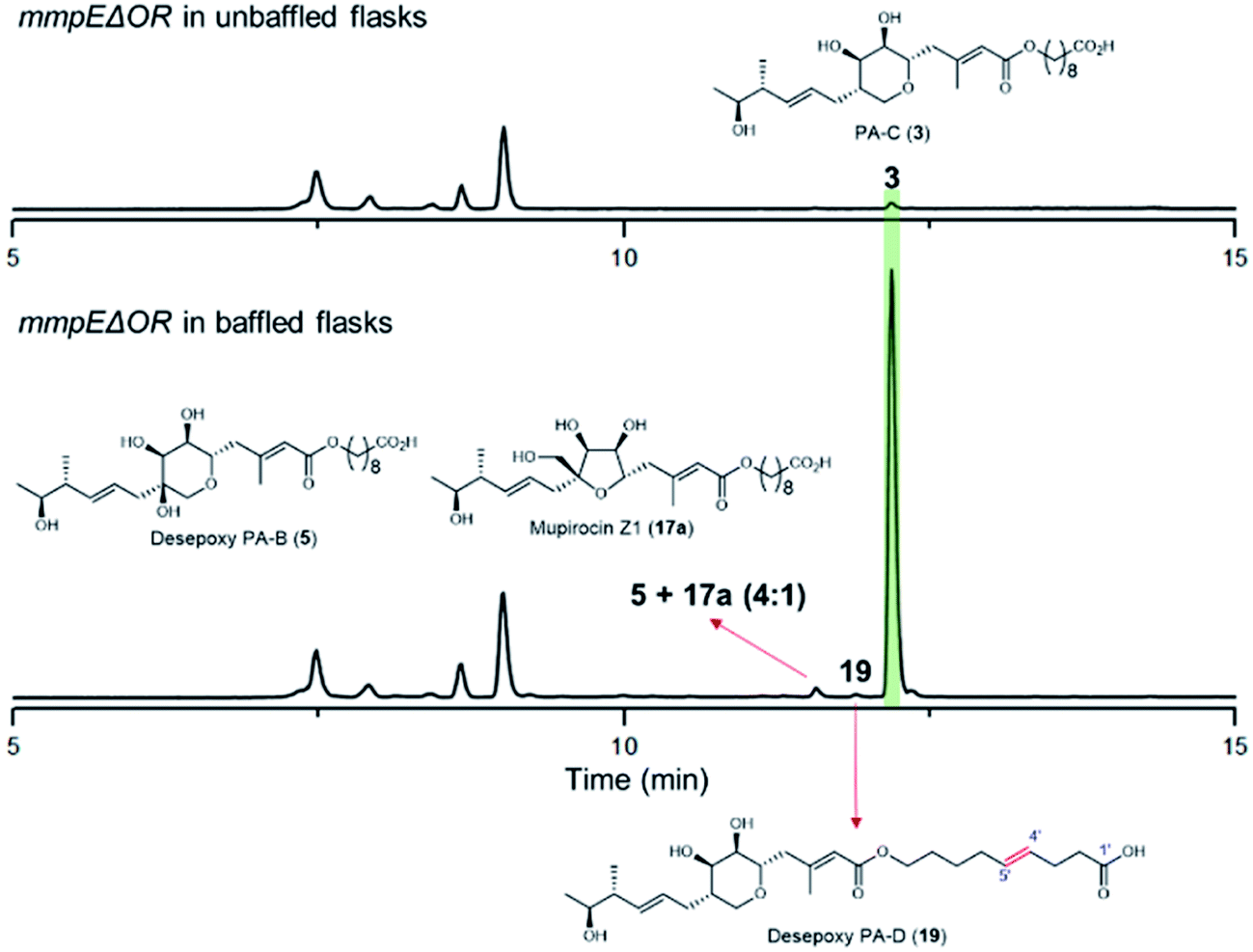 | ||
| Fig. 5 HPLC trace of the mmpEΔOR mutant cultured in baffled flasks compared to in unbaffled flasks. | ||
With significant quantities of PA-C now available, antibiotic activity screening of PA-C, along with PA-A and PA-B, against a range of Gram-positive bacteria (Table 1) was undertaken. The more chemically stable PA-C (3) exhibited comparable antibiotic activity to PA-A, with, notably, similar activity against the four Staphylococcus aureus strains tested, including the Mu50 strain that both exhibits a methicillin-resistant (MRSA) phenotype and reduced susceptibility to glycopeptides including vancomycin. These data confirm the effectiveness of monic acid-based agents against Gram-positive pathogens, including strains challenging to treat with first-line antibiotics, and that antibacterial activity is not adversely affected by substitution of the 10,11-epoxide of mupirocin for the 10,11-alkene. Notably, PA-B displayed substantially lower activity in all susceptibility assays. As PA-B and desepoxy-PA-B are normally intermediates towards the mature antibiotics PA-A and PA-C, respectively, this may be of physiological significance in terms of self-resistance.
| Strains | PA-A (1) | PA-B (2) | PA-C (3) |
|---|---|---|---|
| Bacillus subtilis | 0.06 | 1 | <0.03 |
| S. aureus ATCC 29213 | 0.125 | 4 | 0.25 |
| S. aureus Newman | 0.125 | 2 | 0.125 |
| S. aureus NCTC 6571 | 0.125 | 2 | 0.125 |
| S. aureus Mu50 | 0.125 | 2 | 0.125 |
| S. epidermidis NCTC 11047 | 0.125 | 4 | 0.5 |
Conclusions and discussion
In conclusion we have demonstrated that the late-stage processing in mupirocin biosynthesis of the 10,11-alkene is as efficient as the 10,11-epoxide and that transformations leading to the removal of the 8-hydroxyl groups do not account for the very different levels of PA-A and PA-C production in WT P. fluorescens. Epoxidation catalysed by MmpE is presumably very efficient. The mupirocin pathway was engineered with heterologous genes from the thiomarinol pathway thereby demonstrating that genes from the marine organism Pseudoalternomonas sp. SANK 73390 can be expressed in functional form in the soil bacterium P. fluorescens. Furthermore, it is revealed that both pathways employ the same unusual mechanism of tetrahydropyran (THP) ring formation and that the enzymes responsible for this activity are cross compatible.Whilst the mupirocin and thiomarinol gene clusters show high homology, there are significant differences.12 Genetic engineering of the thiomarinol pathway in the marine organism has, however, proved remarkably difficult (only four mutants have been reported to date8), leading to significant challenges in elucidating the biosynthesis of these marine natural products. We envisage that heterologous expression of components from the thiomarinol pathway in the mupirocin producer will provide an alternative approach elucidating their biosynthesis.
Access to engineered strains of P. fluorescens in which the major product is the more stable PA-C, rather than PA-A in wild type, has potential for industrial scale production of PA-C and subsequent further development as a more stable antibiotic with wider applications than mupirocin.
The plug-and-play approach for the assembly of heterologous genes in the mupirocin biosynthetic gene cluster will be useful for further engineering of polyketide biosynthesis. By building on existing polyketide engineering and characterisation strategies, harnessing current synthetic biology technologies, and utilising advances in metabolic/host engineering, we anticipate future successes in engineering PKSs capable of producing designer polyketides for applications in medicine, fuels, and industrial products.16
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the BBSRC and EPSRC for funding BB/R007853/1 and through the Bristol Centre for Synthetic Biology (BB/L01386X/1) and the EPSRC BristolBridge Award (EP/M027546/1).Notes and references
- M. F. Chellat, L. Raguž and R. Riedl, Angew. Chem., Int. Ed., 2016, 55, 6600–6626 CrossRef CAS PubMed.
- E. Kim, B. S. Moore and Y. J. Yoon, Nat. Chem. Biol., 2015, 11, 649–659 CrossRef CAS PubMed.
- R. Breitling and E. Takano, Cold Spring Harbor Perspect. Biol., 2016, 8, a023994 CrossRef PubMed.
- C. M. Thomas, J. Hothersall, C. L. Willis and T. J. Simpson, Nat. Rev. Microbiol., 2010, 8, 281–289 CrossRef CAS PubMed.
- (a) J. Piel, Nat. Prod. Rep., 2010, 27, 996–1047 RSC; (b) E. J. Helfrich and J. Piel, Nat. Prod. Rep., 2016, 33, 231–316 RSC.
- (a) A. T. Fuller, G. Mellows, M. Woolford, G. T. Banks, K. D. Barrow and E. B. Chain, Nature, 1971, 234, 416–417 CrossRef CAS PubMed; (b) E. B. Chain and G. Mellows, J. Chem. Soc., Perkin Trans. 1, 1977, 294–309 RSC; (c) E. B. Chain and G. Mellows, J. Chem. Soc., Perkin Trans. 1, 1977, 318–322 RSC.
- (a) P. G. Mantle, M. De Langen and V. K. Teo, J. Antibiot., 2001, 54, 166–174 CrossRef CAS PubMed; (b) J. Hothersall, J. e. Wu, A. S. Rahman, J. A. Shields, J. Haddock, N. Johnson, S. M. Cooper, E. R. Stephens, R. J. Cox, J. Crosby, C. L. Willis, T. J. Simpson and C. M. Thomas, J. Biol. Chem., 2007, 282, 15451–15461 CrossRef CAS PubMed.
- S.-S. Gao, L. Wang, Z. Song, J. Hothersall, E. R. Stevens, J. Connolly, P. J. Winn, R. J. Cox, M. P. Crump, P. R. Race, C. M. Thomas, T. J. Simpson and C. L. Willis, Angew. Chem., Int. Ed., 2017, 56, 3930–3934 CrossRef CAS PubMed.
- S.-S. Gao, J. Hothersall, J. Wu, A. C. Murphy, Z. Song, E. R. Stephens, C. M. Thomas, M. P. Crump, R. J. Cox, T. J. Simpson and C. L. Willis, J. Am. Chem. Soc., 2014, 136, 5501–5507 CrossRef CAS PubMed.
- (a) H. Shiozawa, T. Kagasaki, T. Kinoshita, H. Haruyama, H. Domon, Y. Utsui, K. Kodama and S. Takahashi, J. Antibiot., 1993, 46, 1834–1842 CrossRef CAS PubMed; (b) H. Shiozawa, T. Kagasaki, A. Torikata, N. Tanaka, K. Fujimoto, T. Hata, Y. Furukawa and S. Takahashi, J. Antibiot., 1995, 48, 907–909 CrossRef CAS PubMed; (c) H. Shiozawa, A. Shimada and S. Takahashi, J. Antibiot., 1997, 50, 449–452 CrossRef CAS PubMed.
- (a) aA. C. Murphy, D. Fukuda, Z. Song, J. Hothersall, R. J. Cox, C. L. Willis, C. M. Thomas and T. J. Simpson, Angew. Chem., Int. Ed., 2011, 50, 3271–3274 CrossRef CAS PubMed; (b) A. C. Murphy, S.-S. Gao, L.-C. Han, S. Carobene, D. Fukuda, Z. Song, J. Hothersall, R. J. Cox, J. Crosby, M. P. Crump, C. M. Thomas, C. L. Willis and T. J. Simpson, Chem. Sci., 2014, 5, 397–402 RSC.
- (a) A. K. El-Sayed, J. Hothersall, S. M. Cooper, E. Stephens, T. J. Simpson and C. M. Thomas, Chem. Biol., 2003, 10, 419–430 CrossRef CAS PubMed; (b) D. Fukuda, A. S. Haines, Z. Song, A. Murphy, J. Hothersall, E. R. Stephens, R. Gurney, C. Riemer, R. Marshall, R. J. Cox, J. Crosby, C. L. Willis, T. J. Simpson and C. M. Thomas, PLoS One, 2011, 6, e10831 Search PubMed.
- S. M. Cooper, R. J. Cox, J. Crosby, M. P. Crump, J. Hothersall, W. Laosripaiboon, T. J. Simpson and C. M. Thomas, Chem. Commun., 2005, 1179–1181 RSC.
- L. Wang, A. Parnell, C. Williams, N. A. Bakar, M. R. Challand, M. W. van der Kamp, T. J. Simpson, P. R. Race, M. P. Crump and C. L. Willis, Nat. Catal., 2018, 1, 968–976 CrossRef CAS.
- L. R. Hmelo, B. R. Borlee, H. Almblad, M. E. Love, T. E. Randall, B. S. Tseng, C. Lin, Y. Irie, K. M. Storek, J. J. Yang, R. J. Siehnel, P. L. Howell, P. K. Singh, T. Tolker-Nielsen, M. R. Parsek, H. P. Schweizer and J. J. Harrison, Nat. Protoc., 2015, 10, 1820–1841 CrossRef CAS PubMed.
- J. F. Barajas, J. M. Blake-Hedges, C. B. Bailey, S. Curran and J. D. Keasling, Synth. Syst. Biotechnol., 2017, 2, 147–166 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc06192d |
| This journal is © The Royal Society of Chemistry 2020 |