
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA solvent-dependent chirality-switchable thia-Michael addition to α,β-unsaturated carboxylic acids using a chiral multifunctional thiourea catalyst†
Noboru
Hayama
ab,
Yusuke
Kobayashi
 a,
Eriko
Sekimoto
b,
Anna
Miyazaki
b,
Kiyofumi
Inamoto
b,
Tetsutaro
Kimachi
b and
Yoshiji
Takemoto
*a
a,
Eriko
Sekimoto
b,
Anna
Miyazaki
b,
Kiyofumi
Inamoto
b,
Tetsutaro
Kimachi
b and
Yoshiji
Takemoto
*a
aGraduate School of Pharmaceutical Sciences, Kyoto University, Yoshida, Sakyo-ku, Kyoto 606-8501, Japan. E-mail: takemoto@pharm.kyoto-u.ac.jp
bSchool of Pharmacy and Pharmaceutical Sciences, Mukogawa Women's University, 11-68, 9-Bancho, Koshien, Nishinomiya, Hyogo 663-8179, Japan
First published on 14th May 2020
Abstract
An asymmetric thia-Michael addition of arylthiols to α,β-unsaturated carboxylic acids using a thiourea catalyst that bears arylboronic acid and tertiary amine moieties is reported. Both enantiomers of the Michael adducts can be obtained in high enantioselectivity and good yield merely by changing the solvent. The origin of the chirality switch in the products was examined in each solvent via spectroscopic analyses.
Introduction
Due to the importance of organosulfur compounds in medicinal chemistry and biochemistry, their asymmetric synthesis has been studied extensively.1 The catalytic asymmetric thia-Michael addition (TMA) to α,β-unsaturated carbonyl compounds (Fig. 1A) is of particular importance in this context on account of their ability to furnish versatile synthetic intermediates for a variety of biologically active compounds such as benzothiazepine derivatives.2,3 Various activated Michael acceptors have been successfully used for TMA,4e.g. α,β-unsaturated oxazolidinones,5 imides,6 nitro alkenyl isoxazoles,7 thioamides,8 acylpyrazoles,9 carboxylic acid anhydrides,10 and enone diesters.11 However, due to their low inherent electrophilicity, catalytic asymmetric TMA to unactivated Michael acceptors such as α,β-unsaturated esters,12 amides, and carboxylic acids13 remains a challenge. In general, thia-Michael adducts need derivatization to produce biologically active compounds, e.g. a conversion of the carbonyl moiety to carboxylic acid via hydrolysis or oxidation.2 Considering atom and step economy, direct catalytic TMA to α,β-unsaturated carboxylic acids would thus be highly desirable.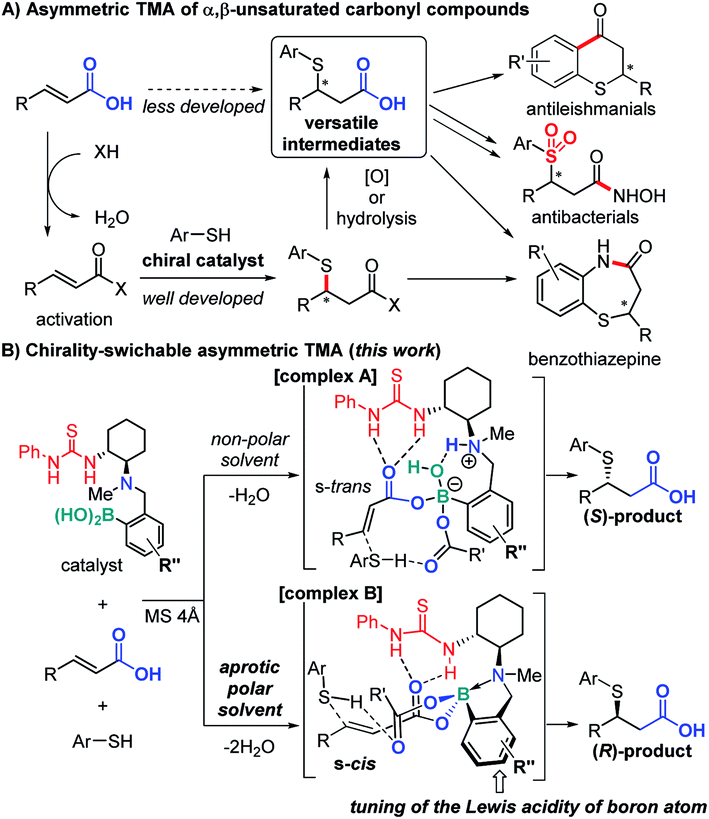 | ||
| Fig. 1 Summary and working hypothesis of this work. | ||
Herein, we report a direct asymmetric TMA to α,β-unsaturated carboxylic acids using a multifunctional organocatalyst, which comprises (1) thiourea as a hydrogen bond (HB) donor, (2) a chiral tertiary amine derived from (R,R)-cyclohexane diamine, and (3) aryl boronic acid moieties (Fig. 1B). Based on our previous work,14 in a non-polar solvent and in the presence of two equivalents of carboxylic acid and molecular sieves (MS), we expect the catalyst to form ternary complex A, which promotes the addition of a nucleophile to the ‘unusual’ s-trans-form of the α,β-unsaturated carboxylate to generate the corresponding (S)-adduct.
Furthermore, we propose that a conformational change of the catalyst15via further dehydration may be possible in an aprotic polar solvent, producing the ‘usual’ (R)-adduct16via addition to the s-cis-form of the α,β-unsaturated carboxylate in complex B. As the acidity of boron influences the strength of N–B dative bonds,17 we altered the structure of the catalyst by modifying the aryl boronic acid moiety. Chirality-switch systems18 that use a catalyst from the same chiral source19 are rare, even though they offer great potential for the construction of chemical libraries for drug discovery.
Results and discussion
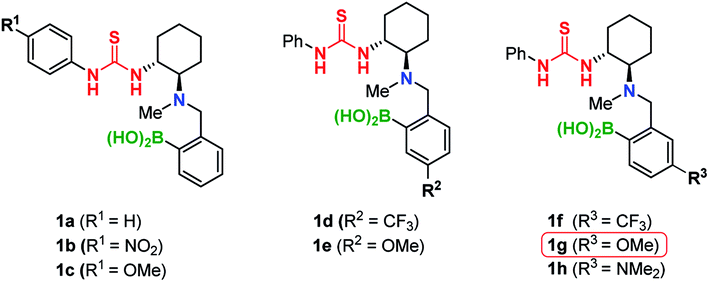
Initially, we explored the TMA of thiophenol 2a to crotonic acid 3a using 10 mol% of catalyst 1a and 4 Å MS in CCl4 (Table 1).|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Catalyst | Yieldb (%) | eec (%) |
| a Unless otherwise noted, the reactions were carried out using 3a (0.1 mmol), 2a (1.0 equiv.), 1a (0.1 equiv.), and 4 Å MS (50 mg) in the specified solvent (1.0 mL) at room temperature for 24 h. b Isolated yield after treatment with TMSCHN2. c Estimated using chiral HPLC after treatment with TMSCHN2. The absolute configuration is indicated in parentheses. d Without 4 Å MS. e CCl4 (50 μL) and 4 Å MS (20 mg). f 4 Å MS (100 mg). | ||||
| 1 | CCl4 | 1a | 90 | 41 (S) |
| 2 | CH2Cl2 | 1a | 21 | 22 (S) |
| 3 | n-Hexane | 1a | 28 | 48 (S) |
| 4 | CH3CN | 1a | 36 | 39 (R) |
| 5 | Acetone | 1a | 68 | 82 (R) |
| 6 | MeOH | 1a | 0 | — |
| 7d | CCl4 | 1a | 0 | — |
| 8d | Acetone | 1a | 11 | 15 (R) |
| 9e | CCl4 | 1a | 91 | 81 (S) |
| 10 | Acetone | 1b | 35 | 33 (R) |
| 11 | Acetone | 1c | 67 | 78 (R) |
| 12 | Acetone | 1d | 57 | 81 (R) |
| 13 | Acetone | 1e | 61 | 68 (R) |
| 14 | Acetone | 1f | 35 | 45 (R) |
| 15 | Acetone | 1g | 60 | 92 (R) |
| 16 | Acetone | 1h | 53 | 45 (R) |
| 17f | Acetone | 1g | 80 | 92 (R) |
| 18e | CCl4 | 1g | 78 | 75 (S) |
|
||||
This reaction furnished Michael adduct (S)-4a, which exhibits the same chirality as that of the aza-Michael addition (Table 1, entry 1).14a In the presence of non-polar solvents such as CCl4, CH2Cl2, and n-hexane, the major isomer of the Michael adduct is (S)-4a (ref. 20) (Table 1, entries 1–3). In contrast, in the presence of aprotic polar solvents such as acetonitrile and acetone, the major product is (R)-4a (Table 1, entries 4 and 5). The reaction did not proceed in MeOH, presumably due to its coordination to the boron atom of the catalyst (Table 1, entry 6). Notably, the catalytic activity of 1a is significantly reduced in the absence of MS; this result suggests that the formation of a boron complex via dehydration is essential for the successful TMA in both polar and non-polar solvents (Table 1, entries 7 and 8). The (S)-selectivity (81% ee) is significantly improved at higher concentrations in CCl4 (Table 1, entry 1 vs. 9). This is presumably due to the rapid formation of complex A (11B NMR: 4 ppm; Fig. S6†), which suppresses the undesired formation of the (R)-adduct (for further details, see the ESI†). Subsequently, we investigated the effect of substituents (R1, R2, R3) on the catalyst in order to improve the yield and (R)-selectivity in acetone. Thioureas 1b–c with different R1 substituents (OMe, NO2) neither improve the yield nor the enantioselectivity (Table 1, entries 10 and 11). Then, we examined the electronic effect of the boronic acid moiety by introducing electron-withdrawing and donating groups (R2, R3) into the aromatic ring of the arylboronic acid moieties. We found that catalysts 1d and 1e, which contain substituents at the meta-position relative to the boron atom (R2), only have a marginal effect on the results (Table 1, entries 12 and 13). However, when substituents are at the para-position relative to the boron atom (R3), a significant improvement of the reactivity and enantioselectivity was observed (Table 1, entries 14–16).This is demonstrated by the excellent results from catalyst 1g, which bears a methoxy group.21 In addition, increasing the amount of MS provides the (R)-adduct in 80% yield with 92% ee (Table 1, entry 17).
With the optimized conditions in hand, we investigated the electronic and steric effects of thiols22 on the asymmetric TMA in two different solvents (Fig. 2). In both solvents, electron-rich aryl thiols generally produce the corresponding adducts (4b–4d) in good yield (64–88%) with high enantioselectivity (80–91% ee). However, a slight decrease in reaction rate was observed for ortho-substituted thiols (4d). Similarly, tert-butyl-substituted benzenethiol produced both enantiomers of 4e with high yield and enantioselectivity. In contrast, the use of thiols with electron-withdrawing substituents, such as chlorine (Cl) and trifluoromethyl (CF3) groups on the aromatic ring, dramatically decreases the enantioselectivity of the adducts 4f and 4g, which was partially ascribed to background reactions.23 The enantioselectivity of 4g was not improved even at lower temperature, and the chemical yield of 4g significantly dropped presumably due to the decreased solubility of the substrates.
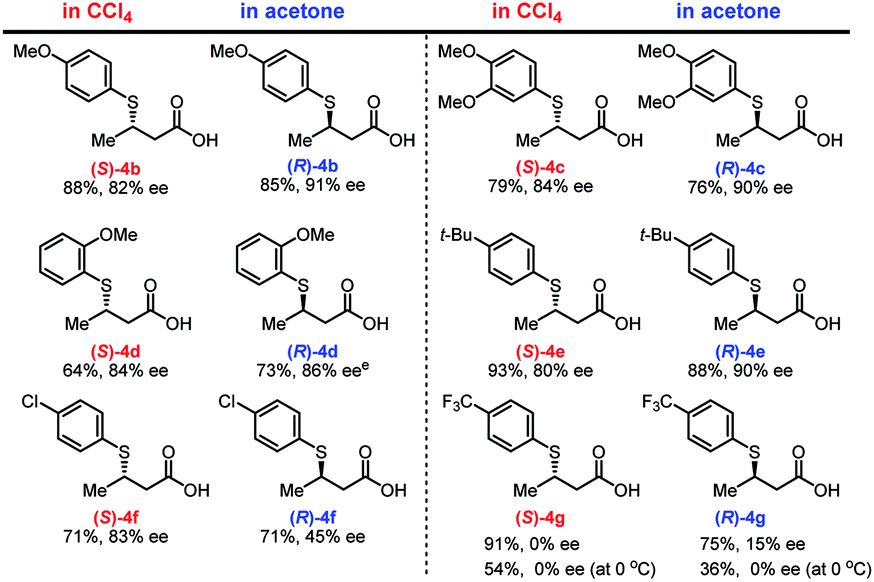 | ||
| Fig. 2 Substrate scope with respect to thiolsa–d. aThe reaction was carried out using 3a (0.1 mmol), 2a (1.0 equiv.), 1a (0.1 equiv.), and 4 Å MS (20 mg) in CCl4 (50 μL) at room temperature for 24 h. bThe reaction was carried out using 3a (0.1 mmol), 2a (1.0 equiv.), 1g (0.1 equiv.), and 4 Å MS (100 mg) in acetone (1.0 mL) at room temperature for 24 h. cIsolated yield after treatment with TMSCHN2. dee values were estimated using chiral HPLC analysis after treatment with TMSCHN2. eThe reaction was performed for 48 h. | ||
Using the optimized conditions, we then investigated the substrate scope with respect to α,β-unsaturated carboxylic acids 3 (Fig. 3). Several different aliphatic α,β-unsaturated carboxylic acids furnish the corresponding adducts (4h–4q) in good yield with good to high ee values in both solvents. An efficient TMA was observed for the linear alkyl Michael acceptors producing ee values of 81–91% (4o–4q). Notably, a γ-branched α,β-unsaturated carboxylic acid generates adducts (R)-4r (88% ee) and (S)-4r (91% ee) in CCl4 and acetone, respectively; however, the yield is somewhat decreased due to steric hindrance. Although enantioselectivity was not observed for adduct 4s using highly reactive trifluoromethyl-substituted α,β-unsaturated carboxylic acids as Michael acceptors, our solvent-dependent chirality-switchable TMA successfully produces both enantiomers of 4t–x in combination with substrates bearing ether, ester, and various aryl groups. One of the limitations of this method is the addition to cinnamic acid derivatives, which resulted in recovery of the starting materials. It is worth mentioning that, using benzoic acid as an additive, a slight increase in ee (ca. 10%) is observed, especially when bulky substrates are employed to obtain (R)-4r and (S)-4x in CCl4. A similar effect was observed in the asymmetric aza-Michael addition to α,β-unsaturated carboxylic acids in CCl4.14c
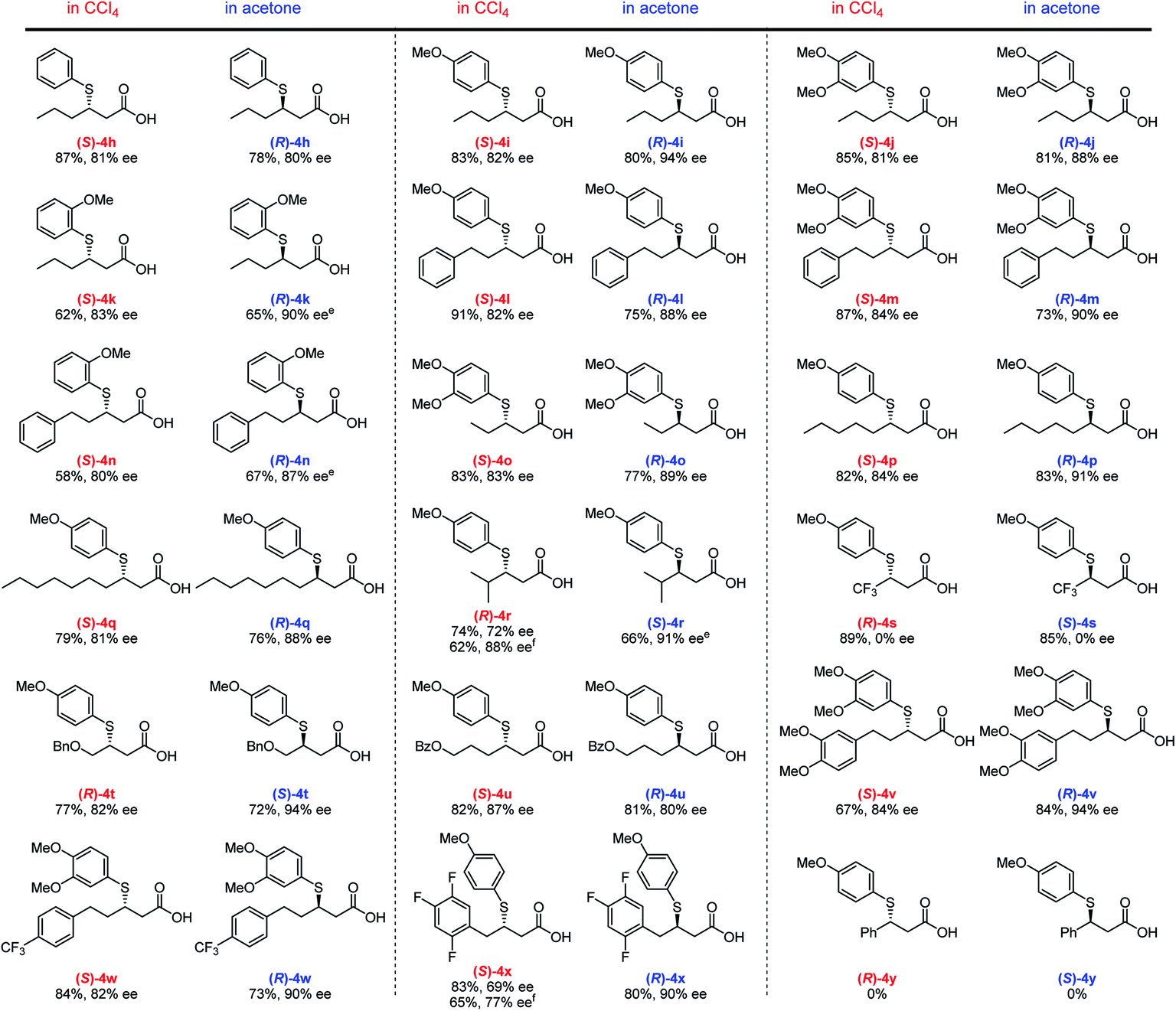 | ||
| Fig. 3 Substrate scope with respect to α,β-unsaturated carboxylic acidsa–d. aUnless otherwise noted, the reaction was carried out using 3 (0.1 mmol), 2 (1.0 equiv.), 1a (0.1 equiv.), and 4 Å MS (20 mg) in CCl4 (50 μL) at room temperature for 24 h. bThe reaction was carried out using 3 (0.1 mmol), 2 (1.0 equiv.), 1g (0.1 equiv.), and 4 Å MS (100 mg) in acetone (1.0 mL) at room temperature for 24 h. cIsolated yield after treatment with TMSCHN2. dee values were estimated using chiral HPLC analysis after treatment with TMSCHN2. eThe reaction was performed for 48 h. fOne equivalent of benzoic acid was added. | ||
Mechanistic insight into the solvent-dependent chirality-switchable TMA was obtained using NMR spectroscopic analysis (Fig. 4) in CDCl3 and CD2Cl2 as substitutes for CCl4 (Fig. S1–S2†), as well as in acetone-d6 (Fig. S3†).
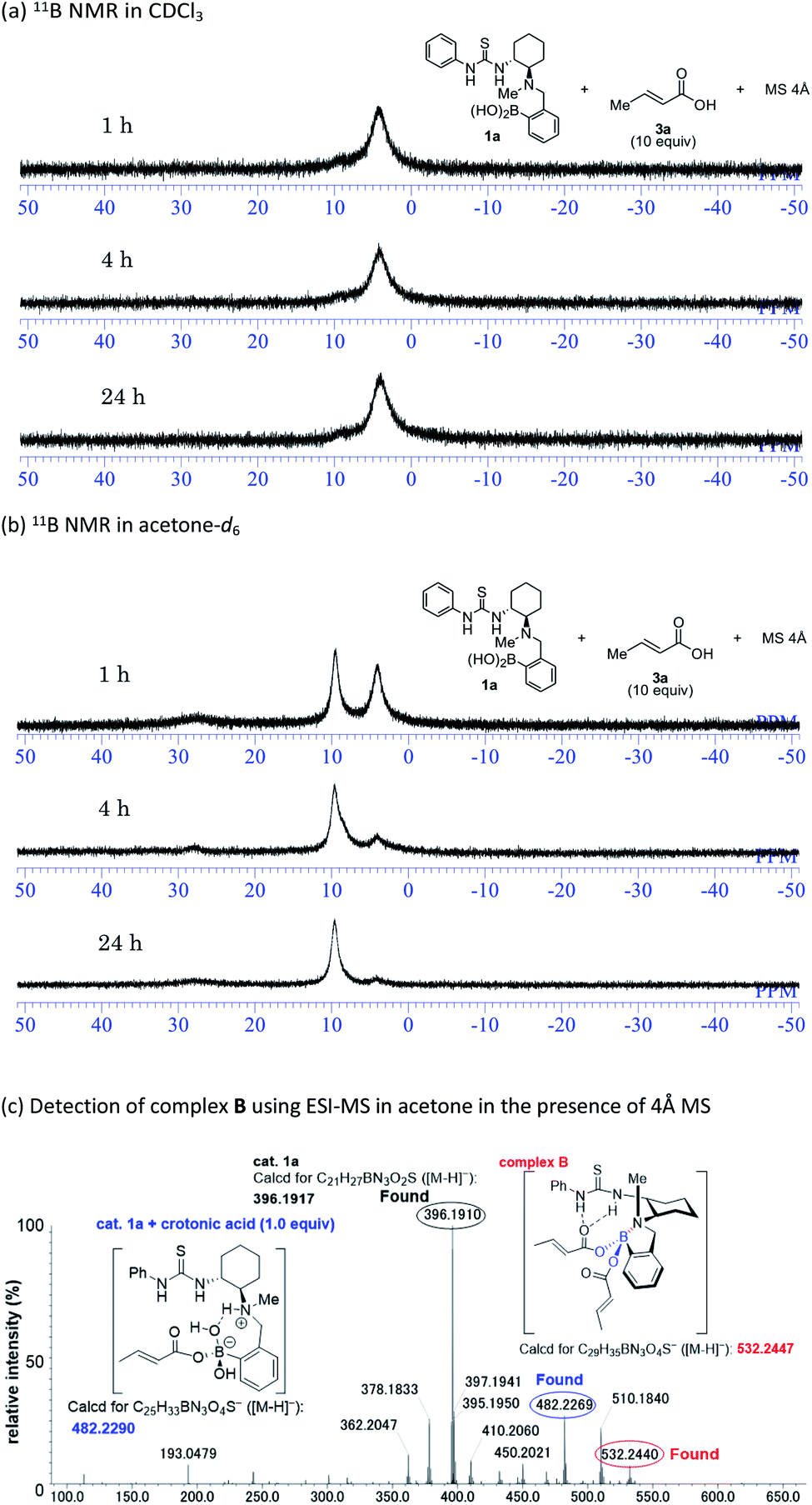 | ||
| Fig. 4 Spectral data in support of the proposed mechanism. | ||
In the presence of thiol 2a (10 equiv.), carboxylic acid 3a (10 equiv.), and 4 Å MS in CDCl3, the 11B NMR resonances for 1a converge at 4 ppm after 1 h (Fig. 4a). This result indicates the formation of the tetrahedral boron complex A and is consistent with previous work on aza-Michael additions. The addition of a nucleophile to the s-trans form of α,β-unsaturated carboxylate A (Fig. 1B) is favored over the addition to the s-cis form, which is due to steric repulsion between the s-cis form and the aromatic ring of the catalyst.14c Interestingly, in acetone-d6, the peaks of 1a gradually converge at 10 ppm in the presence of thiol 2a (10 equiv.), carboxylic acid 3a (10 equiv.), and 4 Å MS (Fig. 4b). We assume that the 10 ppm peak is derived from an N–B dative bond in e.g. complex B, as the N–B dative bond signals shift ca. 4–7 ppm downfield relative to the signals of tetrahedral borate complexes coordinated by water.15 An ESI-MS analysis of mixtures of 1a, 2a, and 4 Å MS in acetone further support the formation of an N–B dative bond (Fig. 4c). The exact mass peak of complex B (m/z calculated for C29H35BN3O4S [M − H]−: 532.2447) was detected at 532.2440 with an error of no more than 5 ppm. It should be mentioned here that such a peak was not detected in CCl4 or CHCl3 (Fig. S4†). Complex B is allegedly generated by the dehydration of complex A or its dimer,14c which is assisted by coordination of acetone to the boron atom. Preliminary computational studies suggest that nucleophilic addition to the s-cis form of complex B (Fig. 1B) is by 2.0 kcal mol−1 more favorable than addition to the s-trans form (Fig. S6–S7†),24 as the N–B chelation forces the carboxylate moiety away from the aromatic ring of the catalyst. The formation of an N–B bond is further supported by experimental results using catalyst 1g; the methoxyl group at the para-position relative to the boron atom on the aromatic ring of 1g facilitates dehydration via the mesomeric effect to form complex B, resulting in high enantioselectivity in acetone (Table 1).25
Finally, using ‘the same’ catalyst, we demonstrate the efficient production of both enantiomers of biologically active compounds2b (Scheme 1). For example, β-sulfonylhydroxamic acid derivatives show potent inhibitory activity towards peptide deformylase and matrix metalloproteases, whereby the activity of one enantiomer is by two orders of magnitude higher than that of the other.2b,c The construction of a library of both enantiomers can be expected to aid clarifying the exact biological activity and to suppress adverse effects caused by these compounds. The chirality-switchable system based on catalyst 1a allows the production of enantiomers 4b,c from thiols 2 and α,β-unsaturated carboxylic acids 3 simply by changing the solvent from CCl4 to acetone.
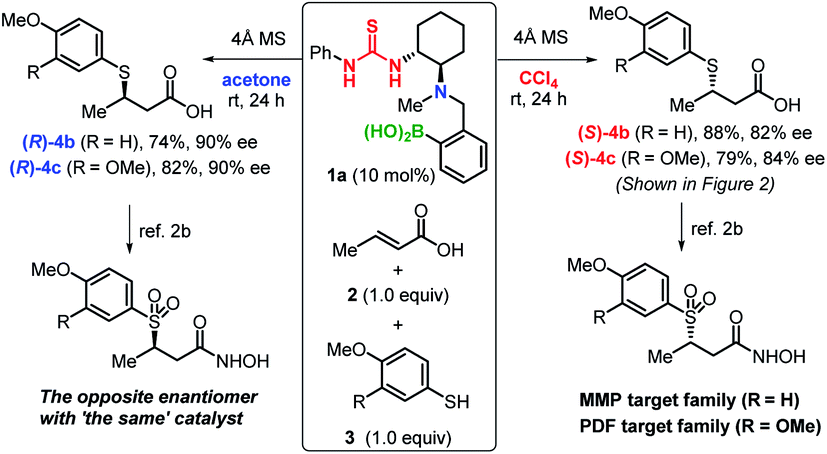 | ||
| Scheme 1 An efficient approach to generate both target enantiomers of 4b,c using catalyst 1a. | ||
Conclusions
We have developed a solvent-dependent asymmetric thia-Michael addition (TMA) of thiols to α,β-unsaturated carboxylic acids, wherein both (S)- and (R)-adducts can be obtained in good yield and high enantioselectivity. Using 11B NMR spectroscopy and ESI-MS analyses, we found that the coordination state of boron in the catalyst depends on the coordinating nature of the aprotic solvent. These findings can be expected to lead to the development of new organoboron catalysts and the construction of chemical libraries. Studies to extend the synthetic applications of this catalytic system are currently in progress in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI grant 16H06384.Notes and references
- (a) J. Clayden and P. MacLellan, Beilstein J. Org. Chem., 2011, 7, 582–595 CrossRef CAS PubMed; (b) P. Chauhan, S. Mahajan and D. Enders, Chem. Rev., 2014, 114, 8807–8864 CrossRef CAS PubMed.
- (a) C. J. Burns, R. D. Groneberg, J. M. Salvino, G. McGeehan, S. M. Condon, R. Morris, M. Morrissette, R. Mathew, S. Darnbrough, K. Neuenschwander, A. Scotese, S. W. Djuric, J. Ullrich and R. Labaudiniere, Angew. Chem., Int. Ed., 1998, 37, 2848–2850 CrossRef CAS; (b) C. Apfel, D. W. Banner, D. Bur, M. Dietz, T. Hirata, C. Hubschwerlen, H. Locher, M. G. P. Page, W. Pirson, G. Rossé and J.-L. Specklin, J. Med. Chem., 2000, 43, 2324–2331 CrossRef CAS PubMed; (c) J. M. Salvino, R. Mathew, T. Kiesow, R. Narensingh, H. J. Mason, A. Dodd, R. Groneberg, C. J. Burns, G. McGeehan, J. Kline, E. Orton, S.-Y. Tang, M. Morrisette and R. Labaudininiere, Bioorg. Med. Chem. Lett., 2000, 10, 1637–1640 CrossRef CAS PubMed; (d) M. Sani, G. Candiani, F. Pecker, L. Malpezzi and M. Zanda, Tetrahedron Lett., 2005, 46, 2393–2396 CrossRef CAS.
- J. B. Bariwal, K. D. Upadhyay, A. T. Manvar, J. C. Trivedi, J. S. Singh, K. S. Jain and A. K. Shah, Eur. J. Med. Chem., 2008, 43, 2279–2290 CrossRef CAS PubMed.
- (a) D. Enders, K. Lüttgen and A. A. Narine, Synthesis, 2007, 959–980 CrossRef CAS; (b) P. Wadhwa, A. Kharbanda and A. Sharma, Asian J. Org. Chem., 2018, 7, 634–661 CrossRef CAS.
- (a) S. Kanemasa, Y. Oderatoshi and E. Wada, J. Am. Chem. Soc., 1999, 121, 8675–8676 CrossRef CAS; (b) S. Kobayashi, C. Ogawa, M. Kawamura and M. Sugiura, Synlett, 2001, 983–985 CrossRef CAS; (c) K. Matsumoto, A. Watanabe, T. Uchida, K. Ogi and T. Katsuki, Tetrahedron Lett., 2004, 45, 2385–2388 CrossRef CAS; (d) S. J. K. Sauerland, E. Kiljunen and A. M. P. Koskinen, Tetrahedron Lett., 2006, 47, 1291–1293 CrossRef CAS; (e) S. Lauzon, H. Keipour, V. Gandon and T. Ollevier, Org. Lett., 2017, 19, 6324–6327 CrossRef CAS PubMed.
- B.-J. Li, L. Jiang, M. Liu, Y.-C. Chen, L. S. Ding and Y. Wu, Synlett, 2005, 603–606 CAS.
- Q.-L. Pei, H.-W. Sun, Z.-J. Wu, X.-L. Du, X.-M. Zhang and W.-C. Yuan, J. Org. Chem., 2011, 76, 7849–7859 CrossRef CAS PubMed.
- T. Ogawa, N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2012, 51, 8551–8554 CrossRef CAS PubMed.
- (a) X.-Q. Dong, X. Fang, H.-Y. Tao, X. Zhou and C.-J. Wang, Adv. Synth. Catal., 2012, 354, 1141–1147 CrossRef CAS; (b) G. Wang, Y. Tang, Y. Zhang, X. Liu, L. Lin and X. Feng, Chem.–Eur. J., 2017, 23, 554–557 CrossRef CAS PubMed; (c) S. Meninno, C. Volpe and A. Lattanzi, Chem.–Eur. J., 2017, 23, 4547–4550 CrossRef CAS PubMed; (d) S. Meninno, S. Naddeo, L. Varricchio, A. Capobianco and A. Lattanzi, Org. Chem. Front., 2018, 5, 1967–1977 RSC.
- (a) Y. Fukata, K. Asano and S. Matsubara, J. Am. Chem. Soc., 2015, 137, 5320–5323 CrossRef CAS PubMed; (b) Y. Fukata, K. Yao, R. Miyaji, K. Asano and S. Matsubara, J. Org. Chem., 2017, 82, 12655–12668 CrossRef CAS PubMed.
- (a) R. Wang, J. Liu and J. Xu, Adv. Synth. Catal., 2015, 357, 159–167 CrossRef CAS; (b) J. L. Fulton, M. A. Horwitz, E. L. Bruske and J. S. Johnson, J. Org. Chem., 2018, 83, 3385–3391 CrossRef CAS PubMed.
- (a) K. Nishimura, M. Ono, Y. Nagaoka and K. Tomioka, J. Am. Chem. Soc., 1997, 119, 12974–12975 CrossRef CAS; (b) X.-Q. Dong, X. Fang and C.-J. Wang, Org. Lett., 2011, 13, 4426–4429 CrossRef CAS PubMed; (c) X. Fang, J. Li and C.-J. Wang, Org. Lett., 2013, 15, 3448–3451 CrossRef CAS PubMed; (d) P. Yuan, S. Meng, J. Chen and Y. Huang, Synlett, 2016, 27, 1068–1072 CrossRef CAS; (e) J. Yang, A. J. M. Farley and D. J. Dixon, Chem. Sci., 2017, 8, 606–610 RSC.
- To the best of our knowledge, there is only one report of a catalytic asymmetric thia-Michael addition involving α,β-unsaturated carboxylic acids; for details, see: P. N. Kalaria, J. R. Avalani and D. K. Raval, Tetrahedron: Asymmetry, 2016, 27, 947–953 CrossRef CAS.
- (a) N. Hayama, T. Azuma, Y. Kobayashi and Y. Takemoto, Chem. Pharm. Bull., 2016, 64, 704–717 CrossRef CAS PubMed; (b) T. Azuma, A. Murata, Y. Kobayashi, T. Inokuma and Y. Takemoto, Org. Lett., 2014, 16, 4256–4259 CrossRef CAS PubMed; (c) N. Hayama, R. Kuramoto, T. Földes, K. Nishibayashi, Y. Kobayashi, I. Pápai and Y. Takemoto, J. Am. Chem. Soc., 2018, 140, 12216–12225 CrossRef CAS PubMed; (d) K. Michigami, H. Murakami, T. Nakamura, N. Hayama and Y. Takemoto, Org. Biomol. Chem., 2019, 17, 2331–2335 RSC.
- (a) L. Zhu, S. H. Shabbir, M. Gray, V. M. Lynch, S. Sorey and E. V. Anslyn, J. Am. Chem. Soc., 2006, 128, 1222–1232 CrossRef CAS PubMed; (b) B. E. Collins, S. Sorey, A. E. Hargrove, S. H. Shabbir, V. M. Lynch and E. V. Anslyn, J. Org. Chem., 2009, 74, 4055–4060 CrossRef CAS PubMed; (c) I. Georgiou, G. Ilyashenko and A. Whiting, Acc. Chem. Res., 2009, 42, 756–768 CrossRef CAS PubMed; (d) J. D. Larkin, J. S. Fossey, T. D. James, B. R. Brooks and C. W. Bock, J. Phys. Chem. A, 2010, 114, 12531–12539 CrossRef CAS PubMed; (e) A. Sakakura, T. Ohkubo, R. Yamashita, M. Akakura and K. Ishihara, Org. Lett., 2011, 13, 892–895 CrossRef CAS PubMed; (f) X. Sun, B. M. Chapin, P. Metola, B. Collins, B. Wang, T. D. James and E. V. Anslyn, Nat. Chem., 2019, 11, 768–778 CrossRef CAS PubMed.
- (a) Y. Kobayashi, Y. Taniguchi, N. Hayama, T. Inokuma and Y. Takemoto, Angew. Chem., Int. Ed., 2013, 52, 11114–11118 CrossRef CAS PubMed; (b) Y. Kobayashi, S. Li and Y. Takemoto, Asian J. Org. Chem., 2014, 3, 403–407 CrossRef CAS.
- B. G. Janesko, J. Chem. Theory Comput., 2010, 6, 1825–1833 CrossRef CAS PubMed.
- For reviews, see: (a) G. Romanazzi, L. Degennaro, P. Mastrorilli and R. Luisi, ACS Catal., 2017, 7, 4100–4114 CrossRef CAS . For examples of solvent-controlled chirality-switchable systems, see: ; (b) Y. Sohtome, S. Tanaka, K. Takada, T. Yamaguchi and K. Nagasawa, Angew. Chem., Int. Ed., 2010, 49, 9254–9257 CrossRef CAS PubMed; (c) J. Flores-Ferrándiz and R. Chinchilla, Tetrahedron: Asymmetry, 2014, 25, 1091–1094 CrossRef; (d) R. J. Chew, X.-R. Li, Y. Li, S. A. Pullarkat and P.-H. Leung, Chem.–Eur. J., 2015, 21, 4800–4804 CrossRef CAS PubMed . For an example of solvent-controlled diastereoselectivity, see:; (e) X. Tian, C. Cassani, Y. Liu, A. Moran, A. Urakawa, P. Galzerano, E. Arceo and P. Melchiorre, J. Am. Chem. Soc., 2011, 133, 17934–17941 CrossRef CAS PubMed.
- Y. Fukata, K. Asano and S. Matsubara, J. Am. Chem. Soc., 2013, 135, 12160–12163 CrossRef CAS PubMed.
- The absolute configuration of 4a was determined by comparing the specific optical rotation to that of an authentic sample. For further details, see the ESI.†.
- Presumably, these moderate results were caused by the poor solubility 1h in acetone.
- Benzyl mercaptan (BnSH) did not produce the corresponding adduct in both solvents, presumably due to the insufficient acidity of S–H proton.
- For example, the TMA of 4-trifluoromethylbenzenthiol with crotonic acid proceeded in ca. 50% yield in acetone (in the presence of MS without a catalyst).
- Alternative pathways for the formation of the (R)-adduct, including the coordination of thiol to the boron atom, cannot be ruled out at this point.
- In the presence of acetone, the reaction via complex A was unlikely to proceed. See the ESI† for the effect of acetone in CCl4 (Scheme S3a†). The effect of pre-mixing of catalyst and 3a in acetone was also described in Schemes S3b and c.†.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data, copies of 1H and 13C NMR spectra, and HPLC chromatograms. See DOI: 10.1039/d0sc01729a |
| This journal is © The Royal Society of Chemistry 2020 |