
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultiple reactivities of flavonoids towards pathological elements in Alzheimer's disease: structure–activity relationship†
Geewoo
Nam‡
a,
Mannkyu
Hong‡
 bc,
Juri
Lee
b,
Hyuck Jin
Lee
d,
Yonghwan
Ji
a,
Juhye
Kang
b,
Mu-Hyun
Baik
*bc and
Mi Hee
Lim
*b
bc,
Juri
Lee
b,
Hyuck Jin
Lee
d,
Yonghwan
Ji
a,
Juhye
Kang
b,
Mu-Hyun
Baik
*bc and
Mi Hee
Lim
*b
aDepartment of Chemistry, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea
bDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: miheelim@kaist.ac.kr
cCenter for Catalytic Hydrocarbon Functionalizations, Institute for Basic Science (IBS), Daejeon 34141, Republic of Korea. E-mail: mbaik2805@kaist.ac.kr
dDepartment of Chemistry Education, Kongju National University, Gongju 32588, Republic of Korea
First published on 8th September 2020
Abstract
Amyloid-β (Aβ) accumulation, metal ion dyshomeostasis, oxidative stress, and cholinergic deficit are four major characteristics of Alzheimer's disease (AD). Herein, we report the reactivities of 12 flavonoids against four pathogenic elements of AD: metal-free and metal-bound Aβ, free radicals, and acetylcholinesterase. A series of 12 flavonoids was selected based on the molecular structures that are responsible for multiple reactivities including hydroxyl substitution and transfer of the B ring from C2 to C3. Our experimental and computational studies reveal that the catechol moiety, the hydroxyl groups at C3 and C7, and the position of the B ring are important for instilling multiple functions in flavonoids. We establish a structure–activity relationship of flavonoids that should be useful for designing chemical reagents with multiple reactivities against the pathological factors of AD.
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disorder responsible for a majority of dementia cases.1,2 Despite extensive research aimed at developing therapeutics, there is no cure and our capability of controlling the development and progression of AD is severely limited.2,3 Currently available symptomatic treatments offer temporary relief using acetylcholinesterase (AChE) inhibitors and N-methyl-D-aspartic acid receptor antagonists.3 AChE terminates cholinergic transmission at the synapse by catalyzing the hydrolysis of the neurotransmitter, acetylcholine (ACh).4 The cholinergic hypothesis identifies the reduced level of ACh as a primary culprit in the pathogenesis of AD.5 In recent years, however, the inability of AChE inhibitors to effectively halt the progressive neurodegeneration in AD has led to a re-evaluation of this hypothesis.6 The prevailing perception of AD pathology reflects the multifaceted quality of the disease.2Identification of additional pathological contributors of AD such as amyloid-β (Aβ),7 metal ions,8 metal-bound Aβ (metal–Aβ),9–12 and reactive oxygen species (ROS)13 corroborates the complex nature of the disease. Aβ, the major component of senile plaques, is an aggregation-prone peptide composed of 36–43 amino acid residues.7 Aβ aggregation is a subject of intensive research and Aβ oligomers were recently identified as potential toxic species capable of disrupting neuronal homeostasis.14 Metal ions present two neurochemical implications: (i) they function as essential cofactors and structural anchors in enzymatic reactions and protein folding and (ii) they exhibit neurotoxicity by catalyzing the generation of ROS.10 For these reasons, metal ions are tightly regulated in biological systems. Dyshomeostasis and miscompartmentalization of metal ions such as Cu(II) and Zn(II) are observed in the brains of AD patients and linked to neurodegeneration.10 Moreover, metal ions can affect the aggregation and conformation of Aβ by directly binding to the peptide.9,10,15 Based on their physiological roles and reactivity with Aβ, metal ions are considered an important part of AD pathogenesis.16 Research regarding the pathological relationship between metal ions and Aβ introduced the concept of metal–Aβ as a pathogenic factor based on its potential toxicity and involvement in producing ROS.10,16,17 Lastly, oxidative stress, characterized by the imbalance between the formation and removal of ROS, has been indicated in a spectrum of diseases including AD for detriments such as lipid peroxidation and peptide oxidation.18,19
Attempts to exploit these pathological elements for the development of single target-based therapeutics have been made, but they were largely shown to be clinically ineffective.20,21 As a result, research efforts have shifted towards understanding the connections among the various pathogenic pathways in AD. There is growing recognition that chemical reagents capable of simultaneously targeting and modulating multiple pathological features are necessary as potential therapeutic candidates and chemical tools in elucidating the pathology of AD at the molecular level. In this work, we evaluated the reactivities of flavonoids against the pathological elements of AD such as metal-free and metal-bound Aβ, free radicals, and AChE and determined the structural features responsible for their versatile reactivities, as depicted in Fig. 1.
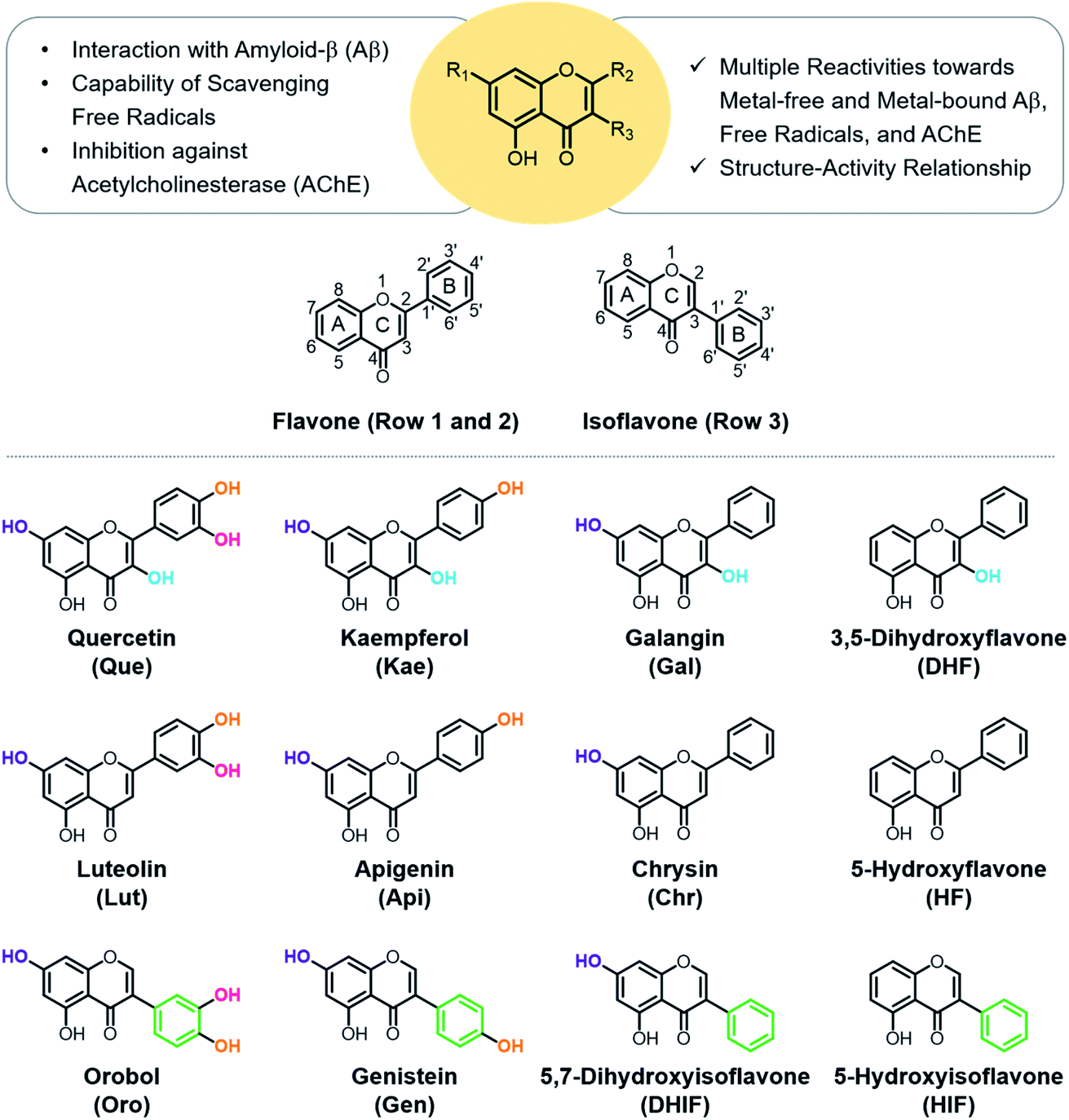 | ||
| Fig. 1 Rational selection of 12 flavonoids. The presented library of flavonoids was chosen based on structural variations including the number and position of hydroxyl groups and the location of the B ring to identify the structural features responsible for reactivities against multiple pathological factors found in AD [i.e., metal-free Aβ, metal–Aβ, free radicals, and AChE]. | ||
Flavonoids are a family of phytochemicals exhibiting low toxicity,22 anti-/pro-oxidant activity,23 and numerous utilitarian biological activities (e.g., anti-cancer,24 anti-viral,25 and anti-bacterial26). The direct administration of flavonoids, however, presents limitations in practical applications. For example, the solubility and stability of flavonoids often present challenges. Therefore, understanding the molecular structures of flavonoids and identifying their pharmacophores could be valuable in designing small molecules with multiple reactivities for investigating and treating AD. In this work, the modulative reactivities of 12 flavonoids enumerated in Fig. 1 towards metal-free Aβ, metal–Aβ, free radicals, and AChE were assessed and analyzed based on their molecular structures to identify the structural moieties critical for their reactivities. Moreover, computational studies were carried out to obtain a more in-depth perspective of their activities against free radicals and AChE.
Results and discussion
Rational selection
Quercetin (Que), luteolin (Lut), and orobol (Oro) were included in our study because they have been previously tested against several pathogenic elements of AD. Both Que and Lut exhibited a variety of biological activities23,24,26 including the ability to modulate the aggregation of metal–Aβ and inhibit AChE activity.27,28Oro was found to interact with metal ions, alter the aggregation of both metal-free and metal-bound Aβ, scavenge free radicals, and inhibit the catalytic activity of AChE.29,30 The 12 flavonoids enumerated in Fig. 1 include three different classes, namely flavonols (row 1), flavones (row 2), and isoflavones (row 3), that offer significant structural variance. These flavonoids were chosen according to three structural criteria: (i) inclusion of the chromone framework containing the 4-oxo group and the double bond between C2 and C3 on the C ring; (ii) variation in hydroxyl groups at C3 (row 1 vs. row 2), C3′, C4′, and C7 (columns 1, 2, and 3, respectively); (iii) change in the position of the B ring from C2 to C3 (row 2 vs. row 3). Previous studies identified the 4-oxo group and the unsaturated bond between C2 and C3 on the C ring to be important in various chemical and biological properties such as their metal-binding ability and antioxidant activity.28,31,32 Moreover, the unsaturated C2–C3 bond may contribute to interactions of the compounds with the hydrophobic regions of Aβ such as the self-recognition site and the C-terminal region that are essential for peptide aggregation.33–36 The 4-oxo functionality can form 5- and 6-membered metal chelation sites with the 3-OH or 5-OH group, respectively. The catechol moiety on the B ring can also chelate metal ions. In addition, the number and location of electron-donating hydroxyl groups can impact the flavonoids' redox potentials that direct their capacity to quench free radicals.37 These 12 flavonoids were obtained through commercial sources or prepared in our laboratory in the cases of Oro29 and 5-hydroxyisoflavone (HIF). The synthetic procedure and characterization for HIF are presented in Scheme 1 and the ESI (Fig. S1†).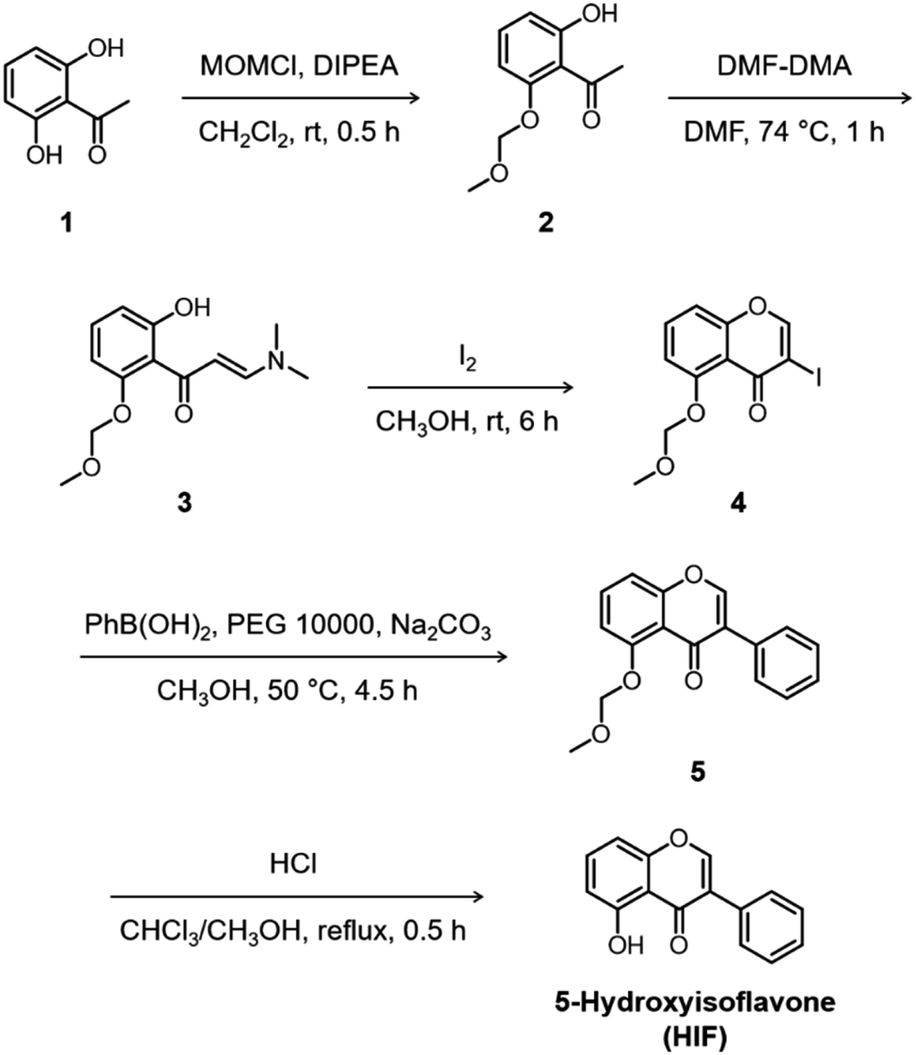 | ||
| Scheme 1 Synthetic routes to 5-hydroxyisoflavone (HIF). | ||
Interaction with Cu(II)
To verify that the flavonoids interact with metal ions in solution, UV-Vis spectra were monitored. Significant spectral changes in the UV-Vis range were observed upon adding Cu(II) to flavonoids, as illustrated in Fig. 2. Two characteristic π–π* transition bands were noted: band I associated with the B ring of flavonoids (λ = ca. 300–400 nm) and band II associated with the A ring (λ = ca. 240–280 nm).38 Incubation with Cu(II) showed optical shifts of either or both band I and II, e.g., bathochromic/hypsochromic and hyper/hypochromic shifts, indicative of interactions between Cu(II) and the oxygen (O) donor atoms in the A, B, and C rings of the flavonoids such as 4-oxo/5-OH, 3-OH/4-oxo, and 3′-OH/4′-OH. An absorption band was observed at ca. 400–440 nm with the addition of Cu(II). This band is associated with Cu(II) binding to flavonoids with implications at the extended resonance structure encompassing the B and C rings, as previously reported.38 The aforementioned band of Que and kaempferol (Kae), however, was diminished at greater Cu(II) concentrations, indicative of their oxidation.39 Together, these observations confirm that all of the selected flavonoids interact with Cu(II) in solution. Unfortunately, quantitative measurements for metal-binding affinities of the flavonoids were not possible due to the limited stability of their corresponding complexes.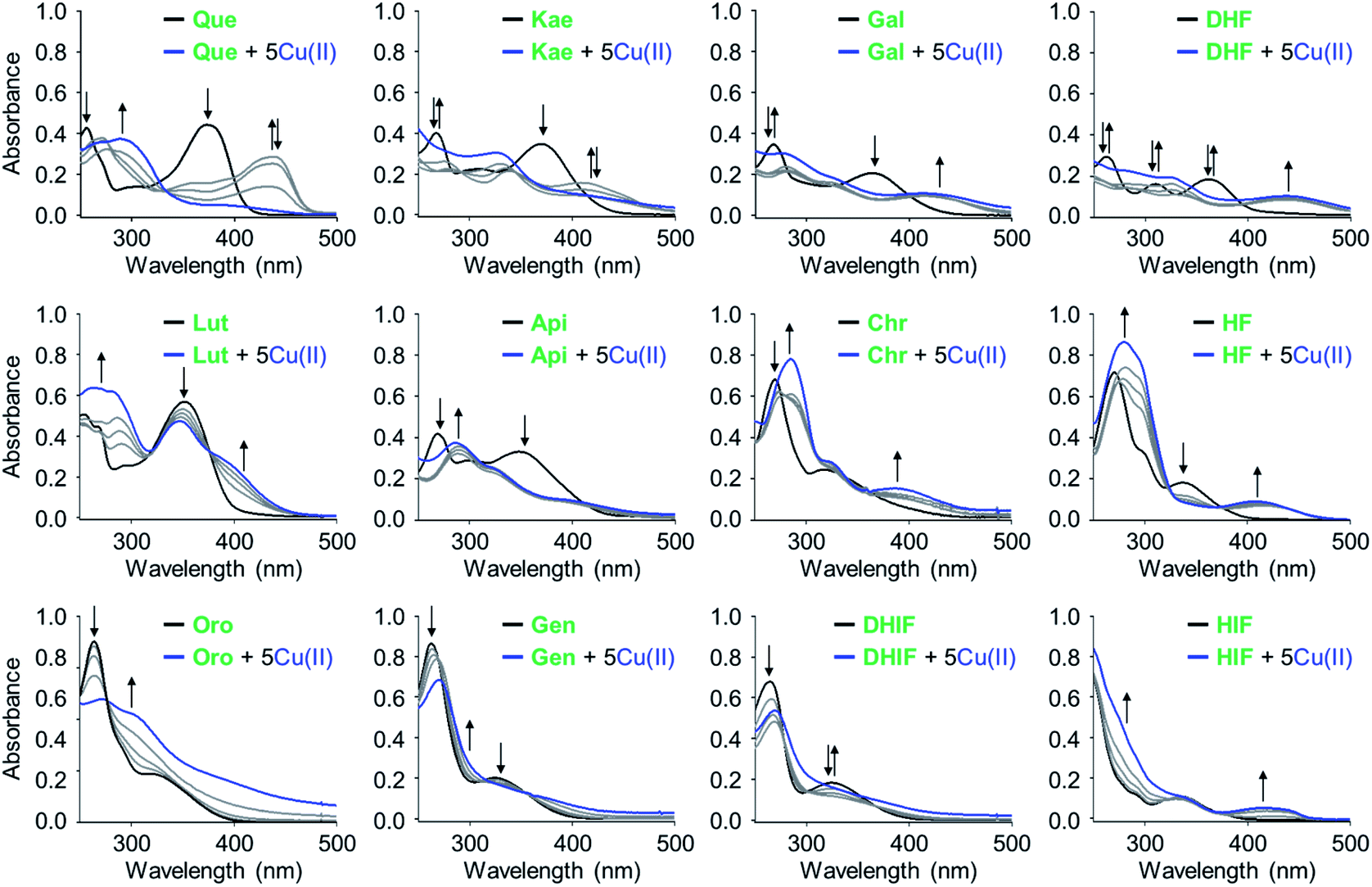 | ||
| Fig. 2 Interaction of the flavonoids with Cu(II) monitored by UV-Vis spectroscopy. Conditions: [flavonoid] = 25 μM; [CuCl2] = 0, 12.5, 25, 50, and 125 μM; 20 mM HEPES, pH 6.6, 150 mM NaCl or EtOH (for Chr and HF). The grey spectra were obtained by addition of 0.5, 1, and 2 equiv of CuCl2. | ||
Modulation on the aggregation of metal-free Aβ42 and metal–Aβ42
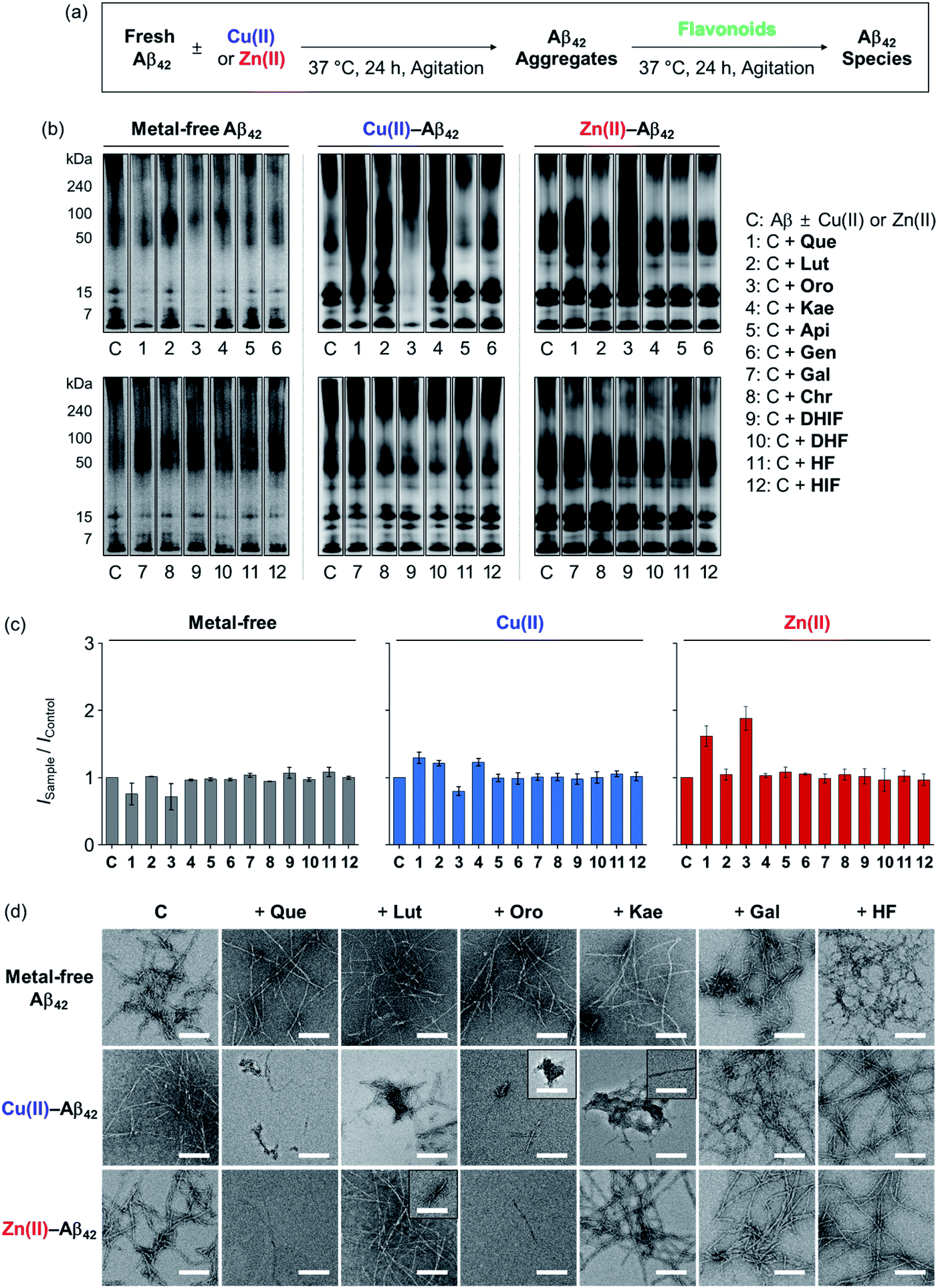
To determine the effects of the flavonoids on the aggregation of metal-free and metal-treated Aβ42, the molecular weight (MW) distribution and morphology of the resultant Aβ42 species upon incubation with the compounds were analyzed by gel electrophoresis with Western blotting (gel/Western blot) and transmission electron microscopy (TEM), respectively. The amount of β-sheet-rich aggregates generated through metal-free and metal-induced Aβ42 aggregation with treatment of the flavonoids could not be quantitatively determined due to the optical interference from the compounds within the analysis window of the assays (e.g., thioflavin-T assay). Two types of Aβ aggregation experiments were performed: inhibition and disaggregation experiments, as summarized by Fig. 3 and 4. For the inhibition experiments, freshly prepared metal-free or metal-treated Aβ42 was incubated with the flavonoids for 24 h to assess whether the compounds could inhibit the formation of peptide aggregates. In the disaggregation experiments, the samples containing Aβ42 with and without metal ions were pre-incubated for 24 h to produce peptide aggregates and then treated with the flavonoids for an additional 24 h. The disaggregation studies determined whether the flavonoids could disassemble preformed peptide aggregates or modulate their aggregation. Under our experimental conditions, Aβ42 alone spontaneously aggregated to yield a heterogeneous mixture of Aβ ensembles including large aggregates that were too big to penetrate the gel matrix and be visualized by gel/Western blot. Such large aggregates were probed by TEM.40,41 On the other hand, smaller Aβ42 species such as monomers and low MW oligomers indicated visible bands in gel/Western blot, but they were not detected by TEM.42,43 To further examine the gel/Western blot data, the band intensities of the gel images were analyzed using the ImageJ software.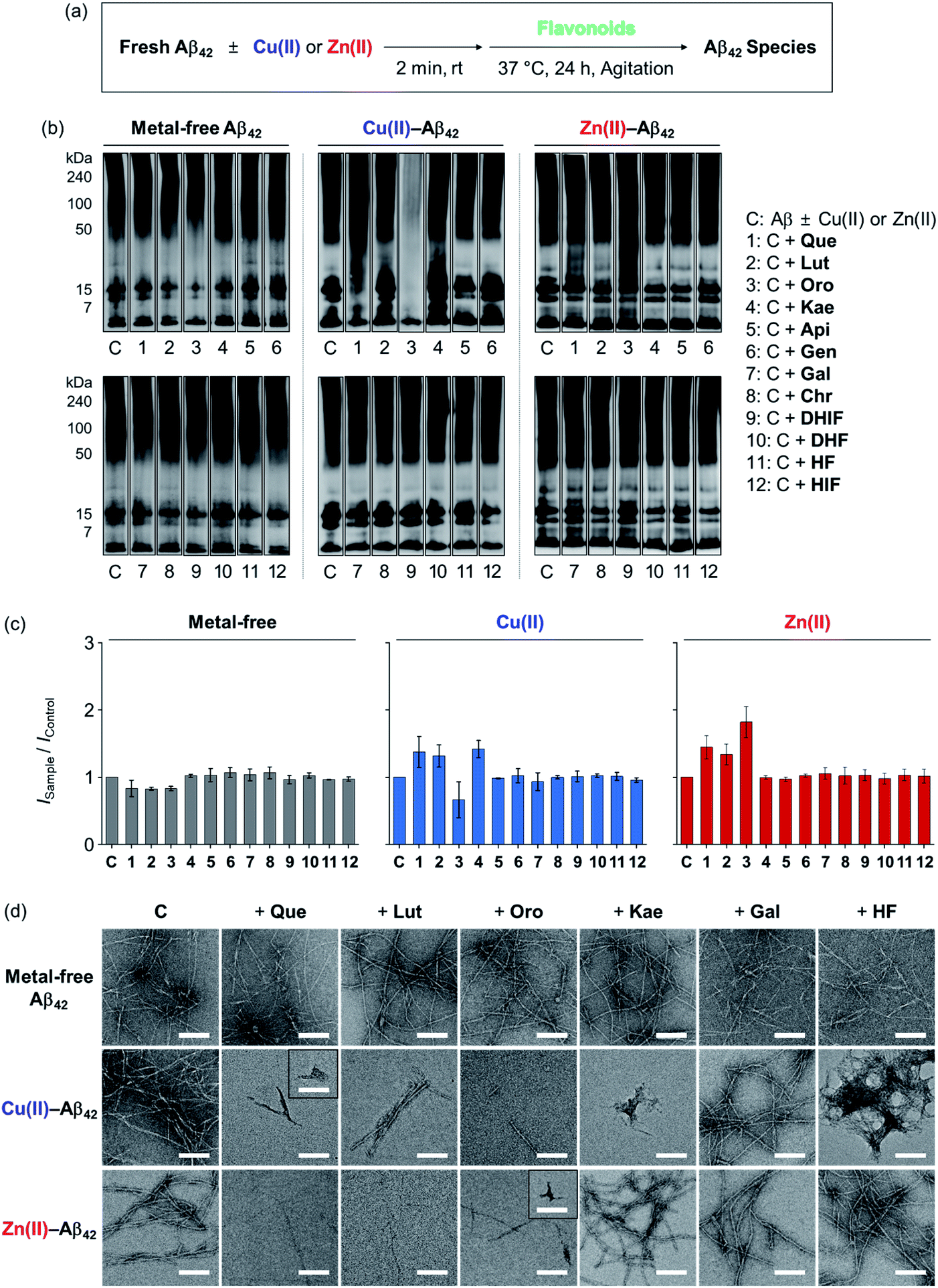 | ||
| Fig. 3 Influence of the flavonoids on the aggregation of metal-free and metal-treated Aβ42. (a) Scheme of the inhibition experiments. (b) Analysis of the MW distribution of the resultant Aβ42 species by gel/Western blot with an anti-Aβ antibody (6E10). The original gel images are presented in Fig. S2a.† (c) Quantification of Aβ42 species visualized in the gel by the ImageJ software. The intensity of the gel from the sample was normalized to that of the corresponding control (ISample/IControl). (d) TEM images of the samples from (b). Conditions: [Aβ42] = 25 μM; [CuCl2 or ZnCl2] = 25 μM; [flavonoid] = 50 μM; pH 7.4 [for metal-free and Zn(II)-containing samples] or pH 6.6 [for Cu(II)-added samples]; 37 °C; 24 h incubation; constant agitation. | ||
 | ||
| Fig. 4 Impact of the flavonoids on the disassembly and aggregation of preformed metal-free and metal-added Aβ42 aggregates. (a) Scheme of the disaggregation experiments. (b) Analysis of the MW distribution of the resultant Aβ42 species by gel/Western blot with an anti-Aβ antibody (6E10). The original gel images are presented in Fig. S2b.† (c) Quantification of Aβ42 species visualized in the gel by the ImageJ software. The intensity of the gel from the sample was normalized to that of the corresponding control (ISample/IControl). (d) TEM images of the samples from (b). Conditions: [Aβ42] = 25 μM; [CuCl2 or ZnCl2] = 25 μM; [flavonoid] = 50 μM; pH 7.4 [for metal-free and Zn(II)-containing samples] or pH 6.6 [for Cu(II)-added samples]; 37 °C; 24 h incubation; constant agitation. | ||
In the inhibition experiments, as shown in Fig. 3b, the introduction of Que, Lut, and Oro showed a minor change in the MW distribution of metal-free Aβ42. The signal intensities of the bands corresponding to smaller oligomeric Aβ42 species (from 7 to 15 kDa) were slightly reduced upon treatment of Que, Lut, and Oro. In the case of Cu(II)–Aβ42, Que, Lut, Oro, and Kae modified the aggregation of the peptide: Que increased the smearing band intensity at ca. 7–270 kDa, Lut and Kae increased the signal in the range of ca. 7–50 kDa, and Oro significantly decreased the band intensity of the entire range (7–270 kDa). Moreover, Que, Lut, and Oro altered the aggregation of Zn(II)–Aβ42, resulting in diverse MW distributions from ca. 15 to 50 kDa. Kae did not significantly influence the aggregation of Aβ42 in the absence and presence of Zn(II). Apigenin (Api), genistein (Gen), galangin (Gal), chrysin (Chr), 5,7-dihydroxyisoflavone (DHIF), 3,5-dihydroxyflavone (DHF), 5-hydroxyflavone (HF), and HIF did not exhibit significant reactivity against metal-free or metal-induced Aβ42 aggregation. As summarized in Fig. 3c, the quantification of the gel/Western data further indicated the degree of the flavonoids' reactivities towards the formation of metal-free and metal-added Aβ42 aggregates. The changes in the band intensity in the gel for the metal-free conditions were definitively smaller relative to those of the metal-treated conditions.
As depicted in Fig. 3d, TEM images corroborated the effect of the flavonoids on Cu(II)-induced Aβ42 aggregation. The flavonoids exhibiting noticeable modulative reactivity towards the aggregation of Cu(II)–Aβ42 in gel/Western blot led to notable morphological changes in the peptide aggregates: (i) Que gave significantly shorter fibrillar aggregates; (ii) Lut decreased the degree of branching in the fibrils; (iii) Oro afforded thinner and shorter aggregates; (iv) Kae generated amorphous assemblies distinct from native Aβ42 fibrils. As expected, Gal and HF did not significantly alter the morphologies of Aβ42 aggregates regardless of the presence of Cu(II) or Zn(II). When Zn(II)–Aβ42 was incubated with Que, Lut, or Oro that modified Zn(II)–Aβ42 aggregation in the gel/Western blot studies, thinner chopped fibrils were detected. In the presence of Kae, Gal, and HF, fibrillar aggregates similar to those from the compound-free Zn(II)–Aβ42 sample were produced. None of the flavonoids studied via TEM exhibited the ability to particularly change the aggregate morphology of metal-free Aβ42.
In the disaggregation experiments, as illustrated in Fig. 4b, Que and Oro very mildly varied the MW distribution of preformed metal-free Aβ42 aggregates. In the case of Cu(II)–Aβ42, Que, Lut, Oro, and Kae exhibited reactivity towards preformed Aβ42 aggregates: (i) Que increased the intensity of the bands larger than ca. 15 kDa; (ii) Lut increased the signal corresponding to Aβ species in the range of ca. 15–100 kDa while decreasing that of the species larger than 100 kDa; (iii) Oro reduced the signal intensity of all MW species below ca. 50 kDa; (iv) Kae increased smearing in the MW range from ca. 15 to 270 kDa. The size distribution of preformed Zn(II)–Aβ42 aggregates upon treatment with Que and Oro differed from that of the compound-free peptide sample. Que led to the enhanced intensity of the bands in the range ca. 15–240 kDa, while Oro resulted in a more significant change in the smearing band of the overall range (ca. 7–270 kDa). Api, Gen, Gal, Chr, DHIF, DHF, HF, and HIF did not exhibit notable reactivity with preformed metal-free and metal-induced Aβ42 aggregates. Demonstrating modulative reactivity towards preformed Cu(II)–Aβ42 aggregates, Lut and Kae did not impact the disaggregation samples of metal-free Aβ42 and Zn(II)–Aβ42. The gel quantification data further supported the significant effects of the reactive flavonoids on the aggregation of metal-free Aβ42 and metal–Aβ42, as presented in Fig. 4c.
As illustrated in Fig. 4d, morphological changes in preformed metal-free and metal-treated Aβ42 aggregates in the presence of the flavonoids were monitored by TEM. As for preformed Cu(II)–Aβ42 aggregates, Que, Lut, Oro, and Kae exhibiting modulative reactivity in the gel/Western blots altered the morphologies of the resultant aggregates with indication of a mixture of amorphous aggregates and thinner chopped fibrils. Treatment of Que and Oro to preformed Zn(II)–Aβ42 aggregates produced shorter fibrils. As expected from the gel/Western blot experiments, the flavonoids lacking modulative reactivity in the disaggregation experiments indicated no significant variation in the morphology of preformed peptide aggregates.
In general, increasing the number of hydroxyl substituents on the backbone promotes the ability of the flavonoids to modify the aggregation pathways of metal–Aβ42. Among the flavonoids, the isoflavone Oro carrying four hydroxyl groups with two potential metal chelation sites, i.e. 4-oxo/5-OH on the A/C rings and 3′-/4′-OH on the B ring, demonstrated notable modulation on the aggregation of both metal-free and metal-added Aβ42. The pertinence of the 3-OH group in the flavonoids' modulative reactivity towards Aβ42 was denoted by the distinctions between (i) Kae and Api and (ii) Que and Lut. Kae displayed notable reactivity with Cu(II)–Aβ42 in both inhibition and disaggregation experiments, while Api did not show such reactivity. Moreover, Que altered the MW distributions of Zn(II)–Aβ42 in both inhibition and disaggregation experiments, but Lut only notably inhibited the generation of Zn(II)–Aβ42 aggregates. Structurally comparing Kae, Gal, and Api, a connection between the 3-OH and 4′-OH groups on the C and B rings, respectively, can be recognized regarding the flavonoid's influence on the aggregation of metal–Aβ42. The presence of the 3-OH or 4′-OH functionality alone (Gal and Api, respectively) does not result in the reactivity towards metal–Aβ42 aggregation; however, the presence of both hydroxyl groups on the B and C rings (Kae) leads to noticeable reactivity. The catechol moiety on the B ring (Que, Lut, and Oro) fostered their interactions with metal ions and Aβ. Catechol-type flavonoids were reported to undergo oxidation to form o-quinones that in turn covalently bind to the lysine residues of Aβ.44 Our results confirm the importance of the catechol moiety in the flavonoids' modulative reactivity towards Aβ in the absence and presence of metal ions and further support that such reactivity could be maintained and may be altered by changing the location of the B ring catechol group, as observed with Oro. Taken together, the aggregation and disaggregation studies reveal three major structural features of flavonoids associated with their ability to modify the aggregation pathways of metal-free Aβ and metal–Aβ: (i) the 3-OH group on the C ring, (ii) the catechol moiety on the B ring, and (iii) the position of the B ring.
Scavenging free organic radicals
To evaluate the capacity of the flavonoids for quenching free organic radicals relative to the vitamin E analog Trolox, the Trolox equivalent antioxidant capacity (TEAC) assay was conducted with the cationic radical form of 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid) as the organic radical substrate.45 As presented in Fig. 5a, the flavonols Que, Kae, Gal, and DHF (Fig. 1, row 1) exhibited notable scavenging capacity against free organic radicals, suggesting the importance of the 3-OH group on the C ring for this activity. As expected, Que, Lut, and Oro carrying a catechol moiety on the B ring showed significant antioxidant activity. Among the selected flavonoids, Que embodying both the 3-OH functionality and the catechol moiety was the most effective scavenger against free organic radicals. In contrast, Api, Chr, DHIF, HF, and HIF presented negligible radical scavenging activity, compared to Trolox. The antioxidant properties of the flavonoids are reportedly affected by the 4-oxo functionality and the double bond between C2 and C3.46 Conjugation of the A, B, and C rings resulting in the stabilization of the flavonoid radical through resonance may be critical for their scavenging activity against free radicals.46 Our results employing Chr, DHIF, HF, and HIF, however, indicate that the 4-oxo group and the unsaturated C2–C3 bond do not guarantee a molecule's ability to quench free radicals effectively. The difference in the scavenging capacity against free radicals between flavonols and flavones, as shown in Fig. 1 (row 1 and 2), respectively, may be prompted by the presence of electron-donating groups, especially at C3, that could lower their redox potentials (vide infra).47 These observations suggest that the 3-OH group on the C ring is an important structural feature for the free radical scavenging capability of flavonoids. Que, Lut, and Oro containing a catechol moiety on the B ring displayed the best inhibitory activity against free organic radicals according to the TEAC assay. Catechol and “catechol-type flavonoids” are recognized for their notable antioxidant activity and mechanistic studies further support their significance.48,49 Conversely, the flavonoids possessing the 4′-OH group without 3′-OH, as depicted in Fig. 1 (column 2), except Kae, did not demonstrate noticeably great antioxidant activity, further emphasizing the significance of the catechol moiety and the 3-OH group for the ability to scavenge free radicals.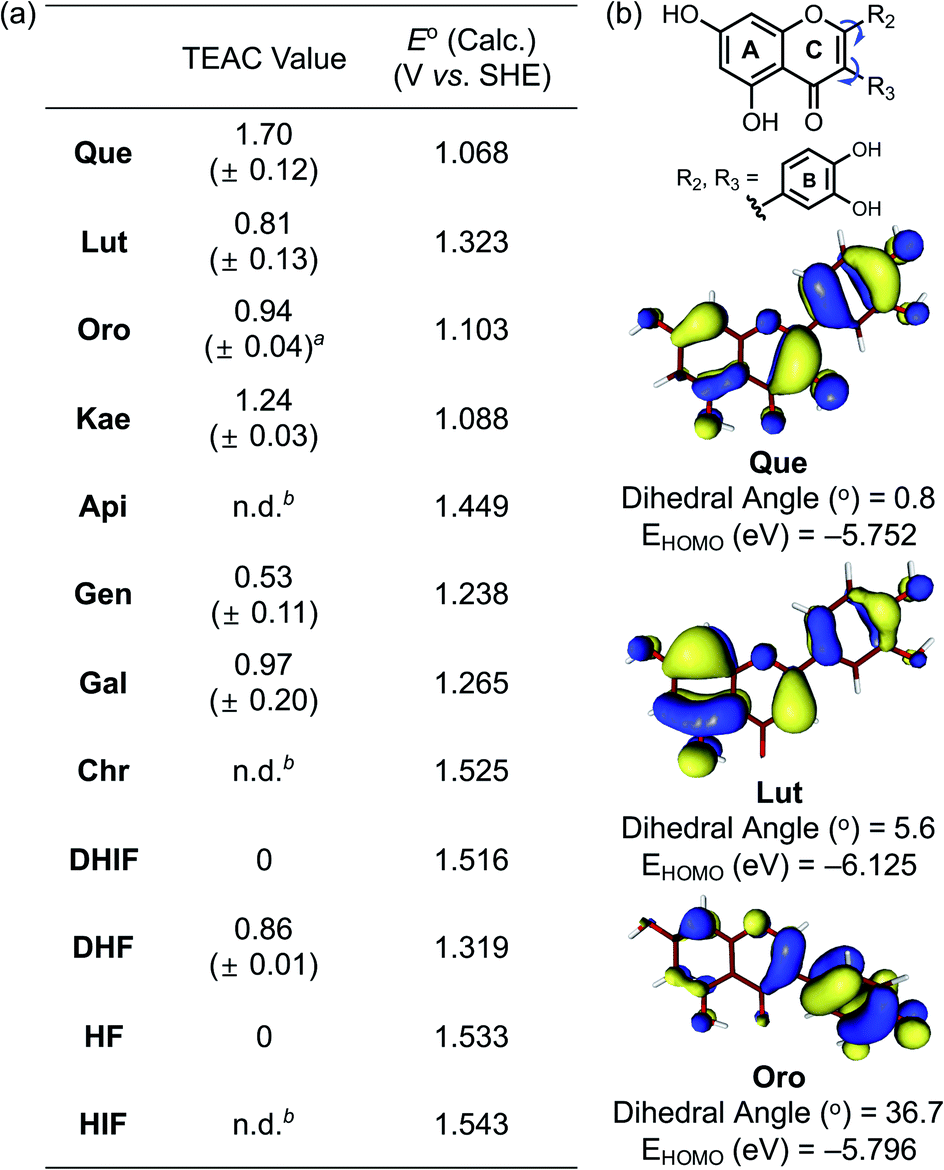 | ||
| Fig. 5 Scavenging activity of the flavonoids against free organic radicals determined by the TEAC assay and their computed redox potentials. (a) Summary of the TEAC values for the flavonoids and their computed redox potentials (E° vs. SHE). Conditions: EtOH; 25 °C; λabs = 734 nm. a This value was obtained from ref. 30. b n.d., not determined. The TEAC values of Api, Chr, and HIF could not be obtained due to limited solubility or marginal antioxidant activity levels undetected under our experimental conditions. (b) Isosurface plots (isodensity value = 0.03 a.u.) of the HOMO energy for Que, Lut, and Oro and their dihedral angles between two planes calculated from carbon coordinates of the A/C rings and the B ring, respectively. | ||
Redox potentials
To verify the antioxidant activity of our flavonoid series, the redox potentials of all compounds were computed following a previously reported method.50 Attempts to experimentally determine the redox potentials were made, but their instability and limited solubility in both aqueous media and organic solvents hampered electrochemical measurements. As summarized in Fig. 5a, the computed redox potentials (E° vs. SHE) of the flavonoids support that the incorporation of electron-donating hydroxyl groups into the molecular framework lowers the redox potentials. Furthermore, the redox potentials depended not only on the number of electron-donating hydroxyl groups but also on their positions affecting the degree of π-conjugation between the core skeleton of the molecule and the functional group. Directly comparing the changes in the calculated E° suggests that the impact of a hydroxyl group on the B or C ring is more prominent than that on the A ring. The 4′-OH (column 2 vs. 3 shown in Fig. 1), 3′-OH (column 1 vs. 2), and 3-OH (row 1 vs. 2) groups on the B and C rings decreased the E° values by ca. 0.18, 0.09, and 0.27 V, respectively, but the 7-OH group on the A ring (column 3 vs. 4) gave a difference of only ca. 0.03 V in the computed E°.Fig. 5b illustrates the calculated dihedral angles and the highest occupied molecular orbital (HOMO) levels of the neutral forms of Que, Lut, and Oro. Lut exhibited the lowest HOMO energy (−6.13 eV) out of the three compounds of interest, which is in accord with the more positive E° (1.32 V vs. SHE). The HOMO level of Que that embodies an additional hydroxyl group at C3 on the framework of Lut with a relatively planar structure was elevated to −5.75 eV. On the other hand, the HOMO level of Oro, a regioisomer of Lut, was at −5.80 eV, which was higher than that of Lut. The better antioxidant ability of Oro can be rationalized by the distinctions in the conjugated π-system of the HOMO for Lut and Oro. Considering the intrinsic electronic property of the C ring enone, the α-carbon of the enone (C3) possesses a δ− partial charge while the β-carbon (C2) has a partial δ+ charge. In the case of Lut, the electron-rich catechol is attached to the δ+ charged β-carbon C2, preferring the π-conjugation throughout the B and C rings. On the other hand, in the case of Oro, the catechol moiety is attached to the δ− charged α-carbon C3 of the enone; thus, the resonance between the B and C rings is weakened. In the optimized structure of Oro, the dihedral angle between the A/C rings and the B ring was 36.7°, which is notably greater than that of Lut. The frontier orbitals showed that the HOMO of Lut was composed of a conjugated π-system throughout the A/C rings and the B ring. In contrast, the orbital lobes in the HOMO of Oro displayed a biased localization towards the B ring resulting in a less electronically stable HOMO. This distorted alignment of π-orbitals and the consequential disruption of π-conjugation between the two π-ring planes elevated the HOMO levels of Oro, making its redox potential more negative. Therefore, our experimental and computational data support that three structural components including the 3-OH group, the catechol moiety, and the position of the B ring considerably influence redox potentials of the flavonoids and, subsequently, their antioxidant activity.
Inhibition against AChE
The 12 flavonoids were tested for their ability to inhibit the catalytic activity of AChE following a previously reported fluorometric assay with slight modifications employing Electrophorus electricus AChE (eeAChE).51 As a reference, tacrine, a potent inhibitor against AChE,52 was tested under our experimental conditions (IC50 = 38.7 ± 5.0 nM). Thereafter, the inhibitory activity of all flavonoids towards AChE was determined under the same conditions. As shown in Fig. 6a, Que, Lut, Oro carrying a catechol moiety displayed notable inhibition against the activity of AChE, exhibiting IC50 values in the nanomolar range. In the case of Lut and Oro, their inhibitory activity against AChE was comparable to that of tacrine. Kae and Gal indicated high nanomolar IC50 values similar to that of Que, while those of Gen, DHIF, and DHF were within the micromolar range. Lastly, the activity of AChE was not inhibited by Api, Chr, HF, and HIF under our experimental settings.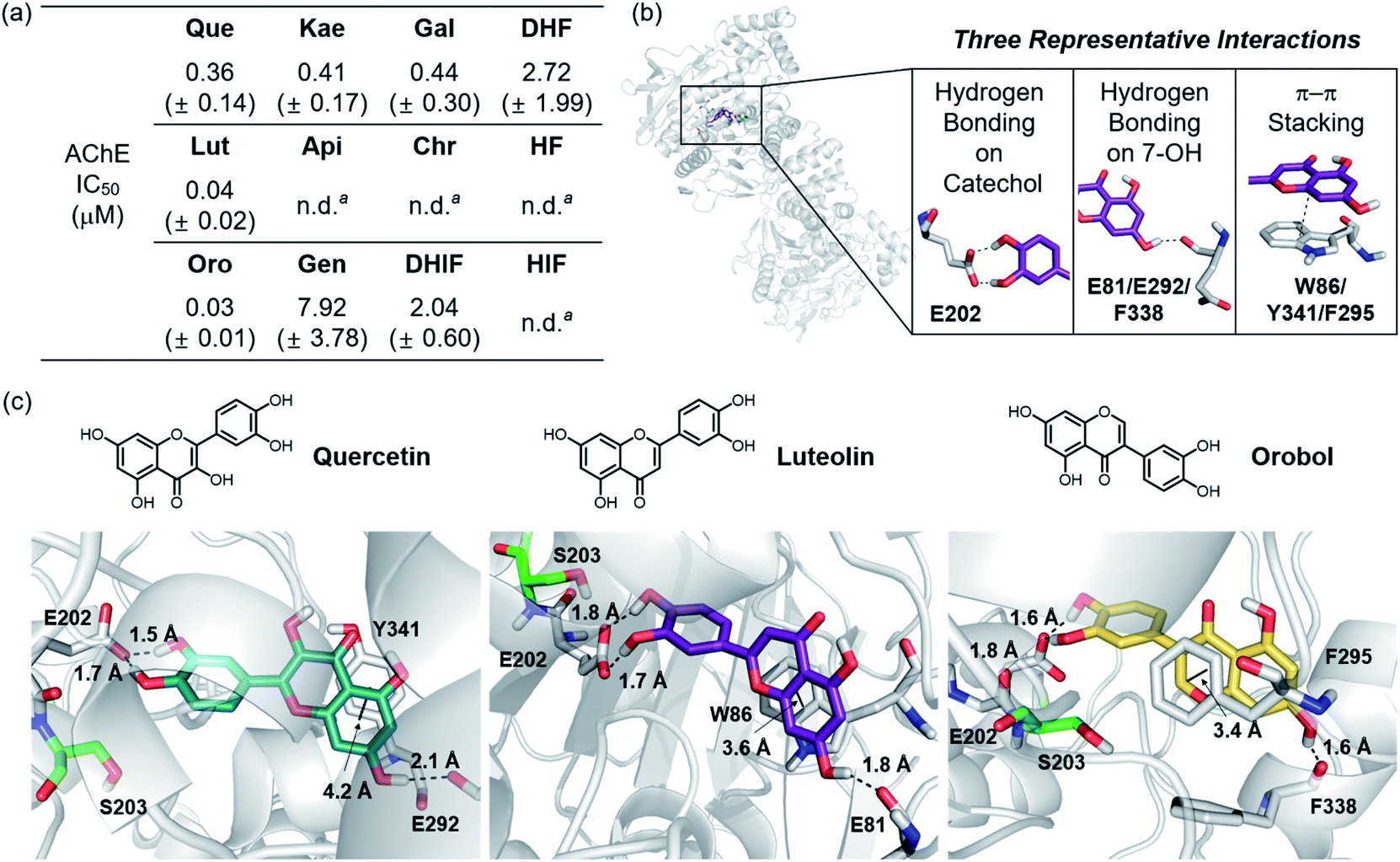 | ||
| Fig. 6 Inhibitory activity of the flavonoids against AChE. (a) Summary of the IC50 values of the flavonoids against eeAChE determined by a fluorometric assay. (b) Intermolecular interactions between the flavonoids and AChE (PDB 1C2O54) observed by aMD simulations. (c) Visualization of the flavonoid–AChE interactions modeled through aMD simulations. N, O, and H (from hydroxyl groups) atoms in the flavonoids are depicted in blue, red, and white, respectively. a n.d., not determined. Inhibitory activity of Api, Chr, HF, and HIF against AChE was too low to be detected under our experimental conditions and, thus, an accurate IC50 value could not be determined. | ||
Relating these experimental results to the molecular structures, several features stand out. The catechol functionality is important for the inhibitory activity against AChE (Que, Lut, and Oro). DHF exhibited an IC50 value in the low micromolar range and the incorporation of the 7-OH group led to an increase in the inhibitory activity as observed with Gal. The distinct inhibitory activity between DHIF and HIF further supports the involvement of the 7-OH group, whereas the pertinence of the 3-OH functionality can be inferred from the general enhancement of the inhibitory capacity from Api, Chr, and HF to Kae, Gal, and DHF, respectively. Lastly, the enhanced AChE inhibitory effects of Gen and DHIF, relative to Api and Chr, implicate the potential influence of the B ring position on their ability to inhibit the catalytic activity of AChE.
Computational studies for interactions with AChE
Based on their distinct structural characteristics and IC50 values against AChE, Que, Lut, Oro, HF, and DHF were chosen for a detailed computational evaluation of their interactions with AChE. Specifically, the binding configurations and interactions of the selected flavonoids towards the AChE dimer were analyzed by accelerated molecular dynamics (aMD) simulations. Details of these simulations are given in the ESI.† This method has been shown to improve the fidelity of the simulated poses in studies of ligand–protein interactions by enhancing the sampling efficiencies.53 The structural clustering analysis of three potent flavonoids, Que, Lut, and Oro, against AChE identified three representative interactions between the compounds and AChE (PDB 1C2O54), as depicted in Fig. 6b and c. (i) The most dominant interaction observed by the aMD simulations is the hydrogen bonding between the catechol moiety on the B ring and neighboring amino acid residues, where the E202 residue in AChE is the dominant hydrogen bond accepting partner. (ii) The 7-OH group on the A ring serves as a hydrogen bond donor to interact with a carbonyl moiety from a backbone amide group of E81/G82, E292/S293, or F338/L339 in the gorge region of the pocket. (iii) A π–π stacking interaction between the chromone framework of the flavonoids on the A and C rings and the amino acid residues containing a π-ring such as W86, Y341, and F295 within the hydrophobic pocket of AChE was detected.Based on the initial identification of these flavonoid–AChE interactions, a more detailed computational analysis was performed on the representative binding modes of the three potent inhibitors, Que, Lut, and Oro, against AChE. As shown in Fig. 6b and c, a closer inspection of the binding configurations between the flavonoids and AChE revealed the hydrogen bonding between the catechol moiety on the B ring and the carboxylate group of E202. The presence of such hydrogen bonding interactions in close proximity to S203 could limit the accessibility of the substrate ACh to the catalytic triad responsible for catalyzing the hydrolysis of the neurotransmitter. Such anchoring of the flavonoids near S203 through hydrogen bonding may explain the relatively high inhibitory activity of Que, Lut, and Oro towards AChE. These observations emphasize the pertinent role of the catechol moiety on the B ring in controlling the activity of AChE. Other binding modes, e.g. where the A ring is arranged towards S203, were also sampled from the clustering analysis, as illustrated in Fig. S3.† They represent a relatively small population, suggesting that they play only a minor role in the overall binding characteristic. Moreover, the π–π stacking interactions between the A/C rings and the tryptophan, tyrosine, and phenylalanine residues also provided additional stability for binding of the flavonoids to AChE. The chromone moieties of Lut and Oro interact with W86 and F295, respectively, while Que displayed a slightly longer π–π distance to Y341. The additional 3-OH functionality on Que, distinguishing it from Lut, did not show notable interactions with any amino acid residues in the binding pocket of AChE, as presented in Fig. S4a.† The hydrogen bond mediated by the 7-OH group indicated longer distances with a large standard deviation than those formed between the catechol moiety and E202.
The binding modes of DHF and HF, whose inhibitory potency towards AChE was relatively weak (IC50 > 2.0 μM), were also analyzed, as visualized in Fig. S5.† The two compounds do not possess the catechol moiety on the B ring or the 7-OH group on the A ring. The clustering analysis of the aMD trajectories indicated distinct binding characteristics of DHF and HF towards AChE compared to Que, Lut, and Oro. DHF did not interact with E202, but it was still compact enough to position itself within the hydrophobic pocket by preserving the hydrogen bonding between the 3-OH group and the side chain of G122. In the case of HF, we were not able to obtain an aMD cluster for the compound populating more than 5% of the trajectories, in accord with its low inhibitory activity against AChE. Nevertheless, we considered the most clustered conformation of HF for further exploration and analysis.
Upon determining the binding configurations between the selected flavonoids and AChE, statistical analysis regarding four specific physical parameters including the distance from S203, the number of hydrogen bonds, the solvent accessible surface area (SASA) of the hydrophobic residues lining the active site pocket, and the number of water molecules in the active site pocket were conducted to provide more in-depth details of the flavonoid–AChE interactions, as shown in Fig. 7 and S4b.† The minimum distance between the flavonoid and S203 was measured. On average, aMD simulations with Que, Lut, Oro, and DHF presented relatively short distances (below 2.5 Å) to S203, as illustrated in Fig. 7a, implying that they can remain in the binding site interior. HF was positioned away (5.1 Å) from the catalytic residue, which is consistent with its minimal inhibitory potency. The distance to S203 alone is not sufficient to discern more potent inhibitors, however, as DHF was also found to maintain close contact with S203 despite its weak inhibitory activity towards AChE. The number of hydrogen bonds between the flavonoids and the binding pocket residues was calculated to assess the relative stabilities of the identified binding poses, as presented in Fig. 7b. The lack of adequately positioned hydrogen bond donors or acceptors on HF and DHF led to poorly established hydrogen bonding with AChE, as shown in Fig. S5.†Que, Lut, and Oro exhibited a significantly greater number of hydrogen bonds than HF and DHF, which supports again the notion that the catechol moiety on the B ring and the 7-OH group on the A ring are crucial structural features for interacting with AChE.
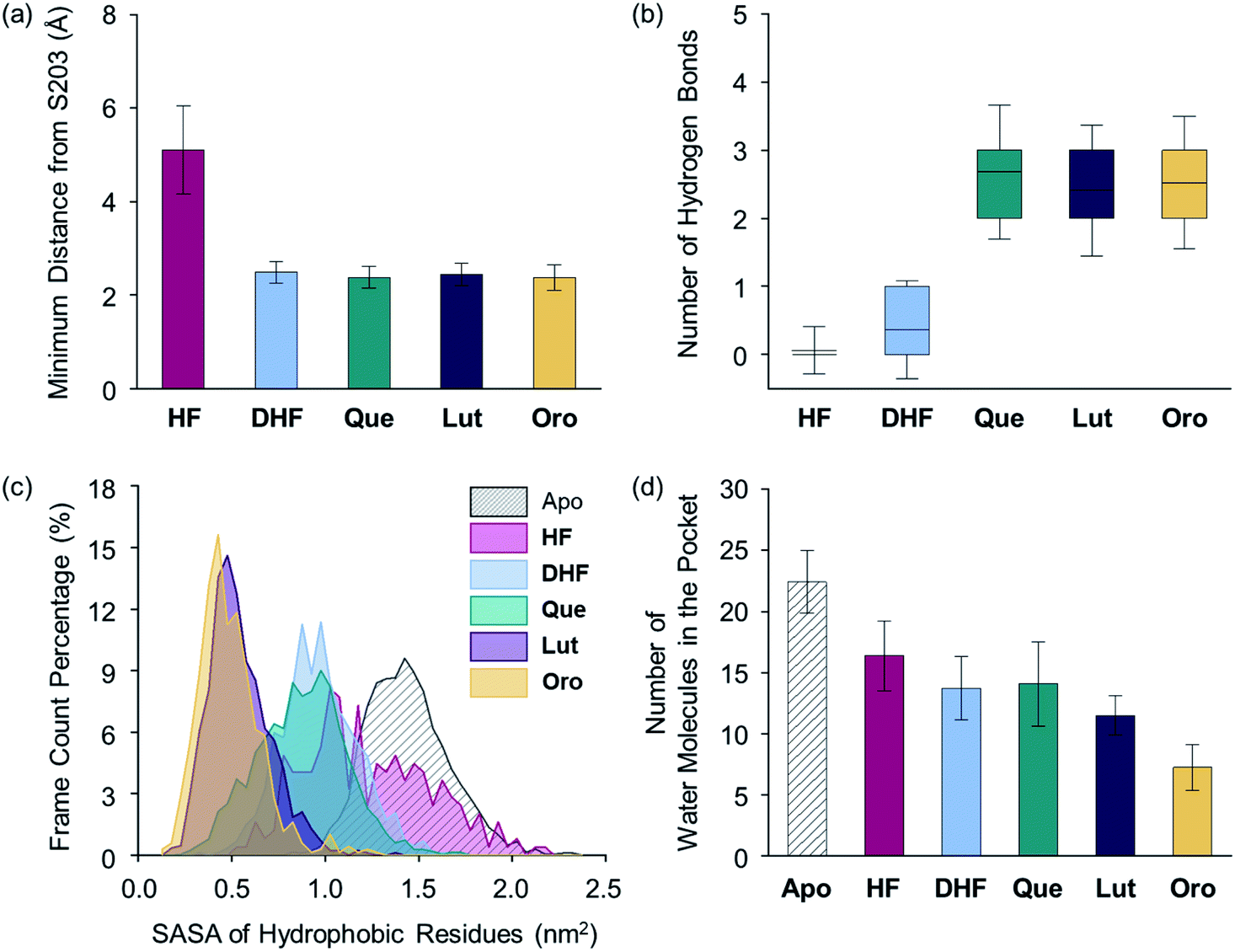 | ||
| Fig. 7 Evaluation of flavonoid-binding determinants against AChE (PDB 1C2O54). (a) Closest distance between the flavonoid and S203 of the catalytic triad. (b) Box plot of the number of hydrogen bonds between the flavonoid and AChE. The box extends to the top 25% and bottom 75% of the clustered data. The black line represents the mean value of each computed case. (c) SASA distribution of the hydrophobic residues in the active site. (d) Average count of water molecules in the binding pocket for the apo and holo cases. Error bars represent the standard deviation. | ||
The SASA values of the hydrophobic residues residing within 6.0 Å from S203 were examined as a measure of the binding pocket stability in the presence of a ligand.55 As displayed in Fig. 7c, the SASA values of the hydrophobic residues in the presence of Lut and Oro were found to be 0.54 and 0.49 nm2, respectively, suggesting that the hydrophobic residues effectively minimized unfavorable contacts with the polar solvent medium. In addition, the relatively low level of fluctuations in the computed SASAs indicated the increased stability of the binding pocket with Lut and Oro. The SASA alone, however, cannot be considered a computational parameter in evaluating the potency of compounds as Que and DHF showed comparable SASA values (0.88 and 0.97 nm2, respectively), while their ability to inhibit AChE activity varied significantly. The HF-bound structure indicated a mean SASA value of 1.23 nm2 with significantly large fluctuations. This implies the unstable organization of the hydrophobic residues upon binding of HF to the active site.
Finally, the dynamics of the water molecules in the binding pocket is another critical factor influencing the binding properties of ligands.56 We analyzed the number of water molecules in the active site gorge within 8.0 Å from the Cα atom of S203 for simulated flavonoid–AChE binding modes, relative to the number of water molecules in the apo state, as illustrated in Fig. 7d. The number of water molecules in holo states decreased by more than 5 in the presence of the flavonoids, while 22 water molecules were present in the apo state. In line with the computational findings described above, the HF-bound conformation contained the most water molecules in the binding pocket (16 on average). In contrast, Que and DHF disclosed lower numbers of water molecules within the active site than that of HF. Lut and Oro exhibiting stronger inhibitory activity against AChE led to the least amount of water molecules in the hydrophobic pocket (on average, 12 and 7, respectively). Combining the experimental and computational results, several structural features of the flavonoids can be connected to their inhibitory activity against AChE. Our structure–activity relationship study regarding the inhibition towards AChE underscores the importance of (i) the catechol moiety on the B ring to facilitate hydrogen bonding with E202, (ii) the 7-OH group for hydrogen bonding, and (iii) the chromone framework for π–π stacking with the hydrophobic residues lining the active site gorge. The aMD simulations also provide a detailed representation of the possible interactions between the flavonoids and AChE at the active site with multiple key parameters related to the strength of their interactions.
Conclusions
The complexity of the multifaceted AD pathology has led to increased interest in the development of chemical reagents with multiple functions. Considering the versatile reactivity of the three naturally occurring flavonoids, a series of 12 flavonoids was selected and investigated with respect to their reactivities towards four pathogenic elements that are implicated in AD including metal-free Aβ42, metal–Aβ42, free radicals, and AChE. Through our detailed investigations, several structural features were identified to be connected to the reactivities towards the aforementioned targets (Fig. 8). First, the catechol moiety on the B ring of the flavonoids notably promoted the molecules' modulative reactivities against metal-free Aβ, metal–Aβ, free organic radicals, and AChE. The two ortho-hydroxyl substituents on the B ring of flavonoids presented the ability to (i) chelate metal ions in a bidentate manner forming a 5-membered ring, (ii) undergo oxidation to produce an ortho-quinone that can covalently bind to the lysine residues in Aβ,44 (iii) effectively scavenge free organic radicals via radical stabilization through hydrogen bonding,57,58 and (iv) inhibit the catalytic activity of AChE by sterically blocking off the catalytic active site by interacting with the amino acid residues lining the active site gorge. Second, the hydroxyl group at C3 contributed towards the (i) chelation of metal ions in a bidentate manner manifesting a 5-membered metal-binding site, (ii) modulation of metal-free or metal-induced Aβ aggregation when accompanied by hydroxyl substituents on the B ring, (iii) scavenging free organic radicals, and (iv) inhibition of AChE activity in conjunction with the hydroxyl group at C7. Third, the isoflavone variation accompanied by the presence of the catechol moiety on the B ring impacted the molecule's reactivities by altering the molecule's thermodynamic properties such as HOMO energy. As for the scavenging activity of the flavonoids against free radicals, the structural features lowering the computed redox potentials were observed to increase the molecule's antioxidant capacity. In addition to the catechol functionality on the B ring, the 7-OH group on the A ring was associated with inhibition against AChE through hydrogen bonding. The chromone framework of the flavonoids also demonstrated supplemental interactions with the active site gorge of AChE through π–π stacking. Overall, our structure–activity relationship study employing a series of 12 flavonoids demonstrates that alterations in the number and location of hydroxyl groups, the presence of a catechol moiety, and the location of the B ring substantially contribute towards the versatile reactivities of flavonoids with multiple pathogenic elements of AD.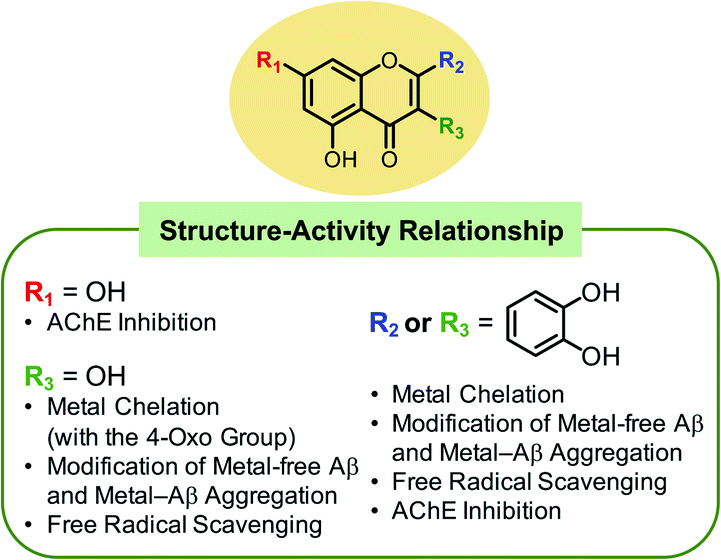 | ||
| Fig. 8 Summary of the findings from the structure–activity relationship studies regarding the flavonoids' modulative reactivities towards multiple pathogenic elements found in AD. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government [NRF-2017R1A2B3002585 and NRF-2016R1A5A1009405 (M. H. L.)] and the Institute for Basic Science (IBS-R10-A1) in Korea (M.-H. B.). We thank Dr Jiyong Park for fruitful discussions on computational analysis.Notes and references
- L. Zhong, Y. Xu, R. Zhuo, T. Wang, K. Wang, R. Huang, D. Wang, Y. Gao, Y. Zhu, X. Sheng, K. Chen, N. Wang, L. Zhu, D. Can, Y. Marten, M. Shinohara, C.-C. Liu, D. Du, H. Sun, L. Wen, H. Xu, G. Bu and X.-F. Chen, Nat. Commun., 2019, 10, 1365 CrossRef.
- M. G. Savelieff, G. Nam, J. Kang, H. J. Lee, M. Lee and M. H. Lim, Chem. Rev., 2019, 119, 1221–1322 CrossRef CAS.
- K. Rajasekhar and T. Govindaraju, RSC Adv., 2018, 8, 23780–23804 RSC.
- T. H Ferreira-Vieira, I. M Guimaraes, F. R Silva and F. M Ribeiro, Curr. Neuropharmacol., 2016, 14, 101–115 CrossRef.
- R. T. Bartus, R. L. Dean III, B. Beer and A. S. Lippa, Science, 1982, 217, 408–414 CrossRef CAS.
- H. Hampel, M. M. Mesulam, A. C. Cuello, A. S. Khachaturian, A. Vergallo, M. R. Farlow, P. J. Snyder, E. Giacobini and Z. S. Khachaturian, J. Prev. Alzheimers Dis., 2019, 6, 2–15 CAS.
- I. W. Hamley, Chem. Rev., 2012, 112, 5147–5192 CrossRef CAS.
- K. P. Kepp, Chem. Rev., 2012, 112, 5193–5239 CrossRef CAS.
- E. Atrián-Blasco, P. Gonzalez, A. Santoro, B. Alies, P. Faller and C. Hureau, Coord. Chem. Rev., 2018, 371, 38–55 CrossRef.
- G. Nam and M. H. Lim, Chem. Lett., 2019, 48, 951–960 CrossRef CAS.
- M. Mold, L. Ouro-Gnao, B. M. Wieckowski and C. Exley, Sci. Rep., 2013, 3, 1256 CrossRef.
- M. G. Savelieff, A. S. DeToma, J. S. Derrick and M. H. Lim, Acc. Chem. Res., 2014, 47, 2475–2482 CrossRef CAS.
- J. Yang, X. Zhang, P. Yuan, J. Yang, Y. Xu, J. Grutzendler, Y. Shao, A. Moore and C. Ran, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 12384–12389 CrossRef CAS.
- S. J. C. Lee, E. Nam, H. J. Lee, M. G. Savelieff and M. H. Lim, Chem. Soc. Rev., 2017, 46, 310–323 RSC.
- M. Mital, N. E. Wezynfeld, T. Frączyk, M. Z. Wiloch, U. E. Wawrzyniak, A. Bonna, C. Tumpach, K. J. Barnham, C. L. Haigh, W. Bal and S. C. Drew, Angew. Chem., Int. Ed., 2015, 54, 10460–10464 CrossRef CAS.
- E. Nam, G. Nam and M. H. Lim, Biochemistry, 2020, 59, 15–17 CrossRef CAS.
- K. P. Kepp, Coord. Chem. Rev., 2017, 351, 127–159 CrossRef CAS.
- F. Collin, Int. J. Mol. Sci., 2019, 20, 2407 CrossRef.
- X. Wang, W. Wang, L. Li, G. Perry, H.-g. Lee and X. Zhu, Biochim. Biophys. Acta, 2014, 1842, 1240–1247 CrossRef CAS.
- C. Berk and M. Sabbagh, Drugs Aging, 2013, 30, 783–792 CrossRef CAS.
- R. M. Anderson, C. Hadjichrysanthou, S. Evans and M. M. Wong, Lancet, 2017, 390, 2327–2329 CrossRef.
- L. McDonald, B. Liu, A. Taraboletti, K. Whiddon, L. P. Shriver, M. Konopka, Q. Liu and Y. Pang, J. Mater. Chem. B, 2016, 4, 7902–7908 RSC.
- S. Kilani-Jaziri, N. Mustapha, I. Mokdad-Bzeouich, D. El Gueder, K. Ghedira and L. Ghedira-Chekir, Tumor Biol., 2016, 37, 6571–6579 CrossRef CAS.
- M. Abotaleb, S. M. Samuel, E. Varghese, S. Varghese, P. Kubatka, A. Liskova and D. Büsselberg, Cancers, 2019, 11, 28 CrossRef CAS.
- N. Mateeva, S. V. K. Eyunni, K. K. Redda, U. Ononuju, T. D. Hansberry, C. Aikens and A. Nag, Bioorg. Med. Chem. Lett., 2017, 27, 2350–2356 CrossRef CAS.
- Y. Xie, W. Yang, F. Tang, X. Chen and L. Ren, Curr. Med. Chem., 2015, 22, 132–149 CrossRef CAS.
- H. J. Lee, R. A. Kerr, K. J. Korshavn, J. Lee, J. Kang, A. Ramamoorthy, B. T. Ruotolo and M. H. Lim, Inorg. Chem. Front., 2016, 3, 381–392 RSC.
- I. Uriarte-Pueyo and M. I Calvo, Curr. Med. Chem., 2011, 18, 5289–5302 CrossRef CAS.
- G. Nam, Y. Ji, H. J. Lee, J. Kang, Y. Yi, M. Kim, Y. Lin, Y.-H. Lee and M. H. Lim, ACS Chem. Neurosci., 2019, 10, 3386–3390 CrossRef CAS.
- G. Nam, Y. Ji, H. J. Lee, J. Kang, Y. Yi, M. Kim, Y. Lin, Y.-H. Lee and M. H. Lim, ACS Chem. Neurosci., 2020, 11, 1372 CrossRef CAS.
- M. D. Engelmann, R. Hutcheson and I. F. Cheng, J. Agric. Food Chem., 2005, 53, 2953–2960 CrossRef CAS.
- S. Cao, X. Jiang and J. Chen, J. Inorg. Biochem., 2010, 104, 146–152 CrossRef CAS.
- S. Sinha, D. H. Lopes and G. Bitan, ACS Chem. Neurosci., 2012, 3, 473–481 CrossRef CAS.
- A. Tiiman, J. Jarvet, A. Gräslund and V. Vukojević, Biochemistry, 2015, 54, 7203–7211 CrossRef CAS.
- L. N. Zhao, Y. Mu and L. Y. Chew, Phys. Chem. Chem. Phys., 2013, 15, 14098–14106 RSC.
- T. A. Enache, A.-M. Chiorcea-Paquim and A. M. Oliveira-Brett, Anal. Chem., 2018, 90, 2285–2292 CrossRef CAS.
- E. Bendary, R. R. Francis, H. M. G. Ali, M. I. Sarwat and S. El Hady, Ann. Agric. Sci., 2013, 58, 173–181 CrossRef.
- L. Zhang, Y. Liu, Y. Wang, M. Xu and X. Hu, Food Chem., 2018, 263, 208–215 CrossRef CAS.
- H. E. Hajji, E. Nkhili, V. Tomao and O. Dangles, Free Radic. Res., 2006, 40, 303–320 CrossRef.
- J. S. Derrick, R. A. Kerr, Y. Nam, S. B. Oh, H. J. Lee, K. G. Earnest, N. Suh, K. L. Peck, M. Ozbil, K. J. Korshavn, A. Ramamoorthy, R. Prabhakar, E. J. Merino, J. Shearer, J.-Y. Lee, B. T. Ruotolo and M. H. Lim, J. Am. Chem. Soc., 2015, 137, 14785–14797 CrossRef CAS.
- J. Han, H. J. Lee, K. Y. Kim, S. J. C. Lee, J.-M. Suh, J. Cho, J. Chae and M. H. Lim, ACS Chem. Neurosci., 2018, 9, 800–808 CrossRef CAS.
- J. Kang, S. J. C. Lee, J. S. Nam, H. J. Lee, M.-G. Kang, K. J. Korshavn, H.-T. Kim, J. Cho, A. Ramamoorthy, H.-W. Rhee, T.-H. Kwon and M. H. Lim, Chem.–Eur. J., 2017, 23, 1645–1653 CrossRef CAS.
- Y. Ji, H. J. Lee, M. Kim, G. Nam, S. J. C. Lee, J. Cho, C.-M. Park and M. H. Lim, Inorg. Chem., 2017, 56, 6695–6705 CrossRef CAS.
- M. Sato, K. Murakami, M. Uno, Y. Nakagawa, S. Katayama, K.-i. Akagi, Y. Masuda, K. Takegoshi and K. Irie, J. Biol. Chem., 2013, 288, 23212–23224 CrossRef CAS.
- V. J. Forrest, Y.-H. Kang, D. E. McClain, D. H. Robinson and N. Ramakrishnan, Free Radicals Biol. Med., 1994, 16, 675–684 CrossRef CAS.
- C. A. Rice-Evans, N. J. Miller and G. Paganga, Free Radicals Biol. Med., 1996, 20, 933–956 CrossRef CAS.
- J. Treml and K. Šmejkal, Compr. Rev. Food Sci. Food Saf., 2016, 15, 720–738 CrossRef CAS.
- I. Tejero, N. González-García, À. González-Lafont and J. M. Lluch, J. Am. Chem. Soc., 2007, 129, 5846–5854 CrossRef CAS.
- J. P. De La Cruz, M. I. Ruiz-Moreno, A. Guerrero, J. A. López-Villodres, J. J. Reyes, J. L. Espartero, M. T. Labajos and J. A. González-Correa, J. Nutr. Biochem., 2015, 26, 549–555 CrossRef CAS.
- M.-H. Baik and R. A. Friesner, J. Phys. Chem. A, 2002, 106, 7407–7412 CrossRef CAS.
- M. F. Santillo and Y. Liu, J. Pharmacol. Toxicol. Methods, 2015, 76, 15–22 CrossRef CAS.
- M. Ahmed, J. B. T. Rocha, M. Corrêa, C. M. Mazzanti, R. F. Zanin, A. L. B. Morsch, V. M. Morsch and M. R. C. Schetinger, Chem.-Biol. Interact., 2006, 162, 165–171 CrossRef CAS.
- D. Hamelberg, J. Mongan and J. A. McCammon, J. Chem. Phys., 2004, 120, 11919–11929 CrossRef CAS.
- Y. Bourne, J. Grassi, P. E. Bougis and P. Marchot, J. Biol. Chem., 1999, 274, 30370–30376 CrossRef CAS.
- E. P. Barros, J. M. Schiffer, A. Vorobieva, J. Dou, D. Baker and R. E. Amaro, J. Chem. Theory Comput., 2019, 15, 5703–5715 CrossRef CAS.
- D. L. Mobley and K. A. Dill, Structure, 2009, 17, 489–498 CrossRef CAS.
- J. S. Wright, E. R. Johnson and G. A. DiLabio, J. Am. Chem. Soc., 2001, 123, 1173–1183 CrossRef CAS.
- D. Procházková, I. Boušová and N. Wilhelmová, Fitoterapia, 2011, 82, 513–523 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section and Fig. S1–S5. See DOI: 10.1039/d0sc02046j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |