
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnlightening advances in polymer bioconjugate chemistry: light-based techniques for grafting to and from biomacromolecules
Rebecca A.
Olson
,
Angie B.
Korpusik
and
Brent S.
Sumerlin
 *
*
George & Josephine Butler Polymer Research Laboratory, Center for Macromolecular Science & Engineering, Department of Chemistry, University of Florida, Gainesville, Florida, USA. E-mail: sumerlin@chem.ufl.edu
First published on 6th May 2020
Abstract
Photochemistry has revolutionized the field of polymer–biomacromolecule conjugation. Ligation reactions necessitate biologically benign conditions, and photons have a significant energy advantage over what is available thermally at ambient temperature, allowing for rapid and unique reactivity. Photochemical reactions also afford many degrees of control, specifically, spatio-temporal control, light source tunability, and increased oxygen tolerance. Light-initiated polymerizations, in particular photo-atom-transfer radical polymerization (photo-ATRP) and photoinduced electron/energy transfer reversible addition–fragmentation chain transfer polymerization (PET-RAFT), have been used for grafting from proteins, DNA, and cells. Additionally, the spatio-temporal control inherent to light-mediated chemistry has been utilized for grafting biomolecules to hydrogel networks for many applications, such as 3-D cell culture. While photopolymerization has clear advantages, there are factors that require careful consideration in order to obtain optimal control. These factors include the photocatalyst system, light intensity, and wavelength. This Perspective aims to discuss recent advances of photochemistry for polymer biomacromolecule conjugation and potential considerations while tailoring these systems.
Introduction
Technologies involving proteins and enzymes comprise a rapidly expanding industrial sector.1 Enzymes serve as catalysts or additives in food processing, animal feedstocks, papermaking, laundry detergents, cosmetics, cleaning products, biofuels, and medicines.1,2 Biomacromolecular therapeutics have garnered particular attention because they exhibit characteristics that small molecules cannot easily replicate. In particular, they often show high target specificity and may assist in protein replacement and augmentation. While drug development is plagued with low success rates from unforeseen side effects in clinical trials, protein therapeutics have often proven more biologically benign and moved through FDA approval processes more quickly.3 Sales of the first FDA-approved therapeutic protein, insulin, began in 1982. Today, there are over 100 FDA approved therapeutic proteins, and a substantial portion of the top 200 prescribed drugs on the market are protein-based (20–30% in 2016–2017).3–5 This industry is still rapidly expanding, with 21 new biologic licenses approved by the FDA in 2018.6Despite their utility, biological macromolecules are often digested rapidly by the body prior to excretion by the kidneys, rendering them ineffective. Also, repeated administration can trigger increased antibody formation, with serious immunogenic effects in some patients.7 Fortunately, the drawbacks of protein therapeutics can often be addressed by affixing a polymer “shield”. The protective attachment of poly(ethylene glycol) (PEG) to proteins has revolutionized the field of bioconjugate chemistry. Abuchowski et al. modified bovine serum albumin (BSA) and bovine liver catalase with PEG using trichloro-s-triazine in 1977.8,9 These seminal works demonstrated that protein conjugates have enhanced aqueous solubility, delayed clearance from the body, and reduced immunological response. Coupling polymers to proteins also increases the storage lifetime by limiting aggregation, a phenomenon that can lead to denaturation.10 Additionally, polymer–protein conjugates tend to have higher retention of activity under biologically taxing conditions: i.e., heating, freezing, lyophilisation, and other harsh but necessary processes.11
The efficacy of polymer–protein conjugates in the field of medicine has been widely recognized, and by 2018 the FDA approved 14 PEG–protein conjugate drugs.4 These pegylated drugs (e.g., Adagen, Neulasta, Pegasys, PEG-Intron, Oncaspar, and Somavert) are used to treat a wide range of issues including immunodeficiencies, hepatitis C, leukemia, and acromegaly.3,12
Increased interest in these conjugates for biomedical applications necessitated advances in the development of polymer–protein conjugation chemistry. Typical conjugation methods are grafting-to, grafting-from, and grafting-through polymerizations (Fig. 1). Grafting-to involves conjugation of a pre-synthesized polymer to a biomolecule. Grafting-from involves chemically modifying the biological entity with a moiety capable of initiation or chain transfer, and then polymerizing directly from the biohybrid macroinitiator. Grafting-through also involves conjugation of a small molecule species to the biomolecule, however, in this case, the molecule (typically vinylic in nature) serves as a comonomer in a subsequent polymerization. This Perspective focuses on grafting-to and grafting-from approaches, but the elementary principles of light-based polymerization techniques discussed in the context of grafting-from reactions also apply to grafting-through reactions.
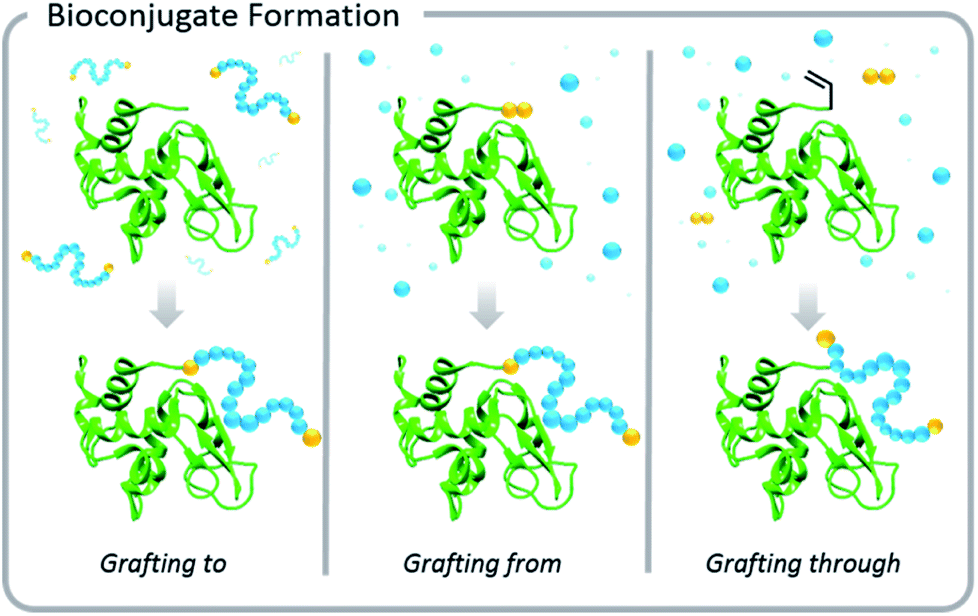 | ||
| Fig. 1 Pictorial representations of conjugation methods: grafting-to entails synthesizing a polymer and attaching it to a biomacromolecule; grafting-from involves polymerizing from a biomolecule that has been modified with a small molecule initiator/chain transfer agent; grafting-through entails modifying the biomacromolecule with a monomer moiety that is incorporated by propagation during subsequent polymerization. | ||
Common conjugation reactions for proteins include carbodiimide coupling (particularly with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC)), N-hydroxysuccinimide coupling (NHS), thiol–maleimide click reactions, and disulfide bond formation (particularly with pyridyl disulfides). There are many exemplary reviews that discuss types of conjugation reactions and the field of bioconjugate synthesis.4,10,12–17
While advances in polymer–protein conjugation chemistry continue, it should be noted that there are inherent challenges associated with synthesizing these materials. Conjugation with and polymerization from biological species comes with a stringent list of requirements: the system must typically rely on no or low metal concentrations, limited organic solvents, low temperatures, and neutral pH values (usually 6–8). Simply, the chemical conditions should be designed to mimic the native environment of the biomacromolecule. In the case of proteins, subsequent polymer grafting must not affect the 3-D structure or block any active binding sites. Furthermore, proteins themselves can interfere with the polymer–protein conjugation process. In grafting-from radical polymerization, free thiols and amines in the amino acid backbone can cause off-target reactions such as aminolysis of chain transfer agents or deleterious chain transfer during polymerization. The aqueous environment can also lead to hydrolysis of reagents used for conjugation. Nonetheless, creative solutions exist to tailor conjugation conditions to make them more selective, mild, water-friendly, oxygen tolerant, and robust.18,19
One innovative solution that has expanded the field of bioconjugation is the exploitation of photochemistry. Light chemistry has been a staple within the biochemistry community for decades. It initially found its place in small molecule applications such as photoaffinity labelling (a technique for identifying active and binding sites)7 and photodynamic therapy.20 More recently, light has been shown to facilitate the synthesis of well-controlled polymers under extremely mild conditions, which serves as a significant benefit for grafting-from polymerization systems. Light also enables spatial and temporal control. Within grafting-to conjugations specifically, these advantages have been exploited to achieve precise placement of polymers, the creation of new biomaterials, and photopatterning to form complex 3-D cell culture environments (Fig. 2).21,22 The use of light to effect polymer–protein bioconjugation is still developing, and the future holds numerous opportunities for study, expansion and refinement. There is a serious push to target conjugations to a larger variety of amino acids, to find more bio-friendly conditions, and to explore grafting with more complex systems, e.g., live cells.23–25 The goal of this Perspective is to critically evaluate the benefits photochemistry brings to polymer–protein conjugation, to serve as a guide when designing such systems, and to elucidate opportunities for further development.
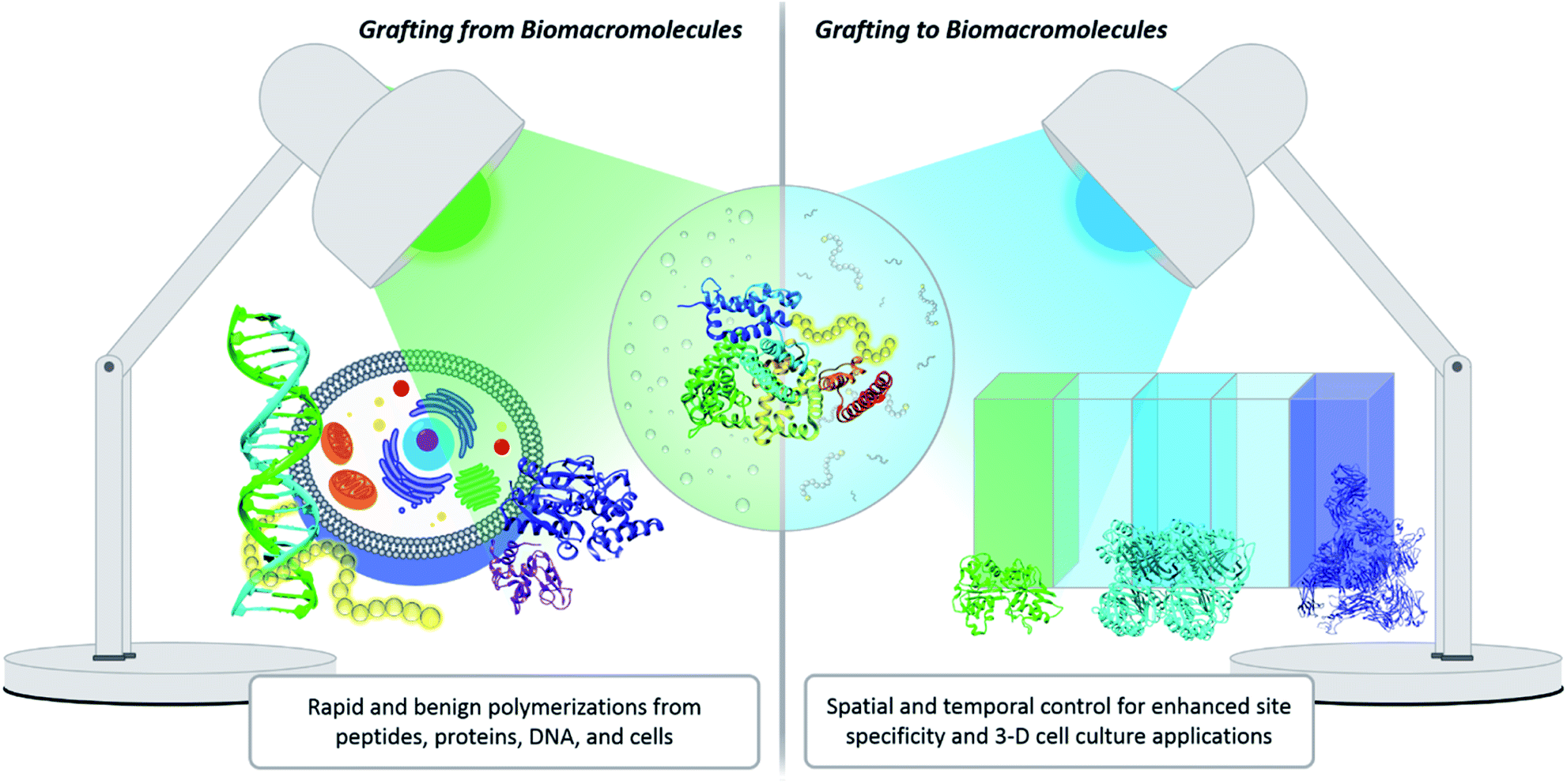 | ||
| Fig. 2 Light has been used for grafting polymers from biological entities such as proteins, cells, and DNA, all of which have potential therapeutic uses. Photochemical reactions for grafting biomacromolecules to structures such as hydrogels have applications in 3-D cell culture, biomedical implants, and drug delivery. | ||
Benefits and caveats of photochemistry
Photopolymerizations are activated through photoexcitation of a chromophore, the light-absorbing moiety of a photocatalyst. During photoexcitation, an electron of the chromophore is promoted to a higher energy orbital.26 This excited chromophore can lose its newly acquired energy through conversion to heat, luminescence emission, homolytic bond cleavage, or energy/electron transfer to another molecule. Both homolytic bond cleavage and energy/electron transfer to another moiety can generate radicals that can initiate polymerization. The probability of a certain photochemical process occurring is proportional to the quantum yield of the reaction, an important parameter in photopolymerizations.26There are several types of commercially available light sources for photopolymerization. The most common are fluorescent lamps, light-emitting diodes (LEDs), and lasers. Fluorescent lights are inexpensive and simple to use; however, their emission profiles are much broader than those of the other light sources.26 Lasers feature monochromatic emission and precision, but their widespread adoption for photopolymerization is limited because of their greater expense. LEDs comprise a new generation of light sources that are favored due to their narrow emission profile, low cost, ease of use, and high performance.27,28 Another benefit of LEDs is their ability to emit light outside the visible region of the electromagnetic spectrum, enabling greater versatility.
Benefits of light
Light confers several significant advantages to bioconjugation chemistry: light reactions (1) often do not require exogenous radical initiators, (2) proceed rapidly at room temperature, (3) exhibit excellent source tunability, and (4) often have enhanced oxygen tolerance (Fig. 3).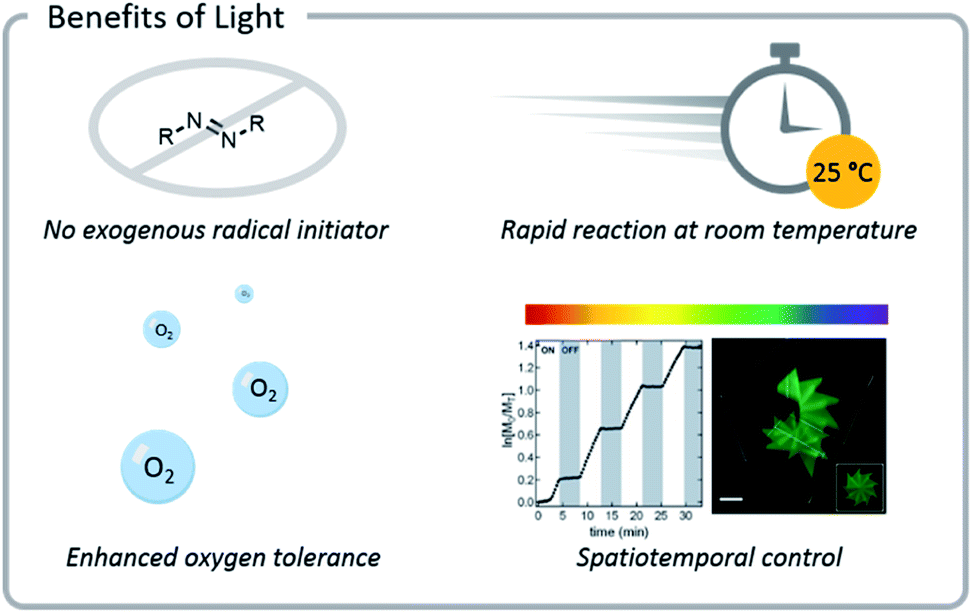 | ||
| Fig. 3 Photochemical reactions often do not need exogenous radical initiators and display enhanced oxygen tolerance. Reactions also proceed rapidly at room temperature and can be tuned easily (light source wavelength and intensity, reaction time and location). Reprinted with permission from ref. 93. Copyright 2017 American Chemical Society. Reprinted by permission from Springer: C. A. DeForest and D. A. Tirrell, A photoreversible protein-patterning approach for guiding stem cell fate in three-dimensional gels, Nat. Mater., 2015.124 | ||
The first advantage of photopolymerization is that while it can be performed using photoinitiators, it does not necessitate the addition of exogenous radical initiators, i.e., radical initiating species that are not bound to the protein. Exogenous radical initiators will inherently initiate and cap a small percentage of chains, resulting in “dead” chains that cannot react further (albeit alternate termination mechanisms such as combination and disproportionation will also result in “dead” chains).29,30 This becomes a particular issue during chain extensions to form block copolymers.31 If a portion of the first block has terminated, then those chains cannot be extended. As the second block is being formed, exogenous radicals can also initiate new chains of the second monomer, leading to a small number of homopolymers of each block. The same phenomenon causes issues when synthesizing bioconjugates through grafting-from polymerizations. When grafting-from a protein, exogenous radical initiators lead to a small number of initiator-derived chains that are free in solution rather than covalently bound to the protein, thus making purification more difficult.32
The second advantage light confers is a rapid rate of polymerization at room temperature, even at relatively low monomer concentrations. At 25 °C, the energy available in a photon at wavelengths relevant for photopolymerization is much greater than the thermal energy available.33 When performing reactions in the presence of proteins, polynucleotides, or cells, it is important to keep the temperature low and the reaction rate rapid to maintain the structural and functional integrity of biological molecules and tissues.34,35 Biologically relevant temperatures are near ambient temperature (30 °C).36 Heating biomacromolecules to higher temperatures may result in aggregation and loss of function, although enhanced tolerance to heat can be achieved by attachment of polymers in some cases.37,38 To compensate for the inability to use the high temperatures needed for most controlled radical polymerizations, higher concentrations of the radical initiator are needed at lower temperatures to offset the slower reaction rates. Alternatively, radical initiators with lower half-life temperatures are used in concentrations 5 to 25 times higher than in a traditional polymerization, which can be problematic due to side reactions that may arise (vide supra).39–41
High concentrations of monomers and other organic compounds can cause protein denaturation and cell death, although altering the identity of the monomer does change the cellular toxicity.23,32 A general guideline suggests that for biomacromolecular conjugation reactions the total organic content should not exceed 20% of the total volume of the reaction.36 Decreasing the monomer concentration typically decreases the rate of the polymerization, but this can be partially offset by the increased radical generation rates associated with light initiation and aqueous conditions.32,42
The third advantage light confers is the ability to tune the irradiation source. Light reactions offer spatio-temporal and reaction rate control by tuning the source directionality, wavelength, and intensity. Temporal control is the ability to reversibly induce propagation by simply toggling the light source on or off. Control of this kind is virtually impossible in thermally-initiated polymerizations due to the limits of heat transfer.26 In radical-based grafting-from bioconjugate polymerizations, temporal control has been used to reversibly halt polymer chain propagation.32,43–45 Temporal control can be important in biological applications for limiting the amount of time sensitive biological entities such as cells are in a radical environment. Spatial control of polymerizations is particularly important for grafting-to systems. For example, when grafting to hydrogel networks, the spatial control of light can create patterns in the gel which guide protein attachment and cell adhesion.21
Polymerizations can also be controlled through light intensity manipulation.28 A higher light intensity correlates with a higher concentration of photons capable of chromophore photoexcitation and subsequent generation of active species.26 If the intensity is sufficient, high monomer conversions can be achieved quickly, often even in the presence of oxygen.46 There are, however, caveats to consider. While more intense light affords faster polymerization rates, it may also increase the chance of denaturing the biological macromolecule and leading to reduced control during radical polymerization.
The wavelength of a light source is another crucial parameter of photopolymerization and provides a means of controlling the polymerization rate without having to alter the temperature. The molecular structure of the photocatalyst affects its molar extinction coefficient – how strongly it absorbs light at a specific wavelength.26 Since photocatalysts have varying degrees of absorption at different wavelengths, the kinetics of a photopolymerization depend on the pairing of a photocatalyst with a wavelength of light.47 Photocatalysts with broad absorption spectra allow for polymerization with light sources spanning several wavelengths.48 In specific cases, wavelength can also govern the mechanism of polymerization, turning reaction pathways on or off.49
NIR-emitting light sources offer the particular advantage of being able to activate photochemical reactions inside the body. When light is irradiated on skin, transmission of the photons competes with absorption and scattering by chromophores that make up the tissue.50 Such scattering disperses the light and reduces its intensity at increasing depths.51 In general, longer wavelengths experience less scattering and penetrate deeper, with NIR light penetrating farther than light in the visible range.27,52 At longer wavelengths than NIR, however, excitation of water molecules becomes non-trivial and causes a heating effect (i.e., microwaves).
The fourth advantage light confers is enhanced oxygen tolerance. Photopolymerizations have the potential to be performed in the presence of oxygen, without experiencing substantial inhibitory effects. Oxygen is a radical scavenger and inhibits traditional radical polymerization by terminating chains.27,53 Oxygen tolerance often arises in photopolymerizations when the photoexcited catalyst chemically converts oxygen into other species that do not interfere with radical polymerizations (vide infra).26 Oxygen tolerance eliminates the need for degassing prior to polymerization. This allows polymerizations to be conducted in extremely low volumes that would normally be difficult to deoxygenate,54 facilitating the synthesis of bioconjugates that are hard to prepare in large quantities.27,55 Additionally, the ability to perform polymerizations directly in multi-well cell culture plates facilitates screening of bioconjugates for biological applications.56 Notably, oxygen tolerance also enables polymerization from live cells, which would not survive rigorous degassing procedures.24
Caveats with light
UV light (<400 nm) is high in energy, which makes it an excellent choice for many photochemical reactions and polymerizations. Unfortunately, UV light can have harmful effects on the structure and function of biomacromolecules. Such high energy light has been associated with deleterious photochemical processes in both proteins and DNA.18 The tertiary structure of a protein is stabilized by disulfide bonds between cysteine residues.57 These covalent crosslinks help constrain the conformation of the polypeptide and, thus, confer thermodynamic stability.58 Prolonged irradiation of proteins with UV light can damage the disulfide linkages in a protein, especially when tyrosine, tryptophan, or phenylalanine (amino acids containing aromatic moieties) are in close proximity to the disulfide bonds.58–60 Each of these amino acids can undergo photoexcitation to the triplet state and then participate in a multitude of degradative reactions. Tryptophan is not abundant, but it is particularly sensitive to photolytic degradation processes because of its high molar extinction coefficient. Upon excitation, it can form a radical cation which can participate in various degradative pathways, including backbone cleavage.60 UV radiation can also harm polynucleotides. This damage can be significant, as DNA and RNA have the highest absorption coefficients among all cellular components, in the 200–300 nm range.61 UV light can photolytically produce radical cations of DNA bases, which initiate single strand breaks in DNA.61 Because typical protein (and polynucleotide) absorption falls in the UV range of 180–305 nm, moving to longer wavelengths which have lower energy, i.e., red-shifting, can reduce these deleterious effects.60 Simply moving from UV or violet light to blue light can prevent damage to the protein structure, although the sensitivity will be protein dependent.42,60Grafting-from
The two types of controlled radical polymerization most commonly used for grafting-from proteins are ATRP62–64 and RAFT65 polymerization. Grafting-from polymerizations are beneficial because purification of the macromolecular bioconjugates from residual monomer is inexpensive and facile. Additionally, grafting-from polymerizations allow for the creation of unique polymeric architectures that can affect the overall hydrodynamic radius and, in some cases, lower the cytotoxicity of the bioconjugate.4 However, when a grafting-from approach is used, the conditions must be gentle enough to not harm the biological entity or induce side reactions. Light-initiated polymerizations have helped to realize this goal.Atom transfer radical polymerization
ATRP was the first controlled radical polymerization technique used for grafting-from biomacromolecules.64,66,67 ATRP utilizes reversible abstraction of a halide from an alkyl compound to generate a radical that propagates through the addition of monomer. This abstraction is typically performed by a metal–ligand activator complex, which is often copper-based. The resulting metal-halide complex (deactivator) can then reinstall the halide on the growing polymer chain-end, imparting control by limiting the rate at which individual chains grow (Fig. 4). Ligands are normally bi- or tri-dentate amines which enhance copper complex solubility and vary the redox potential of the complex.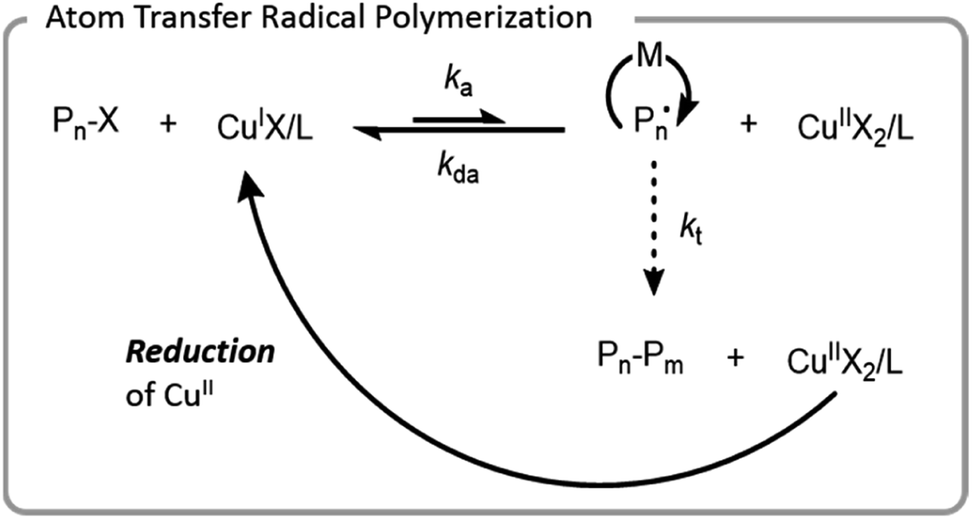 | ||
| Fig. 4 Mechanism of ATRP with a copper catalyst; X denotes a halide, L denotes a ligand, M denotes a monomer, and P denotes a polymer chain. | ||
In early aqueous radical polymerization, there were a few factors that hindered the utility of ATRP in biomacromolecular conjugation applications. Relatively high copper loadings were necessary to achieve well-controlled polymerization with low dispersity and minimal unwanted side reactions. Ligands have a higher rate of dissociation from copper in aqueous systems which led to inefficient deactivation.67 Water also has the potential to hydrolyze the carbon halogen bond.68 Since proteins and living systems can be sensitive to high copper concentrations, the development of lower copper systems advanced the potential for ATRP to be used for the synthesis of more diverse bioconjugates. To achieve comparable polymerization control, these systems generated or regenerated the CuI activation complex. Popular systems for bioconjugation include initiators for continuous activator regeneration (ICAR) ATRP, activators generated by electron transfer (AGET) ATRP, activators regenerated by electron transfer (ARGET) ATRP, and electrochemically mediated ATRP (eATRP).69,70 Each of these systems regenerate CuI which is oxidized to CuII during termination reactions. Typical termination reactions are either combination or disproportionation of two growing radical chains, processes that leave behind the CuII metal-halide–ligand deactivating complex. The higher concentration of deactivator increases the rate of deactivation, thereby increasing the control through a process called the persistent radical effect.69 ICAR ATRP uses organic radicals, and AGET and ARGET use reducing agents for oxidation; AGET and ARGET are differentiated in that AGET starts with a CuII species whereas ARGET starts with a CuI species.71,72 eATRP utilizes an electrical current for the reduction of CuII. All of these techniques have been used for the successful formation of biomacromolecule conjugates.68,73–75 ATRP has even been used for polymerizing from cells.76
Another method for lowering the copper content of aqueous ATRP is using light to either generate or regenerate the active CuI species.77,78 While several operative mechanisms occur simultaneously, the predominant mechanism is excitation of CuII followed by single electron donation from the free amines of the ligand to form a radical cation amine species and an active CuI species.79 One benefit of this polymerization is that no exogenous initiators or reducing agents are needed, simply a higher loading of the ligand. This makes it easy to purify and ideal for grafting from biomaterials. Matyjaszewski et al. demonstrated that blue light (450 nm) could be used for rapid and well-controlled polymerization from both BSA and DNA with CuII and a tris(2-pyridylmethyl)amine (TPMA) ligand (Fig. 5).42 This system had excellent temporal control, evidenced by the ability to halt polymerization when the light source was removed. The blue light was gentle enough to have no substantial impact on the secondary structure of the proteins (BSA and glucose oxidase, GOx) over the course of the reaction. These polymerizations are ideal for high throughput applications due to their temporal control, mild reaction conditions, and moderate oxygen tolerance.80 In fact, photoATRP was performed in a highly automated fashion using a DNA synthesizer; direct synthesis of DNA and polymerization from DNA was achieved without the need to degass.81 This example additionally demonstrated that very low volume reactions can be attained with this system. Anastasaki et al. further demonstrated the versatility and oxygen tolerance of photoATRP for grafting-from proteins.82 (Meth)acryloyl and styrenic polymers were grafted from multiple proteins in polypropylene syringes – the photoATRP system eliminated the need to degas the system.
 | ||
| Fig. 5 PhotoATRP grafting-from polymerization using a BSA macroinitiator under blue light. Reprinted with permission from ref. 42. Copyright 2018 American Chemical Society. | ||
PhotoATRP can be performed with multiple catalyst systems, although copper systems are highlighted in this Perspective because they are the most common. Non-copper-based systems, e.g., FeBr3 and Ru(bpy)3Cl2, may be more desirable for some applications as they may confer enhanced temporal control, lower cost, or a higher degree of tolerance to certain functional groups.43,48,83,84 Additionally, changing the catalyst can allow for polymerization under different wavelengths of light.48 The operative wavelength of the polymerization can also be adjusted by adding a photosensitizer. Strehmel et al. reported the addition of a photosensitizer which allowed photoATRP to be carried out under NIR irradiation.85
With a copper-catalyzed system, the ligand should be chosen to maximize the stabilization of the catalyst complex. Aqueous media exhibits excellent ionic stabilization which increases the dissociation of the halide from the CuII complex, decreasing the amount of deactivator complex and leading to poorly controlled polymerizations. Two of the most common ligands for these aqueous systems are TPMA and tris[2-(dimethylamino)ethyl]amine (Me6TREN). Both compounds coordinate strongly with copper, improving the polymerization control.86 TPMA stabilizes the CuI and CuII complexes more efficiently than Me6TREN, as indicated by the stability constant.86
The presence of salt also plays a role in the stability of the copper catalyst complex. The preference for displacement of halide anions with hydroxyl anions in aqueous systems can be mitigated to some extent by (1) adding halide salts to increase the concentration of halide anions or (2) increasing the concentration of amines in the system (ligands or non-ligating tertiary amines such as pyridine) to push the equilibrium toward the formation of copper(II)-halide species. Consequently, many phosphate buffer solutions for bioconjugations contain sodium chloride (NaCl). These chloride anions replace the bromine polymer chain-ends. A polymer with a chlorine chain-end polymerizes more slowly, but the carbon-chloride bond is also stronger and less susceptible to hydrolysis than the carbon-bromide bond; therefore, it can improve control.68
Choice of catalyst also plays a role in temporal control for photopolymerizations. In a photopolymerization system where KATRP is high (KATRP = kact/kdeact), there is a relatively high number of active species within the system. Higher concentrations of active species require more time to deactivate, and polymer growth can occur even after the light is turned off.87 This can clearly be seen when comparing Me6TREN and TPMA; TPMA has a lower KATRP, so the rate of deactivation is higher. The equilibrium is shifted to the left, resulting in a lower concentration of the active CuI species allowing for the reaction to more effectively be turned “off” immediately when the light source is removed. Me6TREN leads to a higher KATRP, indicating that the deactivation proceeds more slowly allowing for polymer growth in the dark.87
Finally, photocatalyst systems can also impart oxygen tolerance to photoATRP systems under specific conditions, a distinct benefit.88,89 With certain photoATRP systems, polymers can be synthesized without any deoxygenation procedures and maintain monomodal molecular weight distributions and high end-group fidelity, simply by completely filling the reaction vessel (leaving no air above the solvent).88 This chemical degassing is a light-induced reductive scrubbing process. The light excites the copper–ligand photoredox complex, reducing CuII to CuI. The CuI can either (1) reduce the predominant oxygen species, diradicaloid triplet oxygen, to singlet oxygen, which is more zwitterionic in nature and therefore exhibits different reactivity or (2) abstract bromine to form a carbon-centered radical which can react with oxygen in an analogous manner.46,89 The singlet oxygen does not terminate polymer chains, but it does slowly decay back to the lower energy triplet state. This elegant approach has the benefit of avoiding additional reagents (e.g., reducing agents or enzymes) in the reaction mixture, but there are many excellent options for improving oxygen tolerance in reversible-deactivation radical polymerization (RDRP) processes. Amine reducing agents,56 the enzyme glucose oxidase,74,90,91 and other reducing agents,92 including ascorbic acid,93 are quite popular for removing oxygen from both ATRP and RAFT polymerizations. Enhanced oxygen tolerance can be achieved for simple and convenient high throughput polymer synthesis, albeit some methods only have modest control. The oxygen levels in these systems can be measured either with sensors or with absorbance measurements. A common assay uses 9,10-dimethylanthracene to probe conversion of triplet oxygen into singlet oxygen.46,89
Aqueous photoATRP has been refined and uniquely tuned to be effectual in bioconjugation systems. We expect continued development in the area of bioconjugation via photoATRP in the near future. As exciting as these advances are, there are some biomacromolecules whose sensitivity to copper precludes them from participating in bioconjugation reactions using ATRP.94 While the development of metal-free photoATRP may be able to address metal catalyst sensitivity in biological systems in the future,95–98 RAFT polymerizations are an excellent metal-free alternative for us to explore (vide infra).
Reversible addition–fragmentation chain transfer
Conventional RAFT utilizes a chain transfer agent (CTA) which controls the polymerization by a degenerative chain transfer process (Fig. 6). An initiating radical adds to monomer to begin a new polymer chain, Pm. After propagating with monomer, the radical on the end of the propagating chain, Pm, will attack the CTA. A stabilized radical intermediate forms on the thiocarbonylthio-Z group prior to C–S bond cleavage to reform a thiocarbonyl and release a radical moiety which can react with monomer. The radical moiety is either an R group (pre-equilibrium) or a growing polymer chain (main equilibrium in degenerative chain transfer). In the end, the majority of chains should be derived from the R and thiocarbonylthio-Z groups of the CTA, becoming the α- and ω-ends of the chain, respectively.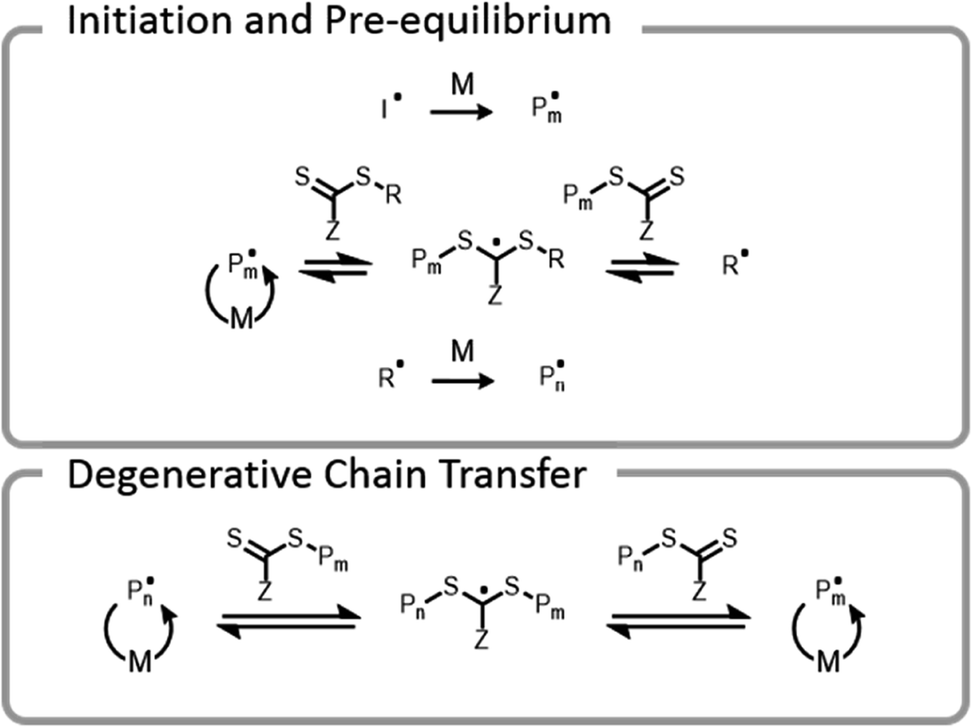 | ||
| Fig. 6 Abridged mechanism of RAFT; I denotes an initiator, M denotes a monomer, and P denotes a polymer chain. | ||
Early RAFT polymer–protein hybrid research39,99 began with a “transfer-to” mechanism that relied on conjugation of the thiocarbonylthio-Z group of the CTA to the protein.16 During polymerization, a polymer chain detaches from a protein, propagates, and then must attack a CTA on the surface of a protein to reattach. With the continuously increasing steric bulk of the chains over the course of the polymerization, the reattachment reaction loses efficiency and can lead to lower monomer conversions and broader molecular weight dispersities. As Sumerlin et al. have shown, these issues can be mitigated by using a grafting-from approach in which the R group of the chain transfer agent is conjugated to the protein.100 In this case, the growing polymer chain remains anchored to the protein throughout the polymerization, and degenerative chain transfer occurs away from the surface of the protein, allowing for high monomer conversion, controlled growth, and narrow dispersities. This technique also results in high end-group fidelity, which is necessary for further reactions or formation of more complex architectures such as block copolymers.40,41
The first bioconjugates formed by RAFT utilized electromagnetic radiation; Bulmus et al. employed RAFT transfer-to polymer–protein conjugation to BSA using gamma radiation.99 Although this method did not significantly decrease the activity of BSA, it can be argued that concerns over the deleterious effects of γ-rays on less robust proteins and the lack of easy accessibility to instrumentation hindered the advancement of this method into mainstream polymer-conjugation chemistry.
The use of visible light for bioconjugation via RAFT did not emerge until 2014 when two groups concurrently developed and introduced light-based grafting-from conjugation techniques.28,45 Haddleton and Chen et al. utilized the water-soluble visible light radical initiator (2,4,6-trimethylbenzoyl)phenyl phosphonic acid sodium (TPO-Na) for polymer–protein conjugate synthesis45 under low-intensity visible light irradiation (λ = 420 nm, I = 0.2 mW cm−2). As previously mentioned, exogenous initiator in solution leads to a small number of undesired free chains not conjugated to the protein, complicating purification after polymerization. Boyer introduced an alternate method, (PET)-RAFT (Fig. 7).28,38 PET-RAFT utilizes a photoredox catalyst which upon excitation can transfer energy/electrons to the CTA, releasing a polymer chain end that can propagate. The relative contributions of energy or electron transfer to CTA activation are photocatalyst and cocatalyst dependent; the electron transfer pathways are described herein.101–103 The photoredox catalyst can be oxidized by a CTA (e.g., trithiocarbonate), forming a trithiocarbonate anion and a propagating chain end. Alternately, if a tertiary amine is present in the mixture, reductive photoelectron transfer can also occur; the excited photocatalyst can obtain an electron from the tertiary amine and then donate it to the CTA, again forming a trithiocarbonate anion and a propagating chain end. If the wavelength of light used overlaps with the absorption of the CTA, then the CTA may be susceptible to direct homolytic bond cleavage, and a photoiniferter mechanism will occur simultaneously (vide infra).49
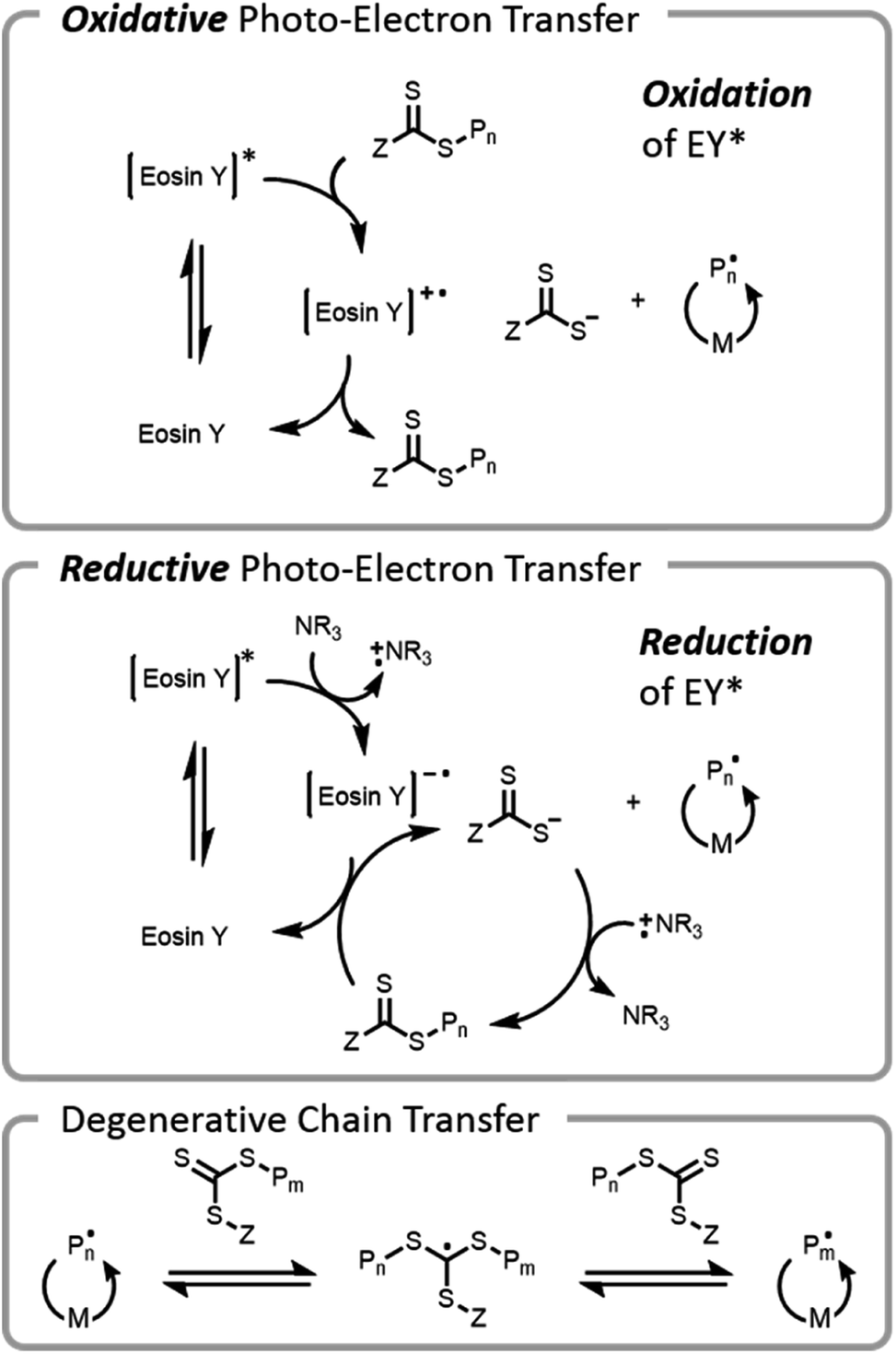 | ||
| Fig. 7 Proposed electron transfer mechanisms operative during PET-RAFT polymerization. M denotes monomer and P denotes growing polymer chain. Oxidative photo-electron transfer processes occur in the presence of a photocatalyst (eosin Y) to (re)generate active chains. Reductive photo-electron transfer processes occur in the presence of both a photocatalyst and an amine. Photolytic cleavage of the chain transfer agent (photoiniferter) is also operative at shorter wavelengths (vide infra). All processes can propagate through degenerative chain transfer. | ||
PET-RAFT can be used to polymerize a large range of monomers (e.g., acrylamides and (meth)acrylates) under biologically friendly conditions with excellent temporal control. If necessary, these reactions can be carried out at low volumes55 and low concentrations (0.1 M monomer).32 Additionally, a successful grafting polymerization from BSA was demonstrated with almost complete retention of activity (96 ± 2%) of the protein. This seminal work opened the door for PET-RAFT grafting-from polymerizations with proteins,32,104 DNA,55 enzymes,37 and lipids.105
The power of photopolymerizations for bioconjugation was further demonstrated by Soh, Hawker, and colleagues using PET-RAFT to grow polymer brushes from the surface of live cells (Fig. 8).24 CTAs were conjugated to the surface of yeast cells via click chemistry. Subsequently, polymers were grown from the surface with reasonable control, chain-end fidelity, and excellent retention of cell function. Poly(ethylene glycol) methyl ether acrylate macromonomers (1 kDa) were used to prevent monomer penetration into the cells, and reaction times were limited to minimize exposure of the cells to the light source. In this case, the grafting-from approach had a significantly enhanced grafting-density compared to the grafting-to approach. While CTA conjugation to the cell wall of yeast could be performed with common coupling chemistry, mammalian cells lack a robust cell wall and therefore needed milder conjugation conditions. To achieve mammalian cell bioconjugates, CTA lipids were synthesized which could insert into the cell membrane, allowing for high cell viability and controlled polymerizations (Fig. 8).105
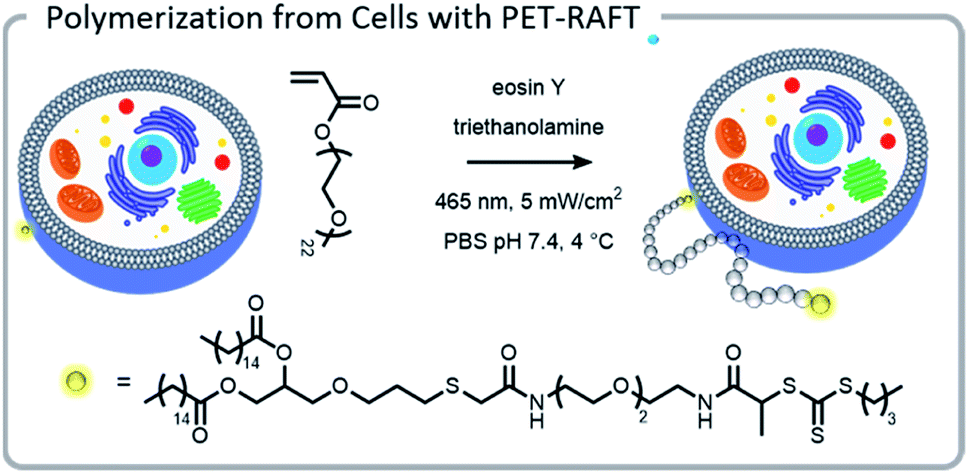 | ||
| Fig. 8 A lipid-mimetic CTA was synthesized and inserted into the membrane of a Jurkat cell prior to a PET-RAFT polymerization under blue light. The reaction did not inhibit normal cell function. Reprinted by permission from Springer: Engineering live cell surfaces with functional polymers via cytocompatible controlled radical polymerization, Nat. Chem., 2017.24 | ||
PET-RAFT utility for bioconjugation reactions is clear, however, careful consideration of the identity of the photocatalyst in conjunction with light wavelength and intensity, type of amine co-catalyst, method of oxygen removal, and CTA is required.
There are numerous organic and inorganic photocatalysts that can be used for PET-RAFT, each of which can be tuned for optimal activity by customizing the light source wavelength and intensity. For bioconjugation applications, catalysts that are operative in low energy visible light are desirable to reduce the likelihood of side reactions that may occur with higher energy UV light (vide supra). Polymerizations with UV light should also be approached carefully to avoid rare but undesirable side reactions due to decomposition of the CTA or self-initiation of monomers.
In particular, dithioester CTAs have been reported to undergo degradation under UV radiation at 365 nm.106,107 Trithiocarbonate CTAs have been used extensively under UV radiation for controlled polymerizations, but in certain environments, they may experience a small degree of decomposition.107 It is also possible that monomers which absorb in the UV region can be self-initiating, resulting in non-ideal kinetic behavior during polymerizations.108
PET-RAFT grafting-from reactions can be performed under blue light with low photocatalyst loadings using photocatalysts such as Ru(bpy)3Cl2.28,38,43,104 However, metal-free organic dyes, particularly eosin Y, are the most commonly applied catalysts for biological applications due to their low toxicity and cost.109,110 Eosin Y has been used for grafting from proteins, DNA, and cells.24,32,37,55 While Ru(bpy)3Cl2 and eosin Y are quite biocompatible, photocatalysts that operate under red light and NIR radiation are favorable since lower energy light has an increased penetration depth and fewer side reactions. NIR initiated PET-RAFT was performed using a chlorophyll catalyst demonstrating that polymerization can occur with the added challenge of an opaque barrier between the reaction mixture and the light source.111 Thus far, most photoconjugation reactions have been performed under blue light, but with the advantages associated with utilizing lower energy light, it is expected that utilizing red or NIR light for grafting-from polymerizations will be a focus area in the future. Given the demonstrated capability of polymerizing from cells, it is foreseeable that NIR radiation may eventually facilitate polymerization from living tissue inside the body.
Photocatalysts are often paired with a tertiary amine co-catalyst, and the nature of the amine can affect the rate of the reaction as well as influence the properties of the final polymer. A tertiary amine can donate an electron to reduce the excited state of the photocatalyst and form a tertiary amine radical cation.49 More stable radical cations are hypothesized to increase the rate of reduction and, consequently, the rate of polymerization.112 There is one caveat to consider, however: using a photocatalyst such as eosin Y and a tertiary amine will result in background initiation, even in systems with no CTA present.112,113 This can lead to a lower end-group fidelity and a higher chain length dispersity. Tertiary amines that have less background initiation are preferable; 4-(dimethylamino)pyridine (DMAP) and tributylamine are particularly efficient reducing agents with low background initiation.87
A final consideration when selecting photocatalysts for PET-RAFT systems is their ability to scrub oxygen. Several photoredox catalysts used for PET-RAFT (notably Ru(bpy)3Cl2,28,38 ZnTPP,46 and eosin Y113) can scrub oxygen from the system by exciting triplet oxygen to singlet oxygen. PhotoRAFT processes with amines also inherently convey some oxygen tolerance because trithiocarbonates are efficient photocatalysts. Upon photoexcitation, the trithiocarbonate can interact with tertiary amines, forming a tertiary amine radical cation and a trithiocarbonate anion which can scrub the system of oxygen.56,114
Of course, in addition to choosing the appropriate photoinitiator catalyst system, the choice of CTA must be carefully considered for PET-RAFT polymerizations. Some CTAs can be susceptible to hydrolysis and aminolysis, resulting in loss of the active CTA in solution and thus a loss of control over the polymerization.115,116 The structure of the CTA also affects the toxicity of the final polymer in cellular systems. For example, poly(N-(2-hydroxypropyl) methacrylamide) (P(HPMA)) synthesized with dithiobenzoates was shown to be highly cytotoxic to three cell lines, unlike comparable polymers synthesized with trithiocarbonates.117 Certain cell lines were also sensitive to other dithiobenzoate end-capped polymers (poly(oligoethylene glycol methyl ether acrylate) (P(OEGA)) and poly(oligoethylene glycol methyl ether methacrylate) (P(OEGMA))), albeit not to the same extent as dithiobenzoate capped P(HPMA).116,117
In summary, the number of factors to be considered in these systems (e.g., photocatalyst systems, chain transfer agents, light source parameters, etc.) and the possible synergistic effects between each demonstrates the vast potential for future exploration, understanding, and refinement. The effects that certain variables will have on diverse proteins are not necessarily known.32 One interesting study compared the activity of two enzymes after they were grafted with N-[3-(dimethylamino)propyl]acrylamide (DMAPA). Both enzymes were lipases, Thermomyces lanuginosa (TL) and Candida antarctica lipase B (CalB), and yet they exhibited opposite responses in their activity after grafting. As this study illustrated, tailoring bioconjugation systems extends beyond consideration of the reaction conditions previously discussed, and beyond monomer identity and molecular weight. Further exploration of these factors and potential coupling reactions for biologically-friendly systems will aid the design of strategically-informed bioconjugation systems.
One final type of grafting-from photopolymerization that should be discussed is the photoiniferter system (Fig. 9). The concept of photoiniferter was introduced by Otsu in 1982, and it was considered the first “living” radical polymerization process.118,119 A photoiniferter is a molecule which upon photolysis can act as an initiator, a transfer agent, and a terminator. While initial systems lacked sufficient control, there has been a recent resurgence of research which showed that this method could be used to synthesize polymers with excellent control, achieving even ultra-high molecular weight species.120 Unlike the PET-RAFT technique, no exogenous electron transfer catalyst is needed because the CTA undergoes homolytic cleavage upon photoexcitation. The lack of photocatalysts or exogenous initiators or catalysts seems attractive for bioconjugate applications. While photoiniferter polymerization has been utilized in a limited number of applications for grafting-from gelatin, these processes typically require shorter wavelength light sources (UV) and higher monomer concentrations.32,121,122 Unfortunately, these two factors can be detrimental to many protein structures.32 There is potential to optimize the system to work efficiently with lower energy light through addition of a photosensitizer or by designing a suitable CTA with an activity that is maximized at longer wavelengths.
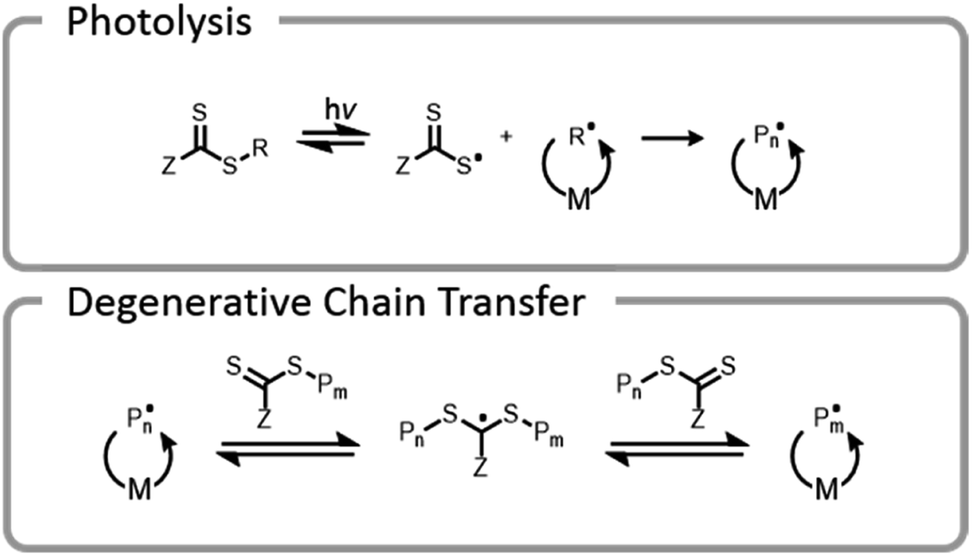 | ||
| Fig. 9 Photoiniferter RAFT mechanism; M denotes monomer and P denotes growing polymer chain; the CTA can undergo homolytic bond cleavage and the R group can initiate polymer chains. The growing chains can either recombine with the radical thiocarbonylthio-Z group (reversible deactivation) or undergo degenerative chain transfer. | ||
Finally, as we have shown, light-based radical grafting-from reactions offer many benefits. Light creates active species in a benign manner and allows for these reactions to be carried out without exogenous initiators. Light-based systems are often more oxygen tolerant than analogous thermally driven systems and confer enhanced user control through light source tunability. Given these advantages, we expect a rise in the utilization of photoATRP and PET-RAFT in grafting-from bioconjugate systems.
Grafting-to
Grafting-to reactions offer the benefit of decoupling the polymerization chemistry from the often sensitive biological entity to which the polymer will be attached, providing a more benign attachment strategy for sensitive proteins or polymers.94 In fact, all of the polymer–protein conjugates that are industrially relevant are synthesized using a grafting-to approach. Nonetheless, there are inherent steric constraints on coupling two macromolecules, and in some cases, purification can be challenging.Light can be used to facilitate grafting-to reactions in a variety of ways: (1) through direct photoactivation and bond formation, (2) through photoactivation of a catalyst, or (3) through photo-unmasking of a reactive moiety. Grafting-to reactions enjoy the same spatial and temporal control that allows site-specificity of labelling to generate unique biological materials and to create complex 3-D cell culture environments that more closely mimic the extracellular matrix (ECM) at different stages of growth and disease. There are several important considerations for these light-based grafting-to reactions: light source wavelength and intensity, oxygen tolerance, and quantum yield. When appropriate conditions are chosen, increased control over the system is clear.
One of the unique benefits of using light is the temporal control that it imparts. This can be utilized to generate highly reactive intermediates in situ that may not have long lifetimes in aqueous conditions, potentially after compounds have preassociated. It is possible that these highly reactive moieties could facilitate more efficient and well-controlled conjugations.7 This potential was explored by synthesizing end-functionalized PEG derivatives with common photoaffinity labelling compounds, aryl azide and diazopyruvate (Fig. 10).7 These photoactive moieties allowed for on-demand conjugation to lysozyme. Aryl azides expelled nitrogen gas leaving behind a nitrene, which rearranged to a seven-membered ring with a highly electrophilic ketenamine. Diazopyruvates also expelled nitrogen gas upon photolysis, leaving a carbene that underwent a Wolff rearrangement. The resultant ketene was highly susceptible to nucleophilic attack.7
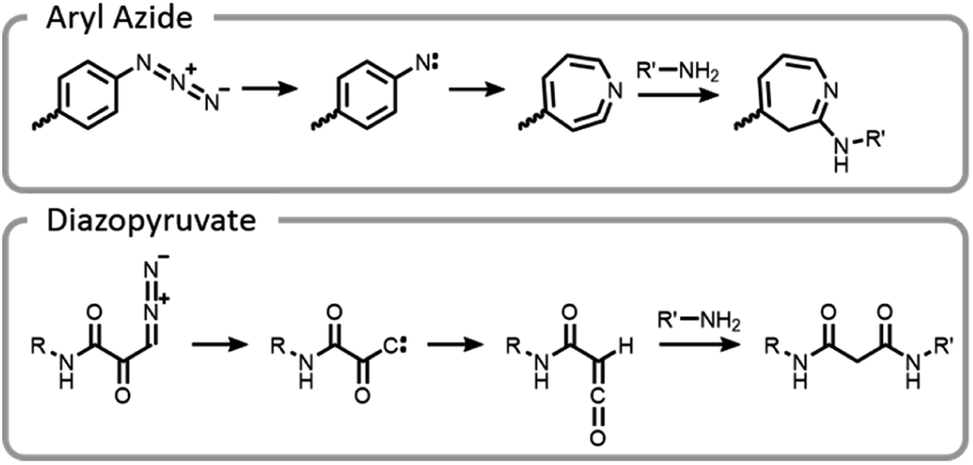 | ||
| Fig. 10 Mechanism of conjugation for aryl azides and diazopyruvates. | ||
Maynard et al. showed how the temporal control light imparts improves the selectivity and yield of grafting-to reactions (Fig. 11).22 A polymer was designed to interact with a protein in a non-covalent host–guest manner, locking the polymer in a specific location, prior to covalent conjugation upon light irradiation. Benzophenone and diazarine are both common photo-initiated covalent bond-forming agents for photoaffinity labelling, so they were chosen for this study (Fig. 11). Upon UV irradiation, the benzophenone formed a triplet state ketone, abstracted a hydrogen radical and then formed a carbon–carbon bond. The other bond-forming agent, diazarine, rearranged to a diazo under UV light, liberating nitrogen gas. The residual carbene rapidly inserted into a carbon–hydrogen bond. While photochemical conjugations are not site-specific, when they are combined with a precise targeting moiety, polymer protein bioconjugation reactions can become more efficient and selective. However, appropriate host–guest pairs must be chosen such that the active site is not blocked.
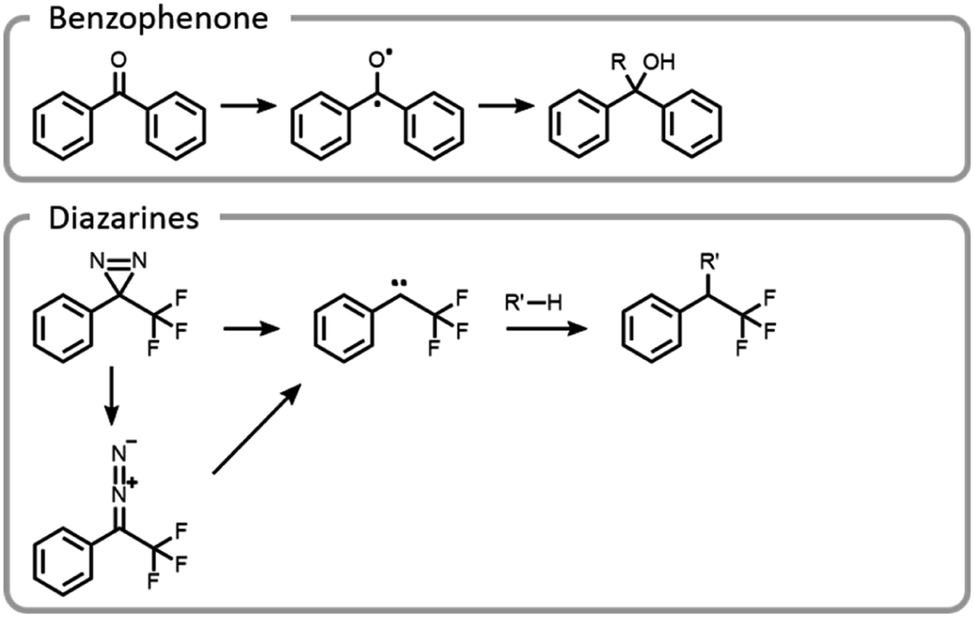 | ||
| Fig. 11 Mechanism of conjugation for benzophenones and diazarines. | ||
A more common conjugation reaction for grafting-to is the thiol–ene reaction, which is often used to modify hydrogel networks for 3-D cell culture. The thiyl radical attacked an ene, forming a carbon-centered radical that abstracted a hydrogen from a thiol, restarting the catalytic process (Fig. 12).123 Networks were formed in the presence of cells using a bio-orthogonal and benign technique, strained alkyne–azide click. Subsequently, a thiol–ene reaction was used to attach peptides to the hydrogels; these peptides promoted cell adhesion to the network. By using light to specifically attach peptides to certain areas, cell morphology and migration patterns were altered.123 The ability to control cell growth is important for cell differentiation and understanding disease states. Control studies where the peptides were not adhered to a hydrogel network did not produce the same changes in cells.124
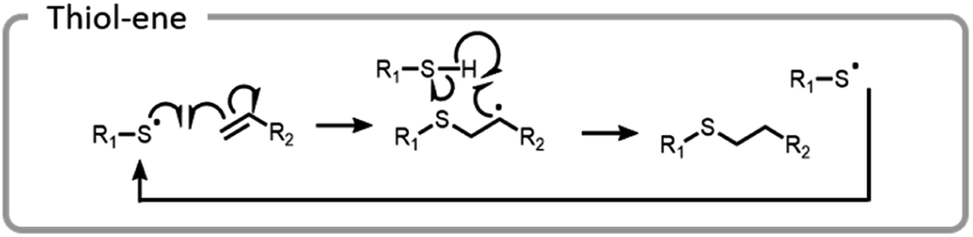 | ||
| Fig. 12 Mechanism of thiol–ene catalytic reaction. | ||
One concern with bioconjugation to thiol–ene networks is the higher photoinitiator concentration that is often needed in these dilute systems where the carbon radical may find another species to terminate with before coming into contact with a thiol, thereby limiting the catalytic cycle. An additional concern is the potential for radical crosslinking of the networks in these processes. Crosslinking increases the modulus of the material, which also has an impact on cell differentiation and growth. Therefore, retention of a constant modulus should be verified during these processes. A final concern is that thiyl radicals may have cross-reactivity with disulfide bonds on proteins or cells.125
Photo-unmasking of reactive moieties for protein or peptide conjugation enables modification of crosslinked materials such as hydrogels. Generating a reactive species after the material is made can allow for attachment of sensitive moieties after harsh processing conditions126 or for the control of minute structures in 3-D cell culture.127 If the reactive moiety is relatively stable under cell culture conditions (such as an aldehyde), the unmasking can potentially be carried out prior to addition of proteins or cells to prevent light from denaturing the sensitive species.128 However, if these moieties are revealed afterward, then the cell penetration will be diffusion-limited, a potentially useful tool for studying the diffusion behavior of cells. o-Nitrobenzyl can be used for photo-unmasking processes because it can undergo a photo-rearrangement to release a moiety and unveil an aldehyde. This aldehyde could then be conjugated to proteins after irradiation. With this spatial control, it was clearly seen that attaching different proteins to the hydrogel can govern cell migration and growth (Fig. 13).
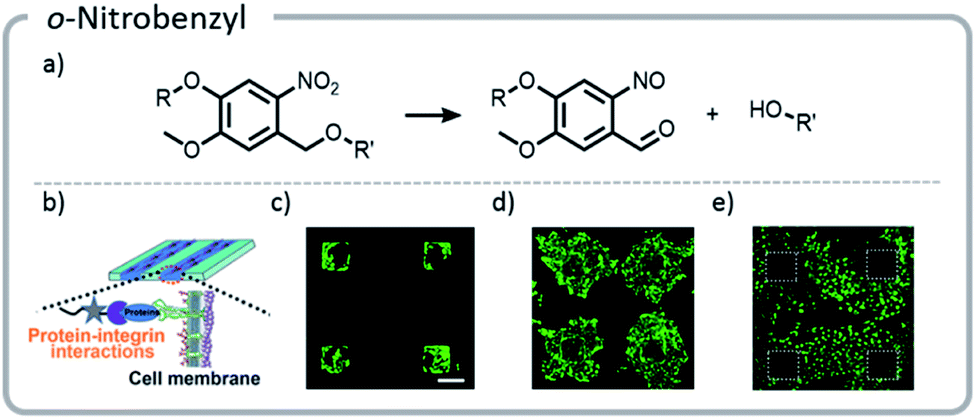 | ||
| Fig. 13 (a) Mechanism of o-nitrobenzyl release; (b) schematic indicating that proteins patterned on the hydrogels will interact with the cell wall, guiding cell movement; (c) cells adhering to the regions that were patterned with gelatin; (d) the hydrogel with cells (c) was irradiated a second time, and gelatin was adhered nonspecifically. Cells migrated out from their initial squares; (e) the area around the squares of cells (c) was irradiated and patterned with fibronectin. The cells preferentially migrated to areas with the second protein. Reprinted with permission from ref. 128. Copyright Wiley, Advanced Functional Materials 2018. | ||
Such an unmasking strategy for patterning gels has been used successfully to study the effects of polypeptide gradients on breast cancer cell lines and cell response to treatment.129 Photomasks were used to create a gradient of the polypeptide epidermal growth factor in a hyaluronic acid hydrogel. The hydrogel was decorated with thiols protected with nitrodibenzofuran. This protecting group was removed with NIR two-photon irradiation, releasing a free thiol that reacted with maleic anhydride moieties via a thiol-Michael addition. Shoichet et al. studied how these gradients affect the movement of three breast cancer cell lines through the material, as well as how these gradients influence the response of the cell lines to drugs; the authors' results indicate these gradients did affect cellular behavior.129 These 3-D culture platforms have the potential to allow for more accurate assessment of how cells will respond to certain drugs.
More recent work has focused not only on creating unique gradients and structures within materials, but also utilizing the inherent temporal control of light to create more dynamic systems.124 In one example, an o-nitrobenzyl derivative, (2-(2-nitrophenyl)propyloxycarbonyl) (NPPOC), was affixed to the hydrogel network. Upon photo-deprotection of the NPPOC with UV light, an alkoxyamine was revealed. A protein with a tethered aldehyde was added, which reacted with the alkoxyamine to form an oxime. The linker between the protein and the hydrogel also contained a photocleavable moiety, nitrobenzyl ester (oNB). The gel was irradiated with light, incubated with protein, and finally, the protein was released from the gel with a second light reaction.
Using this approach, gels were patterned using a photomask. With more advanced multi-photon pulsed laser irradiation, 3-D images were produced with excellent (∼1 μm) resolution, and proteins were adhered in specific patterns within the gel. If the light intensity or irradiation time was carefully chosen, such that only a limited amount of the NPPOC was removed initially, then further irradiation resulted in simultaneous cleavage of the linker between the protein and the hydrogel, deprotecting NPPOC, and revealing reactive alkoxyamines for further protein conjugation. In other words, one protein could be removed while another was attached (Fig. 14).
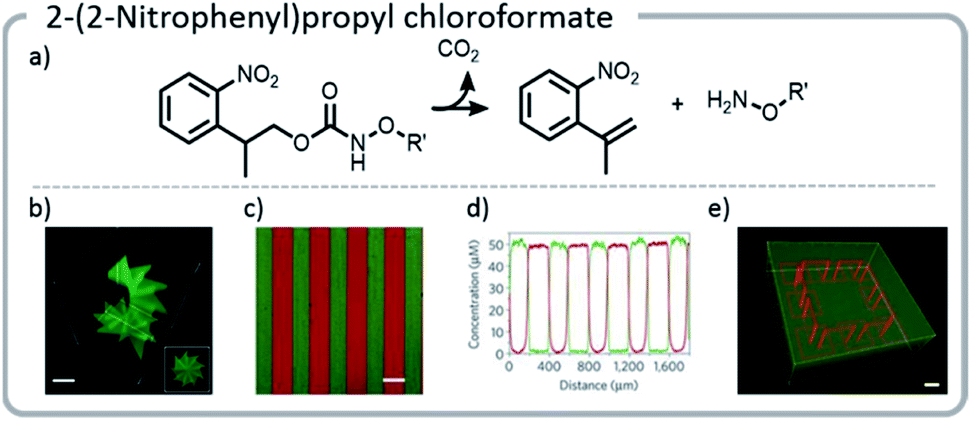 | ||
| Fig. 14 (a) Mechanism of NPPOC release; (b) NPPOC was released from the gel using multiphoton laser scanning, and fluorescently labeled BSA was attached to the liberated alkoxyamine; (c and d) photomasks can be used to selectively irradiate and attach different fluorescently labeled proteins to discrete locations on the hydrogel; (e) a hydrogel was labeled with a green fluorescent protein. Then, further irradiation was used to simultaneously release the green protein in specific areas and create more reactive sites to attach a red protein; an interlocked protein chain was created. Reprinted by permission from Springer: C. A. DeForest and D. A. Tirrell, A photoreversible protein-patterning approach for guiding stem cell fate in three-dimensional gels, Nat. Mater., 2015.124 | ||
The approach above paved the way for dynamic exchange of covalently bound bioconjugates, showing that gels could effectively be erased and rewritten. Anseth et al. took that concept one step further and demonstrated that with the correct choice of light-driven chemistry, the exchange reaction could be cycled indefinitely (Fig. 15).21 With allyl sulfide chemistry, proteins that have lost their activity could be removed and replaced with fresh, active protein. This exchange of biological entities utilizes the temporal control of light to mimic a more realistic ECM environment which continuously changes with time. As cells grow and differentiate within the matrix, various biological cues could be presented. Allyl sulfide moieties participated in degenerative chain transfer reactions with thiyl radicals; an associative exchange took place when a thiyl radical attacked the ene of the allyl sulfide forming a carbon-centered radical. The radical then reacted intramolecularly, cleaving a carbon–sulfur bond, to reform an ene and a thiyl radical (Fig. 15). A potential issue arises from the equilibrium nature of the system: since the thiyl radical on the initiating species and the released protein species are similar in reactivity, the deactivated protein could react again with another allyl sulfide; therefore, an excess of the desired active protein was required to drive the equilibrium toward protein attachment. However, these studies clearly showed that this chemistry could be used cleanly for several conjugation and release cycles at relatively low concentrations. The photopatterning of these proteins could be done in the presence of cells and the patterning of proteins was shown to assist with controlling the phenotype of the cell.
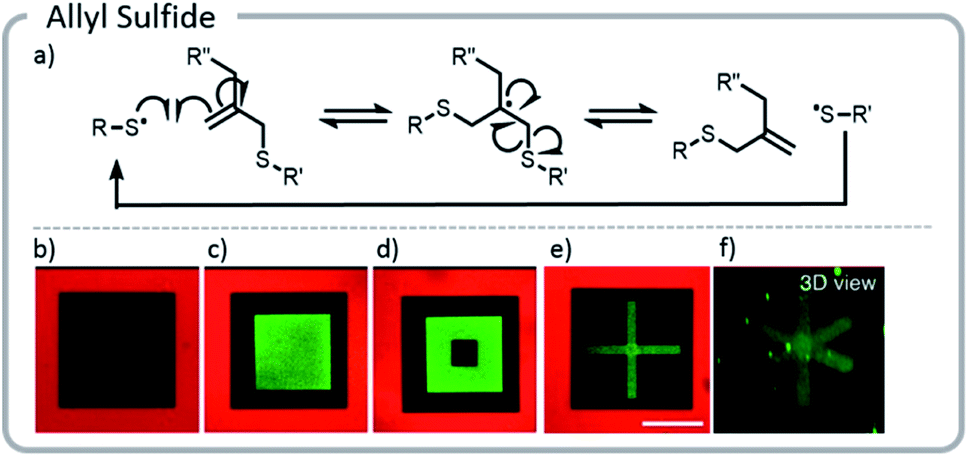 | ||
| Fig. 15 (a) Degenerative chain transfer mechanism with allyl sulfide moieties; (b) two-photon irradiation was used to pattern a hydrogel with a fluorescent red protein. Light was used to release the protein in a square and replace it with PEG-thiol; (c) the same hydrogel was patterned with a green fluorescent protein. (d) The green fluorescent protein was released and replaced with PEG. (e and f) Two viewpoints of a 3-D x created from proteins. Reprinted with permission from ref. 21. Please direct permission requests to the American Chemical Society (copyright 2018) https://pubs.acs.org/doi/10.1021/acscentsci.8b00325. | ||
These examples demonstrate the power light can have for grafting-to reactions. A continued initiative to create more complex and dynamic systems will aid the realization of mimicking biological systems. Light reactions can be used for attaching proteins to polymers in solution; however, it can be argued that the true power of light for grafting-to reactions will lie in the ability to both create 3-dimensional patterns and attach biomolecules to pre-existing materials formed under harsh processing conditions. The push to make these systems viable with longer wavelength light is considerable, and more research will likely move towards NIR systems, despite the cost. The use of associative exchange chemistry130 is a particularly exciting topic in materials chemistry at the moment, and Anseth21 has shown how this associative chemistry can be used for truly dynamic systems. Multi stimuli-responsive systems, particularly using logic gates, can also be used to emulate cellular signalling processes. The advancements observed in recent years, and those to come, will help us better understand healthy and disease states of the body, programming of cellular growth, and the creation of better biomaterials for implantation.
Conclusions and future outlook
Light-based technology has significantly expanded the scope of bioconjugate chemistry and opened the door to target increasingly complex biological systems. Studies of grafting-from polymerizations involving biomacromolecules using photoATRP and PET-RAFT will likely increase since they afford the advantages of benign conditions and higher oxygen tolerance. The biologically friendly nature of these photopolymerizations ultimately allowed for the first controlled polymerization from the surface of live cells.24 While significant ground-work has been laid for these photopolymerization systems, exploration of various commercially viable proteins as well as variations in live cell type is essential, as these unique systems will bring their own challenges and discoveries.The refined spatial and temporal control light confers can be utilized to graft biomacromolecules to networks for the creation of more complex biological materials. These materials can serve as drug release vehicles or mimic the ECM for cell culture conditions. Creation of increasingly intricate systems can help in understanding healthy and diseased tissue states in vivo. Additionally, precisely grafted gels can help control cell differentiation in cell culture platforms, an important step toward personalized medicine. Light also has the potential to impart higher site specificity to reactions, but systems must be carefully chosen such that the specificity does not come at the cost of inadvertently blocking the active site.
These advances have been exploited in a relatively young field with major potential for growth. In both grafting-to and grafting-from reactions, there is a push to design new systems which rely on longer wavelengths of light. However, lower energy irradiation is not sufficient to overcome the activation energy barrier or provide appreciable rates for some reactions. In some cases, this challenge can be overcome by two-photon irradiation in the NIR range, an approach that will likely play an increasingly important role in light-based therapeutics. Both grafting-to and grafting-from systems will also benefit from expanding potential conjugation reactions for polymer–protein attachment. Exploring covalent adaptable bonds, especially reversible bonds that are stimuli-responsive, will allow for on-demand dissociation of biohybrid structures. The benefits light brings to bioconjugation chemistry are vast, and there are many new horizons to be explored.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This material is based on work supported by the National Science Foundation (DMR-1904631). This research was conducted with Government support under and awarded by DoD through the ARO (W911NF-17-1-0326). All protein graphics included in this Perspective were created with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311.131References
- J. Chapman, E. A. Ismail and Z. C. Dinu, Catalysts, 2018, 8, 238 CrossRef.
- R. Singh, M. Kumar, A. Mittal and P. K. Mehta, 3 Biotech, 2016, 6, 174 CrossRef PubMed.
- B. Leader, Q. J. Baca and D. E. Golan, Nat. Rev. Drug Discovery, 2008, 7, 21–39 CrossRef CAS PubMed.
- J. H. Ko and H. D. Maynard, Chem. Soc. Rev., 2018, 47, 8998–9014 RSC.
- J. A. Crommelin, R. D. Sindelar and B. Meibohm, Pharmaceutical Biotechnology: Fundamentals and Applications, Springer, 5th edn, 2019 Search PubMed.
- 2018 Biological License Application Approvals, https://www.fda.gov/vaccines-blood-biologics/development-approval-process-cber/2018-biological-license-application-approvals, accessed October 2019 Search PubMed.
- E. Pedone and S. Brocchini, React. Funct. Polym., 2006, 66, 167–176 CrossRef CAS.
- A. Abuchowski, J. R. McCoy, N. C. Palczuk, T. van Es and F. F. Davis, J. Biol. Chem., 1977, 252, 3582–3586 CAS.
- A. Abuchowski, T. van Es, N. C. Palczuk and F. F. Davis, J. Biol. Chem., 1977, 252, 3578–3581 CAS.
- J. D. Wallat, K. A. Rose and J. K. Pokorski, Polym. Chem., 2014, 5, 1545–1558 RSC.
- G. T. Hermanson, in Bioconjugate Techniques, ed. G. T. Hermanson, Academic Press, San Diego, 1996, pp. xxi–xxiii Search PubMed.
- A. C. Obermeyer and B. D. Olsen, ACS Macro Lett., 2015, 4, 101–110 CrossRef CAS.
- A. J. Russell, S. L. Baker, C. M. Colina, C. A. Figg, J. L. Kaar, K. Matyjaszewski, A. Simakova and B. S. Sumerlin, AIChE J., 2018, 64, 3230–3245 CrossRef CAS.
- S. L. Baker, B. Kaupbayeva, S. Lathwal, S. R. Das, A. J. Russell and K. Matyjaszewski, Biomacromolecules, 2019, 20, 4272–4298 CrossRef CAS PubMed.
- I. Cobo, M. Li, B. S. Sumerlin and S. Perrier, Nat. Mater., 2015, 14, 143–159 CrossRef CAS PubMed.
- B. S. Sumerlin, ACS Macro Lett., 2012, 1, 141–145 CrossRef CAS.
- T. A. Wright, R. C. Page and D. Konkolewicz, Polym. Chem., 2019, 10, 434–454 RSC.
- K. M. Burridge, T. A. Wright, R. C. Page and D. Konkolewicz, Macromol. Rapid Commun., 2018, 39, 1800093 CrossRef PubMed.
- J. Yeow, R. Chapman, A. J. Gormley and C. Boyer, Chem. Soc. Rev., 2018, 47, 4357–4387 RSC.
- B. Yang, Y. Chen and J. Shi, Chem. Rev., 2019, 119, 4881–4985 CrossRef CAS PubMed.
- J. C. Grim, T. E. Brown, B. A. Aguado, D. A. Chapnick, A. L. Viert, X. Liu and K. S. Anseth, ACS Cent. Sci., 2018, 4, 909–916 CrossRef CAS PubMed.
- E.-W. Lin, N. Boehnke and H. D. Maynard, Bioconjugate Chem., 2014, 25, 1902–1909 CrossRef CAS PubMed.
- J. Geng, W. Li, Y. Zhang, N. Thottappillil, J. Clavadetscher, A. Lilienkampf and M. Bradley, Nat. Chem., 2019, 11, 578–586 CrossRef CAS PubMed.
- J. Niu, D. J. Lunn, A. Pusuluri, J. I. Yoo, M. A. O'Malley, S. Mitragotri, H. T. Soh and C. J. Hawker, Nat. Chem., 2017, 9, 537–545 CrossRef CAS PubMed.
- K. A. Davis, P.-J. Wu, C. F. Cahall, C. Li, A. Gottipati and B. J. Berron, J. Biol. Eng., 2019, 13, 5 CrossRef PubMed.
- N. Corrigan, J. Yeow, P. Judzewitsch, J. Xu and C. Boyer, Angew. Chem., Int. Ed., 2019, 58, 5170–5189 CrossRef CAS PubMed.
- J. Phommalysack-Lovan, Y. Y. Chu, C. Boyer and J. T. Xu, Chem. Commun., 2018, 54, 6591–6606 RSC.
- J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef CAS PubMed.
- S. Houshyar, D. J. Keddie, G. Moad, R. J. Mulder, S. Saubern and J. Tsanaktsidis, Polym. Chem., 2012, 3, 1879–1889 RSC.
- J. Vandenbergh and T. Junkers, Macromolecules, 2014, 47, 5051–5059 CrossRef CAS.
- J. Vandenbergh, T. D. Ogawa and T. Junkers, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2366–2374 CrossRef CAS.
- B. S. Tucker, M. L. Coughlin, C. A. Figg and B. S. Sumerlin, ACS Macro Lett., 2017, 6, 452–457 CrossRef CAS.
- S. Chatani, C. J. Kloxin and C. N. Bowman, Polym. Chem., 2014, 5, 2187–2201 RSC.
- H. Shih and C. C. Lin, Macromol. Rapid Commun., 2013, 34, 269–273 CrossRef CAS PubMed.
- S. J. Bryant, C. R. Nuttelman and K. S. Anseth, J. Biomater. Sci., Polym. Ed., 2000, 11, 439–457 CrossRef CAS PubMed.
- S. Averick, A. Simakova, S. Park, D. Konkolewicz, A. J. D. Magenau, R. A. Mehl and K. Matyjaszewski, ACS Macro Lett., 2012, 1, 6–10 CrossRef CAS.
- M. Kovaliov, M. L. Allegrezza, B. Richter, D. Konkolewicz and S. Averick, Polymer, 2018, 137, 338–345 CrossRef CAS.
- J. Xu, K. Jung, N. A. Corrigan and C. Boyer, Chem. Sci., 2014, 5, 3568–3575 RSC.
- C. Boyer, V. Bulmus, J. Liu, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, J. Am. Chem. Soc., 2007, 129, 7145–7154 CrossRef CAS PubMed.
- H. Li, M. Li, X. Yu, A. P. Bapat and B. S. Sumerlin, Polym. Chem., 2011, 2, 1531–1535 RSC.
- M. Li, H. Li, P. De and B. S. Sumerlin, Macromol. Rapid Commun., 2011, 32, 354–359 CrossRef CAS PubMed.
- L. Fu, Z. Wang, S. Lathwal, A. E. Enciso, A. Simakova, S. R. Das, A. J. Russell and K. Matyjaszewski, ACS Macro Lett., 2018, 7, 1248–1253 CrossRef CAS PubMed.
- B. P. Fors and C. J. Hawker, Angew. Chem., Int. Ed., 2012, 51, 8850–8853 CrossRef CAS PubMed.
- N. J. Treat, B. P. Fors, J. W. Kramer, M. Christianson, C. Y. Chiu, J. R. de Alaniz and C. J. Hawker, ACS Macro Lett., 2014, 3, 580–584 CrossRef CAS.
- X. Li, L. Wang, G. Chen, D. M. Haddleton and H. Chen, Chem. Commun., 2014, 50, 6506–6508 RSC.
- N. Corrigan, D. Rosli, J. W. J. Jones, J. Xu and C. Boyer, Macromolecules, 2016, 49, 6779–6789 CrossRef CAS.
- S. Shanmugam, J. T. Xu and C. Boyer, J. Am. Chem. Soc., 2015, 137, 9174–9185 CrossRef CAS PubMed.
- S. Dadashi-Silab, X. Pan and K. Matyjaszewski, Macromolecules, 2017, 50, 7967–7977 CrossRef CAS.
- C. A. Figg, J. D. Hickman, G. M. Scheutz, S. Shanmugam, R. N. Carmean, B. S. Tucker, C. Boyer and B. S. Sumerlin, Macromolecules, 2018, 51, 1370–1376 CrossRef CAS.
- C. Ash, M. Dubec, K. Donne and T. Bashford, Laser Med. Sci., 2017, 32, 1909–1918 CrossRef PubMed.
- M. Clement, G. Daniel and M. Trelles, J. Cosmet. Laser Ther., 2005, 7, 177–189 CrossRef PubMed.
- Z. Zhao and P. W. Fairchild, Dependence of light transmission through human skin on incident beam diameter at different wavelengths, BIOS '98 International Biomedical Optics Symposium, 1998, DOI:10.1117/12.308184.
- C. Decker and A. D. Jenkins, Macromolecules, 1985, 18, 1241–1244 CrossRef CAS.
- J. Yeow, R. Chapman, J. T. Xu and C. Boyer, Polym. Chem., 2017, 8, 5012–5022 RSC.
- T. Lueckerath, T. Strauch, K. Koynov, C. Barner-Kowollik, D. Y. W. Ng and T. Weil, Biomacromolecules, 2019, 20, 212–221 CrossRef CAS PubMed.
- C. Stubbs, T. Congdon, J. Davis, D. Lester, S.-J. Richards and M. I. Gibson, Macromolecules, 2019, 52, 7603–7612 CrossRef CAS PubMed.
- M. Matsumura, G. Signor and B. W. Matthews, Nature, 1989, 342, 291–293 CrossRef CAS PubMed.
- Y. M. E. Fung, F. Kjeldsen, O. A. Silivra, T. W. D. Chan and R. A. Zubarev, Angew. Chem., Int. Ed., 2005, 44, 6399–6403 CrossRef CAS PubMed.
- M. T. Neves-Petersen, S. Pertersen and G. P. Gajula, in Molecular Photochemistry – Various Aspects, ed. S. Saha, InTech, 2012 Search PubMed.
- B. A. Kerwin and R. L. Remmele Jr, J. Pharm. Sci., 2007, 96, 1468–1479 CrossRef CAS PubMed.
- H. Gorner, J. Photochem. Photobiol., B, 1994, 26, 117–139 CrossRef CAS.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- A. L. Lewis and S. W. Leppard, Conjugation Reactions, WO Pat., 2004063237A1, 2004.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- D. Bontempo and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 6508–6509 CrossRef CAS PubMed.
- B. S. Lele, H. Murata, K. Matyjaszewski and A. J. Russell, Biomacromolecules, 2005, 6, 3380–3387 CrossRef CAS PubMed.
- A. Simakova, S. E. Averick, D. Konkolewicz and K. Matyjaszewski, Macromolecules, 2012, 45, 6371–6379 CrossRef CAS.
- K. Matyjaszewski, W. Jakubowski, K. Min, W. Tang, J. Huang, W. A. Braunecker and N. V. Tsarevsky, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15309–15314 CrossRef CAS PubMed.
- A. J. D. Magenau, N. C. Strandwitz, A. Gennaro and K. Matyjaszewski, Science, 2011, 332, 81–84 CrossRef CAS PubMed.
- W. Jakubowski and K. Matyjaszewski, Angew. Chem., 2006, 118, 4594–4598 CrossRef.
- W. Jakubowski and K. Matyjaszewski, Macromolecules, 2005, 38, 4139–4146 CrossRef CAS.
- S. E. Averick, S. K. Dey, D. Grahacharya, K. Matyjaszewski and S. R. Das, Angew. Chem., 2014, 126, 2777–2782 CrossRef.
- A. E. Enciso, L. Fu, A. J. Russell and K. Matyjaszewski, Angew. Chem., Int. Ed., 2018, 57, 933–936 CrossRef CAS PubMed.
- Y. Sun, S. Lathwal, Y. Wang, L. Fu, M. Olszewski, M. Fantin, A. E. Enciso, G. Szczepaniak, S. Das and K. Matyjaszewski, ACS Macro Lett., 2019, 8, 603–609 CrossRef CAS.
- J. Y. Kim, B. S. Lee, J. Choi, B. J. Kim, J. Y. Choi, S. M. Kang, S. H. Yang and I. S. Choi, Angew. Chem., Int. Ed., 2016, 55, 15306–15309 CrossRef CAS PubMed.
- D. Konkolewicz, K. Schröder, J. Buback, S. Bernhard and K. Matyjaszewski, ACS Macro Lett., 2012, 1, 1219–1223 CrossRef CAS.
- X. Pan, N. Malhotra, A. Simakova, Z. Wang, D. Konkolewicz and K. Matyjaszewski, J. Am. Chem. Soc., 2015, 137, 15430–15433 CrossRef CAS PubMed.
- T. G. Ribelli, D. Konkolewicz, S. Bernhard and K. Matyjaszewski, J. Am. Chem. Soc., 2014, 136, 13303–13312 CrossRef CAS PubMed.
- A. Simakova, A. Russell and K. Matyjaszewski, High-throughput synthesis of biomolecule–polymer conjugates, WO Pat., 2018227010A1, 2018.
- X. Pan, S. Lathwal, S. Mack, J. Yan, S. R. Das and K. Matyjaszewski, Angew. Chem., Int. Ed., 2017, 56, 2740–2743 CrossRef CAS PubMed.
- A. Theodorou, E. Liarou, D. M. Haddleton, I. G. Stavrakaki, P. Skordalidis, R. Whitfield, A. Anastasaki and K. Velonia, Nat. Commun., 2020, 11, 1486 CrossRef CAS PubMed.
- G. Zhang, I. Y. Song, K. H. Ahn, T. Park and W. Choi, Macromolecules, 2011, 44, 7594–7599 CrossRef CAS.
- M. Rolland, N. P. Truong, R. Whitfield and A. Anastasaki, ACS Macro Lett., 2020, 459–463 CrossRef CAS.
- C. Kutahya, C. Schmitz, V. Strehmel, Y. Yagci and B. Strehmel, Angew. Chem., Int. Ed., 2018, 57, 7898–7902 CrossRef PubMed.
- M. Fantin, A. A. Isse, A. Gennaro and K. Matyjaszewski, Macromolecules, 2015, 48, 6862–6875 CrossRef CAS.
- N. D. Dolinski, Z. A. Page, E. H. Discekici, D. Meis, I.-H. Lee, G. R. Jones, R. Whitfield, X. Pan, B. G. McCarthy, S. Shanmugam, V. Kottisch, B. P. Fors, C. Boyer, G. M. Miyake, K. Matyjaszewski, D. M. Haddleton, J. R. de Alaniz, A. Anastasaki and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 268–273 CrossRef CAS PubMed.
- E. Liarou, R. Whitfield, A. Anastasaki, N. G. Engelis, G. R. Jones, K. Velonia and D. M. Haddleton, Angew. Chem., Int. Ed., 2018, 57, 8998–9002 CrossRef CAS PubMed.
- E. Liarou, A. Anastasaki, R. Whitfield, C. E. Iacono, G. Patias, N. G. Engelis, A. Marathianos, G. R. Jones and D. M. Haddleton, Polym. Chem., 2019, 10, 963–971 RSC.
- F. Oytun, M. U. Kahveci and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1685–1689 CrossRef CAS.
- R. Chapman, A. J. Gormley, K.-L. Herpoldt and M. M. Stevens, Macromolecules, 2014, 47, 8541–8547 CrossRef CAS.
- S.-i. Yamamoto and K. Matyjaszewski, Polym. J., 2008, 40, 496–497 CrossRef CAS.
- J. Niu, Z. A. Page, N. D. Dolinski, A. Anastasaki, A. T. Hsueh, H. T. Soh and C. J. Hawker, ACS Macro Lett., 2017, 6, 1109–1113 CrossRef CAS.
- M. Kovaliov, D. Cohen-Karni, K. A. Burridge, D. Mambelli, S. Sloane, N. Daman, C. Xu, J. Guth, J. Kenneth Wickiser, N. Tomycz, R. C. Page, D. Konkolewicz and S. Averick, Eur. Polym. J., 2018, 107, 15–24 CrossRef CAS.
- J. C. Theriot, C.-H. Lim, H. Yang, M. D. Ryan, C. B. Musgrave and G. M. Miyake, Science, 2016, 352, 1082–1086 CrossRef CAS PubMed.
- E. H. Discekici, A. Anastasaki, J. Read de Alaniz and C. J. Hawker, Macromolecules, 2018, 51, 7421–7434 CrossRef CAS.
- C. Bian, Y.-N. Zhou, J.-K. Guo and Z.-H. Luo, Macromolecules, 2018, 51, 2367–2376 CrossRef CAS.
- C. Lu, C. Wang, J. Yu, J. Wang and F. Chu, Green Chem., 2019, 21, 2759–2770 RSC.
- J. Liu, V. Bulmus, D. L. Herlambang, C. Barner-Kowollik, M. H. Stenzel and T. P. Davis, Angew. Chem., Int. Ed., 2007, 46, 3099–3103 CrossRef CAS PubMed.
- P. De, M. Li, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2008, 130, 11288–11289 CrossRef CAS PubMed.
- N. Corrigan, J. Xu, C. Boyer and X. Allonas, ChemPhotoChem, 2019, 3, 1193–1199 CrossRef CAS.
- P. Seal, J. Xu, S. De Luca, C. Boyer and S. C. Smith, Adv. Theory Simul., 2019, 2, 1900038 CrossRef.
- L. R. Kuhn, M. L. Allegrezza, N. J. Dougher and D. Konkolewicz, J. Polym. Sci., 2020, 58, 139–144 CrossRef CAS.
- C. Ma, X. Liu, G. Wu, P. Zhou, Y. Zhou, L. Wang and X. Huang, ACS Macro Lett., 2017, 6, 689–694 CrossRef CAS.
- A. Watanabe, J. Niu, D. J. Lunn, J. Lawrence, A. S. Knight, M. Zhang and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1259–1268 CrossRef CAS.
- J. F. Quinn, L. Barner, C. Barner-Kowollik, E. Rizzardo and T. P. Davis, Macromolecules, 2002, 35, 7620–7627 CrossRef CAS.
- L. Lu, H. J. Zhang, N. F. Yang and Y. L. Cai, Macromolecules, 2006, 39, 3770–3776 CrossRef CAS.
- L. Y. Liu and W. T. Yang, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 846–852 CrossRef CAS.
- J. Xu, S. Shanmugam, H. T. Duong and C. Boyer, Polym. Chem., 2015, 6, 5615–5624 RSC.
- Y. Nakayama and T. Matsuda, J. Biomed. Mater. Res., 1999, 48, 511–521 CrossRef CAS PubMed.
- S. Shanmugam, J. T. Xu and C. Boyer, Angew. Chem., Int. Ed., 2016, 55, 1036–1040 CrossRef CAS PubMed.
- B. Nomeir, O. Fabre and K. Ferji, Macromolecules, 2019, 52, 6898–6903 CrossRef CAS.
- H. J. Avens and C. N. Bowman, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6083–6094 CrossRef CAS PubMed.
- Q. Fu, K. Xie, T. G. McKenzie and G. G. Qiao, Polym. Chem., 2017, 8, 1519–1526 RSC.
- D. B. Thomas, A. J. Convertine, R. D. Hester, A. B. Lowe and C. L. McCormick, Macromolecules, 2004, 37, 1735–1741 CrossRef CAS.
- C.-W. Chang, E. Bays, L. Tao, S. N. S. Alconcel and H. D. Maynard, Chem. Commun., 2009, 3580–3582 RSC.
- D. Pissuwan, C. Boyer, K. Gunasekaran, T. P. Davis and V. Bulmus, Biomacromolecules, 2010, 11, 412–420 CrossRef CAS PubMed.
- T. Otsu, M. Yoshida and T. Tazaki, Makromol. Chem., Rapid Commun., 1982, 3, 133–140 CrossRef CAS.
- T. Otsu, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2121–2136 CrossRef CAS.
- R. N. Carmean, T. E. Becker, M. B. Sims and B. S. Sumerlin, Chem, 2017, 2, 93–101 CAS.
- N. Morikawaand and T. Matsuda, J. Biomater. Sci., Polym. Ed., 2002, 13, 167–183 CrossRef PubMed.
- S. Ohya and T. Matsuda, J. Biomater. Sci., Polym. Ed., 2005, 16, 809–827 CrossRef CAS PubMed.
- C. A. DeForest, B. D. Polizzotti and K. S. Anseth, Nat. Mater., 2009, 8, 659–664 CrossRef CAS PubMed.
- C. A. DeForest and D. A. Tirrell, Nat. Mater., 2015, 14, 523–531 CrossRef CAS PubMed.
- E. R. Ruskowitz and C. A. DeForest, Nat. Rev. Mater., 2018, 3, 17087 CrossRef CAS.
- H. Zhang, W. S. Trout, S. Liu, G. A. Andrade, D. A. Hudson, S. L. Scinto, K. T. Dicker, Y. Li, N. Lazouski, J. Rosenthal, C. Thorpe, X. Jia and J. M. Fox, J. Am. Chem. Soc., 2016, 138, 5978–5983 CrossRef CAS PubMed.
- Y. Luo and M. S. Shoichet, Biomacromolecules, 2004, 5, 2315–2323 CrossRef CAS PubMed.
- Z. Ming, J. Fan, C. Bao, Y. Xue, Q. Lin and L. Zhu, Adv. Funct. Mater., 2018, 28, 1706918 CrossRef.
- S. A. Fisher, R. Y. Tam, A. Fokina, M. M. Mahmoodi, M. D. Distefano and M. S. Shoichet, Biomaterials, 2018, 178, 751–766 CrossRef CAS PubMed.
- G. M. Scheutz, J. J. Lessard, M. B. Sims and B. S. Sumerlin, J. Am. Chem. Soc., 2019, 141, 16181–16196 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |