
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMacrocyclization of an all-D linear α-helical peptide imparts cellular permeability†
Srinivasaraghavan
Kannan
 *a,
Pietro G. A.
Aronica
a,
Simon
Ng
b,
Dawn Thean
Gek Lian
b,
Yuri
Frosi
b,
Sharon
Chee
b,
Jiang
Shimin
b,
Tsz Ying
Yuen
c,
Ahmad
Sadruddin
d,
Hung Yi Kristal
Kaan
d,
Arun
Chandramohan
d,
Jin Huei
Wong
a,
Yaw Sing
Tan
a,
Zi Wei
Chang
i,
Fernando J.
Ferrer-Gago
b,
Prakash
Arumugam
a,
Yi
Han
e,
Shiying
Chen
e,
Laurent
Rénia
i,
Christopher J.
Brown
b,
Charles W.
Johannes
c,
Brian
Henry
d,
David P.
Lane
b,
Tomi K.
Sawyer
f,
Chandra S.
Verma
*agh and
Anthony W.
Partridge
*d
*a,
Pietro G. A.
Aronica
a,
Simon
Ng
b,
Dawn Thean
Gek Lian
b,
Yuri
Frosi
b,
Sharon
Chee
b,
Jiang
Shimin
b,
Tsz Ying
Yuen
c,
Ahmad
Sadruddin
d,
Hung Yi Kristal
Kaan
d,
Arun
Chandramohan
d,
Jin Huei
Wong
a,
Yaw Sing
Tan
a,
Zi Wei
Chang
i,
Fernando J.
Ferrer-Gago
b,
Prakash
Arumugam
a,
Yi
Han
e,
Shiying
Chen
e,
Laurent
Rénia
i,
Christopher J.
Brown
b,
Charles W.
Johannes
c,
Brian
Henry
d,
David P.
Lane
b,
Tomi K.
Sawyer
f,
Chandra S.
Verma
*agh and
Anthony W.
Partridge
*d
aBioinformatics Institute, Agency for Science, Technology and Research (A*STAR), 30 Biopolis Street, #07-01 Matrix, Singapore 138671, Singapore. E-mail: raghavk@bii.a-star.edu.sg; chandra@bii.a-star.edu.sg; Fax: +65 6478 9048; Tel: +65 6478 8353 Tel: +65 6478 8273
bp53 Laboratory, Agency for Science, Technology and Research (A*STAR), 8A Biomedical Grove, #06-04/05, Neuros/Immunos, Singapore 138648
cInstitute of Chemical & Engineering Science, Agency for Science, Technology and Research (A*STAR), 8 Biomedical Grove, #07, Neuros Building, Singapore 138665
dMSD International, Translation Medicine Research Centre, 8 Biomedical Grove, #04-01/05 Neuros Building, Singapore, 138665, Singapore. E-mail: anthony_partridge@merck.com
eMerck & Co., Inc., Kenilworth, New Jersey, USA
fMerck & Co., Inc., Boston, Massachusetts, USA
gSchool of Biological Sciences, Nanyang Technological University, 60 Nanyang Drive, Singapore 637551
hDepartment of Biological Sciences, National University of Singapore, 14 Science Drive 4, Singapore 117543
iSingapore Immunology Network (SIgN), Agency for Science, Technology and Research (A*STAR), 8A Biomedical Grove, #03-06, Immunos, Singapore 138648
First published on 11th May 2020
Abstract
Peptide-based molecules hold great potential as targeted inhibitors of intracellular protein–protein interactions (PPIs). Indeed, the vast diversity of chemical space conferred through their primary, secondary and tertiary structures allows these molecules to be applied to targets that are typically deemed intractable via small molecules. However, the development of peptide therapeutics has been hindered by their limited conformational stability, proteolytic sensitivity and cell permeability. Several contemporary peptide design strategies are aimed at addressing these issues. Strategic macrocyclization through optimally placed chemical braces such as olefinic hydrocarbon crosslinks, commonly referred to as staples, may improve peptide properties by (i) restricting conformational freedom to improve target affinities, (ii) improving proteolytic resistance, and (iii) enhancing cell permeability. As a second strategy, molecules constructed entirely from D-amino acids are hyper-resistant to proteolytic cleavage, but generally lack conformational stability and membrane permeability. Since neither approach is a complete solution, we have combined these strategies to identify the first examples of all-D α-helical stapled and stitched peptides. As a template, we used a recently reported all D-linear peptide that is a potent inhibitor of the p53–Mdm2 interaction, but is devoid of cellular activity. To design both stapled and stitched all-D-peptide analogues, we used computational modelling to predict optimal staple placement. The resultant novel macrocyclic all D-peptide was determined to exhibit increased α-helicity, improved target binding, complete proteolytic stability and, most notably, cellular activity.
Introduction
Protein–protein interactions (PPIs) are central to most biological processes and are often dysregulated in disease.1,2 Thus, PPIs are attractive therapeutic targets but are typically refractory to traditional drug approaches. Specifically, PPI surfaces are generally large and flat, lacking the deep cavities that typically accommodate small molecules. This has contributed to the limited success in developing small molecule inhibitors for PPI targets.3 The realization that ∼40% of all PPIs are mediated by relatively short structural elements prompted the strategy of peptide-based inhibitors as orthosteric competitors of the PPI of interest.4 When taken out of the protein–ligand context and synthesized, such peptides are often unstructured, yet able to adopt their biologically-relevant conformation upon protein target binding.4 However, for intracellular targets, the peptide modality may be confounded by proteolytic sensitivity, low conformational stability (yielding weak affinities and off-target effects), and poor cell permeability (further limiting engagement of intracellular targets and/or oral bioavailability).5–11 To address these issues, macrocyclization and peptide backbone modification strategies are often pursued.5–13 Macrocyclization can improve drug-like properties by constraining the peptides toward their bound conformations. Thus, entropic penalties upon binding are often reduced, binding constants improved, and opportunities for unwanted off-target binding events are decreased. Secondly, macrocyclization may confer varying degrees of proteolytic resistance by modifying key backbone and/or side-chain structural moieties in the peptide. Thirdly, macrocyclization may enhance cell permeability, through increased stability of intramolecular hydrogen bonding with corresponding decreases in the desolvation penalties incurred during the transit of peptides across the apolar cell membrane. Amongst the cyclization techniques described, stapling via olefin metathesis using a non-proteogenic amino acid such as alpha-methyl alkenyl side chains has proven to be very effective,13–18 particularly for stabilizing the helical conformation. Stapling requires incorporation of the appropriate unnatural amino acid precursors placed at appropriate locations along the peptide sequence such that they do not interfere with the binding face of the helix. Linkers can be of different types, and can span different lengths, typically resulting in i, i + 3, i, i + 4, or i, i + 7 staples. In addition, the scope of applications for RCM strategies has recently expanded to include non-helical peptides.19,20The stapled peptide strategy has been successfully applied to inhibit several PPIs of therapeutic potential including, BCL-2/Mcl-1 family,21–24 β-catenin–TCF,25 Rab–GTPase-effector,26 ERα–coactivator protein,27 Cullin3–BTB,28 VDR–coactivator protein,29 eIf4E30 and p53–Mdm2/Mdm4.31–34 Noteworthy, in the case of p53–Mdm2/Mdm4, a dual selective stapled peptide (ALRN-6924) has been further successfully advanced to phase II clinical trials.35–37 Although this example is encouraging for the advancement of stapled peptides into the clinic, challenges still remain. Amongst these, engineering molecules with sufficient proteolytic stability for sustained target binding and cellular activity is critical. Indeed, although stapling L-amino acid peptides can confer resistance to protease-mediated degradation, the effect is often not complete, and residues located outside of the macrocycle may still be susceptible to cleavage.38–40 One way to overcome such degradation is through the introduction of unnatural amino acids. For example Fiacco et al.,41 have incorporated an unnatural amino acid in an mRNA display library and identified peptides that confer resistance to protease-mediated degradation. However, peptides that retain L-amino acids can still be susceptible to proteolytic degradation in a context-specific manner. On the other hand, all-D peptides/mini-proteins are inherently hyperstable against proteolysis and have been engineered with strong binding affinity against a variety of targets including, VEGF–VEGF-receptor,42 PD-1–PD-L1 (ref. 43) and human immunodeficiency virus type 1 (HIV-1) entry.44 More recently, Garton et al.45 have discovered two all D-peptides, GLP1R and PTH1R agonists using a mirror image of the entire PDB. In these cases where the target is extracellular, the hyper-stable properties conferred by all-D molecules give obvious advantages. All-D linear peptide antagonists with inherent proteolytic hyperstability have also been discovered with strong binding affinity against intracellular proteins.46,47 However, similar to their linear all-L amino acid counterparts, these unfortunately lack the ability to permeate cells to engage their targets.
We hypothesized that combining both strategies, i.e. all-D-peptide and helical macrocyclization might provide synergistic results. Thus, we embarked on introducing a hydrocarbon staple into an all-D peptide inhibitor of the p53–Mdm2/Mdm4 interaction. p53 is a key tumour suppressor protein, which primarily functions as a DNA transcription factor, which is commonly inactivated in cancer and normally plays a crucial role in guarding the cell in response to various stress signals through the induction of cell cycle arrest, apoptosis or senescence.48 Mechanisms that frequently result in the inactivation of p53 and tumorigenesis include increased expression of the p53-negative regulators Mdm2 and Mdm4. Both Mdm2 and Mdm4 attenuate p53 function by interacting directly with p53 and preventing its interaction with the relevant activation factors required for transcription e.g. dTAFII, hTAFII. In addition, they are both E3 ligase components and target p53 for proteosomal mediated degradation. Mdm4, unlike Mdm2, has no intrinsic E3 ubiquitin ligase activity. Instead Mdm4 forms heterodimeric complexes with Mdm2 whereby it stimulates the ubiquitin activity of Mdm2. As a result, p53 activity and protein levels are acutely suppressed by Mdm2 and Mdm4 overexpression. Development of inhibitors to disrupt the interactions of p53 with either Mdm2 or Mdm4, or both, are therefore highly desirable as they will prevent p53 degradation and restore a p53 dependent transcriptional anti-tumour response.49,50
The structural interface of the p53–Mdm2/Mdm4 complex is characterized by an α-helix from the N-terminal transactivation domain of p53 which binds into a hydrophobic groove on the surface of the N-terminal domain of both Mdm2 and Mdm4. Three hydrophobic residues, Phe19, Trp23 and Leu26 from p53 are critical determinants of this interaction and project deeply into the Mdm2 interaction groove (Fig. S1A†). The isolated p53 peptide is largely disordered, morphing into an α-helical conformation upon binding. There are several examples of small molecules, peptides and biologics that mimic these interactions and compete for Mdm2/4 binding, with the release of p53.37 However, a large majority of the small molecules developed exhibit little affinity and activity against Mdm4, which possesses several distinct structural differences in the p53 peptide binding groove compared to Mdm2. Although several Mdm2 specific molecules have entered initial clinical trials, they have largely been met with dose limiting toxicities in patients.37 Overexpression of Mdm4 in tumours has been demonstrated to attenuate the effectiveness of Mdm2 specific compounds, presumably through the maintenance of heterodimeric complexes of Mdm2 and Mdm4 that inhibit and target p53 for proteosomal degradation. Mdm2-selective inhibitors may also induce higher levels of Mdm4. This highlights the importance of targeting both proteins simultaneously to achieve efficient activation of p53 for an optimal therapeutic response. ALRN-6924, a dual binding Mdm2/4 L-amino acid stapled peptide, is currently in clinical trials and has been reported to show promise in terms of tumour response and low toxicity.51
dPMI-δ, the all-D linear peptide that served as our starting point, was derived from a mirror image phage display screen reported by Liu et al.46 Specifically, they reported several 12-mer D-peptide antagonists of Mdm2 (termed dPMI-α, β, γ) that bind with affinities as low as 35 nM and are resistant to proteolytic degradation. dPMI-δ is a corresponding analogue that was modified with two unnatural amino acids (6-F-dTrp3 and p-CF3-dPhe7) to improve the Mdm2 binding with a reported Kd of 220 pM.52 Crystal structures52 of the complex between this peptide and the N-terminal domain of Mdm2 showed that the peptide was bound in a conformation similar to that adopted by the wild-type peptide (the all-L amino acid peptide derived from p53). The helix, as expected, was left-handed and projected the side chains of 6-F-dTrp3, p-CF3-dPhe7 and dLeu11 into the hydrophobic pocket of Mdm2 (Fig. S1B†), in conformations similar to those adopted by the side chains of Phe19, Trp23 and Leu26 in the wild type peptide (Fig. S1A†). Unfortunately, this peptide lacked cell permeability, but did activate p53 in cells when delivered using nano-carriers.47 Given our recent success in the cellular inhibition of Mdm2 and activation of p53 by stapled peptides (containing L-amino acids),33,53–55 we questioned whether we could achieve cellular permeation by stapling the D-peptide. We report here the computational design, synthesis and biological evaluation of stapled versions of dPMI-δ. These modifications improved binding and imparted cell permeability to result in disruption of the p53–Mdm2/Mdm4 interaction and ultimately upregulated p53 activity. As an extension of our work, we show that a bicyclic (stitched) version of these peptides demonstrates superior binding and cellular properties relative to the stapled peptide precursors.
Results
Conformational landscape of dPMI-δ peptide in apo and Mdm2-bound states
We sought to rationally design stapled dPMI-δ analogues that would stabilize helical structure and preserve or enhance binding affinities. Accordingly, we applied molecular dynamics (MD) simulations to the published co-crystal structure of the Mdm2–dPMI-δ complex to understand its structural details critical for the maintenance of the binding motif. During the simulation, the bound conformation of the dPMI-δ peptide remained stable with an RMSD of <2 Å relative to its starting conformation (Fig. S2A†). The bound dPMI-δ peptide retained its crystallographic α-helical conformation throughout the simulation (>95% α-helicity). The peptide bound state of Mdm2 also remained stable with an RMSD of <2 Å (Fig. S2B†). The bound conformation of the peptide is stabilized by hydrogen bonds and hydrophobic interactions. A hydrogen bond observed in the crystal structure between the side chain N of 6-F-dTrp3 and the backbone O of Gln72 (Fig. S1B†), is preserved in ∼80% of the simulation. Other hydrogen bonds seen in the crystal structure are reflected in the simulations, but for shorter durations, included those between: (i) the side chains of Gln72 (Mdm2) and dThr1 (dPMI-δ), (ii) the side chains of Lys94/His96 (Mdm2) and dGlu8 (dPMI-δ), (iii) the side chains of His96/Tyr100 (Mdm2) and the backbone carbonyl of dLeu11 (dPMI-δ) (Fig. S1B†). As expected, the three critical residues 6-F-dTrp3, p-CF3-dPhe7 and dLeu11 from dPMI-δ were buried into the hydrophobic binding pocket in Mdm2 (Fig. S2C†) throughout the simulation.Peptide design was also informed by understanding the conformational landscape of the free dPMI-δ peptide in solution. Simulations were carried out starting from the bound conformation of the peptide extracted from the crystal structure of the Mdm2–dPMI-δ complex. Biasing Potential Replica Exchange MD (BP-REMD), a Hamiltonian Replica Exchange Method that has been used successfully to explore peptide landscapes,56,57 was used to enhance the conformational sampling of the peptide. Unsurprisingly, the free peptide exhibited increased flexibility with RMSD ranging between 2–6 Å (Fig. S2D†). The two peaks (3–4 Å and 6 Å) correspond to the partially folded and unfolded states of the peptide, a rapid loss in α-helicity is seen resulting in a state where only ∼21% of the sampled conformations are alpha helical. This prediction was experimentally confirmed by circular dichroism (CD) spectroscopy, which showed that the peptide was ∼20% helical in solution (Fig. S2E†). This was also expected and consistent with the linear peptides derived from the natural p53 sequence.
Design and synthesis of all D-stapled peptides
Relative to the above simulations, we sought to design stapled analogues of dPMI-δ that would maximize helicity in solution and maintain target binding. To identify appropriate positions on the dPMI-δ peptide for the introduction of the hydrocarbon linkers, we sought to determine residues that, upon mutation, would result in minimal perturbation to the peptide–Mdm2 interaction. The overall binding energy of the peptide to Mdm2 during the MD simulations is decomposed into the energetic contributions of each residue of the peptide. Unsurprisingly, 6-F-dTrp3, p-CF3-dPhe7 and dLeu11 are the major contributors to the total binding energy followed by dTyr4 and dLeu10 (Fig. 1A). The contributions from the other 7 residues are either negligible or even slightly destabilizing. We next carried out computational alanine scans of the residues of the peptide by mutating each residue to D-alanine and computing the change in the binding energy for each conformation sampled during the MD simulation and averaging the changes (Fig. 1B). The results mirror the residue-wise contributions (Fig. 1A) in that the D-alanine mutations were most deleterious at positions that contributed most, i.e. 6-F-dTrp3 and p-CF3-dPhe7 of dPMI-δ (>∼10 kcal mol−1) (Fig. 1B) while substitutions at positions dTyr4, dGlu8 and dLeu11 resulted in loss of ∼2–5 kcal mol−1 in the overall binding energy (Fig. 1B). In contrast the other positions were quite tolerant to D-Ala substitutions. Overall, these studies suggested 7 positions where staples could be incorporated without significant perturbations to target binding. The incorporation of staples requires careful selection of sidechains with appropriate stereochemistry. As stapling of the left-handed alpha-helices that are formed by all-D peptide has not been conducted previously, we first needed to select the appropriate stereocenters. We reasoned that the stereo-centers should be a mirror-image of the standard strategies that have proven effective for stapling right-handed alpha-helices (i.e., S5 to S5 for (i, i + 4) linkages, and R8 to S5 for (i, i + 7) linkages). Accordingly, we choose to employ R5 to R5 and S8 to R5 for (i, i + 4) linkages and (i, i + 7) linkages respectively. Using these linkages and the simulations to guide staple placement, we designed several stapled versions of dPMI-δ (details are shown in Fig. 2) and the chemical structures of the dPMI-δ peptides are shown in ESI figure (Fig. S3†).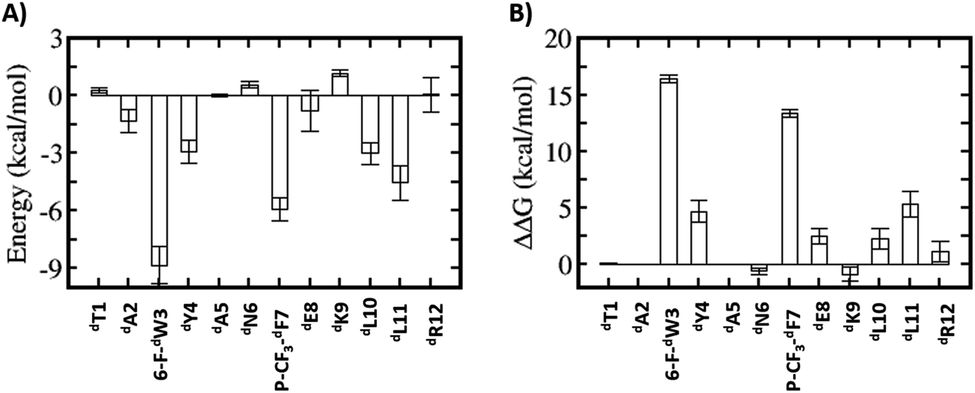 | ||
| Fig. 1 Energetic analysis of the MD simulations of the dPMI-δ–Mdm2 complex. (A) Binding energy contributions of the dPMI-δ peptide residues. (B) Computational alanine (D-Ala) scanning of dPMI-δ peptide residues. | ||
 | ||
| Fig. 2 A helical wheel representation of the dPMI-δ template sequence used for the design of stapled peptides. Residues that are linked through all hydrocarbon linkers i, i + 4 and i, i + 7 are highlighted in blue and red respectively. Sequences of the linear and stapled dPMI-δ peptides are shown on the right. | ||
Peptide stapling increases helicity
BP-REMD simulations suggested that all of the designed stapled dPMI-δ analogues should have increased solution-based helicity. Specifically, we predicted solution helicities between 24–39%; values that were increased compared to the predicted and measured values of ∼20% for the unstapled parent sequence (vide supra). The values for the stapled analogues agreed well with those obtained experimentally via CD spectroscopy (ranging from 24.5% to 40%) (Fig. 3 and Table 1). This increase in helicity upon stapling mirrors what has been reported for stapling all-L amino acid peptides.14,58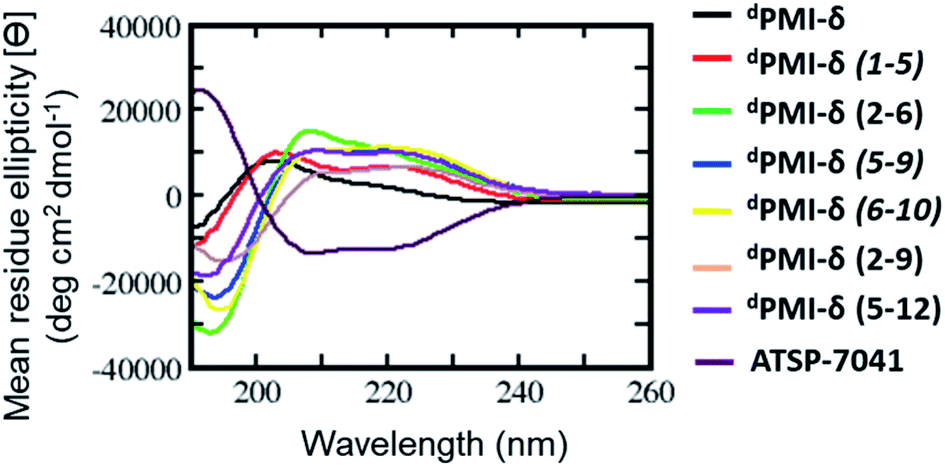 | ||
| Fig. 3 Secondary structure analysis of linear, stapled dPMI-δ and ATSP-7041 peptides determined through circular dichroism (CD). Note that this spectra of linear and stapled dPMI-δ peptides are inverted, as expected for a peptide consisting of D-amino acids only. | ||
| Peptide | dPMI-δ | dPMI-δ(1–5) | dPMI-δ(2–6) | dPMI-δ(2–9) | dPMI-δ(5–9) | dPMI-δ(6–10) | dPMI-δ(5v12) | ATSP-7041 |
| CD (% helicity) | 20.4 | 24.7 | 46.7 | 26.5 | 38.0 | 39.9 | 31.8 | 49.6 |
| FP Mdm2 Kd (nM) | 41.4 (1.2) | 16.8 (1.3) | 7540 (1.2) | 232 (1.4) | 142.3 (1.1) | 118 (1.2) | 33.9 (1.4) | 33.8 (1.6) |
| SPR Kd (nM) | <1 | <1 | >500 | 7.4 | 29 | 59 | <1 | <1 |
Stability and binding affinity are improved upon peptide stapling
We next carried out MD simulations of the stapled dPMI-δ peptides bound to Mdm2. Using the linear dPMI-δ peptide/Mdm2 co-crystal structure as a starting point, staples were modelled into the all-D peptide at 6 sets of residues and subject to MD simulations. The stapled peptides remained stable during the MD simulations and remained largely (∼95%) helical. The three critical residues 6-F-dTrp3, p-CF3-dPhe7 and dLeu11 remained buried in the hydrophobic pocket/binding site of Mdm2 (Fig. 4). The hydrocarbon linkers remained largely exposed to solvent without engaging the Mdm2 surface; this contrasts with some of the L-amino acid stapled peptides where the staples contributed to the binding by engaging with the surface of Mdm2.33,54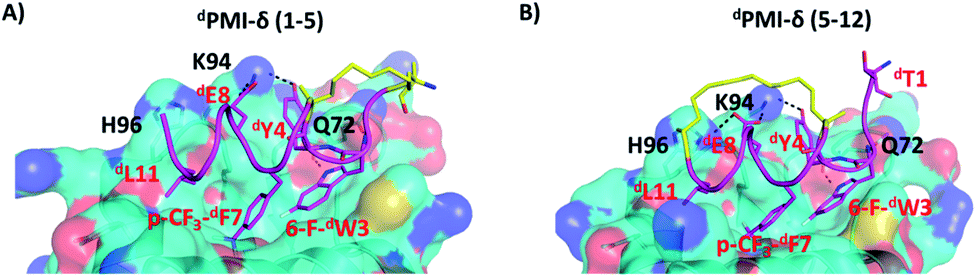 | ||
| Fig. 4 Structural representation of MD snapshot of (A) dPMI-δ(1–5)–Mdm2 and (B) dPMI-δ(5–12)–Mdm2 complex. Mdm2 is shown as surface and bound peptide is shown as cartoon with interacting residues highlighted in sticks. The hydrocarbon linker is highlighted in yellow. Hydrogen bond interactions are shown as dotted lines (black). | ||
Next, the ability of linear and stapled dPMI-δ peptides to bind to Mdm2 was measured using fluorescence polarization (FP) using Mdm2 protein (residues 1–125) and a fluorescently-tagged tracer-peptide. As a positive binding control, we used ATSP-7041,34 a validated Mdm2 binder. Previously, we have demonstrated the importance of including detergent in FP assay buffers59 and accordingly explored two concentrations of Tween-20 (0.1%, Fig. S4 and Table S1;† and 0.001%, Fig. 5A and Table 1). For many peptides, the higher detergent concentration resulted in a large shift to weaker binding constants. We noted that this particular series of peptides is fairly apolar (due to a predominance of apolar residues and the hydrocarbon staple) and hypothesized that the observed affinity shift was due to sequestration of these molecules by the detergent micelles. In the lower detergent condition, the linear dPMI-δ peptide displayed strong affinity for Mdm2 with a Kd of ∼41 nM. Two of the six stapled dPMI-δ peptides displayed strong affinity towards Mdm2, two stapled peptides dPMI-δ(1–5) and dPMI-δ(5–12) binding with Kds of ∼17 nM and ∼34 nM respectively. Three of the stapled peptides, dPMI-δ(2–9), dPMI-δ(5–9) and dPMI-δ(6–10) displayed slightly reduced affinities for Mdm2, with Kds of ∼232 nM, ∼142 nM and ∼118 nM respectively. In contrast, the peptide dPMI-δ(2–6), displayed very weak binding with Kd of 7540 nM (Fig. 5A and Table 1). The SPR binding patterns mirrored the 0.001% Tween-20 FP assay, with the linear and stapled dPMI-δ (1–5 staple, and 5–12 staple) binding strongly with a Kd of <1 nM, whereas the other stapled dPMI-δ peptides displayed reduced affinity (peptides dPMI-δ(2–9), dPMI-δ(5–9), dPMI-δ(6–10) with Kd of 7.4 nM, 29 nM, 59 nM respectively) with dPMI-δ(2–6) as a non-binder with Kd > 500 nM (Fig. 5B and Table 1). Models provided an explanation for the reduced binding of dPMI-δ(2–6) and dPMI-δ(2–9): a key hydrogen bond between the backbone of 6-fluro-dTrp at position 3 and the sidechain of Q72 is lost when the residue at position 2 is replaced with a stapled linker as the alpha-methyl interferes with and prevents the formation of this hydrogen bond. Our models are unable to demonstrate whether this is a kinetic effect or a thermodynamic effect; nevertheless, ATSP-7041 is also known to lose affinity when Thr2 is mutated to either Aib or N-methyl Thr.60
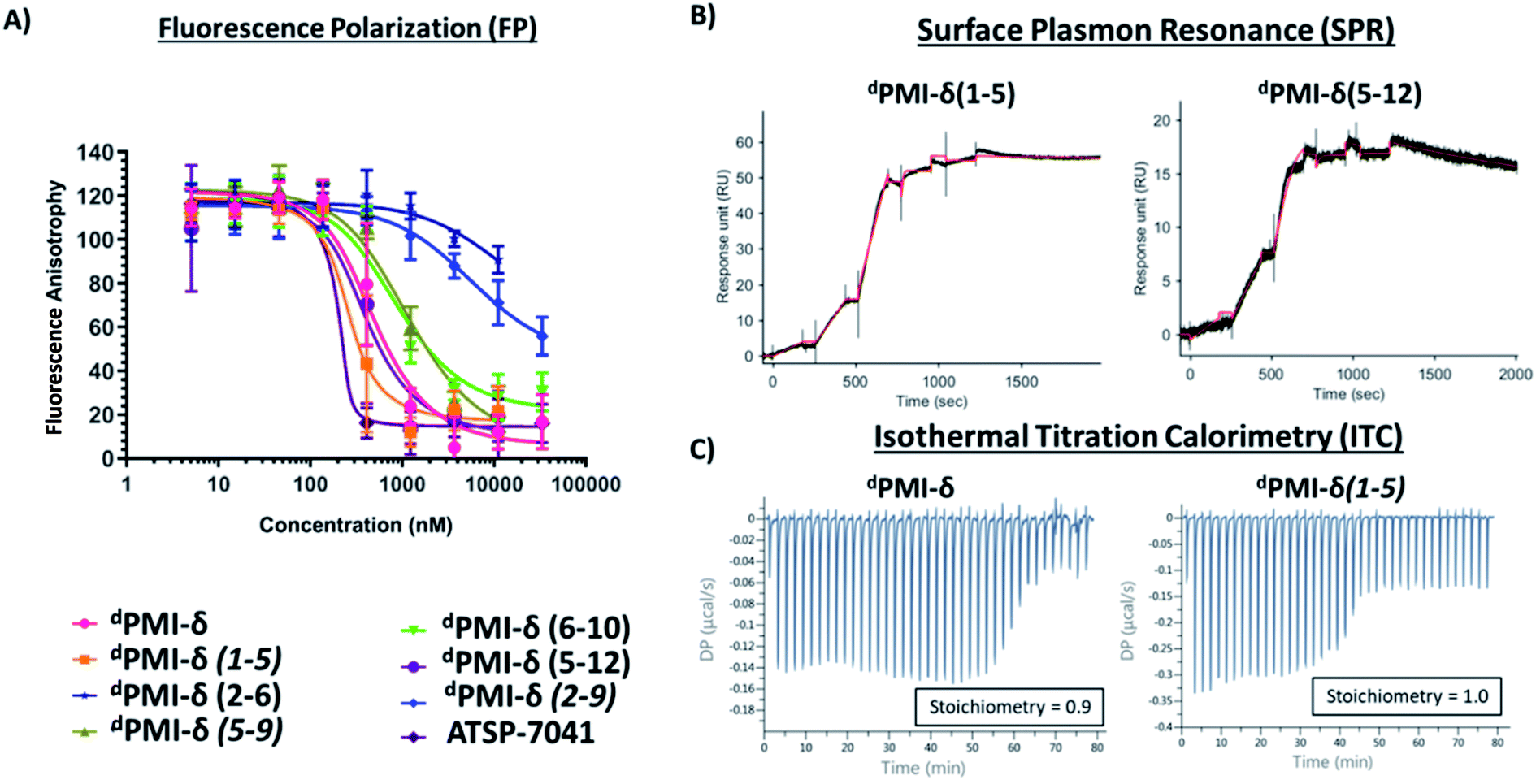 | ||
| Fig. 5 Binding activity of stapled dPMI-δ peptides toward Mdm2 protein determined through various biophysical and biochemical methods. (A) Fluorescence polarization (FP) (0.001% Tween-20) binding analysis of linear, stapled dPMI-δ and ATSP-7041 peptides and Mdm2 protein. Binding activity of peptides toward Mdm2 protein determined through (B) Surface Plasmon Resonance (SPR) and (C) Isothermal Titration Calorimetry (ITC). | ||
The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric binding was further confirmed by ITC experiments (Fig. 5C). Both the linear and stapled dPMI-δ peptides bound to Mdm2 with ΔG = ∼−11 kcal mol−1 (ESI Table 2†), with the enthalpy of binding (ΔH) ranging from −15.8 kcal mol−1 for the dPMI-δ(1–5) to −8.25 kcal mol−1 for the dPMI-δ(5–12). Both the linear dPMI-δ peptide and dPMI-δ(1–5) stapled peptide are less helical in solution, but had a favourable enthalpic contribution to binding. The favourable enthalpy compensates for the entropic penalty paid as the disordered peptide gets ordered during binding to Mdm2. On the other hand, stapled peptides dPMI-δ(5–12) and dPMI-δ(6–10), which had increased helicity (32% and 40% helicity, respectively), make favourable entropic contributions to the binding; this compensates for the loss in favourable enthalpic contributions, with the result that binding is retained.
:1 stoichiometric binding was further confirmed by ITC experiments (Fig. 5C). Both the linear and stapled dPMI-δ peptides bound to Mdm2 with ΔG = ∼−11 kcal mol−1 (ESI Table 2†), with the enthalpy of binding (ΔH) ranging from −15.8 kcal mol−1 for the dPMI-δ(1–5) to −8.25 kcal mol−1 for the dPMI-δ(5–12). Both the linear dPMI-δ peptide and dPMI-δ(1–5) stapled peptide are less helical in solution, but had a favourable enthalpic contribution to binding. The favourable enthalpy compensates for the entropic penalty paid as the disordered peptide gets ordered during binding to Mdm2. On the other hand, stapled peptides dPMI-δ(5–12) and dPMI-δ(6–10), which had increased helicity (32% and 40% helicity, respectively), make favourable entropic contributions to the binding; this compensates for the loss in favourable enthalpic contributions, with the result that binding is retained.
Next the proteolytic stabilities of the stapled and unstapled dPMI-δ peptides were investigated by incubating these molecules in whole cell homogenate and human plasma. As expected, the all-D amino acids composition rendered all peptides (linear and stapled) resistant to proteolytic degradation. Specifically, >90% of each peptide remained detectable in the homogenate during the 4 hour incubation (Fig. S5†). Small decreases in peptide concentrations over time were attributed to sample loss due to binding to Labware and instrument surfaces rather than through proteolysis. Similarly both the linear and stapled all-D peptides remained stable in human plasma, with >90% of each peptide detectable during the 4 hour incubation (Fig. S5†). Consistent with related analogues60 the ATSP-7041 peptide was found to be resistant to proteolytic degradation with ∼90% of the peptide detectable in the homogenate and human plasma during the 4 hour incubation. Thus, stapled all-L amino acid peptides can achieve protease stability but typically only after extensive and time-consuming optimization for drug-like properties. Indeed, not all peptides (including stapled peptides) made up of L-amino acids are resistant to proteolysis, and stability remains a significant hurdle in the development of these peptides into drug-like molecules.39,42 In contrast, all-D peptides (both linear and stapled) have inherent stability, thus allowing researchers to focus on other attributes required for a clinic-ready molecule.
Cellular uptake of all-D stapled peptides
To investigate the effect of peptide stapling in the cellular context, linear and stapled dPMI-δ peptides were added to HCT116 cells with a stably transfected p53-responsive β-lactamase reporter gene. After 16 hours of peptide incubation, no p53 activation was observed for the linear dPMI-δ peptide, even at the highest concentration tested (50 μM). In contrast, three of the six stapled dPMI-δ peptides showed dose responsive increases in p53 activity, while the other three were inactive across the range of peptide concentrations tested (Fig. 6A).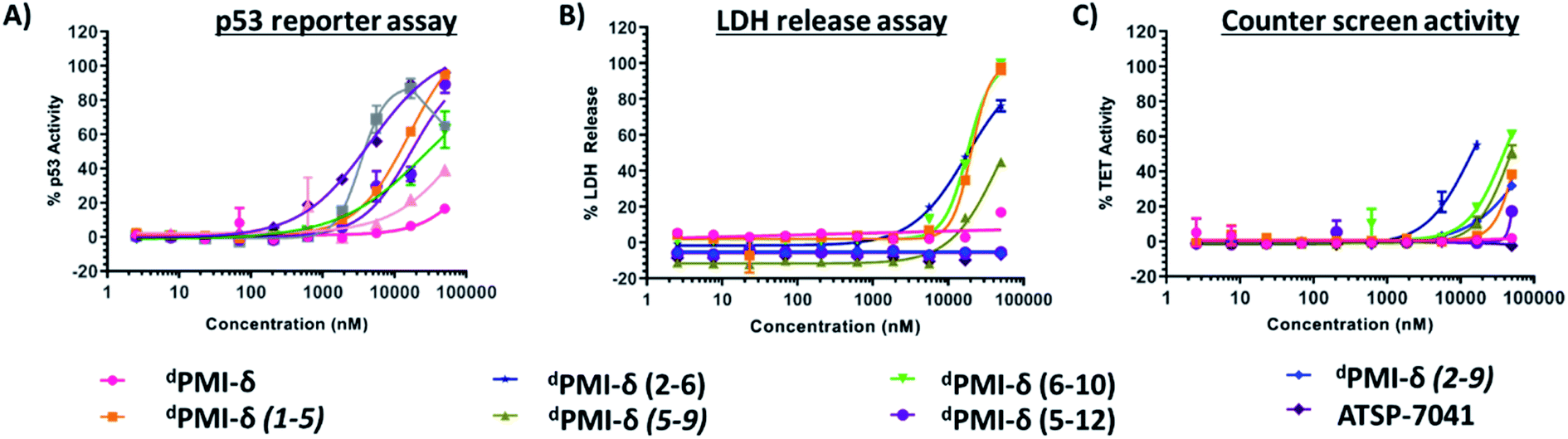 | ||
| Fig. 6 (A) Linear, stapled dPMI-δ and ATSP-7041 peptides titrated on to HCT116 p53 reporter cells and p53 transcriptional activation assessed in the presence of 2% of serum. (B) Linear, stapled dPMI-δ and ATSP-7041 peptides titrated on to HCT116 cells and LDH release measured. (C) Activity of linear, stapled dPMI-δ and ATSP-7041 peptides measured in a counter screen. | ||
Cellular activity correlated well with the biophysical and biochemical data. Stapled peptides dPMI-δ(1–5), dPMI-δ(5–12) and dPMI-δ(6–10) bound Mdm2 well (with Kds of 17 nM, 34 nM and 118 nM respectively from the FP assay) and also entered cells, demonstrating measurable cellular activation of p53 with EC50s of 13.7 μM, 21.3 μM and 30.3 μM respectively (Table 2). While these peptides clearly cross the cell membrane and activate p53, in contrast, the linear peptide (dPMI-δ) and the stapled analogues, dPMI-δ(5–9) and dPMI-δ(2–9), do not appear to enter cells as they have sub-micromolar Mdm2 affinity but are inactive in the p53 cell reporter assay. We were not able to ascertain whether dPMI-δ(2–6) enters cells since it does not bind Mdm2.
| Peptide | dPMI-δ | dPMI-δ(1–5) | dPMI-δ(2–6) | dPMI-δ(2–9) | dPMI-δ(5–9) | dPMI-δ(6–10) | dPMI-δ(5–12) | ATSP-7041 |
| P53 activity EC50 (μM) (2% FBS, 16 h) | >50 | 13.7 (1.2) | >50 | >50 | >50 | 30.3 (3.1) | 21.3 (2.6) | 4.2 (2.1) |
| LDH release EC50 (μM) (2% FBS, 16 h) | >50 | 24 (3) | 17.7 (1.9) | >50 | 46.5 (1.2) | 18.5 (0.7) | >50 | >50 |
| Counter screen EC50 (μM) (2% FBS, 16 h) | >50 | >50 | 13.9 (4.2) | >50 | 47 (3.0) | 39 (4.0) | >50 | >50 |
Membrane distribution and counterscreen activity of all-D stapled peptides
Macrocyclic peptides that are cell permeable are often hydrophobic in nature,60,61 a property that can impart an ability to disrupt the outer membrane and result in cellular leakage.61,62 To assess whether the results from our p53 reporter assay were potentially compromised by membrane damage, we carried out a membrane integrity assay (lactate dehydrogenase (LDH) release) under identical conditions to our p53 cellular assay. The linear dPMI-δ peptide which did not show any p53 cellular activity also did not show any LDH leakage at concentrations as high as 50 μM (Fig. 6B and Table 2). The stapled peptides, dPMI-δ(2–9) and dPMI-δ(5–9) also did not cause LDH leakage, even at concentrations as high as 50 μM. The stapled peptides dPMI-δ(2–9) and dPMI-δ(5–9) which were weaker binders in biochemical assays and did not result in any cell activity also did not cause LDH release, suggesting that these two stapled peptides are cell impermeable. Interestingly the most cell active stapled peptide dPMI-δ(5–12), didn't cause any LDH leakage, suggesting that the activity observed in the p53 receptor activation assay is through intracellular target engagement. Peptide dPMI-δ(2–6), a non-binder of Mdm2 and without any measurable cell activity, induced LDH leakage with EC50 17.7 μM. Peptides dPMI-δ(1–5) and dPMI-δ(6–10), binders of Mdm2 with cell efficacy, also resulted in considerable LDH leakage with EC50 of 24 μM, and 18.5 μM respectively. In fact, the EC50 observed in the p53 reporter activity assays is similar to the EC50 observed for the LDH leakage assays, indicating that these two stapled peptides cause membrane disruption and the readout of the p53 reporter assay may not be due to intracellular target engagement but instead due to the nonspecific cytotoxicity resulting from plasma membrane lysis. Importantly, although LDH leakage can also be a consequence of p53-mediated apoptosis, this effect is cell line dependent. In this case, we have excluded this possibility as ATSP-7041, our validated control peptide, did not result in LDH leakage. Overall, we have demonstrated that dPMI-δ(5–12) enters the cells without membrane disruption, engages the Mdm2–p53 complex, resulting in p53 reporter activity.To further validate intracellular target engagement, we carried out a counterscreen assay with an identical reporter gene but one whose expression is independent of p53 activation. Most of the stapled dPMI-δ peptides including the linear peptide had EC50 values > 50 μM (Fig. 6C). For the linear dPMI-δ peptide and stapled peptides dPMI-δ(5–9) and dPMI-δ(2–9), this result was unsurprising as these molecules appear to be cell impermeable. Interestingly, stapled peptides dPMI-δ(1–5) and dPMI-δ(6–10) each demonstrated significant activity in the reporter assay and LDH leakage assay, and didnot exhibit activity or were weakly active (dPMI-δ(6–10) 39 μM) in the counterscreen assay. Stapled peptide dPMI-δ(2–6) which had no activity in the reporter assay, demonstrated significant activity in the counterscreen assay and is similar to the EC50 observed for the LDH leakage assays. dPMI-δ(5–12) did not exhibit any activity in the counterscreen assay, with EC50 > 50 μM.
Design, synthesis, binding and cellular activity of double staple and stitched dPMI-δ peptides
Encouraged by the results of the stapled dPMI-δ peptides, with particular interest in dPMI-δ(5–12) which showed on-target cellular activity without confounding activities in the LDH release or counterscreen assays, we hypothesized whether incorporation of an additional staple would confer further improvements in binding and cellular activity. Recent studies have highlighted the limitations of peptides carrying single staples including low cell permeability, low proteolytic stability and low cellular activity and have shown that these can be overcome with the introduction of an additional staple.39,40,63 In such bicyclic arrangements, two pairs of hydrocarbon stapling residues are incorporated into a single peptide sequence. To avoid any cross reactivity during olefin metathesis, sufficient spacing between the two pairs of non-natural amino acid staple precursors are required, often resulting in a longer peptide sequence.Several double-stapled peptides have been shown to successfully inhibit pathways in Rab8a GTP-ase,38 HIV-1,39 respiratory syncytial virus entry,40,63 Ral GTP-ase,64 estrogen receptor-α65 and BCL9.66 All these peptides exhibited increased helicity, increased proteolytic resistance and increased binding as compared to the corresponding single stapled peptides. Some of these double-stapled peptides even demonstrated enhanced cell permeability. Double-stapled peptides can also be designed with a common attachment/anchoring point and peptides with such contiguous hydrocarbon staples are also referred to as “stitched” peptides.67 Recently Hilinski et al.68 reported the synthesis of stitched peptides using RCM reactions that exhibited improvements in thermal and chemical stability, proteolytic stability and cell permeability. From the 6 stapled peptides designed we found that two, dPMI-δ(1–5) and dPMI-δ(5–12), both exhibited improved binding and cellular properties. We introduced an addition staple between positions 9 and 12 (i + 3) in dPMI-δ(1–5) resulting in a dPMI-δ(1–5,9–12) double stapled peptide (Fig. 7A and B). Combining dPMI-δ(1–5) and dPMI-δ(5–12) resulted in a stitched peptide, dPMI-δ(1–5–12), with the common attachment point for the two staples localised at residue 5 (Fig. 7A). As expected the CD spectra of the stitched dPMI-δ(1–5–12) showed increased helicity (52% helicity, Fig. 7C and Table 3). This agrees with reports on other peptides showing that the stitched and double stapled peptides often display increased helicity compared to the single stapled peptides.38–40,63–67 Curiously, the stitched scrambled peptide had only 28% helicity. In contrast the double stapled peptide dPMI-δ(1–5,9–12) did not show enhanced helicity (20% helicity, Fig. 7C and Table 3). The binding of the stitched (dPMI-δ(1–5–12)) and double stapled (dPMI-δ(1–5,9–12)) peptides to Mdm2 was first confirmed by SPR experiments. Both these peptides displayed tight binding with Kd of <1 nM, suggesting that the addition of a second staple was well tolerated (Fig. 7D and Table 3); no binding was observed with the scrambled version of the stitched peptide.
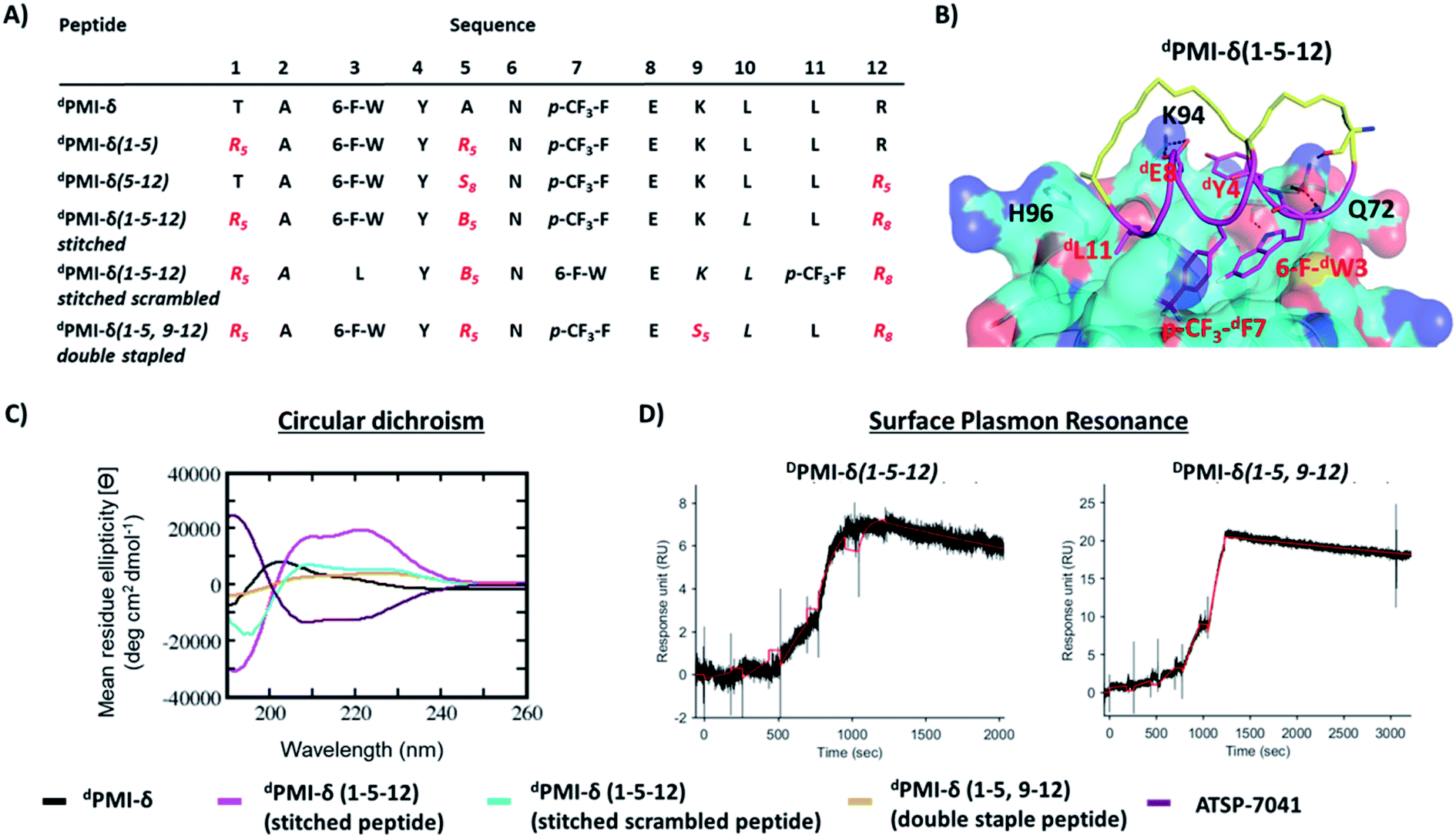 | ||
| Fig. 7 (A) Sequences of linear, stapled and stitched dPMI-δ peptides are shown. (B) MD snapshot of stitched dPMI-δ(1–5–12)–Mdm2 complex. Mdm2 is shown as surface and bound peptide (magenta) is shown as cartoon with interacting residues highlighted in sticks. The hydrocarbon stitch (linker) is highlighted in yellow. Hydrogen bond interactions are shown as dotted lines (black). (C) Secondary structure analysis of linear, double stapled, stitched dPMI-δ and ATSP-7041 peptides determined through circular dichroism (CD). Note that this CD spectra of linear, double stapled and stitched dPMI-δ peptides is inverted, as expected for a peptide consisting of D-amino acids only. (D) Binding activity of double stapled and stitched dPMI-δ peptides toward Mdm2 protein determined through SPR. | ||
| Peptide | dPMI-δ | dPMI-δ(1–5) | dPMI-δ(5–12) | dPMI-δ(1–5–12) stitched | dPMI-δ(1–5–12) stitched scrambled | dPMI-δ(1–5,9–12) double staple | ATSP-7041 |
| CD (% helicity) | 20.4 | 24.7 | 31.8 | 52.1 | 28 | 20 | 49.6 |
| FP Mdm2 Kd (nM) | 41.4 (1.2) | 16.8 (1.3) | 33.9 (1.4) | 62.9 (1.1) | >10000 |
1.5 (1.5) | 33.8 (1.6) |
| SPR Kd (nM) | <1 | <1 | <1 | <1 | >500 | <1 | <1 |
| P53 activity EC50 (μM) (2% FBS, 16 h) | >50 | 13.7 (1.2) | 21.3 (2.6) | 4 (0.4) | >50 | 34.6 (7.6) | 4.2 (2.1) |
| LDH release EC50 (μM) (2% FBS, 16 h) | >50 | 24 (3) | >50 | >50 | >50 | >50 | >50 |
| Counter screen EC50 (μM) (2% FBS, 16 h) | >50 | >50 | >50 | >50 | >50 | >50 | >50 |
Both the stitched dPMI-δ(1–5–12) and double-stapled dPMI-δ(1–5,9–12) peptides bound Mdm2 with Kd of 63 nM and 1.5 nM in FP assay (0.001% Tween-20) (Fig. 8A and Table 3). This confirms that the additional staple retains (in the case of stitched dPMI-δ(1–5–12)) and enhances (in the case of double-stapled dPMI-δ(1–5,9–12)) the target engagement by these peptides. dPMI-δ(1–5–12) displayed enhanced cellular activity with EC50 of 4 μM (Fig. 8B and Table 3), at 16 h, a three to five-fold increase compared to the single stapled peptides (dPMI-δ(1–5), dPMI-δ(5–12)). Similar increases in cell permeability for a stitched peptide have been reported earlier.67 The stitched scrambled peptide did not show any binding or cellular activity (Fig. 8B). However the double-stapled peptide dPMI-δ(1–5,9–12) didn't have significant cellular activity with an EC50 of 34.6 μM, at 16 h (Fig. 8B and Table 3). Although recent studies have reported that the double-stapled peptides appear to follow the same trend as their single-stapled counterparts,38–40,63–65 lack of cell activity observed for the double-stapled peptide here, demonstrate that enhanced cellular activity is not uniform. No detectable LDH leakage was observed, even at concentrations as high as 50 μM (Fig. 8C and Table 3), and there was negligible counter screen activity, confirming that the designed stitched peptide dPMI-δ(1–5–12) enter cells without membrane disruption and result in the activation of p53 by inhibiting the Mdm2–p53 complex (Fig. 8D and Table 3). The intracellular target engagement of stitched dPMI-δ(1–5–12) peptide was further validated using western-blot analysis. Stabilisation of Mdm2 and activation of p53 was observed for dPMI-δ(1–5–12) and ATSP-7041, whereas the dPMI-δ linear peptide failed to do so (Fig. S6†).
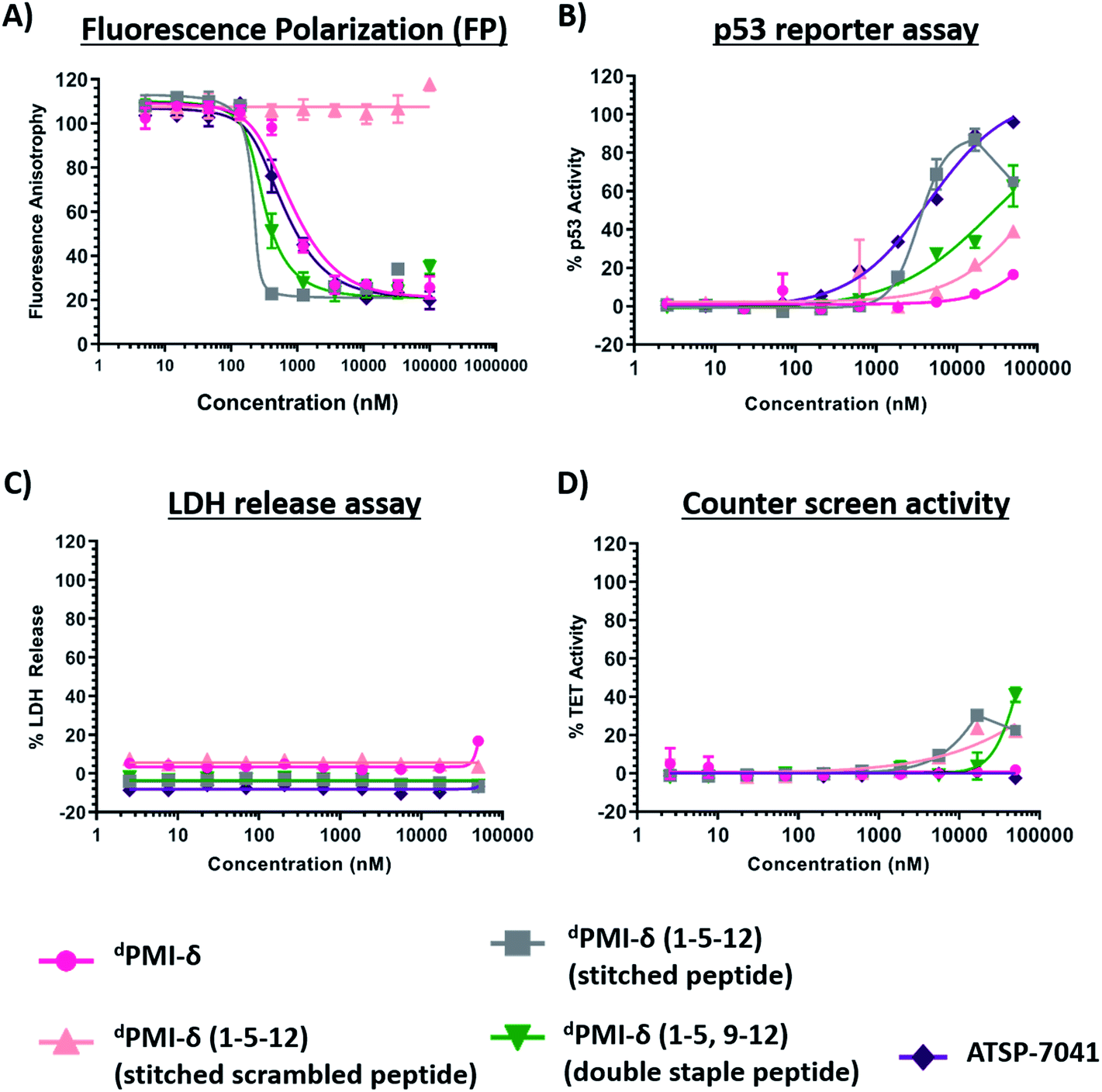 | ||
| Fig. 8 Binding activity of linear, double stapled, stitched dPMI-δ and ATSP-7041 peptides toward Mdm2 protein determined through (A) fluorescence polarization (FP) (0.001% Tween-20) assay. (B) Linear, double stapled, stitched dPMI-δ and ATSP-7041 peptides were titrated on to HCT116 p53 reporter cells and p53 transcriptional activation assessed in the presence of 2% serum. (C) Linear, double stapled, stitched dPMI-δ and ATSP-7041 peptides were titrated on to HCT116 cells and LDH release measured. (D) Activity of linear, double stapled, stitched dPMI-δ and ATSP-7041 peptides were measured in a counter screen. | ||
We also tested the ability of dPMI-δ, dPMI-δ(1–5,9–12), dPMI-δ(1–5–12) and scrambled dPMI-δ(1–5–12) peptides to inhibit the p53 (1–52)–Mdm2 interaction in a yeast 2-hybrid (Y2H) assay optimized for detecting small-molecule inhibitors.68 In this assay, inactivation of p53(1–52)/Mdm2 interaction inhibits growth of yeast cells in a nutrient medium lacking histidine and adenine.68 Consistent with our observations in mammalian cells, the linear dPMI-δ peptide and the double stapled dPMI-δ(1–5,9–12) peptide had no effect on the p53–Mdm2 interaction (Fig. 9A and B). In contrast, the dPMI-δ(1–5–12) peptide completely prevented the growth of yeast cells expressing p53/Mdm2 at 12.5 μM (Fig. 9C, left panel). This inhibition was specific as the dPMI-δ(1–5–12) peptide had no effect on interaction between two yeast kinetochore proteins Csm1 and Dsn1 (Fig. 9C, right panel). Moreover, a scrambled version of dPMI-δ(1–5–12) had no effect on the p53–Mdm2 interaction (Fig. 9D). Our results indicate that dPMI-δ(1–5–12) crosses both the cell wall and plasma membrane and specifically inhibits the p53(1–52)/Mdm2 interaction in yeast cells.
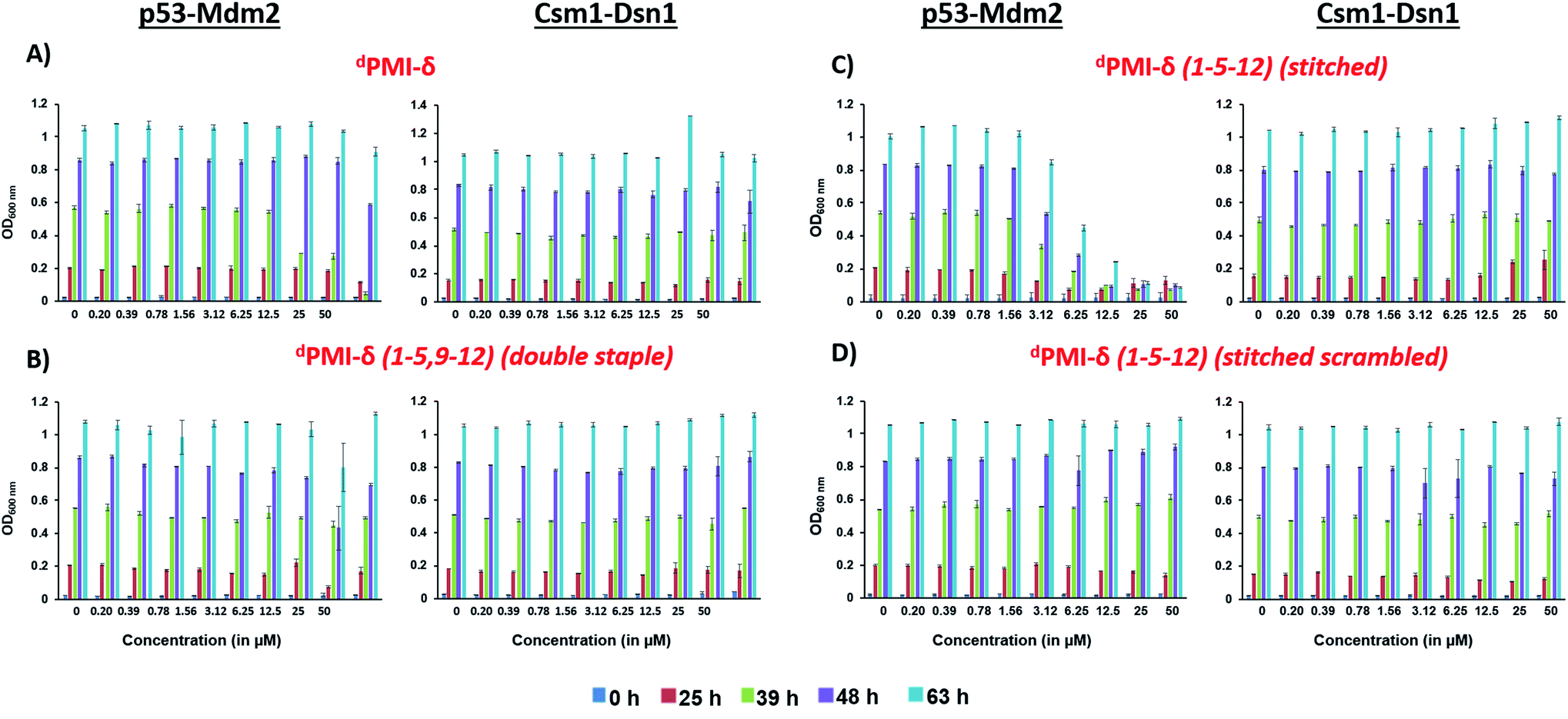 | ||
| Fig. 9 Overnight cultures of ABC9Δ strain containing plasmids encoding either Gal4 AD-p53 (1–52)/Gal4 BD-Mdm2 or Gal4 AD-Csm1/Gal4 BD-Dsn1 were inoculated at OD600 = 0.2 into selective medium containing DMSO or dPMI-δ or dPMI-δ(1–5,9–12), dPMI-δ(1–5–12) and scrambled dPMI-δ(5–12) peptides at the indicated concentrations in duplicate. For each strain, growth as measured by average OD600 of duplicate cultures is plotted at different time points following inoculation (0, 25, 39, 48, and 63 h). Ends of the vertical bar indicate the OD600 values of the duplicate cultures. Data for the dPMI-δ, dPMI-δ(1–5,9–12), dPMI-δ(1–5–12) and scrambled dPMI-δ(1–5–12) peptides are shown in panels (A), (B), (C) and (D) respectively. For each peptide, data showing their effect on p53–Mdm2 and Csm1–Dsn1 interactions are presented on the left and right respectively. | ||
Dual inhibition by dPMI-δ peptides
Mdm4 is homologous to Mdm2 and is also a negative regulator of p53, often found overexpressed in some cancer cells. Studies have shown that dual inhibition (of Mdm2 and Mdm4) appears to be critical for full activation of p53-dependent tumor suppression.34,35,37 Thus, we were interested to know if the all-D peptides had dual-inhibitory properties. As a control, the single stapled peptide ATSP-7041, a validated Mdm2/Mdm4 binder, was observed to bind to Mdm4 with Kd 9.6 nM. The N-terminal domains of Mdm2 and Mdm4 share high sequence similarity (∼75%) (Fig. 10A) and structural similarity (RMSD: ∼1 Å) with most of the residues in the binding pocket conserved (Fig. 10A). Therefore we generated a model of the dPMI-δ peptide bound to a structure of Mdm4 using the structure of the complex between dPMI-δ and Mdm2 as template. Models of the stapled/stitched dPMI-δ peptide bound to Mdm4 were generated by incorporating appropriate linkers in the dPMI-δ–Mdm4 structure. We next carried out MD simulations of the (un)stapled/stitched dPMI-δ peptides bound to Mdm4. The stapled peptides remained stable during the MD simulations and remained largely (∼95%) helical. The three critical residues 6-F-dTrp3, p-CF3-dPhe7 and dLeu11 remained buried in the hydrophobic pocket/binding site of Mdm4 (Fig. 10B). The hydrocarbon linkers remained largely exposed to solvent without engaging the Mdm4 surface. The binding of dPMI-δ peptides with Mdm4 was further confirmed by FP assay.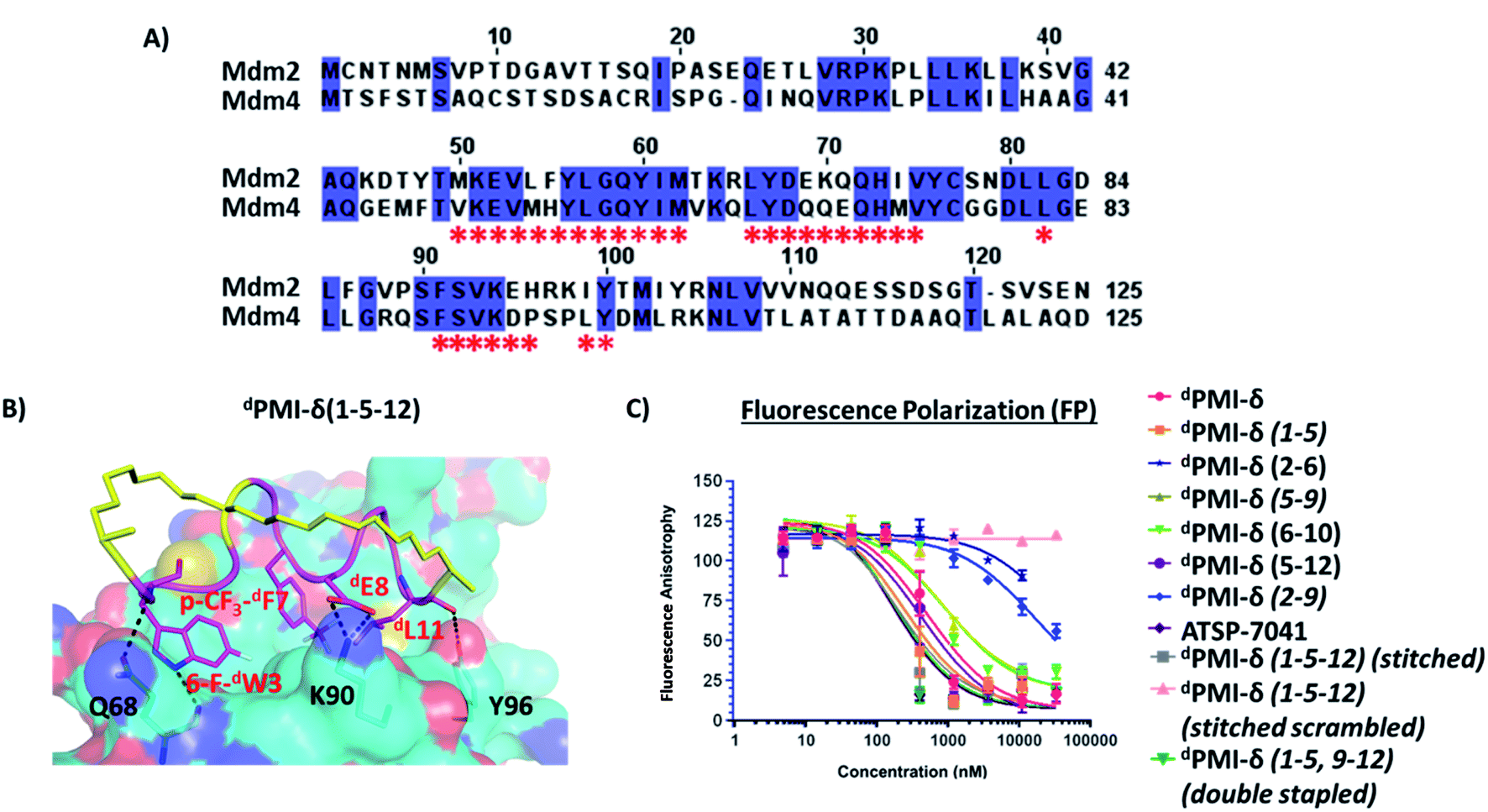 | ||
| Fig. 10 (A) Sequence comparison of N-terminal domains of Mdm2 and Mdm4. Identical residues are highlighted and binding pocket residues (residues that are within 6 Å of bound peptide) are highlighted as *. (B) MD snapshot of stitched dPMI-δ(1–5–12)–Mdm4 complex. Mdm4 is shown as surface and bound peptide is shown as cartoon with interacting residues highlighted as sticks. The hydrocarbon stitch (linker) is highlighted in yellow. Hydrogen bond interactions are shown as dotted lines (black). (C) Fluorescence polarization (FP) (0.001% Tween-20) binding analysis of linear, stapled, stitched dPMI-δ and ATSP-7041 peptides and Mdm4 protein. | ||
In the FP assay, the Mdm4 binding data mirrored that of Mdm2, in both assay buffers. Similar to Mdm2, the higher detergent concentration led to weaker binding affinities, something we attribute to micellar sequestration (Fig. S4 and Table S1† for 0.1% Tween-20; Fig. 10 and Table 4 for 0.001% Tween-20). In the presence of the lower detergent concentration, the linear and stapled dPMI-δ(1–5), dPMI-δ(5–9), dPMI-δ(6–10), dPMI-δ(5–12) peptides bound strongly with Kds of 32 nM, 15 nM, 49 nM, 59 nM and 27 nM, respectively. The other two stapled peptides dPMI-δ(2–6), dPMI-δ(2–9), displayed weaker binding (Fig. 10 and Table 4) with Kds of 2355 nM and 1037 nM, respectively in the FP assay. Both the stitched dPMI-δ(1–5–12) and double-stapled dPMI-δ(1–5,9–12) peptides displayed strong affinity towards Mdm4, with Kd of 12 nM and 10.2 nM respectively (Fig. 10 and Table 4). In conclusion, the stitched dPMI-δ(1–5–12) and double-stapled dPMI-δ(1–5,9–12) peptides are high affinity dual inhibitors of Mdm2 and Mdm4.
| Peptide | dPMI-δ | dPMI-δ(1–5) | dPMI-δ(2–6) | dPMI-δ(2–9) | dPMI-δ(5–9) | dPMI-δ(6–10) | dPMI-δ(5–12) | dPMI-δ(1–5–12) stitched | dPMI-δ(1–5–12) stitched scrambled | dPMI-δ(1–5,9–12) double stapled | ATSP-7041 |
| FP Mdm4 Kd (nM) | 32.2 (10.9) | 15.1 (6.5) | 2355 (577) | 1037 (165.6) | 49.0 (10.8) | 58.8 (18.8) | 27.0 (14.7) | 11.8 (4.1) | >10000 |
10.2 (4.2) | 9.6 (3.8) |
Crystal structure of Mdm2–dPMI-δ(1–5,9–12) complex
We tried to obtain crystal structures of the stitched peptide dPMI-δ(1–5–12) in complex with Mdm2 (6–125) and of the double stapled peptide dPMI-δ(1–5,9–12) in complex with Mdm2 (6–125). We could only obtain crystals for the latter and were able to resolve its structure (Fig. 11A and B). The elucidated structure possessed only a single copy of the Mdm2–dPMI-δ(1–5,9–12) complex in the asymmetric unit. The p53 peptide binding groove on Mdm2 was occupied by a single molecule of dPMI-δ(1–5,9–12), which interacted with Mdm2 by projecting the following three residues 6-F-dTrp3, p-CF3-dPhe7 and dLeu11 deep into the hydrophobic peptide binding site on Mdm2. The Mdm2–dPMI-δ(1–5,9–12) complex was overlaid with the Mdm2–dPMI-δ structure (PDB ID: 3TPX), which demonstrated that the double stapling maintained the overall fold of the bound linear peptide dPMI-δ and its critical interactions with Mdm2 (Fig. 11C). The computationally modelled structure of the Mdm2–dPMI-δ(1–5,9–12) complex is very similar to the co-crystal structure of the Mdm2–dPMI-δ(1–5,9–12) complex; the peptide RMSD is <1 Å between the two, and the overall fold along with the protein–peptide interactions is in good agreement (Fig. 11D). Further, as predicted, neither hydrocarbon staple linkers engage in any contacts with the surface of Mdm2 and both are exposed into the solvent.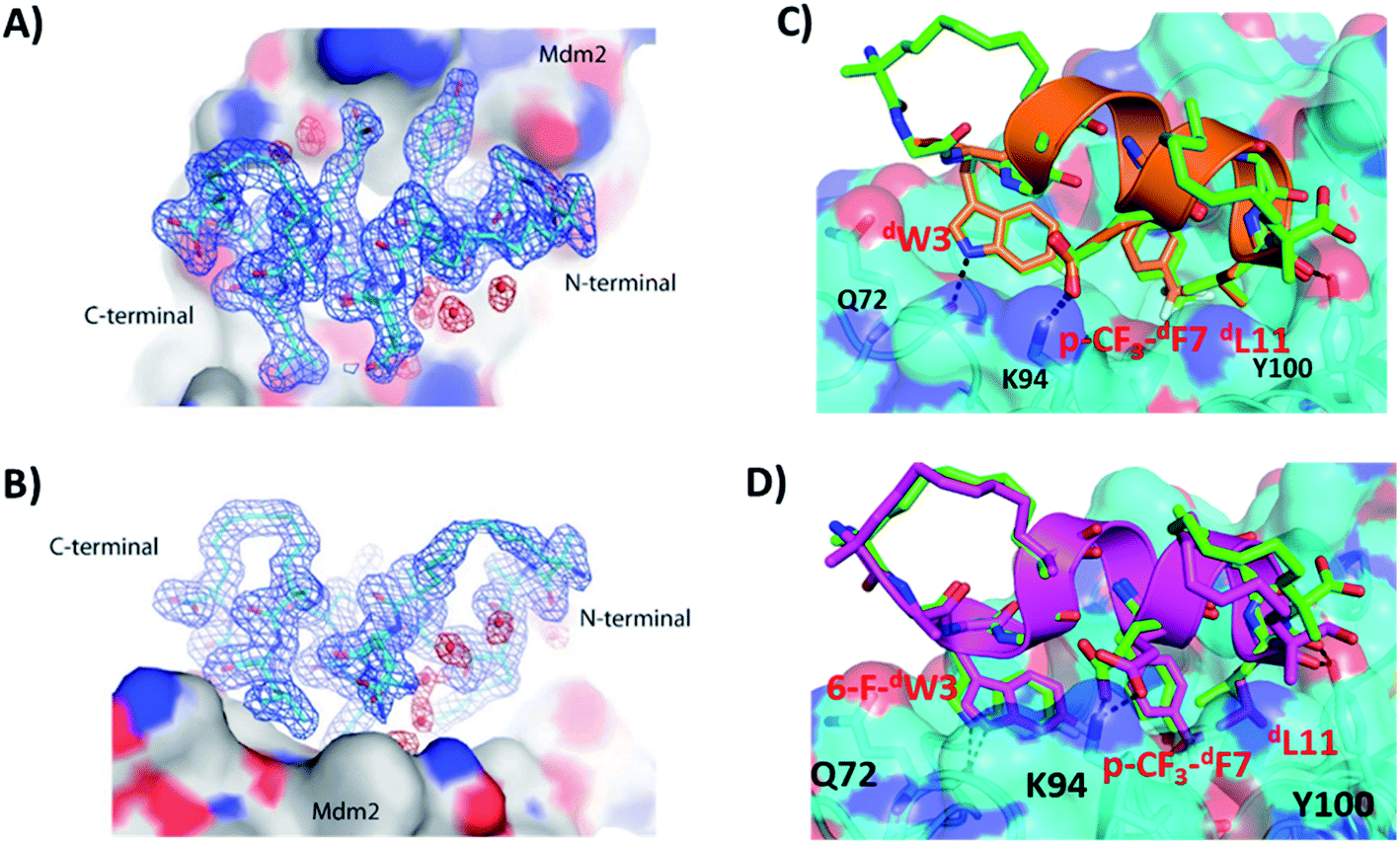 | ||
| Fig. 11 (A) Top down view of dPMI-δ(1–5,9–12) bound to Mdm2. (B) 90° rotation of the dPMI-δ(1–5,9–12)–Mdm2 complex orientation shown in panel (A), which clearly depicts the presence of both hydro-carbon staples in dPMI-δ(1–5,9–12). The 2Fo–Fc electron density map (shown with the blue mesh) for dPMI-δ(1–5,9–12) was contoured at 1.0σ. Interfacial structured waters involved in Mdm2–dPMI-δ(1–5,9–12) complex formation are shown using the red mesh. (C) Comparison of crystal structure of the dPMI-δ(1–5,9–12)–Mdm2 complex with the dPMI-δ–Mdm2 structure (PDB ID: 3TPX). Mdm2 is shown as surface (cyan) with the bound dPMI-δ(1–5,9–12) (green) and dPMI-δ (orange) peptides shown as cartoon. The three critical residues (dW3, p-CF3-dF7 and dL11 in dPMI-δ, 6-F-dW3, p-CF3-dF7 and dL11 in dPMI-δ(1–5,9–12)) and residues involved in protein–peptide interactions are shown as sticks and the interactions are highlighted (black dashed lines). (D) Comparison of the computationally modelled structure of the dPMI-δ(1–5,9–12)–Mdm2 complex with the crystal structure of the dPMI-δ(1–5,9–12)–Mdm2 complex. Mdm2 is shown as surface (cyan) with the bound modelled dPMI-δ(1–5,9–12) (magenta) and experimental dPMI-δ(1–5,9–12) (green) peptides shown as cartoon. The three critical residues (6-F-dW3, p-CF3-dF7 and dL11) and residues involved in protein–peptide interactions are shown as sticks and the interactions are highlighted (black dashed lines). | ||
Discussion
Peptide based inhibitors are emerging as next generation therapeutic modalities because of their high target specificity, high biocompatibility and low toxicity. However, liabilities such as conformational instability, proteolytic sensitivity and lack of cell permeability hinder their potential. Stapling peptides to constrain them in their active/bound conformations results in several benefits such as improved stability, target binding and cell permeability. Although stapling L-amino acid peptides can confer resistance to protease-mediated degradation, the effect is often not complete, especially for residues located outside of the staple. On the other hand, D-amino acid peptides show complete resistance to proteolysis, increased stability and bioavailability, hence appear to be suitable for oral administration. Unfortunately, all-D linear peptides, similar to their all-L linear counterparts, generally lack membrane permeability and therefore cellular activity.We hypothesized that a combination of the two strategies (i.e., all D-amino acids and stapling) might provide a robust strategy resulting in molecules that possess all the required properties for intracellular target engagement. Accordingly, we embarked on introducing a hydrocarbon staple into an all-D peptide inhibitor of the p53–Mdm2/Mdm4 interaction that had been discovered using mirror-image phage display.46 Guided by the available crystal structure of dPMI-δ bound to the N-terminal domain of Mdm2, we designed six stapled dPMI-δ peptides using a combination of modelling and molecular simulations. All 6 stapled peptides displayed helicities ranging from 24% to 40% which compared with 21% for the linear counterpart. Five of the six stapled peptides retained binding to Mdm2 while two of the peptides demonstrated increased affinity for Mdm2 in the biophysical and biochemical experiments. One of the peptides also showed enhanced cellular uptake without detectable membrane disruption and disrupted the Mdm2–p53 complex, leading to activation of p53. No correlation was apparent between helicity and binding or with cellular activity, as has also been reported for L-amino acid peptides.
We next decided to introduce a second staple generating a double stapled peptide and a stitched peptide. The stitched peptide displayed the highest helicity (52%) while the double stapled peptide remained unchanged at 20%, similar to that adopted by the linear peptide. Nevertheless, both these peptides displayed increased binding to Mdm2, suggesting that although these molecules bind to the same Mdm2 pocket, their binding mechanisms are different from each other. Specifically, we speculate that conformational selection dominates the binding of the stitched peptide to Mdm2, while induced fit is the dominant mechanism of binding of the double stapled peptide. However only the stitched peptide displayed increased cellular activity, probably resulting from increased cell permeability; the double stapled peptide appears unable to cross the cell membrane. Although stitching resulted in increased helicity, increased affinity and more importantly enables cell permeability, it is not clear how this latter is achieved. Increased hydrophobicity resulting from the hydrocarbon linker of the stitched peptide could be a major driving force, however the lack of permeability for the double stapled peptide which has a longer hydrophobic linker casts doubt on the hydrophobicity–permeability link. Therefore, understanding cell permeability warrants further studies to systematically investigate the factors enabling cell permeability of these peptides.
While stapling appeared to impart cell permeability, some of the stapled peptides caused membrane disruption. Curiously, while all the stapled and stitched peptides displayed reporter activity at 4 h, only 4 peptides (dPMI-δ(1–5), dPMI-δ(5–12), dPMI-δ(6–10), dPMI-δ(1–5–12)) continued to show activity at 16 hours. At the same time, peptides dPMI-δ(6–10) and dPMI-δ(1–5), both binders of Mdm2, cause LDH leakage and hence it is unclear what results in p53 activation: target engagement or membrane disruption, likely some combination of the two.
A counterscreen assay, with a readout that is identical to that in our primary cellular screen, but that is independent of p53 activation, was carried out to find peptides with off-target effects. We found that the peptide dPMI-δ(2–6), a non-binder of Mdm2, and dPMI-δ(6–10), a binder of Mdm2, both disrupt the membrane, and are found to activate p53 even at 16 hours, showing counter screen activity; this could result from membrane disruption or off-target activity, or both. In contrast dPMI-δ(5–12) and the stitched peptide dPMI-δ(1–5–12) showed no membrane disruption or off-target activity, activating p53 through intracellular target engagement. The on-target engagement of stapled and stitched peptides was further validated in western blot assays. Thus, it is important to use a combination of LDH leakage, counter screen assays and target engagement/reporter activation to rule out false positives (that result from off-target engagement and membrane disruption). The yeast assay confirmed the on target activity of the dPMI-δ(1–5–12) peptide adding support to the favourable effects of this approach to peptide stapling on cell permeability. It is remarkable that this peptide has the ability to cross even thick cell walls such as in yeast. Although the molecular mechanisms behind the increased cell permeability of the stitched D-peptide remain unclear, it could be attributed to the increased conformational rigidity and/or increased hydrocarbon content of the peptide.
While we were unable to obtain crystals of the stitched peptide in complex with Mdm2, we successfully resolved the structure of the double stapled peptide in complex with Mdm2. The latter showed that the critical interactions of the peptide with Mdm2 were retained and the hydrocarbon staple linker points into solvent, without engaging in any contacts with Mdm2. Given that our computationally predicted structure matched the crystal structure to within 1 Å, we are confident that our model of the stitched peptide complexed to Mdm2 would also closely mirror the actual complex; the key interactions with Mdm2 are maintained and the staple points into solvent without engaging in any contacts with Mdm2 (in any case the 1–5 staple is common to both structures). Stapling also enabled the peptide to bind to Mdm4 with high affinity, The Mdm4 binding data mirrored the Mdm2 data, resulting in a cell permeable dual inhibitor of Mdm2/Mdm4. It is possible that in the activation assays, binding to Mdm4 likely contributes to p53 activation. Several studies have shown that dual inhibition of Mdm2 and Mdm4 appears to be critical for full activation of p53-dependent tumour suppression.
We finally decided to explore the potential immunogenicity of dPMI-δ(1–5–12) or ATSP-7041, for which we immunised mice with these peptide together with a strong adjuvant, Freund complete Adjuvant. We did not observe any significant induction of peptide-specific IgG or IgM antibodies between the dPMI-δ(1–5–12) and ATSP-7041 immunized versus control mice (Fig. S7†). In contrast, mice immunized with AMA-1, a malaria immunogenic protein, had substantial levels of AMA-1-specific IgG or IgM antibodies. These observations suggest that dPMI-δ(1–5–12) and ATSP-7041 are non-immunogenic, and provide further promise for developing these compounds as potential therapeutics.
The three key properties that are required during the early discovery phase for peptide therapeutics against intracellular targets are (I) binding, (II) stability, and (III) permeability. Simultaneously trying to optimize for multiple properties can be challenging as making an improvement to one property can often compromise another. Choosing an all-D peptide as a lead series simplifies the task and potentially shortens discovery time-lines since such molecules are hyperstable to protease degradation. Furthermore, this work demonstrates that macrocyclization of an all-D linear peptide can enhance both binding and cellular activities, as has been reported for L-amino acid peptides. In this specific case, this workflow has resulted in a molecule that has significant therapeutic potential for p53-WT cancers.
Materials and methods
Peptide design and molecular dynamics simulations were carried out with the AMBER18 package69 following a protocol used earlier.60 Protocols for peptide synthesis, Mdm2/Mdm4 protein production, SPR experiments, ITC experiment, FP assays and cellular studies were taken from a similar study;60 in the current study, we used HCT116 cells. For a more detailed description, please refer to the ESI.†Conflicts of interest
The authors declare no competing financial interest. S. K. and C. S. V. are founder directors of SiNOPSEE Therapeutics.Acknowledgements
We would like to acknowledge Lin Yan and Jerome Hochman for critical reading of the manuscript. The authors thank National Super Computing Centre (NSCC) for computing facilities. This work was supported by MSD and A*STAR (grant IDs, H17/01/a0/010, IAF111213C, H18/01/a0/015, IAF-ICP(I19/01/E0/039)).References
- I. Petta, S. Lievens, C. Libert, J. Tavernier and K. De Bosscher, Modulation of Protein–Protein Interactions for the Development of Novel Therapeutics, Mol. Ther., 2015, 24, 707–718 CrossRef PubMed.
- S. J. Y. Macalino, et al., Evolution of In silico Strategies for Protein-Protein Interaction Drug Discovery, Molecules, 2018, 23, 1963 CrossRef PubMed.
- D. E. Scott, A. R. Bayly, C. Abell and J. Skidmore, Small molecules, big targets: drug discovery faces the protein–protein interaction challenge, Nat. Rev. Drug Discovery, 2016, 15, 533–550 CrossRef CAS.
- L. Nevola and E. Giralt, Modulating protein–protein interactions: the potential of peptides, Chem. Commun., 2015, 51, 3302–3315 RSC.
- J. L. Lau and M. K. Dunn, Therapeutic peptides: historical perspectives, current development trends, and future directions, Bioorg. Med. Chem., 2017, 26, 2700–2707 CrossRef PubMed.
- M. Bakail and F. Ochsenbein, Targeting protein–protein interactions, a wide open field for drug design, C. R. Chim., 2016, 19, 19–27 CrossRef CAS.
- A. Henninot, J. C. Collins and J. M. Nuss, The current state of peptide drug discovery: back to the future, J. Med. Chem., 2018, 61, 1382–1414 CrossRef CAS.
- C. Morrison, Constrained peptides' time to shine?, Nat. Rev. Drug Discovery, 2018, 17, 531–533 CrossRef CAS.
- E. Valeur, et al., New Modalities for Challenging Targets in Drug Discovery, Angew. Chem., Int. Ed., 2017, 56, 10294–10323 CrossRef CAS.
- D. R. Cary, M. Ohuchi, P. C. Reid and K. Masuya, Constrained Peptides in Drug Discovery and Development, J. Synth. Org. Chem., Jpn., 2017, 75, 1171–1178 CrossRef CAS.
- A. Vinogradov, et al., Macrocyclic Peptides as Drug Candidates: Recent Progress and Remaining Challenges, J. Am. Chem. Soc., 2019, 141, 4167–4181 CrossRef CAS PubMed.
- T. K. Sawyer, et al., Macrocyclic α helical peptide therapeutic modality: a perspective of learnings and challenges, Bioorg. Med. Chem., 2018, 26, 2807–2815 CrossRef CAS PubMed.
- L. Perarro, et al., Diversity-Oriented Stapling Yields Intrinsically Cell-Penetrant Inducers of Autophagy, J. Am. Chem. Soc., 2017, 139(23), 7792–7802 CrossRef PubMed.
- L. D. Walensky and G. H. Bird, Hydrocarbon-stapled peptides: principles, practice, and progress, J. Med. Chem., 2014, 57, 6275–6288 CrossRef CAS PubMed.
- Y. S. Tan, D. P. Lane and C. S. Verma, Stapled peptide design: principles and roles of computation, Drug Discovery Today, 2016, 21, 1642–1653 CrossRef CAS PubMed.
- A. M. Ali, et al., Stapled Peptides Inhibitors: A New Window for Target Drug Discovery, Comput. Struct. Biotechnol. J., 2019, 17, 263–281 CrossRef CAS.
- M. Klein, Stabilized helical peptides: overview of the technologies and its impact on drug discovery, Expert Opin. Drug Discovery, 2017, 12, 1117–1125 CrossRef CAS.
- J. Lerge, et al., Stapled peptides as a new technology to investigate protein–protein interactions in human platelets, Chem. Sci., 2018, 9, 4638–4643 RSC.
- W. Xu, Macrocyclized Extended Peptides: Inhibiting the Substrate-Recognition Domain of Tankyrase, J. Am. Chem. Soc., 2017, 139, 2245–2256 CrossRef CAS.
- M. M. Wiedmann, et al., Development of Cell-Permeable, Non-Helical Constrained Peptides to Target a Key Protein–Protein Interaction in Ovarian Cancer, Angew. Chem., Int. Ed., 2017, 56, 524–529 CrossRef CAS.
- L. D. Walensky, et al., A Stapled BID BH3 Helix Directly Binds and Activates BAX, Mol. Cell, 2006, 24, 199–210 CrossRef CAS PubMed.
- T. Okamoto, et al., Stabilizing the Pro-Apoptotic BimBH3 Helix (BimSAHB) does not Necessarily Enhance Affinity or Biological Activity, ACS Chem. Biol., 2013, 8, 297–302 CrossRef CAS.
- G. H. Bird, et al., Distinct BimBH3 (BimSAHB) Stapled Peptides for Structural and Cellular Studies, ACS Chem. Biol., 2014, 9, 831–837 CrossRef CAS.
- R. R. Araghi, et al., Iterative optimization yields Mcl-1-targeting stapled peptides with selective cytotoxicity to Mcl-1-dependent cancer cells, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E886–E895 CrossRef CAS.
- L. Dietrich, et al., Cell permeable stapled peptide inhibitor of Wnt signaling that targets β-catenin protein–protein interactions, Cell Chem. Biol., 2017, 24, 958–968 CrossRef CAS PubMed.
- J. Spiegel, et al., Direct targeting of Rab-GTPase-effector interactions, Angew. Chem., Int. Ed. Engl., 2014, 53, 2498–2503 CrossRef CAS PubMed.
- M. Xie, et al., Structural Basis of Inhibition of ERα-Coactivator Interaction by High-Affinity N-Terminus Isoaspartic Acid Tethered Helical Peptides, J. Med. Chem., 2017, 60, 8731–8740 CrossRef CAS PubMed.
- I. Paola de, et al., Cullin3-BTB interface: a novel target for stapled peptides, PLoS One, 2015, 10(4), 0121149 CrossRef.
- T. Misawa, et al., Structural development of stapled short helical peptides as vitamin D receptor (VDR)-coactivator interaction inhibitors, Bioorg. Med. Chem., 2015, 23, 1055–1061 CrossRef CAS.
- D. Lama, et al., Structural insights reveal a recognition feature for tailoring hydrocarbon stapled-peptides against the eukaryotic translation initiation factor 4E protein, Chem. Sci., 2019, 10, 2489–2500 RSC.
- F. Bernal, A. F. Tyler, S. J. Korsmeyer, L. D. Walensky and G. L. Verdine, Reactivation of the p53 tumor suppressor pathway by a stapled p53 peptide, J. Am. Chem. Soc., 2007, 129, 2456–2457 CrossRef CAS PubMed.
- L. K. Henchey, J. R. Porter, I. Ghosh and P. S. Arora, High specificity in protein recognition by hydrogen-bond-surrogate a-helices: selective inhibition of the p53/MDM2 complex, Chembiochem, 2010, 11, 2104–2107 CrossRef CAS PubMed.
- C. J. Brown, et al., Stapled peptides with improved potency and specificity that activate p53, ACS Chem. Biol., 2013, 8, 506–512 CrossRef CAS PubMed.
- Y. S. Chang, et al., Stapled a-helical peptide drug development: a potent dual inhibitor of MDM2 and MDM4 for p53-dependent cancer therapy, Proc. Natl. Acad. Sci. U.S.A., 2013, 110, E3445–E3455 CrossRef CAS PubMed.
- A. Burgess, et al., Clinical overview of MDM2/X-targeted therapies, Front. Oncol., 2016, 6, 1–7 Search PubMed.
- K. Kojima, J. Ishizawa and M. Andreeff, Pharmacological activation of wild-type p53 in the therapy of leukemia, Exp. Hematol., 2016, 44, 791–798 CrossRef CAS PubMed.
- V. Tisato, R. Voltan, A. Gonelli, P. Secchiero and G. Zauli, MDM2/X inhibitors under clinical evaluation: perspectives for the management of hematological malignancies and pediatric cancer, J. Hematol. Oncol., 2017, 10, 133 CrossRef PubMed.
- P. M. Cromm, et al., Protease-Resistant and Cell-Permeable Double-Stapled Peptides Targeting the Rab8a GTPase, ACS Chem. Biol., 2016, 11, 2375–2382 CrossRef CAS.
- G. H. Bird, et al., Hydrocarbon double-stapling remedies the proteolytic instability of a lengthy peptide therapeutic, Proc. Natl. Acad. Sci. U.S.A., 2010, 107, 14093–14098 CrossRef CAS PubMed.
- V. Gaillard, et al., A Short Double-Stapled Peptide Inhibits Respiratory Syncytial Virus Entry and Spreading, Antimicrob. Agents Chemother., 2017, 61, e02241–e02216 CrossRef CAS.
- S. V. Fiacco, et al., Directed Evolution of Scanning Unnatural-Protease-Resistant (SUPR) Peptides for in vivo Applications, ChemBioChem, 2016, 17, 1643–1651 CrossRef CAS PubMed.
- M. Uppalapati, et al., A Potent d-Protein Antagonist of VEGF-A is Nonimmunogenic, Metabolically Stable, and Longer-Circulating in vivo, ACS Chem. Biol., 2016, 11, 1058–1065 CrossRef CAS.
- H. N. Chang, et al., Blocking of the PD-1/PD-L1 Interaction by a D-Peptide Antagonist for Cancer Immunotherapy, Angew. Chem., Int. Ed., 2015, 54, 11760–11764 CrossRef CAS PubMed.
- B. D. Welch, et al., Design of a potent D-peptide HIV-1 entry inhibitor with a strong barrier to resistance, J. Virol., 2010, 84, 11235–11244 CrossRef CAS PubMed.
- M. Garton, et al., Method to generate highly stable D-amino acid analogs of bioactive helical peptides using a mirror image of the entire PDB, Proc. Natl. Acad. Sci. U.S.A., 2018, 115, 1505–1510 CrossRef CAS PubMed.
- M. Liu, et al., A left handed solution to peptide inhibition of the p53-MDM2 interaction, Angew. Chem., Int. Ed., 2010, 49, 3649–3652 CrossRef CAS PubMed.
- M. Liu, et al., D-peptide inhibitors of the p53-MDM2 interaction for targeted molecular therapy of malignant neoplasms, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14321–14326 CrossRef CAS.
- D. P. Lane, p53, guardian of the genome, Nature, 1992, 358, 15–16 CrossRef CAS PubMed.
- O. karni-Schmidt, M. Lokshin and C. Prives, The roles of MDM2 and MDM4 in cancer, Annu. Rev. Pathol., 2016, 11, 617–644 CrossRef CAS PubMed.
- D. Pei, Y. Zhang and J. Zheng, Regulation of p53: a collaboration between Mdm2 and Mdm4, Oncotarget, 2012, 3, 228–235 CrossRef PubMed.
- F. Funda Meric-Bernstam, et al., Phase I trial of a novel stapled peptide ALRN-6924 disrupting MDM4- and MDM2- mediated inhibition of WT p53 in patients with solid tumors and lymphomas, J. Clin. Oncol., 2017, 35, 2505 Search PubMed.
- C. Zhan, et al., An ultrahigh affinity d-peptide antagonist of MDM2, J. Med. Chem., 2012, 55, 6237–6241 CrossRef CAS.
- T. Y. Yuen, et al., Stereoisomerism of stapled peptide inhibitors of the p53-Mdm2 interaction: an assessment of synthetic strategies and activity profiles, Chem. Sci., 2019, 10, 6457–6466 RSC.
- Y. S. Tan, et al., Benzene Probes in Molecular Dynamics Simulations Reveal Novel Binding Sites for Ligand Design, J. Phys. Chem. Lett., 2016, 7, 3452–3457 CrossRef CAS PubMed.
- S. Kannan, et al., Inhibiting S100B(ββ) for Activating Wild-Type p53: Design of Stapled Peptides, ACS Omega, 2019, 4, 5335–5344 CrossRef CAS.
- S. Kannan and M. Zacharias, Enhanced sampling of peptide and protein conformations using replica exchange simulations with a peptide backbone biasing-potential, Proteins, 2007, 66, 697–706 CrossRef CAS.
- K. Ostermeir and M. Zacharias, Hamiltonian replica-exchange simulations with adaptive biasing of peptide backbone and side chain dihedral angles, J. Comput. Chem., 2014, 35, 150–158 CrossRef CAS.
- C. E. Schafmeister, J. Po and G. L. Verdine, An all-hydrocarbon cross-linking system for enhancing the helicity and metabolic stability of peptides, J. Am. Chem. Soc., 2000, 122, 5891–5892 CrossRef CAS.
- S. Ng, et al., De-risking drug discovery of intracellular targeting peptides: screening strategies to eliminate false-positive hits, ACS Med. Chem. Lett., 2020 Search PubMed , in press.
- A. Patridge, et al., Incorporation of Putative Helix-Breaking Amino Acids in the Design of Novel Stapled Peptides: Exploring Biophysical and Cellular Permeability Properties, Molecules, 2019, 24, 2292 CrossRef PubMed.
- A. Furukawa, et al., Passive membrane permeability in cyclic peptomer scaffolds is robust to extensive variation in side chain functionality and backbone geometry, J. Med. Chem., 2016, 59, 9503–9512 CrossRef CAS.
- Y. C. Li, et al., A versatile platform to analyze low-affinity and transient protein-protein interactions in living cells in real time, Cell Rep., 2014, 9, 1946–1958 CrossRef CAS.
- G. H. Bird, et al., Mucosal delivery of a double-stapled RSV peptide prevents nasopulmonary infection, J. Clin. Invest., 2014, 124, 2113–2124 CrossRef CAS.
- J. C. Thomas, et al., Inhibition of Ral GTPases using a stapled peptide approach, J. Biol. Chem., 2016, 291, 18310–18325 CrossRef CAS.
- T. E. Speltz, et al., A “cross-stitched” peptide with improved helicity and proteolytic stability, Org. Biomol. Chem., 2018, 16, 3702–3706 RSC.
- S. A. Kawamoto, et al., Design of triazole-stapled BCL9 α-helical peptides to target the β-catenin/B-cell CLL/lymphoma 9 (BCL9) protein-protein interaction, J. Med. Chem., 2012, 55, 1137–1146 CrossRef CAS.
- G. J. Hilinski, et al., Stitched α-helical peptides via bis ring-closing metathesis, J. Am. Chem. Soc., 2014, 136, 12314–12322 CrossRef CAS.
- J. H. Wong, et al., A yeast two-hybrid system for the screening and characterization of small-molecule inhibitors of protein-protein interactions identifies a novel putative Mdm2-binding site in p53, BMC Biol., 2017, 15, 108 CrossRef PubMed.
- D. Case, D. A. Pearlman, J. W. Caldwell, et al., Amber 18, University of California, San Francisco, 2018 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc06383h |
| This journal is © The Royal Society of Chemistry 2020 |