
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSmall-molecule fluorescent probes for imaging gaseous signaling molecules: current progress and future implications
Mingwang
Yang
,
Jiangli
Fan
 *,
Jianjun
Du
and
Xiaojun
Peng
*
*,
Jianjun
Du
and
Xiaojun
Peng
*
State Key Laboratory of Fine Chemicals, Dalian University of Technology, No. 2 Linggong Road, Dalian 116024, P. R. China. E-mail: fanjl@dlut.edu.cn; pengxj@dlut.edu.cn
First published on 20th April 2020
Abstract
Endogenous gaseous signaling molecules including nitric oxide (NO), carbon monoxide (CO) and hydrogen sulfide (H2S) have been demonstrated to perform significant physiological and pharmacological functions and are associated with various diseases in biological systems. In order to obtain a deeper insight into their roles and mechanisms of action, it is desirable to develop novel techniques for effectively detecting gaseous signaling molecules. Small-molecule fluorescent probes have been proven to be a powerful approach for the detection and imaging of biological messengers by virtue of their non-invasiveness, high selectivity, and real-time in situ detection capability. Based on the intrinsic properties of gaseous signaling molecules, numerous fluorescent probes have been constructed to satisfy various demands. In this perspective, we summarize the recent advances in the field of fluorescent probes for the detection of NO, CO and H2S and illustrate the design strategies and application examples of these probes. Moreover, we also emphasize the challenges and development directions of gasotransmitter-responsive fluorescent probes, hoping to provide a general implication for future research.
 Mingwang Yang | Mingwang Yang received his M.S. degree from the Dalian University of Technology in 2013. He is currently a PhD candidate under the supervision of Prof. Jiangli Fan in the State Key Laboratory of Fine Chemicals, Dalian University of Technology. His research interests focus on fluorescent and chemiluminescent probes and their biological applications. |
 Jiangli Fan | Jiangli Fan received her PhD from the Dalian University of Technology in 2005. In 2010, she attended the University of South Carolina (USA) as a visiting scholar. She is currently a professor at the State Key Laboratory of Fine Chemicals, Dalian University of Technology. Her research is focused on fluorescent dyes and molecular probes, fluorescence biological imaging, nanoparticle coating and fluorescence tracing drug release studies. |
 Jianjun Du | Jianjun Du is a professor at the State Key Laboratory of Fine Chemicals at the Dalian University of Technology. He received his B.S. and PhD degrees in Applied Chemistry from the Dalian University of Technology in 2004 and 2010, respectively. After his postdoctoral fellowship work at Nanyang Technological University (Singapore) in 2010–2013, he started his independent research career at the Dalian University of Technology. His research is focused on organic/inorganic nanoparticles with functional organic molecules. |
 Xiaojun Peng | Xiaojun Peng received his PhD in 1990 at the Dalian University of Technology. After completing postdoctoral research at Nankai University (China), he has worked at the Dalian University of Technology since 1992. In 2001 and 2002 he was a visiting scholar at Stockholm University and Northwestern University (USA). Currently he is the director of the State Key Laboratory of Fine Chemicals at the Dalian University of Technology. His research interests cover dyes for fluorescence bioimaging/labeling and digital printing/recording. |
1. Introduction
Nitric oxide (NO), carbon monoxide (CO) and hydrogen sulfide (H2S) are well known for their toxic effects and environmental hazards. However, emerging evidence has suggested that they display a range of physiological and pathological functions as well as participating in various signaling pathways in biological systems (they are called gaseous signaling molecules or gasotransmitters).1 For example, NO possesses the functions of regulating neurotransmission and vascular tone, inhibiting blood platelet activation, and relaxing blood vessels; CO has important physiological functions in the cardiovascular, nervous, and gastrointestinal systems; the functions involving H2S include vasorelaxation regulation, apoptosis, neuromodulation and inhibition of insulin signaling.2 Meanwhile, abnormal metabolism of gasotransmitters may lead to various physiological diseases, including cardiovascular disease, hypertension, Alzheimer's disease, diabetes, ischemia reperfusion, heart failure, etc.3 Recently, studies also showed that these gasotransmitters represent potential therapeutic agents in clinical applications.4 For instance, NO is routinely applied as a therapeutic agent in infants suffering from pulmonary hypertension, H2S is in early-stage clinical development as an intravenous pharmaceutical agent, and CO is an agent against various diseases associated with oxidative stress.5 Although the gasotransmitters are clearly involved in numerous diseases and have multitudinous efficacy, their roles and mechanism of action are still ambiguous. Considering the significant physiological roles and promising therapeutic actions of gaseous signaling molecules, it is essential to develop a highly efficient technique for tracking them in biological systems.So far, various methods have been reported for the detection of signaling molecules. These methods, including gas chromatography (GC), colorimetry, electron paramagnetic resonance and electrochemistry, require complex procedures to handle samples that are detrimental to biological samples and it is difficult to monitor the state of gasotransmitters in real-time.6–8 Owing to the characteristics of excellent sensitivity and selectivity, simple operation and real-time monitoring and noninvasive observation of the detected object in situ, fluorescence detection technology has gained much attention.9 Accordingly, based on the intrinsic properties of the gaseous signaling molecules, abundant specific responsive fluorescent probes have been designed and reported.
To the best of our knowledge, there are several reviews that describe one of the gaseous signaling molecules (e.g. NO,10 CO,11 and H2S).12–15 However, a systematic and comprehensive overview of all gaseous signaling molecules, especially based on the small-molecule fluorescent probe category, is still rare.16,17 In the last few years, gasotransmitter-responsive fluorescent probes, especially those for detecting CO, have been developed and reported rapidly. Therefore, a systematic and timely survey of the progress in this active research field is urgent. This review systematically and specifically summarizes the recent development of fluorescent probes for the detection and imaging of NO, CO and H2S in biological systems. This review is divided into three parts according to the analytes including NO-responsive fluorescent probes, CO-responsive fluorescent probes and H2S-responsive fluorescent probes. In each subsection, the design concept, recognition mechanisms and application of the representative examples of fluorescent probes for NO, CO and H2S are discussed in detail. Finally, a discussion regarding the potential challenges and directions of gasotransmitter-responsive fluorescent probes for future development is also provided briefly.
2. Nitric oxide (NO)-responsive fluorescent probes
NO is synthesized using L-arginine, NADPH and oxygen with three different NO synthase (neuronal, inducible, and endothelial NOS) catalysts. In addition to endogenous NO, scientific researchers have built various types of exogenous NO donors such as the NOC series and light-controlled release NO series. However, excess intracellular NO may react with superoxide radicals to generate peroxynitrite anions (ONOO−), which are a robust oxidant that can destroy proteins, lipids, DNA and so on.2,3 Therefore, the development of various effective fluorescent probes is desirable to investigate the ambiguous roles and disease-associated biochemical pathways of NO in biosystems. Based on the recognition mechanisms of NO, fluorescent probes are divided into the following four categories.2.1 o-Phenylenediamine (OPD)-based reactions
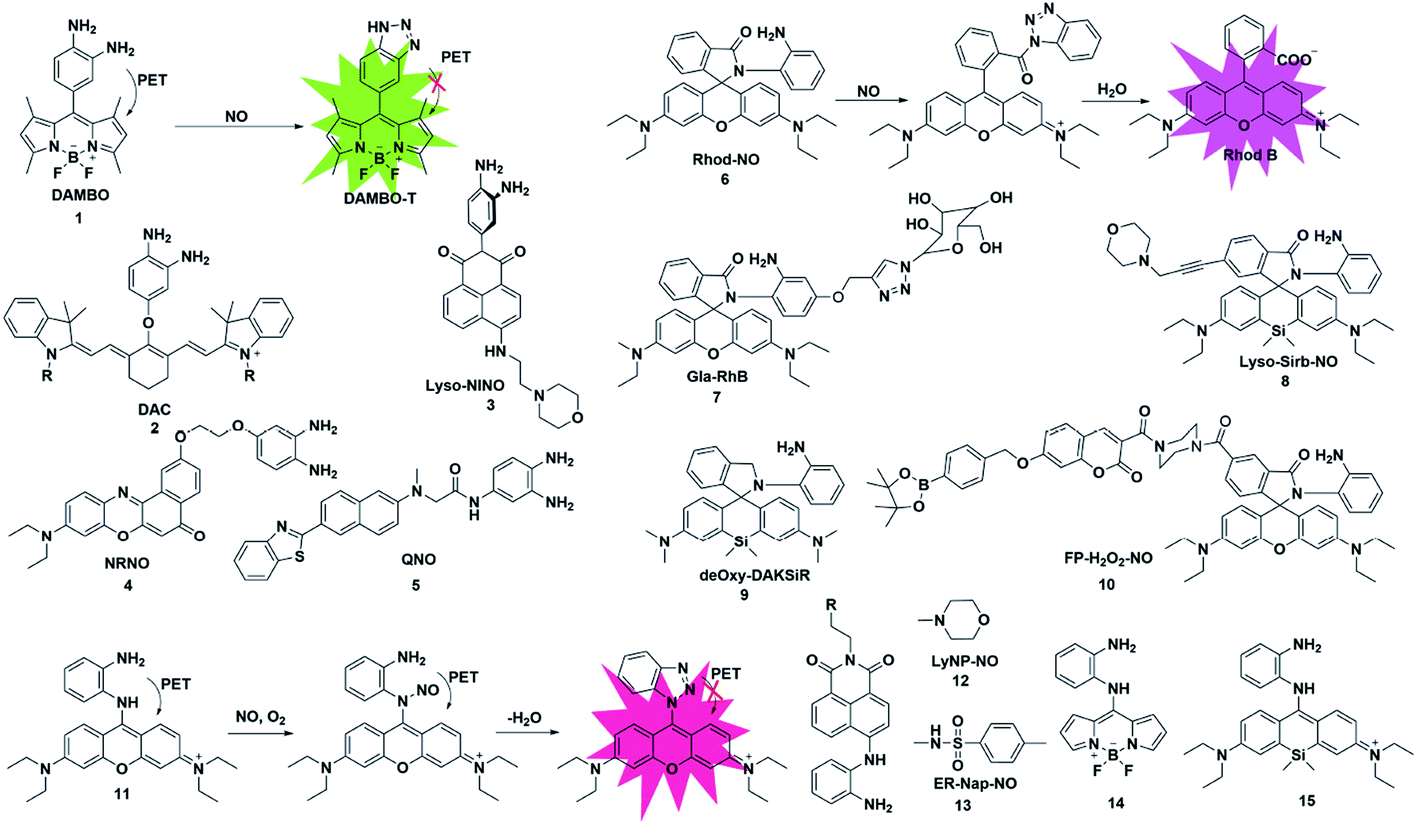
o-Phenylenediamine (OPD) has a strong electron-donating ability, and its electron will be transferred to the HOMO of the excited state fluorophores, which will make the excited state fluorophore electron unable to return to its ground state (PET process), resulting in fluorescence quenching. Due to the strong nucleophilicity of amino nitrogen, it reacts with NO to form benzotriazole in the presence of O2, which possesses a weaker electron-donating ability than OPD, thereby inhibiting the PET action and recovering the fluorescence signal.Based on this strategy, Nagano reported NO-responsive fluorescent probe 1 (ref. 18) with BODIPY as the fluorophore and OPD as the recognition group. Due to the presence of PET, the probe had a low quantum yield (0.02); however, its fluorescence quantum yield sharply increased to 0.74 after reaction with NO (the PET process was suppressed).
Fluorescent probes with excitation and emission in the near-infrared region (NIR, 650–900 nm) have attracted more and more attention due to their low light damage, strong tissue penetration and low background fluorescence. Subsequently, Nagano constructed an example of NIR fluorescent probe 2 by introducing OPD in the middle position of the cyanine dye skeleton.19 The maximum absorption and emission wavelengths of the probe were at 767 and 785 nm, respectively. It reacted rapidly with NO and was used to detect NO in rat kidneys. Considering the important role of lysosomes in cells, a lysosome-targeted and two-photon NO fluorescent probe 3 was designed for the first time by Xiao et al.20 with 1,8-naphthimide as the fluorophore due to its outstanding two-photon (TP) properties, easy modification and stable structure and 4-(2-aminoethyl)morpholine as the lysosomal targeting group. Probe 3 specifically recognized NO under acidic conditions (pH 5.0). In the presence of NO, the fluorescence intensity of the probe exhibited a 16-fold enhancement. Co-localization experiments showed that the probe can specifically localize in lysosomes. For the first time, endogenous NO has been captured in the lysosomes of macrophage cells. Liu's group reported a far-red emissive two-photon excitable probe 4, using Nile Red as the fluorophore for the detection of NO in biological systems.21 The probe can specifically detect NO with a fast response time (180 s) and a limit of detection as low as 46 nM and could be used to visualize the NO generated in a lipopolysaccharide (LPS)-induced inflammation process for the first time. The same group reported a quinoline-based two-photon fluorescent probe (probe 5)22 which has a large two-photon absorption cross section value of 52 GM. By using two-photon microscopy, probe 5 can detect NO in living cells and rat hippocampal slices at a depth of 90–180 μm.
Different from that of the PET-based fluorescent probes above, the fluorescence sensing mechanism of several probes is based on the “open-loop” response of the rhodamine lactam spiral ring. As shown in Fig. 1, Zeng and Xu reported a rhodamine spirolactam based probe for NO using the spiro ring opening reaction.23 First, diazotization of the amino group in probe 6 was caused by attack by NO, resulting in the opening of the spiro ring to generate the rhodamine B acylbenzotriazole intermediate. The rhodamine B tertiary amide exhibited strong absorption and emission characteristics and was further hydrolyzed to form rhodamine B and benzotriazole due to its instability in aqueous solution. In 2018, Zhang et al. fabricated a hepatocyte-targeting fluorescent probe 7 for the first time.24 In this work, a galactose unit was selected not only as the hepatocyte targeting moiety, but also to improve the water solubility of probe 7. Taking advantage of the meritorious properties, the probe was successfully applied to visualize hepatocellular NO in both HepG2 cells and zebrafish.
 | ||
| Fig. 1 Sensing mechanism of o-phenylenediamine-based fluorescent probe 1 and the structure of representative examples. | ||
It is known that the fluorescence properties of traditional rhodamine dyes are sensitive to the pH of the solution. To overcome this problem, Wang and co-workers prepared a Si-rhodamine (SiR)-NO probe which featured excellent NIR emission and a pH-independent spiro ring.25 Probe 8 showed almost no fluorescence signal in a buffer solution with a pH range from 2.0 to 12.0, and an approximate 800-fold fluorescence enhancement of the probe toward NO was observed at pH 5.0. Thanks to the low background fluorescence, probe 8 exhibited higher sensitivity than probe 6, especially in an acidic environment. To achieve lysosome targeting, a morpholine group was incorporated into the probe.
However, the rhodamine B acylbenzotriazole intermediate, generated from the initial reaction of probe 6 with NO, suffers from slow hydrolysis, which results in a relatively long fluorescence response time of the probe to NO (about 30 min). And the intermediate may encounter cysteine-induced native-chemical-ligation and cyclization cascade reactions, generating non-fluorescent rhodamine lactam. To address this issue, Guo et al. reported an OPD locked Si-rhodamine deoxylactam as a NIR fluorescent probe for detecting NO in vitro and in vivo (probe 9).26 The probe can effectively avoid the interference of intracellular Cys in the detection of NO. The meritorious advantages enabled the probe to visualize endogenous NO in living cells and mouse models.
Multiple reactive species (e.g. H2O2 and NO) may play interrelated roles in complex physiological processes. A single fluorescent probe 10 that is capable of simultaneously detecting endogenously generated NO and H2O2 in living cells was presented by Lin's group.27 The boronate group was introduced in the fluorescent dye which consisted of 7-hydroxycoumarin and rhodamine through a rigid piperazine linker and served as a H2O2 reactive site. By observing the changes in the independent fluorescence emission channels, the situation of the analytes can be effectively observed. This work established a strategy for monitoring multiple biological species using a single fluorescent probe with multiple recognition sites.
However, there are still some issues that need our attention. Generally, a benzotriazole product is formed upon reacting OPD with NO. As benzotriazole is sensitive to pH, its deprotonation at physiological pH may result in the formation of an electron-rich triazolate, which may lead to fluorescence quenching through the acceptor-excited photo induced electron transfer (a-PET) mechanism.28
To overcome this deficiency, Guo developed a mitochondria-specific fluorescent probe for NO through conjugating a pyronin dye with one of the amino groups of OPD (probe 11).28 In the process of detecting NO, the benzotriazolate-induced fluorescence quenching action via the PET process under physiological conditions was suppressed due to the absence of the NH protons in the generated triazole. Surprisingly, the triazole produced by the reaction of the probe with NO will further serve as a tool to detect Cys and GSH through green and red channels. With the help of intracellular Cys and GSH, the probe can detect the exogenous and endogenous NO of mitochondria in cells through two different emission channels.
Subsequently, two fluorescent probes were reported for specifically detecting NO by adopting a similar construction strategy to that reported by Guo. In 2016, Kumar developed a lysosomal-targeting fluorescent probe 12 for the visualization of exogenously and endogenously generated NO in living cells.29 The probe was also applied to monitor the NO levels in the hippocampus region of the rat brain after stimulating the tissue with N-methyl-D-aspartate (NMDA). Similarly, by using methyl sulfonamide as an endoplasmic reticulum (ER)-targeting group, Li and co-workers constructed an effective fluorescent probe for imaging intracellular NO (probe 13).30 More importantly, probe 13 was applied for investigating the relationship between tunicamycin-induced ER stress and the level of NO in living cells and tissues, which was beneficial for exploring the physiological role of NO during ER-stress and its associated diseases.
In 2017, Bhuniya et al. designed and synthesized a Raman-active fluorescent probe 14 based on the BODIPY fluorophore.31 In the presence of NO, the probe was transferred to benzotriazole which showed an increased emission signal at 527 nm and a characteristic intense Raman band centred at 1440 cm−1 for –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–. And in order to prevent NO probes from being affected by thiols, especially Cys and GSH, Zhu developed a fast and selective NIR fluorescent probe 15 for real-time and in vivo targeted tracing of endogenous NO.32 The probe was constructed by directly conjugating a silicon-substituted xanthene NIR unit and OPD group and had significant features for selective NO detection, including a fast NIR response at 710 nm within 1 min and specific localization in mitochondria. This excellent probe may provide the possibility to explore more physiological functions of NO in biosystems.
N–. And in order to prevent NO probes from being affected by thiols, especially Cys and GSH, Zhu developed a fast and selective NIR fluorescent probe 15 for real-time and in vivo targeted tracing of endogenous NO.32 The probe was constructed by directly conjugating a silicon-substituted xanthene NIR unit and OPD group and had significant features for selective NO detection, including a fast NIR response at 710 nm within 1 min and specific localization in mitochondria. This excellent probe may provide the possibility to explore more physiological functions of NO in biosystems.
2.2 N-Nitrosation of aromatic secondary amine
The nitrosamine detection mechanism of NO is mainly based on the nitration of an aromatic secondary amine to form a stable nitrosated compound. Generally, the initial fluorescence state of this type of probe is quenched due to the PET process. In the presence of NO, the inhibition of the PET effect resulted in the turn-on of probe fluorescence. In 2016, Guo used the N-nitrosation mechanism of electron-rich aromatic secondary amines under aerobic conditions to construct two fluorescent probes for the detection of NO for the first time (probes 16 and 17).33 The two probes also displayed a fast fluorescence response time (within seconds), excellent sensitivity (nM level) and selectivity toward NO over others analytes (ROS, thiols, methylglyoxal and so on). Using the developed probes, the stimulation-induced NO in RAW264.7 murine macrophages and the endogenous NO in endothelial cells after oxygen–glucose deprivation (OGD) was imaged (Fig. 2).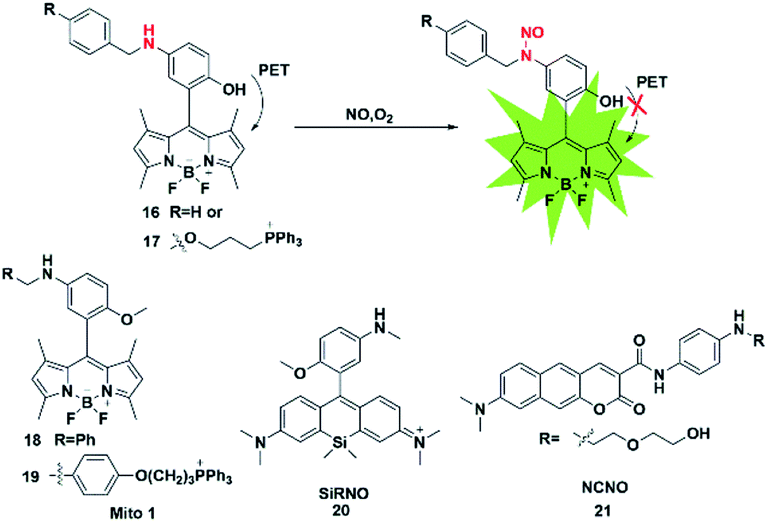 | ||
| Fig. 2 Sensing mechanism of N-nitrosation of aromatic secondary amine-based fluorescent probe 16 and the structure of representative examples. | ||
Subsequently, in 2018, the same group reported two novel fluorescent probes 18 and 19 which possessed the ability to detect NO and ONOO− simultaneously through replacing the reactive group N-benzyl-4-hydroxyaniline in probe 17 with N-benzyl-4-methoxyaniline.34 These probes can distinguish M1 macrophages from M2 macrophages in terms of their difference in inducible NO synthase (iNOS) levels. More importantly, the authors used probe 18 as a tool to perform repolarization assessments of tumor-associated macrophages from the pro-tumoral M2 phenotype to the anti-tumoral M1 phenotype and image the NO communication in immune-mediated M1 macrophages which phagocytose cancer cells. These results showed that these fluorescent probes may play an important role in cancer immunotherapy research and anticancer drug screening.
The first “NIR-to-NIR” probe based on a silicon-rhodamine (SiR) scaffold for the detection of NO with 4-methoxy-N-methylaniline as the NO recognition group was designed by Liu in 2017 (probe 20).35 The main absorption band of the probe was centered at 651 nm and the emission wavelength after reaction with NO was 672 nm. The absorption and emission of the probe were in the NIR region, which enabled the probe to visualize NO in situ in a mouse model of xenograft tumor and reveal the process of NO production in tumor development. To gain a deeper imaging depth and higher signal-to-noise (S/N) ratio in fluorescence imaging, a benzo[g]coumarin derivative (8-(dimethylamino)-2-oxo-2H-benzo[g]chromene-3-carboxylic acid) was used as a red-emissive TP fluorophore by Liu (probe 21).36 The probe was successfully applied to monitor the changes of the NO content in a mouse kidney ischemia-reperfusion injury (IRI) model for the first time. Most of these fluorescent probes are of the “off-on” type because of their PET response mechanism.
2.3 2-Amino-3-dialkylaminobiphenyl (AD)-based cyclization
In addition to the above strategies, researchers have found that the reaction of NO with mono-substituted biphenyl compounds to generate diazoles can also be applied for detecting NO. Anslyn et al. introduced a novel sensing mechanism for NO detection.37 As shown in Fig. 3, 2-dimethylaminophenyl-5-cyano-α-naphthylamine (probe 22) serves as both the fluorophore and NO receptor. It combined with NO to form an electron-deficient nitrosamine, followed by an electrophilic aromatic substitution reaction to produce a hydroxyhydrazine derivative. Finally, the derivative eliminated H2O to generate a stable six-membered cyclic compound AZO550. Upon addition of NO, the absorption wavelength red-shifted from 352 nm to 450 nm and the fluorescence intensity was significantly enhanced with a 1500-fold turn-on signal from a dark background. The high specificity, facile synthesis and cell membrane permeability make it a superior probe for both intra- and extra-cellular NO detection.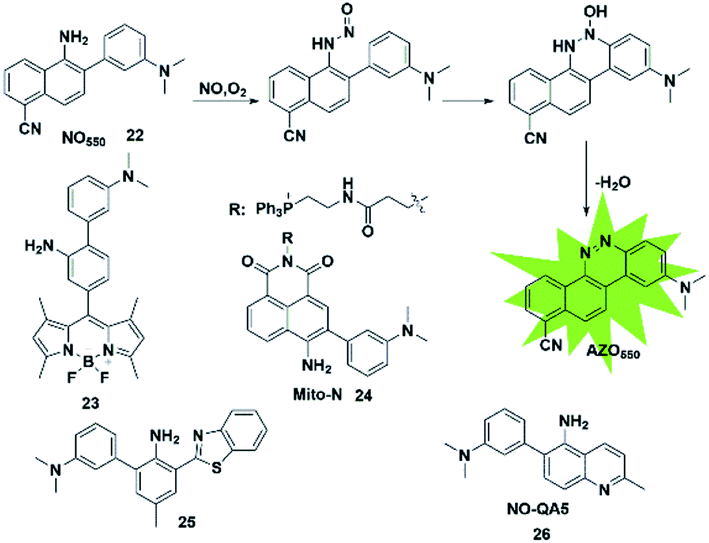 | ||
| Fig. 3 Sensing mechanism of 2-amino-3-dialkylaminobiphenyl (AD)-based fluorescent probe 22 and the structure of representative examples. | ||
Similarly, Guo et al. synthesized a novel fluorescent probe for detecting NO by using 2-amino-3′-dimethylaminobiphenyl (AD) as the NO-binding site and fluorescence quencher via the PET mechanism (probe 23).38 Once the probe reacts with NO, the above-mentioned PET process is blocked due to the absence of an electron-donating amino group in the product, resulting in fluorescence recovery. This probe can quickly and selectively detect NO with a detection limit as low as 30 nM and can be applied to the visualization of NO in cells.
Ratiometric probes provide a built-in correction function to reduce experimental artifacts caused by factors such as instrumentation, concentration and photobleaching. Utilizing this strategy, Zhang developed a ratiometric two-photon fluorescent probe for imaging NO (probe 24).39 Through the Suzuki reaction, 3-dimethylaminobiphenyl was incorporated into the naphthalimide scaffold as a NO-recognizing group. It can successfully detect the NO level in living cells, tissues and an inflammatory mouse model without interference from other active oxygen and nitrogen species. Yoon's group also reported an excited-state intramolecular proton transfer (ESIPT) based fluorescent probe (probe 25)40 for ratiometric monitoring of NO in vitro and in vivo. Different from probe 22 reported by Anslyn,37 Song constructed a two-photon fluorescent probe 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 41 by 3-dimethylaminophenyl linking at 6-position of 5-aminoquinoline for rapid detection of both NO and NO2−. In the presence of analytes, the major azoic regioisomer displays ICT emission (540 nm) and a two-photon absorption section (δΦ = 57 GM), resulting in a turn-on fluorescence response. All these advantages of the probe make it suitable for imaging exogenous and endogenous NO in RAW 264.7 cells under one- and two-photon excitation. This type of fluorescent probes can effectively resist some common interferences such as dehydroascorbic acid (DHA), ascorbic acid (AA), glutathione (GSH) and certain phenolic compounds.
41 by 3-dimethylaminophenyl linking at 6-position of 5-aminoquinoline for rapid detection of both NO and NO2−. In the presence of analytes, the major azoic regioisomer displays ICT emission (540 nm) and a two-photon absorption section (δΦ = 57 GM), resulting in a turn-on fluorescence response. All these advantages of the probe make it suitable for imaging exogenous and endogenous NO in RAW 264.7 cells under one- and two-photon excitation. This type of fluorescent probes can effectively resist some common interferences such as dehydroascorbic acid (DHA), ascorbic acid (AA), glutathione (GSH) and certain phenolic compounds.
2.4 Metal–ligand complex-based fluorescent probes
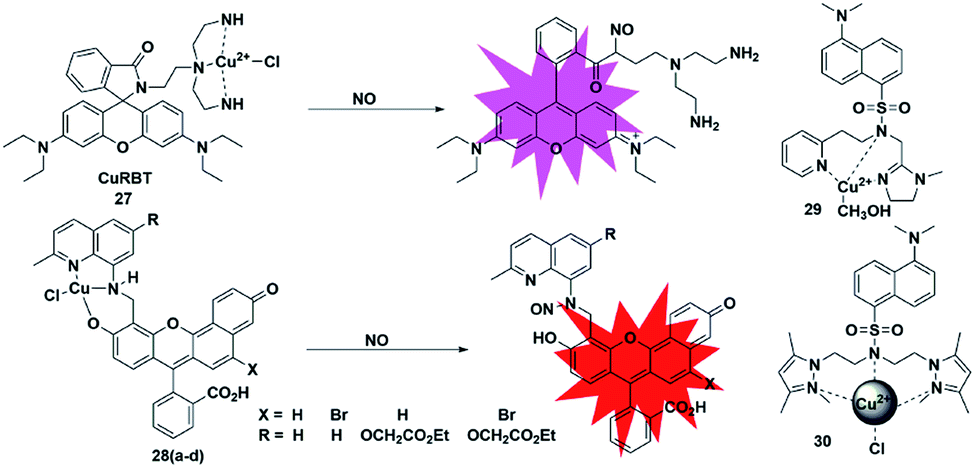
Metal–ligand complex-based fluorescent probes are another widely used type for detecting NO. In these probes, paramagnetic or heavy atom effects of transition metals can quench the fluorescence signal of fluorophores. After the probes react with NO, the reduction and/or release of the transition metal may prevent fluorescence quenching, thereby restoring the fluorescence emission of the probes.A rhodamine–Cu(II) complex, probe 27, was designed by Duan et al. for detecting NO.42 It was synthesized by incorporating a tris(2-aminoethyl) amine(tren) moiety which served as an efficient Cu2+ chelator to rhodamine B dye. The Cu(II) center was coordinated to three nitrogen donors and one chloride anion in planar square geometry. One possible sensing mechanism is that NO may react with Cu2+ to generate Cu+ and NO+, which then caused the spiral ring to open and liberate the rhodamine fluorophore (Fig. 4). It featured a 700-fold fluorescent enhancement and a detection limit of 1 nM toward NO without being affected by ONOO−, NO2−, NO3−, ClO− and H2O2. This probe can be applied for monitoring intracellular NO.
 | ||
| Fig. 4 Sensing mechanism of metal–ligand complex-based fluorescent probe 27 and the structure of representative examples. | ||
Lippard et al. reported a series of seminaphthofluorescein-based fluorescent probes 28a–d for imaging NO in living cells.43 In the presence of NO under anaerobic conditions, the Cu(II) complex fluorescent probes will generate about 20–45 fold enhancement in their fluorescent intensity when the excitation wavelengths are in the range of 535 to 575 nm and achieve emission maxima between 550 and 625 nm. The mechanism of this detection may be that the Cu2+ complex of the probes was reduced to Cu+ in the presence of NO, followed by the elimination reaction and then nitrosation of the secondary amine moiety. The high selectivity and low cytotoxicity of these fluorescent probes has enabled them to be successfully used to image NO in Raw 264.7 macrophages. Another two Cu(II) complexes with a tridentate N-donor ligand (probes 29 and 30)44,45 were reported by Mondal and Ali, respectively. These complexes can also be applied as turn-on NO tools.
High sensitivity is an important index for fluorescence detection. The detection limits (LODs) of NO probes discussed in the subsections are summarized and shown in Table 1.
| Probe | LOD | Probe | LOD | Probe | LOD | Probe | LOD |
|---|---|---|---|---|---|---|---|
| 1 | No | 8 | 32.6 nM | 15 | No | 24 | 21 × 103 nM |
| 2 | No | 9 | 0.12 nM | 16(17) | 4.8 nM | 25 | 17 nM |
| 3 | 5 nM | 10 | No | 18(19) | 0.4 nM | 26 | 15 nM |
| 4 | 46 nM | 11 | 12 nM | 20 | 14 nM | 27 | 1 nM |
| 5 | 84 nM | 12 | 3.63 nM | 21 | 37 nM | 28 | No |
| 6 | 3 nM | 13 | 3.3 nM | 22 | 30 nM | 29 | 10 nM |
| 7 | 1.62 nM | 14 | 9.5 nM | 23 | 30 nM | 30 | 1.6 nM |
3. Carbon monoxide (CO)-responsive fluorescent probes
Carbon monoxide (CO) is endogenously generated in mammalian tissues during the oxidative catabolism of heme by the heme oxygenase enzymes (HO-1, HO-2 and HO-3) and is thought to be the second gaseous messenger. In a similar way to NO, CO may also act as a neurotransmitter by regulating cyclic guanosine monophosphate (cGMP).46,47 Exogenous CO delivered at low concentrations has shown therapeutic potential as an anti-inflammatory agent. Thus, it is necessary to develop highly effective and sensitive fluorescent probes for detecting CO in biological samples. At present, novel carbon monoxide releasing molecules (CORMs) are usually utilized to investigate CO in biological systems. According to the detection mechanism, the design of CO fluorescent probes can be divided into the following strategies.3.1 Transition metal-mediated carbonylation reaction
Palladium and rhodium ions can coordinate with various ligands (e.g. fluorophores) to form complexes due to their strong coordination ability. Generally, palladium and rhodium ions may quench the fluorescence of the ligand dyes via heavy-atom electronic effects. In 2012, Chang et al. reported a reaction-based fluorescent probe for selective imaging of CO in living cells using palladium-mediated carbonylation (probe 31).48 The cyclopalladated species displayed low fluorescence quantum yield (Φ = 0.01) due to the heavy atom quenching effect of palladium ions. Once the probe was bound to CO, it underwent a carboxylation reaction to release Pd(0), accompanied by the fluorescence recovery of the BODIPY ligand (Φ = 0.44). Moreover, it showed higher selectivity for CO than for other interfering species and was successfully used to image changes of CO in HEK293T cells. Notably, this was the first fluorescent probe for the detection of CO which provided possibilities for investigating the function of CO and guidance for developing other CO probes in the future.In 2014, Lin's group described the first carbazole–coumarin (CC)-based two-photon turn-on fluorescent probe 32,49 which was capable of detecting CO in living tissues. By fusing carbazole with coumarin, the authors synthesized a novel platform, which exhibited a push–pull nature and remarkable two-photon properties. Probe 32 showed very weak fluorescence in PBS buffer because of the heavy atom quenching effect of palladium. Upon bonding with CO, a maximal fluorescence enhancement (11-fold) was observed, which was superior to that reported previously. However, unlike the palladium-mediated carbonylation of the probe reported by Chang, this probe's response to CO was based on the protonolysis of its Pd–C bond. It was successfully applied for imaging exogenous CO in mouse liver tissue with a penetration depth of 180 μm for the first time.
Studies have shown that HO-1 expression is upregulated under hypoxic conditions to protect the cells from hypoxia-induced damage. And as one of the products of the catabolism of heme by HO-1, CO will suppress hypoxia-induced ROS by increasing its level in cells. Tang et al. developed two fluorescent probes 33a and 33b based on azobenzene-cyclopalladium for imaging of endogenous CO under hypoxic conditions (probes 33).50 Compared with probe 33a, probe 33b displayed more advantages including better sensitivity, faster response time and higher fluorescence enhancement. Encouraged by these merits, probe 33b was used to directly visualize the intracellular CO upregulation under hypoxic stress and oxygen–glucose deprivation/reperfusion (OGD/R) conditions for the first time. This research provides a theoretical and practical approach to investigate endogenous CO in physiological processes.
Similarly, Wang et al. designed a benzimidazole functionalized palladacycle for the detection of CO (probe 34).51 The carboxyl group was incorporated into the benzimidazole moiety to improve the water solubility of the probe. The experiments demonstrated that probe 34 showed a high cellular uptake speed and can be applied for imaging CO (Fig. 5).
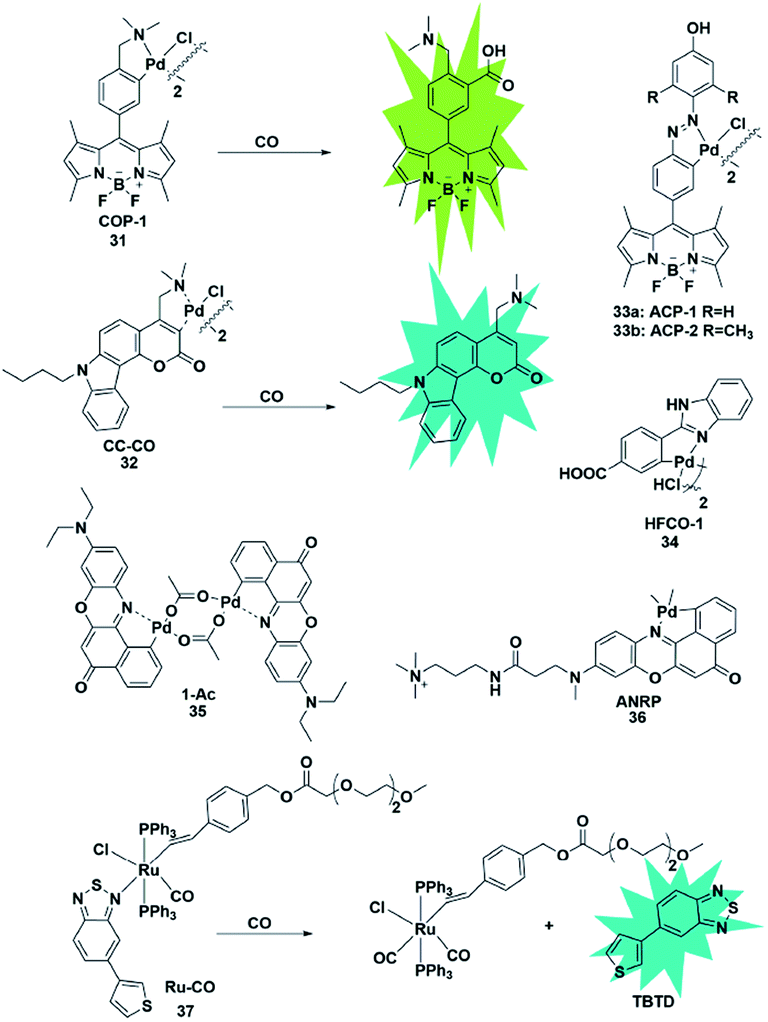 | ||
| Fig. 5 Sensing mechanism of transition metal-mediated fluorescent probe 31 and the structure of representative examples. | ||
The aforementioned fluorescent probes suffered from short emission wavelengths and are usually used to detect CO in living cells. The detection of endogenous CO in living animals remains an unmet challenge. More importantly, these probes possess relatively small flexible ligands and unstable coordination structures, which may result in high background fluorescence. In 2017, Lin et al. reported a novel NIR CO fluorescent probe 35 based on the Nile Red dye.52 Probe 35 exhibited extremely low background fluorescence (Φ = 4.69 × 10−3) at 660 nm mainly due to the strong quenching effect of Pd in the complex and its high stability in aqueous solution. Compared with reported Pd-based fluorescent probes showing a 10-fold increase in fluorescence, probe 35 exhibited about a 60-fold fluorescence turn-on response. It was applied for tracking exogenous CO in HeLa cells and endogenous CO induced by hypoxic conditions in Raw 264.7 cells and zebrafish embryos in one- and two-photon modes. Encouraged by the NIR emission wavelength of the probe, the authors investigated the CO changes in living organs that were treated with different stimulants. Notably, it was successfully used to monitor CO in living mice for the first time. This work provides a NIR tool for CO exploration in vivo.
Subsequently, Zhang et al. constructed a cell-membrane-anchored fluorescent probe 36 by introducing a long hydrophobic chain containing a polar head into traditional Nile Red dye.53 It rapidly bound to the cell membrane (within 1 min) and displayed a long retention time. CO released from living cells under LPS and heme-stimulated conditions and during the drug-induced hepatotoxicity process could be effectively visualized by using this probe for fluorescence bioimaging. The imaging experiments of the probe also proved that cancer cells release more CO than normal cells and the liver was the main organ for CO production in mice.
Different from the palladium complexes, Wilton-Ely reported a ruthenium(II) vinyl complex that was capable of selectively detecting CO in vitro and ex vivo (probe 37).54 The weak fluorescence emission properties of this probe were attributed to the quenching of 5-(3-thienyl)-2,1,3-benzothiadiazole (TBTD) by the ruthenium(II) atom (the heavy atom effect). In the presence of CO, it undergoes a displacement with the TBTD in the complex, so that the fluorophore is separated from the complex, thereby generating an obvious enhancement in the fluorescence intensity. Notably, the CO in cells that were collected from the exudates of an air pouch inflammation mouse model was also successfully detected by using this probe as an imaging tool.
3.2 Tsuji–Trost reaction
The CO fluorescent probes mentioned above had rigorous requirements on the structure and coordination ability of fluorophores. In the presence of palladium, allyl compounds are attacked by nucleophiles (such as compounds containing N and O atoms) followed by a nucleophilic substitution reaction, which is named the Tsuji–Trost reaction.55 Utilizing this reaction as a design strategy, researchers have developed a large number of CO-responsive fluorescent probes in recent years (Fig. 6).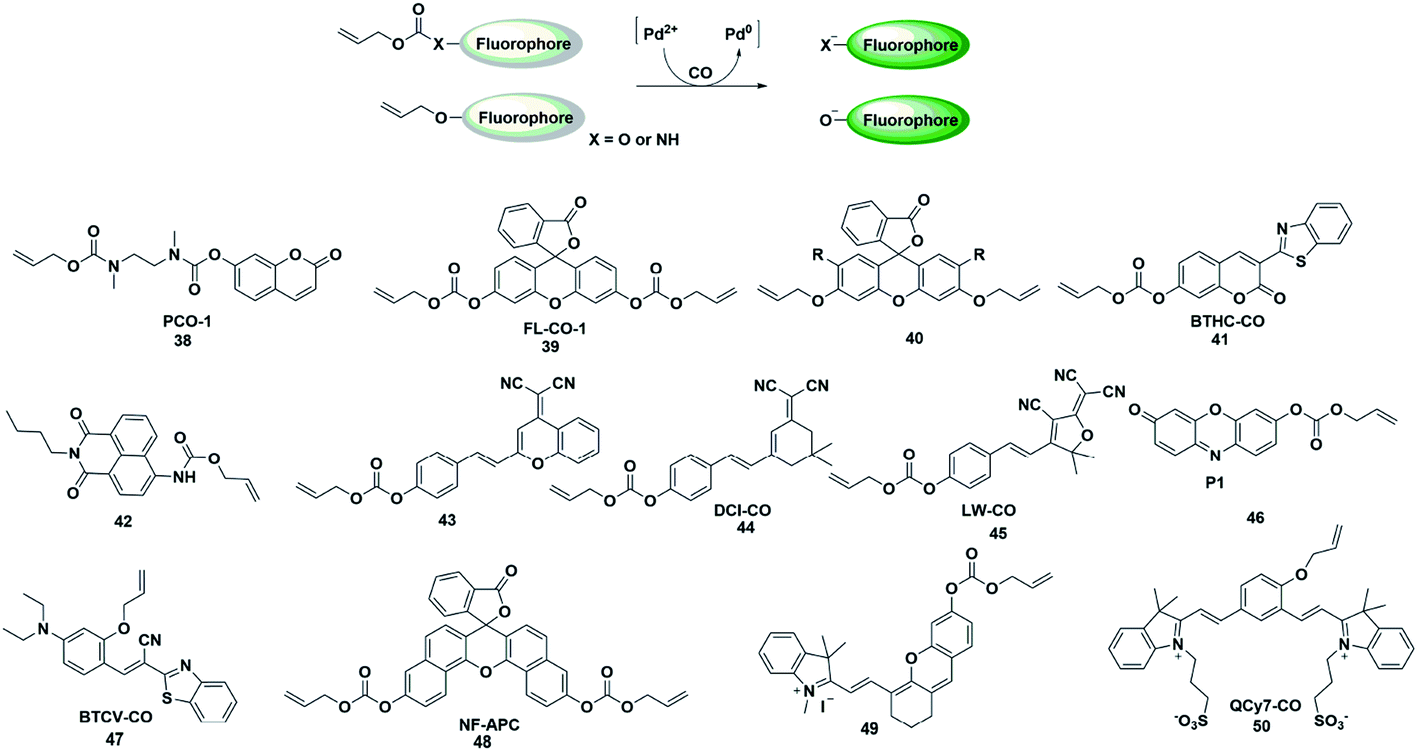 | ||
| Fig. 6 Sensing mechanism of fluorescent probes based on the Tsuji–Trost reaction and the structure of representative examples. | ||
In 2015, Dhara et al. reported a coumarin-based fluorescent probe 38 for the detection of CO through a Pd(0)-mediated intramolecular cyclization–elimination reaction.56 The weak fluorescence intensity of the probe was obtained by protecting the hydroxyl group of coumarin via allyl-functionalized carbamate. The proposed detection mechanism may involve the following processes. The allyl of probe 38 was first eliminated through a Pd(0)-initiated nucleophilic substitution reaction, resulting in a secondary amine structure. Then an entire cyclization–elimination occurred spontaneously to generate the 7-hydroxycoumarin dye and release N,N′-dimethylimidazolidinone, which ultimately resulted in enhanced fluorescence. In the titration experiments, PdCl2 and a CO donor (CORM-3) were added simultaneously into the testing buffer to in situ form the Pd(0) species. This was used for detecting CO in vitro and imaging exogenous CO in A 549 cells. This research provided a novel approach and a general strategy for developing CO-responsive fluorescent probes.
In the next few years, Feng's group reported various fluorescent probes based on the Pd(0)-mediated Tsuji–Trost reaction.57–60 In 2016, they reported a readily available fluorescent probe 39 (ref. 57) based on the allyl chloroformate functionalized fluorescein for the detection of CO. The probe showed near-zero background fluorescence. This was the first fluorescent probe that could be used to visualize the CO produced by heme stimulation in A 549 cells. However, the fluorescence intensity increased spontaneously over time in the testing system and probe 39 was sensitive to pH and UV light. The main reason was that the allyl-carbonate group lacked sufficient stability in buffer. Subsequently, by using allyl ether instead of allyl-carbonate as the protective unit and recognition site, Feng et al. reported a second generation fluorescein-based CO-responsive fluorescent probe (probe 40).58 This probes exhibited superior stability compared to probe 39 and excellent sensing performance for CO. It is worth noting that this was the first fluorescent probe that utilized allyl ether as the reaction site for CO, which will provide more options for developing effective CO-detecting fluorescent probes. By functionalizing 3-benzothiazolyl-7-hydroxycoumarin (BTHC) with an allyl chloroformate group, Feng et al. also constructed another CO signaling molecule (probe 41).59 And it can be applied in a low dosage (1 μM level) for bioimaging CO in cells.
Thanks to these results, they reported the first colorimetric and ratiometric fluorescent probe 42 for effectively detecting CO in vitro and in cells.60 Attachment of a CO-specific carbamate derivative to the amino unit of naphthalimide will restrict its intramolecular charge transfer (ICT) effect and eventually lead to a blue shift in emission wavelength (472 nm). When CO was added, the allyl acetate group was eliminated and amino-naphthalimide was released simultaneously through the Pd(0)-initiated Tsuji–Trost reaction; as a result, the ICT process was restored and the emission wavelength was red-shifted to 545 nm. The probe could colorimetrically and ratiometrically detect CO in solution with high selectivity and sensitivity. The test paper system made with this probe could detect CO in the air with the naked eye.
In recent decades, dicyanomethylene-4H-pyran (DCM) and 2-dicyanomethylene-3-cyano-4,5,5-trimethyl-2,5-dihydrofuran (TCF) derivatives have been widely used for developing donor–π–acceptor (D–π–A) structured fluorescent probes which exhibit large Stokes shifts from the ultrafast ICT process and long emission in the NIR region due to their efficient strong electron-withdrawing ability. By regulating the ICT process, some fluorescent probes have been constructed by researchers to detect CO (probes 43–45).61–63 These probes exhibit significant colorimetric changes, high selectivity and sensitivity toward CO, which can be effectively used to visualize CO in vitro, in living cells or in vivo.
In 2019, Mondal et al. reported a water-soluble fluorogenic molecular platform for imaging CO in biofluids (probe 46).64 Resorufin was selected as the fluorophore based on its excellent water solubility, good biocompatibility and easy availability. Instead of using CO release molecules (CORMs), the author used CO gas (dissolved in solution and living cells) to investigate the detection capability of the probe. The results demonstrated that it showed admirable selectivity and sensitivity over other ROS, RNS and fatty acids. The nanomolar detection limit (62 nM) implies the ability of the probe to detect CO in physiological concentrations (i.e., 0.0019–0.1 μM).
Since most of the reported CO-responsive fluorescent probes accumulate in biological samples, they suffered from the undesirable aggregation-caused quenching (ACQ) effect, which makes their fluorescence intensity much weaker than that in solution. Thus, Tang et al. designed the Tsuji–Trost reaction-based fluorescent probe 47 (ref. 65) with aggregation-induced emission (AIE) characteristics for detecting and imaging CO. The relatively weak fluorescence emission was induced by fast nonradiative decay promoted by the cis–trans isomerization of the C–C bond of the alkene linker. Upon bonding to CO, the probe would be converted to a phenolate intermediate followed by a rapid cyclization reaction, thus restricting the rotation of the C–C bond, resulting in an enhancement in the fluorescence intensity. The excellent selectivity, sensitivity, and fast response of the probe toward CO made it capable of real-time detection and visualization of CO in vitro and in living systems. This is probably the first AIE fluorescent probe to detect CO.
Zhang et al. reported an example of a colorimetric and NIR fluorescent probe by functionalizing naphthofluorescein with an allyl chloroformate group for the detection of CO in living cells (probe 48).66 In 2017, Feng's group also synthesized a colorimetric and NIR fluorescent probe 49 for CO detection.67 The hydroxyl group of the modified hemicyanine dye was protected by a carbonate unit, which reduced the electron donating ability of the hydroxyl group and the ICT process of the entire molecule, resulting in a weak fluorescence emission of the probe. When the protective group was removed by the Pd(0)-activated Tsuji–Trost reaction, the probe showed a significant fluorescence enhancement. The NIR fluorescence signal response made it superiorly suitable for bioimaging CO in living animals. Recently, they also designed a novel NIR fluorescent probe based on a unique cyanine fluorophore for rapidly detecting CO (probe 50).68 When the allyl ether of the probe was eliminated by a CO-activated reaction, a unique conjugated π-electron system was generated which has a similar conjugation pattern and NIR emission properties to Cy7. The cell imaging results showed that more endogenous CO was produced under oxidative stress conditions.
3.3 Nitro reduction
In biological imaging, using heavy atoms or additives is still an alternative. In order to overcome the above issues, the following methods for CO detection were developed. Because CO has a strong reducing ability, it can reduce the nitro group to an amino group under certain conditions. The main difference between this strategy and the abovementioned methods for detecting CO is that it does not require palladium ions or the addition of a third substance. Based on this strategy, a series of fluorescent probes have been reported for sensing CO in solution and in biological samples (Fig. 7).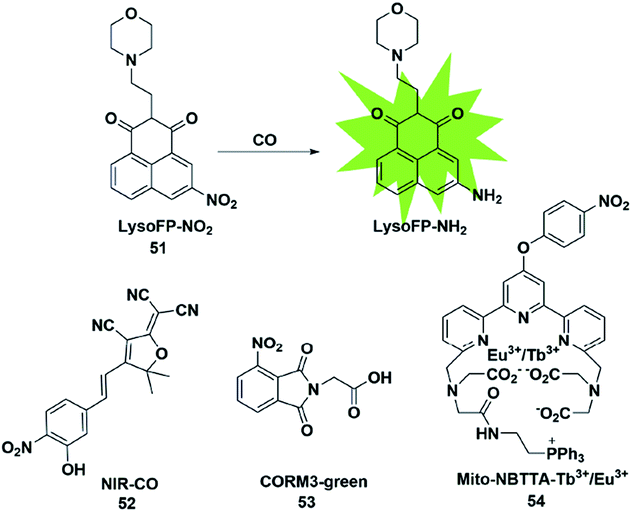 | ||
| Fig. 7 Sensing mechanism of fluorescent probe 51 based on nitro reduction and the structure of representative examples. | ||
In 2017, Dhara et al. reported a lysosome-targeting fluorescent probe 51 for the detection of CO based on a new recognition site.69 The probe consisted of the following three parts: 1,8-naphthalimide (acted as an ideal fluorophore), a morpholine unit (served as the lysosome-targeting moiety) and a nitro group (a novel CO-responsive site). Probe 51 exhibited very weak fluorescence intensity (Φ = 0.0016) due to the PET quenching effect of the nitro group on the 1,8-naphthalimide. Once it reacted with CO, the nitro group was reduced to a strong electron-donating amino group which made LysoFP-NH2 exhibit a strong ICT process (Φ = 0.1025); as a result, the fluorescent intensity at about 525 nm was increased 75 times. The probe was mainly located in the lysosome and could monitor the changes of the CO level in MCF-7 cells.
Then two examples of fluorescent probes, NIR-CO and CORM3-green, based on the nitro group reduction reaction were reported by Zhu and Feng, respectively (probes 52 and 53).70,71 Probe 52 was synthesized with TCF derivatives and designed through regulating the ICT process of the probe. When the nitro group of the probe 52 was directly reduced by CO to an amino group, the inhibited ICT process was released, which caused the probe to generate an off-on NIR fluorescence signal (665 nm). More importantly, the down-regulation of HO-1 suppressed by high glucose (HG) in zebrafish and the up-regulation of HO-1 induced by transient glucose deprivation (TGD) in living cells were visualized for the first time by imaging the glucose-caused changes of CO levels. Probe 53 was based on the ESIPT mechanism. When the nitro group of phthalimide was transformed into an amino group in the presence of CORM-3, the ESIPT process was activated to generate a fluorescence signal. Utilizing it as tool, the authors investigated CORM-3 in solution, in living cells and in animals.
In 2019, Yuan et al. reported a mitochondria-targeting ratiometric time-gated luminescence probe for CO based on lanthanide complexes (probe 54).72 When the probe reacted with CO, the nitro group was reduced to an amino group followed by the 1,6-rearrangement–elimination reaction and liberation of the 4-nitrobenzyl moiety, resulting in an enhancement of Tb3+ emission at 540 nm and reduction of Eu3+ emission at 610 nm. Triphenylphosphonium served as a mitochondria-anchoring unit to enable the probe to be accumulated in the mitochondria of cells. The exogenous and endogenous CO in living cells, liver tissue slices, Daphnia magna and mice was successfully visualized by fluorescence imaging of probe 54.
However, a recent study by Wang et al.73 found that the CO fluorescence probes based on the nitro-reduction reaction were not responsive to CO from all sources. The ruthenium carbonyl moiety (ruthenium core for activating the reduction action) was essential to causing the signal response of nitro reduction-based fluorescent probes. As a result, this type of probe can only detect CO derived from ruthenium-based CO releasing molecules (CORM-2 or CORM-3) and not other general CO sources (e.g. CO gas or metal-free organic CO donors). Accordingly, it is necessary to use multiple CO sources to evaluate the signal response of fluorescent probes for CO.
3.4 Other mechanisms
In addition to the three mechanisms for detecting CO described above, Zhou et al. reported a novel recognition site in 2019.74 As shown in Fig. 8, the probe 55 was designed by incorporating 2-(hydrazonomethyl)-pyridine as a CO-responsive group in lactam form. Rhodamine B was selected as the fluorophore due to its high fluorescence quantum yield, good biocompatibility and accessibility. The possible mechanism proposed was that the probe first reacted with CO to generate an intermediate I1 with a three-membered ring, followed by isomerization after combining with a proton to form I2 bearing a four-membered ring, which was finally converted into rhodamine B and by-product P2via interaction with water. In addition, according to the energy distribution diagram, the Gibbs free energies of all reactions were negative except for the first step, which indicated that all the reactions from probe 55 to RhB were thermodynamically feasible. Various biologically relevant species including ROS, RNS and heavy metal ions (Hg2+, Cu2+ and Fe2+) could lead to negligible fluorescence changes. With satisfactory results, it was used to image both exogenous and endogenous CO in HepG2 cells. Notably, this work provided a novel recognition mechanism for constructing more effective and CO-reactive fluorescent probes. Besides, the detection limits (LODs) of CO probes discussed in the subsections are summarized and shown in Table 2.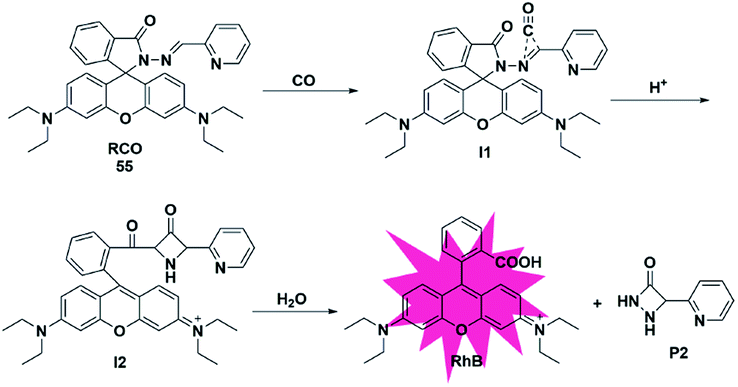 | ||
| Fig. 8 Sensing mechanism of fluorescent probe 55. | ||
| Probe | LOD | Probe | LOD | Probe | LOD | Probe | LOD |
|---|---|---|---|---|---|---|---|
| 31 | 1000 nM | 38 | 7.77 nM | 45 | 3.2 nM | 52 | 6.1 nM |
| 32 | 653 nM | 39 | 37 nm | 46 | 62 nM | 53 | 16 nM |
| 33 | 720 nM | 40 | 46 nM | 47 | 30.8 nM | 54 | 440 nM |
| 34 | 60 nM | 41 | 25 nM | 48 | 255 nM | 55 | 10 nM |
| 35 | 50 nM | 42 | 58 nM | 49 | 3.2 nM | ||
| 36 | 230 nM | 43 | 57 nM | 50 | 15.8 nM | ||
| 37 | No | 44 | 38 nM | 51 | 600 nM |
4. Hydrogen sulfide (H2S)-responsive fluorescent probes
Hydrogen sulfide (H2S) is confirmed as the third endogenous gas signal molecule after NO and CO. Endogenous H2S is mainly derived from both the enzymatic hydrolysis of L-cysteine and non-enzymatic production. The related enzymes for H2S synthesis in mammalian systems are as follows: cystathionine beta synthase (CBS), cystathionine gamma lyase (CSE), cysteine aminotransferase and mercaptopyruvate sulfurtransferase (CAT/MST). In biosystems, H2S can undergo thiolation with cysteine derived from the translation of downstream proteins and then attach to the iron centre of heme.2,3,5 Owing to the biological significance of H2S, it is important to develop effective detection methods for the study of H2S. According to the detection mechanism, the design of H2S fluorescent probes can be divided into the following strategies.4.1 Azide reduction
Organic azide units have been extensively applied as an important functional group in biorthogonal reactions, referring to those chemical reactions that can be performed in living cells or tissues without interfering with the native biochemical reactions in biosystems. In the synthesis of complex organic molecules, the electron withdrawing azide groups can be converted into electron donating amino moieties via the H2S-mediated chemoselective reduction reaction. Accordingly, incorporation of the azide trigger into various fluorophore scaffolds may provide a novel strategy for detecting H2S by changing the electronic distribution of the whole molecule. Cho and co-workers reported a ratiometric TP fluorescent probe for mitochondrial H2S by using 4-azidobenzyl carbamate as the response site and 6-(benzo[d]thiazol-2′-yl)-2-(methylamino) naphthalene as the fluorophore (probe 56).75 Upon addition of H2S, the aryl azide was reduced to aniline that underwent intramolecular elimination to release azaquinone methide and the fluorophore (Fig. 9), resulting in the change of the fluorescence emission wavelength. The difference of endogenous H2S levels which were generated by different expression levels of cystathionine β-synthase (CBS) in wild-type and DJ-1-knockout astrocytes and brain slices was effectively visualizing through two-photon microscopy ratiometric imaging of probe 56.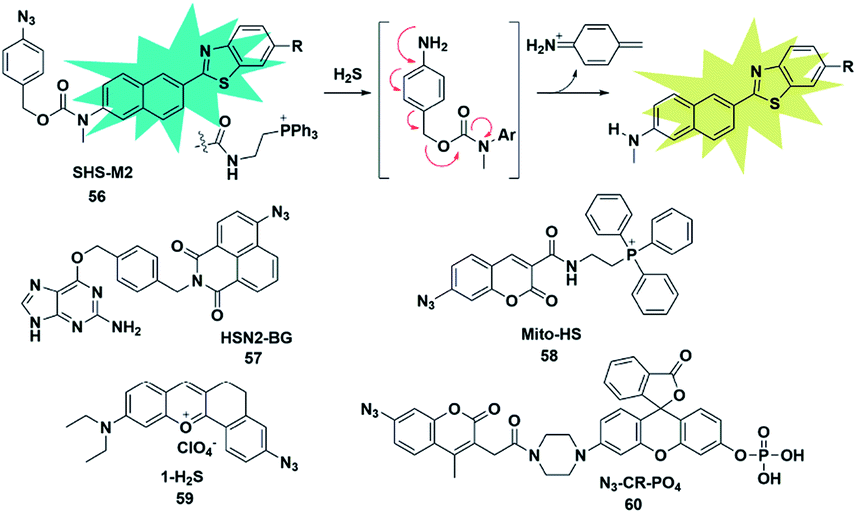 | ||
| Fig. 9 Sensing mechanism of fluorescent probe 56 based on azide reduction and the structure of representative examples. | ||
In 2016, Pluth designed an organelle-targeted fluorescent probe for detecting differential subcellular donation of H2S by incorporating SNAP-tag (benzylguanine-ligated substrates in combination with AGT fusion proteins to cause subcellular localization ability) into naphthalimide dye.76 In addition to imaging endogenous H2S in Chinese hamster ovary cells, probe 57 was also applied to investigating the subcellular H2S distributions supplied by various H2S donors such as ADT-OH, diallyltrisulfide (DATS), AP39 and GYY4137. A recent study has shown that H2S was overexpressed in cancer cells compared to normal cells due to the higher level of H2S related synthetases CBS and CSE in cancer cells. Bhuniya et al. constructed a turn-on biorthogonal fluorescent probe 58 for imaging endogenous H2S generation in cancer cells.77 Notably, Mito-HS could quickly detect (15 min) the formation of endogenous H2S in cancer cells (HeLa, MDA-MB-231 and DU145) for the first time without any external stimulants. Due to the ability of probe 58 to track endogenous H2S in cancer cells, it may be a potential effective tool to distinguish cancer cells from normal cells. By extending the conjugated system of the naphthalene coumarin analogue, Lin et al. synthesized a novel two-photon fluorescent probe 59 for sensing H2S near the nucleolus region of living cells.78 The probe was also successfully used for fluorescence imaging of H2S in zebrafish and mouse models.
The activity of phosphatase, an important hydrolase, is dynamically regulated by other upstream signal molecules such as ROS and gasotransmitters (e.g. H2S). In 2019, Zhang constructed a two-response site fluorescent probe for simultaneously sensing H2S and phosphatase via two independent fluorescence signal channels (probe 60).79 The H2S-responsive fluorophore coumarin derivative (emission at 445 nm) was linked to rhodol (emission at 545 nm) which served as a phosphate-reactive dye. More importantly, through combination with the FRET signal between rhodol and coumarin fluorescence signals, the authors established a three-channel strategy to detect and image H2S and phosphatase activity and investigate the correlation between them in living cells. The results revealed that intracellular H2S was vital for maintaining the high activity of phosphatase.
4.2 Nitro reduction
Similar to azide reduction, H2S can also reduce nitro groups to amino groups, which was used as a new strategy to construct fluorescent probes for the detection of H2S. However, unlike the ICT process of azide reduction, this approach of probes is generally based on the PET quenching mechanism due to the strong electron-withdrawing properties of nitro groups. Cheng et al. developed a NIR fluorescent probe 61 (ref. 80) for specifically detecting H2S in cells via equipping a heptamethine cyanine fluorophore with m-nitrophenol (a strong electron-withdrawing group). Probe 61 displayed a low fluorescence signal due to the donor-excited PET process from the fluorophore to m-nitrophenol. In the presence of H2S, the nitro group was reduced to an amino group (Cy-NH2) under mild conditions, inhibiting the PET process and resulting in a significant enhancement in the fluorescence signal of the probe. The probe exhibited high selectivity and sensitivity for H2S and was utilized to monitor the H2S changes in RAW 264.7 cells. In 2017, Hua et al. reported a series of N-annulated perylene-based colorimetric and ratiometric NIR fluorescent probes for the detection of H2S in biological samples (probe 62).81 By incorporating different functional groups into the fluorophore, the targeted probes (Mito-NPNM and Lyso-NPNM) were successfully applied to ratiometrically visualize endogenous H2S in the mitochondria and lysosomes of cells, respectively (Fig. 10).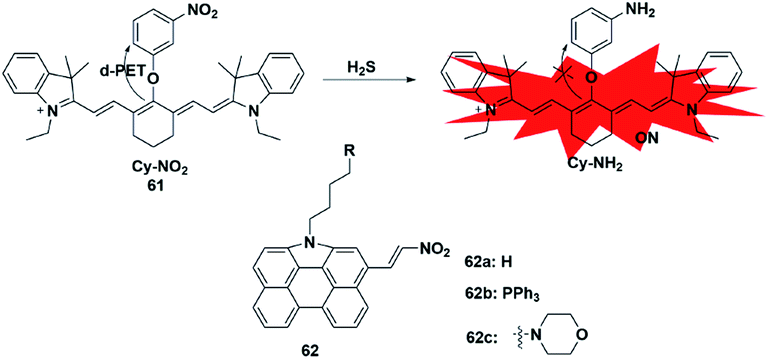 | ||
| Fig. 10 Sensing mechanism of fluorescent probe 61 based on nitro reduction and the structure of representative examples. | ||
4.3 Nucleophilic attack approach
Since the pKa of H2S is 7, it mainly exists in the form of anionic HS− under physiological pH conditions, which endows it with excellent nucleophilicity and allows it to participate in chemical reactions as a nucleophile. Taking advantage of the strong nucleophilic nature of H2S, several types of fluorescent probes have been reported for the detection of H2S. The main challenge of this strategy is the selective detection of H2S without interference from other nucleophilic thiols found in biological systems, especially cysteine and glutathione (at millimolar levels). Generally, these probes are designed with functionalized electrophilic units that are used to change the fluorescence properties of the entire molecules and serve as H2S recognition sites. We will focus on the following detection mechanisms.The 2,4-dinitrophenyl ether group has been extensively employed as both the responsive moiety for H2S and the fluorescence mediator. In 2019, Wu et al. reported a turn-on optoacoustic probe 63 based on the benzothiazole–xanthene dyad for imaging metformin-induced generation of H2S and liver injury.82 The fluorescence signal of NR-OH was quenched by 2,4-dinitrophenyl ether due to its strong electron-withdrawing ability. Upon addition of H2S, 2,4-dinitrophenyl ether was eliminated and transformed into electron-donating hydroxyl, generating the turn-on fluorescence and optoacoustic signals. The overdosed metformin-induced liver injury in the mouse model was visualized using probe 63via the detection of endogenous hepatic H2S. More importantly, with the help of orthogonal-view 3D multispectral optoacoustic tomography (MSOT) images, the probe was successfully employed to distinguish, accurately locate and visualize the volume of liver injury.
In addition, another nitro group, 7-nitro-1,2,3-benzoxadiazole (NBD), also has the ability to serve as a H2S-responsive site through thiolysis induced by H2S via the nucleophilic aromatic substitution mechanism. Utilizing this group, Zhang et al. synthesized a NIR (emission at 796 nm) fluorescent probe 64 (ref. 83) for selectively detecting H2S in cells and in vivo. By using this probe as an imaging tool, the endogenous H2S in colon cancer cells (HCT116, HT29) and tumors could be imaged successfully. Meanwhile, the fluorescence experiments indicated that higher levels of H2S were produced in the liver of mice.
As a robust nucleophile, HS− can undergo a nucleophilic addition reaction at the electrophilic centre, which mainly disturbs the extended π conjugate structure and then causes changes in the fluorescence signal of probes. Utilizing a merocyanine derivative as the H2S reaction group, He et al. reported a novel fluorescent probe 65 for selective ratiometric imaging of H2S in the mitochondria of living cells.84 As designed, the indolenium C-2 atom of the probe would be attacked by H2S as an electrophilic agent which may eliminate the properties of merocyanine but retain the fluorescence signal of coumarin. This research provided a reliable and effective principle for designing H2S ratio fluorescent probes through the nucleophilic addition of HS− to merocyanine derivatives.
Unlike conventional thiols, H2S can theoretically be considered an unsubstituted thiol, which may perform nucleophilic reactions twice. However, other (e.g. Cys and GSH) thiols only undergo nucleophilic reaction once. Using this difference, Xian et al. reported a novel recognition group containing bis-electrophilic enters for specifically reacting with H2S without being affected by thiols.85 As shown in Fig. 11, probe 66 (disulfide-containing probe) first underwent nucleophilic attack by H2S to generate a free SH containing intermediate followed by a spontaneous secondary nucleophilic cyclization to release the fluorophore. This probe could effectively and quickly sense H2S in aqueous solution and living cells.
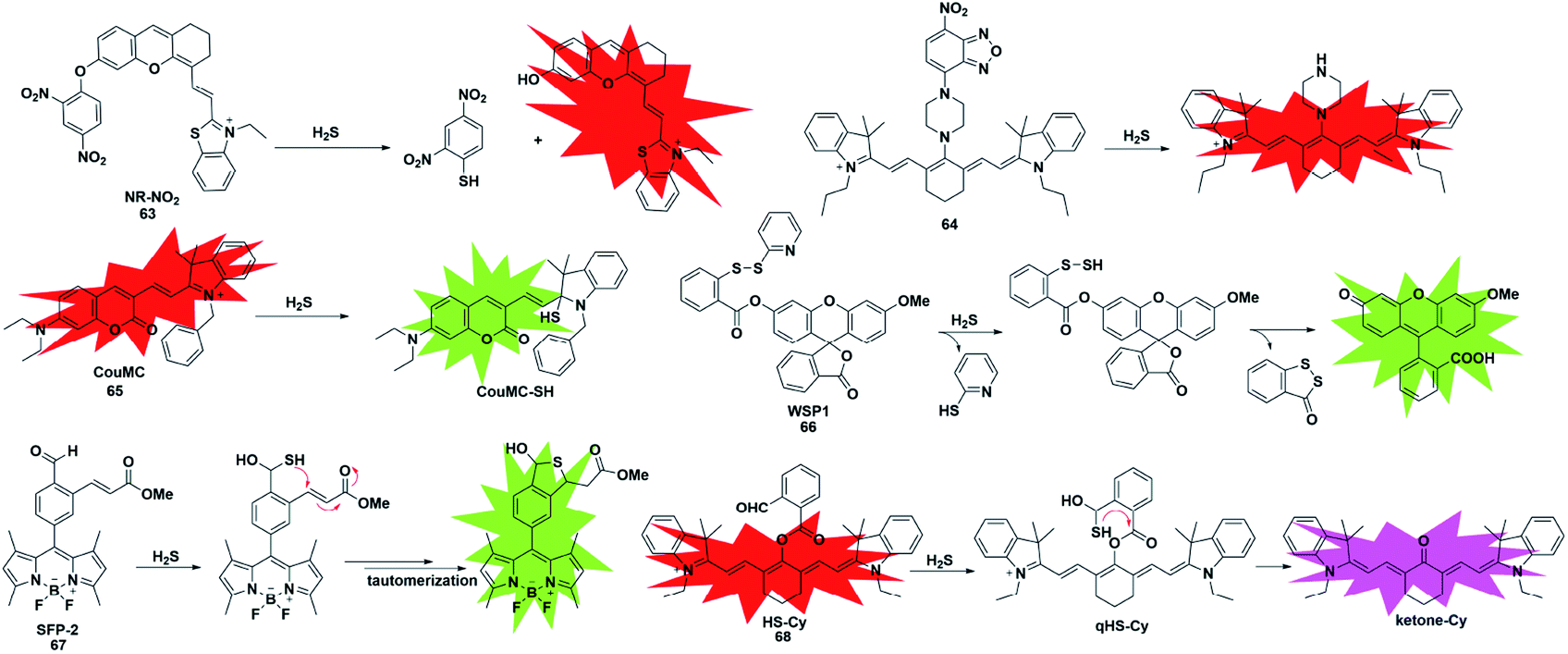 | ||
| Fig. 11 Sensing mechanism and structure of fluorescent probes 63–68 based on the nucleophilic reaction. | ||
Another strategy that utilizes the dual nucleophilic capabilities of H2S is the tandem Michael addition mechanism, which was originally reported by He's group (probe 67).86 During molecular design, the probe was functionalized by introducing an aldehyde group which was employed in the first nucleophilic reaction of H2S to generate a hemithioacetal intermediate containing an exposed thiol. Subsequently, the thiol will undergo a Michael addition to the proximal α, β-unsaturated ester to form a thioacetal, leading to a turn-on increase in the fluorescence signal. Although the first step could be performed when thiol was added, the formed intermediate could not undergo Michael addition and reversibly yielded the original probe. It showed high selectivity for H2S and was applied to investigate the function of H2S in biological systems. A similar approach was used by Tang et al. to construct a cyanine-based NIR ratiometric fluorescent probe for H2S detection (probe 68).87 As shown in Fig. 11, it was synthesized by linking 2-carboxybenzaldehyde and heptamethine cyanine to form an ester which supplied an effective reactive group. Similar to that reported by He, the formed sulfhydryl group (qHS-Cy) would attack the carbonyl to yield hydroxy cyanine, which transformed into its keto form under physiological conditions (ketone-Cy). The endogenously produced H2S in human A549 cells was detected and imaged by HS-Cy.
4.4 CuS precipitation
The selective detection of H2S is also achieved by the reaction with Cu2+ chelated by fluorophores to generate a CuS precipitate. Generally, the paramagnetism of Cu2+ in the fluorescent probe will cause fluorescence quenching. Based on this approach, Nagano et al. reported an azamacrocyclic Cu2+ complex (probe 69)88 for highly selective and sensitive detection and imaging of H2S in live cells. The fluorescein was functionalized by incorporating a cyclen azamacrocycle that will bind Cu2+ to cause low background fluorescence. Upon addition of H2S, the paramagnetic Cu2+ will be released from the azamacrocyclic ring by generating CuS, resulting in fluorescence enhancement. Cyclen was selected as the chelator because it had a stronger stable complex with Cu2+. As a result, high concentration of GSH could not induce a distinct response in the fluorescence signal (Fig. 12).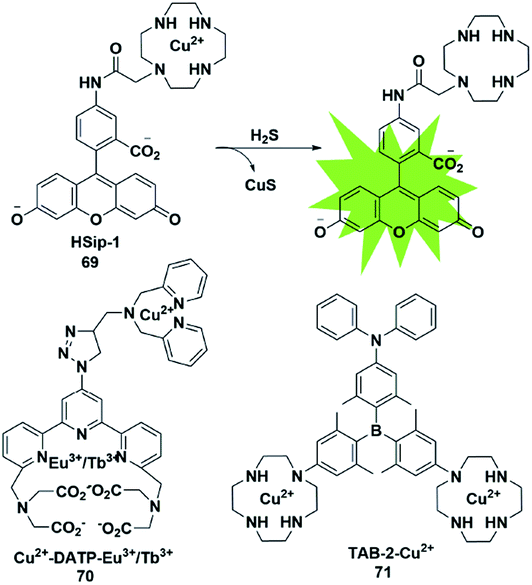 | ||
| Fig. 12 Sensing mechanism of fluorescent probe 69 based on CuS precipitation and the structure of representative examples. | ||
Based on the CuS precipitation strategy, Yuan designed a novel Cu2+-coupled lanthanide complex (probe 70)89 for the ratiometric time-gated luminescence detection of H2S in vitro and in vivo. By functionalizing triarylboron with cyclen and diphenylamine, Yang et al. constructed a two-photon fluorescent probe for sensing H2S (probe 71).90 Notably, the H2S-induced finite aggregation of the probe would increase its photostability and lead to changes of the fluorescence lifetime. Meanwhile, the H2S distribution in NIH/3T3 cells was successfully visualized by utilizing fluorescence lifetime microscopy.
4.5 Coordinative-based strategy
Reaction-based H2S fluorescent probes are mostly irreversible and can only be used as one-pot tools, which is undesirable for the measurement of real biological samples since it may upset the balance of the analytes under physiological conditions. In recent years, transition metal complexes characterized by H2S or HS− binding to the metal center have been gradually developed as effective sensors for detecting HS− or H2S.91–96 The process of H2S detection and release was realized by coordination or discoordination with a metal complex, constructing a reversible detection approach. For example, Pellecchia et al. reported a copper porphyrin complex for sensing H2S in aqueous solution (probe 72) (Fig. 13).92 The verification experiments demonstrated that the fluorescence response in the presence of H2S was attributed to the combination of the analyte with the copper center. In 2018, they also synthesized a fluorescent pyridoxal-based Zn(II) receptor (probe 73)93 for detecting HS−. This strategy is highly desirable and may become a promising approach to construct more reversible probes for sensing H2S in biological samples. Besides, the detection limits (LODs) of H2S probes discussed in the subsections are summarized and shown in Table 3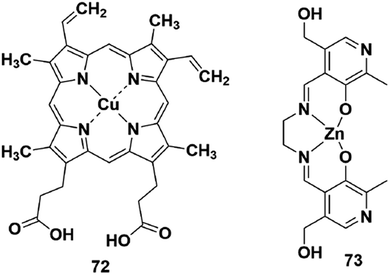 | ||
| Fig. 13 Representative fluorescent probes based on the coordination-based approach. | ||
| Probe | LOD | Probe | LOD | Probe | LOD | Probe | LOD |
|---|---|---|---|---|---|---|---|
| 56 | 400 nM | 61 | No | 66 | 60 nM | 71 | 47 nM |
| 57 | No | 62 | 139 nM | 67 | No | 72 | 1000 nM |
| 58 | 24.3 nM | 63 | 40 nM | 68 | 5–10 nM | 73 | No |
| 59 | 47 × 103 nM | 64 | 39.6 nM | 69 | No | ||
| 60 | 1 nM | 65 | 1000 nM | 70 | 190 nM |
5. Conclusions and perspectives
In this perspective, we systematically summarized the recent progress in fluorescent probes for bioimaging gaseous signalling molecules including NO, CO and H2S. The detection strategies, respective response mechanisms and imaging applications of representative fluorescent probes for each gas molecule were also discussed in detail. Great achievements have been made in this research field. For instance, in order to avoid the influence of MGO in NO detection, single-substituted OPD and N-nitrosation of aromatic secondary amines were successively reported in the molecular designing. Utilizing the reduction capacity of CO, researchers developed various CO-recognition probes through introducing the PdCl2-mediated Tsuji–Trost reaction and Ru-based nitro reduction. By designing a reasonable electrophilic structure, such as 2,4-dinitrophenyl ether, tandem Michael addition or NBD group, many reported probes could effectively detect H2S without interference from thiols.Despite the rapid development and application of fluorescent probes for the detection of gasotransmitters, there are still many issues to be addressed in future work. Firstly, it is well known that gaseous signalling molecules usually suffer from ultralow concentration, short lifetime, complex physical environments, distribution in specific regions, etc. Therefore, there is an urgent need to develop fluorescent probes with ultra-high sensitivity and selectivity, ultra-fast response, and efficient positioning capabilities. Secondly, studies have shown that while a gas molecule exerts a physiological role alone, it also works in coordination with other gasotransmitters or regulates the level of others. For example, CO and H2S will interact to generate a new mediator nitroxyl (HNO), which has important functions in signaling pathways and regulates vascular tone. Thus, the development of multifunctional, multi-recognition site fluorescent probes is essential to investigate the mutual regulation and interaction between three gaseous signalling molecules. Thirdly, compared to the visible (400–700 nm) and NIR-I (700–900 nm) emission, the second near-infrared (NIR-II) window (1000–1700 nm) possesses intrinsic properties such as deeper penetration, a higher S/N ratio and excellent imaging temporal and spatial resolution. The development of effective and gasotransmitter-responsive fluorescent probes in the NIR-II region is still a promising strategy for investigating more physiological processes. Finally, due to its ability to reduce bacterial infections and regulate inflammation, NO has the potential to be a promising wound therapeutic agent. And CO has emerged as another therapeutic agent against various diseases associated with oxidative stress. Since traditional NO and CO donors are non-fluorescent, effective monitoring of gas-based therapy is a challenging task. Thus, combining the fluorescence detection of NO and CO with gasotransmitter-mediated disease treatment is of great significance for the future. We hope that the principles summarized in this perspective will give useful insights into the future development of gasotransmitter probes for bioimaging applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21925802, 21878039, 21822804, and 21421005), NSFC-Liaoning Joint Fund (U1608222 and U1908202) and National Key Research and Development Plan (2018AAA0100301).Notes and references
- L. Li and P. K. Moore, Biochem. Soc. Trans., 2007, 35, 1138–1141 CrossRef CAS PubMed
.
- B. Olas, Clin. Chim. Acta, 2015, 445, 115–121 CrossRef CAS
- K. R. Olson, J. A. Donald, R. A. Dombkowski and S. F. Perry, Respir. Physiol. Neurobiol., 2012, 184, 117–129 CrossRef CAS
- A. Papapetropoulos, R. Foresti and P. Ferdinandy, Br. J. Pharmacol., 2015, 172, 1395–1396 CrossRef CAS PubMed
- Y. Qian and J. B. Matson, Adv. Drug Delivery Rev., 2017, 110, 137–156 CrossRef PubMed
- E. M. Hetrick and M. H. Schoenfisch, Annu. Rev. Anal. Chem., 2009, 2, 409–433 CrossRef CAS PubMed
- M. Plaza, S. Santoyo, L. Jaime, G. Garcia-Blairsy Reina, M. Herrero, F. J. Senorans and E. Ibanez, J. Pharmaceut. Biomed. Anal., 2010, 51, 450–455 CrossRef CAS PubMed
- Y. Lee and J. Kim, Anal. Chem., 2007, 79, 7669–7675 CrossRef CAS PubMed
- D. Wu, A. C. Sedgwick, T. Gunnlaugsson, E. U. Akkaya, J. Yoon and T. D. James, Chem. Soc. Rev., 2017, 46, 7105–7123 RSC
- Z. Liu, W. He and Z. Guo, Chem. Soc. Rev., 2013, 42, 1568–1600 RSC
- X. Liu, N. Li, M. Li, H. Chen, N. Zhang, Y. Wang and K. Zheng, Coord. Chem. Rev., 2019, 404 Search PubMed
- V. S. Lin and C. J. Chang, Curr. Opin. Chem. Biol., 2012, 16, 595–601 CrossRef CAS PubMed
- Z. Guo, G. Chen, G. Zeng, Z. Li, A. Chen, J. Wang and L. Jiang, Analyst, 2015, 140, 1772–1786 RSC
- R. Kaushik, A. Ghosh and D. A. Jose, Coord. Chem. Rev., 2017, 347, 141–157 CrossRef CAS
- A. R. Lippert, J. Inorg. Biochem., 2014, 133, 136–142 CrossRef CAS
- M. Strianese and C. Pellecchia, Coord. Chem. Rev., 2016, 318, 16–28 CrossRef CAS
- N. Kumar, V. Bhalla and M. Kumar, Coord. Chem. Rev., 2013, 257, 2335–2347 CrossRef CAS
- Y. Gabe, Y. Urano, K. Kikuchi, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2004, 126, 3357–3367 CrossRef CAS PubMed
- E. Sasaki, H. Kojima, H. Nishimatsu, Y. Urano, K. Kikuchi, Y. Hirata and T. Nagano, J. Am. Chem. Soc., 2005, 127, 3684–3685 CrossRef CAS PubMed
- H. Yu, Y. Xiao and L. Jin, J. Am. Chem. Soc., 2012, 134, 17486–17489 CrossRef CAS PubMed
- Z. Mao, W. Feng, Z. Li, L. Zeng, W. Lv and Z. Liu, Chem. Sci., 2016, 7, 5230–5235 RSC
- X. Dong, C. H. Heo, S. Chen, H. M. Kim and Z. Liu, Anal. Chem., 2014, 86, 308–311 CrossRef CAS PubMed
- H. Zheng, G.-O. Shang, S.-Y. Yang, X. Gao and J.-G. Xu, Org. Lett., 2008, 10, 2357–2360 CrossRef CAS
- P. Zhang, Y. Tian, H. Liu, J. Ren, H. Wang, R. Zeng, Y. Long and J. Chen, Chem. Commun., 2018, 54, 7231–7234 RSC
- B. Wang, S. Yu, X. Chai, T. Li, Q. Wu and T. Wang, Chem.–Eur. J., 2016, 22, 5649–5656 CrossRef CAS PubMed
- Y. Huo, J. Miao, L. Han, Y. Li, Z. Li, Y. Shi and W. Guo, Chem. Sci., 2017, 8, 6857–6864 RSC
- L. Yuan, W. Lin, Y. Xie, B. Chen and S. Zhu, J. Am. Chem. Soc., 2012, 134, 1305–1315 CrossRef CAS PubMed
- Y.-Q. Sun, J. Liu, H. Zhang, Y. Huo, X. Lv, Y. Shi and W. Guo, J. Am. Chem. Soc., 2014, 136, 12520–12523 CrossRef CAS PubMed
- N. Gupta, S. I. Reja, V. Bhalla, M. Gupta, G. Kaur and M. Kumar, Chem.–Asian J., 2016, 11, 1020–1027 CrossRef CAS PubMed
- S.-J. Li, D.-Y. Zhou, Y. Li, H.-W. Liu, P. Wu, J. Ou-Yang, W.-L. Jiang and C.-Y. Li, ACS Sens., 2018, 3, 2311–2319 CrossRef CAS PubMed
- K. N. Bobba, G. Saranya, S. M. Alex, N. Velusamy, K. K. Maiti and S. Bhuniya, Sens. Actuators, B, 2018, 260, 165–173 CrossRef CAS
- J. Tang, Z. Guo, Y. Zhang, B. Bai and W.-H. Zhu, Chem. Commun., 2017, 53, 10520–10523 RSC
- J. Miao, Y. Huo, X. Lv, Z. Li, H. Cao, H. Shi, Y. Shi and W. Guo, Biomaterials, 2016, 78, 11–19 CrossRef CAS PubMed
- Y. Huo, J. Miao, J. Fang, H. Shi, J. Wang and W. Guo, Chem. Sci., 2019, 10, 145–152 RSC
- Z. Mao, H. Jiang, X. Song, W. Hu and Z. Liu, Anal. Chem., 2017, 89, 9620–9624 CrossRef CAS PubMed
- Z. Mao, H. Jiang, Z. Li, C. Zhong, W. Zhang and Z. Liu, Chem. Sci., 2017, 8, 4533–4538 RSC
- Y. Yang, S. K. Seidlits, M. M. Adams, V. M. Lynch, C. E. Schmidt, E. V. Anslyn and J. B. Shear, J. Am. Chem. Soc., 2010, 132, 13114–13116 CrossRef CAS PubMed
- X. Lv, Y. Wang, S. Zhang, Y. Liu, J. Zhang and W. Guo, Chem. Commun., 2014, 50, 7499–7502 RSC
- X. Zhu, J.-Q. Chen, C. Ma, X. Liu, X.-P. Cao and H. Zhang, Analyst, 2017, 142, 4623–4628 RSC
- L. Chen, D. Wu and J. Yoon, Sens. Actuators, B, 2018, 259, 347–353 CrossRef CAS
- C.-G. Dai, J.-L. Wang, Y.-L. Fu, H.-P. Zhou and Q.-H. Song, Anal. Chem., 2017, 89, 10511–10519 CrossRef CAS PubMed
- X. Hu, J. Wang, X. Zhu, D. Dong, X. Zhang, S. Wu and C. Duan, Chem. Commun., 2011, 47, 11507–11509 RSC
- M. D. Pluth, M. R. Chan, L. E. McQuade and S. J. Lippard, Inorg. Chem., 2011, 50, 9385–9392 CrossRef CAS PubMed
- B. Mondal, P. Kumar, P. Ghosh and A. Kalita, Chem. Commun., 2011, 47, 2964–2966 RSC
- R. Alam, T. Mistri, P. Mondal, D. Das, S. K. Mandal, A. R. Khuda-Bukhsh and M. Ali, Dalton Trans., 2014, 43, 2566–2576 RSC
- S. W. Ryter and A. M. K. Choi, Transl. Res., 2016, 167, 7–34 CrossRef CAS PubMed
- S. W. Ryter and L. E. Otterbein, Bioessays, 2004, 26, 270–280 CrossRef CAS
- B. W. Michel, A. R. Lippert and C. J. Chang, J. Am. Chem. Soc., 2012, 134, 15668–15671 CrossRef CAS PubMed
- K. Zheng, W. Lin, L. Tan, H. Chen and H. Cui, Chem. Sci., 2014, 5, 3439–3448 RSC
- Y. Li, X. Wang, J. Yang, X. Xie, M. Li, J. Niu, L. Tong and B. Tang, Anal. Chem., 2016, 88, 11154–11159 CrossRef CAS
- M. Sun, H. Yu, K. Zhang, S. Wang, T. Hayat, A. Alsaedi and D. Huang, ACS Sens., 2018, 3, 285–289 CrossRef CAS PubMed
- K. Liu, X. Kong, Y. Ma and W. Lin, Angew. Chem. Int. Ed., 2017, 56, 13489–13492 CrossRef CAS PubMed
- S. Xu, H.-W. Liu, X. Yin, L. Yuan, S.-Y. Huan and X.-B. Zhang, Chem. Sci., 2019, 10, 320–325 RSC
- C. de la Torre, A. Toscani, C. Marin-Hernandez, J. A. Robson, M. Carmen Terencio, A. J. P. White, M. J. Alcaraz, J. D. E. T. Wilton-Ely, R. Martinez-Manez and F. Sancenon, J. Am. Chem. Soc., 2017, 139, 18484–18487 CrossRef PubMed
- J. T. Mohr and B. M. Stoltz, Chem.–Asian J., 2007, 2, 1476–1491 CrossRef CAS PubMed
- S. Pal, M. Mukherjee, B. Sen, S. K. Mandal, S. Lohar, P. Chattopadhyay and K. Dhara, Chem. Commun., 2015, 51, 4410–4413 RSC
- W. Feng, D. Liu, S. Feng and G. Feng, Anal. Chem., 2016, 88, 10648–10653 CrossRef CAS PubMed
- S. Feng, D. Liu, W. Feng and G. Feng, Anal. Chem., 2017, 89, 3754–3760 CrossRef CAS PubMed
- W. Feng, D. Liu, Q. Zhai and G. Feng, Sens. Actuators, B, 2017, 240, 625–630 CrossRef CAS
- W. Feng, J. Hong and G. Feng, Sens. Actuators, B, 2017, 251, 389–395 CrossRef CAS
- L. Yan, D. Nan, C. Lin, Y. Wan, Q. Pan and Z. Qi, Spectrochim. Acta, Part A, 2018, 202, 284–289 CrossRef CAS PubMed
- S. Gong, J. Hong, E. Zhou and G. Feng, Talanta, 2019, 201, 40–45 CrossRef CAS PubMed
- Z. Wang, Z. Zhao, C. Liu, Z. Geng, Q. Duan, P. Jia, Z. Li, H. Zhu, B. Zhu and W. Sheng, Photochem. Photobiol. Sci., 2019, 18, 1851–1857 RSC
- B. Biswas, M. Venkateswarulu, S. Sinha, K. Girdhar, S. Ghosh, S. Chatterjee, P. Mondal and S. Ghosh, ACS Appl. Bio Mater., 2019, 2, 5427–5433 CrossRef CAS
- J. Wang, C. Li, Q. Chen, H. Li, L. Zhou, X. Jiang, M. Shi, P. Zhang, G. Jiang and B. Z. Tang, Anal. Chem., 2019, 91, 9388–9392 CrossRef CAS PubMed
- J.-w. Yan, J.-y. Zhu, Q.-f. Tan, L.-f. Zhou, P.-f. Yao, Y.-t. Lu, J.-h. Tan and L. Zhang, RSC Adv., 2016, 6, 65373–65376 RSC
- W. Feng and G. Feng, Sens. Actuators, B, 2018, 255, 2314–2320 CrossRef CAS
- E. Zhou, S. Gong and G. Feng, Sens. Actuators, B, 2019, 301, 127075 CrossRef CAS
- K. Dhara, S. Lohar, A. Patra, P. Roy, S. K. Saha, G. C. Sadhukhan and P. Chattopadhyay, Anal. Chem., 2018, 90, 2933–2938 CrossRef CAS PubMed
- Z. Wang, C. Liu, X. Wang, Q. Duan, P. Jia, H. Zhu, Z. Li, X. Zhang, X. Ren, B. Zhu and W. Sheng, Sens. Actuators, B, 2019, 291, 329–336 CrossRef CAS
- W. Feng, S. Feng and G. Feng, Anal. Chem., 2019, 91, 8602–8606 CrossRef CAS PubMed
- Z. Tang, B. Song, H. Ma, T. Luo, L. Guo and J. Yuan, Anal. Chem., 2019, 91, 2939–2946 CrossRef CAS PubMed
- Z. Yuan, X. Yang, L. K. De La Cruz and B. Wang, Chem. Commun., 2020, 56, 2190–2193 RSC
- C. Zhang, H. Xie, T. Zhan, J. Zhang, B. Chen, Z. Qian, G. Zhang, W. Zhang and J. Zhou, Chem. Commun., 2019, 55, 9444–9447 RSC
- S. K. Bae, C. H. Heo, D. J. Choi, D. Sen, E.-H. Joe, B. R. Cho and H. M. Kim, J. Am. Chem. Soc., 2013, 135, 9915–9923 CrossRef CAS PubMed
- L. A. Montoya and M. D. Pluth, Anal. Chem., 2016, 88, 5769–5774 CrossRef CAS PubMed
- N. Velusamy, A. Binoy, K. N. Bobba, D. Nedungadi, N. Mishra and S. Bhuniya, Chem. Commun., 2017, 53, 8802–8805 RSC
- K. Liu, C. Liu, H. Shang, M. Ren and W. Lin, Sens. Actuators, B, 2018, 256, 342–350 CrossRef CAS
- P. Ou, R. Zhang, Z. Liu, X. Tian, G. Han, B. Liu, Z. Hu and Z. Zhang, Angew. Chem. Int. Ed., 2019, 58, 2261–2265 CrossRef CAS PubMed
- R. Wang, F. Yu, L. Chen, H. Chen, L. Wang and W. Zhang, Chem. Commun., 2012, 48, 11757–11759 RSC
- X. Zhang, H. Tan, Y. Yan, Y. Hang, F. Yu, X. Qu and J. Hua, J. Mater. Chem. B, 2017, 5, 2172–2180 RSC
- L. Sun, Y. Wu, J. Chen, J. Zhong, F. Zeng and S. Wu, Theranostics, 2019, 9, 77–89 CrossRef CAS PubMed
- K. Zhang, J. Zhang, Z. Xi, L.-Y. Li, X. Gu, Q.-Z. Zhang and L. Yi, Chem. Sci., 2017, 8, 2776–2781 RSC
- Y. Chen, C. Zhu, Z. Yang, J. Chen, Y. He, Y. Jiao, W. He, L. Qiu, J. Cen and Z. Guo, Angew. Chem. Int. Ed., 2013, 52, 1688–1691 CrossRef CAS PubMed
- B. Peng, W. Chen, C. Liu, E. W. Rosser, A. Pacheco, Y. Zhao, H. C. Aguilar and M. Xian, Chem.–Eur. J., 2014, 20, 1010–1016 CrossRef CAS PubMed
- Y. Qian, J. Karpus, O. Kabil, S.-Y. Zhang, H.-L. Zhu, R. Banerjee, J. Zhao and C. He, Nat. Commun., 2011, 2, 1–7 Search PubMed
- X. Wang, J. Sun, W. Zhang, X. Ma, J. Lv and B. Tang, Chem. Sci., 2013, 4, 2551–2556 RSC
- K. Sasakura, K. Hanaoka, N. Shibuya, Y. Mikami, Y. Kimura, T. Komatsu, T. Ueno, T. Terai, H. Kimura and T. Naganot, J. Am. Chem. Soc., 2011, 133, 18003–18005 CrossRef CAS PubMed
- Z. Tang, B. Song, H. Ma, Y. Shi and J. Yuan, Anal. Chim. Acta, 2019, 1049, 152–160 CrossRef CAS PubMed
- J. Liu, X. Guo, R. Hu, X. Liu, S. Wang, S. Li, Y. Li and G. Yang, Anal. Chem., 2016, 88, 1052–1057 CrossRef CAS PubMed
- E. Galardon, A. Tomas, P. Roussel and I. Artaud, Dalton Trans., 2009, 9126–9130 RSC
- S. Mirra, S. Milione, M. Strianese and C. Pellecchia, Eur. J. Inorg. Chem., 2015, 2272–2276 CrossRef CAS
- M. Strianese, M. Lamberti and C. Pellecchia, Dalton Trans., 2018, 47, 17392–17400 RSC
- M. D. Hartle, M. R. Tillotson, J. S. Prell and M. D. Pluth, J. Inorg. Biochem., 2017, 173, 152–157 CrossRef CAS PubMed
- M. Strianese, M. Lamberti, A. Persico and C. Pellecchia, Inorg. Chim. Acta, 2020, 501 Search PubMed
- M. Strianese, M. Lamberti and C. Pellecchia, Dalton Trans., 2017, 46, 1872–1877 RSC
| This journal is © The Royal Society of Chemistry 2020 |