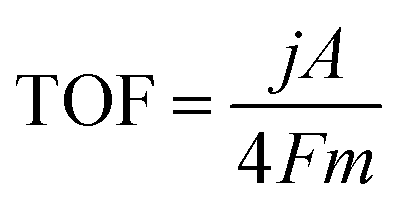DOI:
10.1039/D0SC01348J
(Edge Article)
Chem. Sci., 2020,
11, 6012-6019
Pseudo-atomic-scale metals well-dispersed on nano-carbons as ultra-low metal loading oxygen-evolving electrocatalysts†
Received
5th March 2020
, Accepted 21st May 2020
First published on 22nd May 2020
Abstract
Solving challenges for the scaling-up, high metal loadings and low turnover frequency (TOF, defined as mol O2 per mol metal per second), of FeNi catalysts in water electrolysis, we report the first discovery of pH tunable tannic acid single molecular layer formed on nano-sized carbons (NCs), which promotes the gram-production of pseudo-atomic-scale FeNi oxyhydroxide nanoclusters well-dispersed on NCs. It results in ultra-low metal loading (0.42 μg cm−2) and remarkably large TOF of 14.03 s−1 for the oxygen evolution reaction, which is three orders of magnitude higher than that of state-of-the-art FeNi catalysts. A “volcano”-shaped activity trend in specific activity and TOF was found to depend on the Fe content in FeNi oxyhydroxide. The micro-morphologies from the atomic-level exposure of active sites and surface spectra analyses confirm the model of synergism between Ni and Fe centers.
Introduction
Water electrolysis, the most promising hydrogen source for storing energy from renewable energy sources, such as solar and wind energy, compensates for the intermittency of sunlight as a primary source of power.1–4 The key half-reaction in water electrolysis is the oxygen evolution reaction (OER), caused by a kinetically sluggish process that involves a four-proton-coupled electron transfer. For the OER in acid medium, the best catalysts, offering both high activity and stability, are IrO2 and RuO2. However, their high cost and scarcity could hinder their large-scale applications.5–8 For the alkaline OER, catalysts based on transition metal oxyhydroxides (M(OOH), where M = Fe, Co, Ni, etc.), especially NiFe(OOH), are promising economic alternatives to precious metals.6,9 Different types of NiFe(OOH) have also been much explored, including those in the form of nano-sheets and those supported on nanostructured carbons.10–29 Now, overpotentials are reported to be so close to those of precious metals that further improvements seem needless. However, their large-scale use is limited because of the high metal loadings (the sizes of key metal parts in these materials are mostly larger than 5–10 nm) required to achieve a suitable overpotential (even costlier than using precious metals) and very low TOF, which is a parameter that compares intrinsic activity, i.e., quantifies how many molecules (in the case of water splitting: H2 and O2) are evolved per second per site at a specific overpotential, e.g. η = 300 mV.1 Here, we present a promising strategy to overcome this challenge by preparing well-dispersed pseudo-atomic-scale M(OOH) nanoclusters (1–2 nm) on nano-carbon (NC) supports, including single-wall carbon nanotubes (SWCNTs), multi-wall carbon nanotubes (MWCNTs), and graphene (GE), as new OER electrocatalysts. The resulting decrease in the size of the active sites from the nano- to subnanometer or atomic scale offers the following advantages: (i) significant reduction in catalytic metal usage by maximizing the M(OOH) utilization efficiency, (ii) high activity through low-coordination and unsaturated sites and M(OOH) nanocluster–support interactions, and (iii) well-defined active sites for mechanistic studies.
Tannic acid (TA), a natural product of polyphenol with 10 benzene rings surrounded by numerous phenolic hydroxyl groups (Fig. S1†), was used to assist the dispersion of NCs in an aqueous solution.30–32 To the best of our knowledge, we present in this study the first pH-controllable TA molecule monolayer on a carbon surface. Under alkaline conditions (pH > 8), the H-bonding between TA molecules breaks owing to its pKa of ∼7.4,33 hindering the multilayer formation of TA and leaving only π–π interactions between the TA monolayer and carbon surface. In a 1.0 M KOH aqueous solution (KOHaq, pH ∼ 13) with only <1 fM Ni2+ or Fe2+ (ultra-low solubility product constants for Ni(OH)2 (5.5 × 10−16) and Fe(OH)2 (4.9 × 10−17)),34 surprisingly, the TA monolayer plays a vital role, promoting electrodeposition of Ni(OOH) or NiFe(OOH) on a glassy carbon electrode (GCE) (Ni@GCE or FeNi@GCE). Good OER performance is demonstrated with a metal loading of only <0.0026 μg cm−2, considerably lower than that for the advanced Ni–Fe OER catalysts (1780 to 30 μg cm−2).10,14,20,21,24–26,28,35–44
We extend the pH-controllable TA monolayer on bulk carbon to NCs. The TA monolayer provides excellent metal ion catchability and promotes homogeneous dispersion of pseudo-atomic-scale FeNi(OOH) nanoclusters on NCs. This results in an ultra-low metal loading (0.42 μg cm−2) and remarkably high TOF (14.03 s−1) for the OER, which is three orders of magnitude higher than that of the state-of-the-art FeNi catalysts. Gram-scale production and long-term stability are also demonstrated.
Results and discussion
Preparation of M(OOH) OER electrocatalysts in <1 fM metal ion containing alkaline aqueous solution (pH 13)
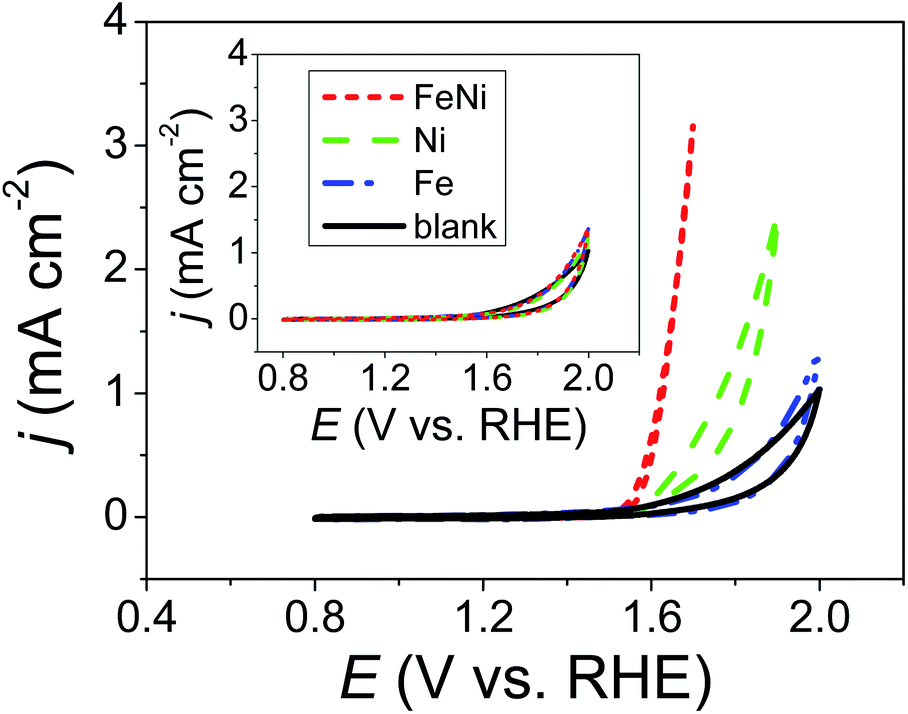
The inset of Fig. 1 shows CVs of a 1.0 M KOHaq saturated with Ni2+, Fe2+, and a mixture of Fe2+ and Ni2+ ions (Niaq, Feaq, and FeNiaq, respectively) recorded on a GCE. Ultra-low metal ion concentrations were expected in the 1.0 M KOHaq (<1 fM) due to the ultra-low solubility product constants of Ni(OH)2 and Fe(OH)2 (Ksp = 5.5 × 10−16 and 4.9 × 10−17, respectively). As expected, there was no difference in the voltammetric behavior between that with the presence of metal ions and that with the lack thereof. Surprisingly, after the addition of 2.0 μM TA, significant oxidative currents began to sharply rise at 1.5 V and 1.6 V vs. RHE in FeNiaq and Niaq, respectively (Fig. 1). With the appearance of these oxidation currents, violently generated oxygen bubbles appeared on the GCE, confirming that the oxidative current comes from the water oxidation and then generates oxygen. Considering the anodic current (∼0.025 μA) for metal oxyhydroxide (M(OOH)) formation between 1.2 and 1.4 V with a scan rate of 0.05 V s−1, the metal loading is evaluated to be <0.0026 μg cm−2, considerably lower than the metal loading (1780 to 30 μg cm−2) in the state-of-the-art Ni–Fe OER catalysts. In the KOHaq (pH ∼ 13) with only <1 fM metal ions, for the first time, Ni(OOH) and Ni–Fe OER catalysts were prepared with 2.0 μM TA playing a vital role.
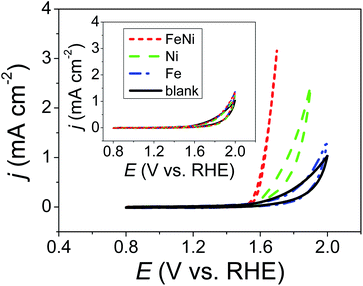 |
| | Fig. 1 CVs of 2.0 μM TA containing 1.0 M KOH aqueous solutions (KOHaq) saturated with (dashed line) Ni2+, (dashed dotted line) Fe2+, and (dotted line) mixture of Fe2+ and Ni2+ ions, respectively, were recorded on a GCE. Inset: the same CVs are recorded in the absence of TA. | |
pH-controllable TA molecule monolayer on a carbon surface
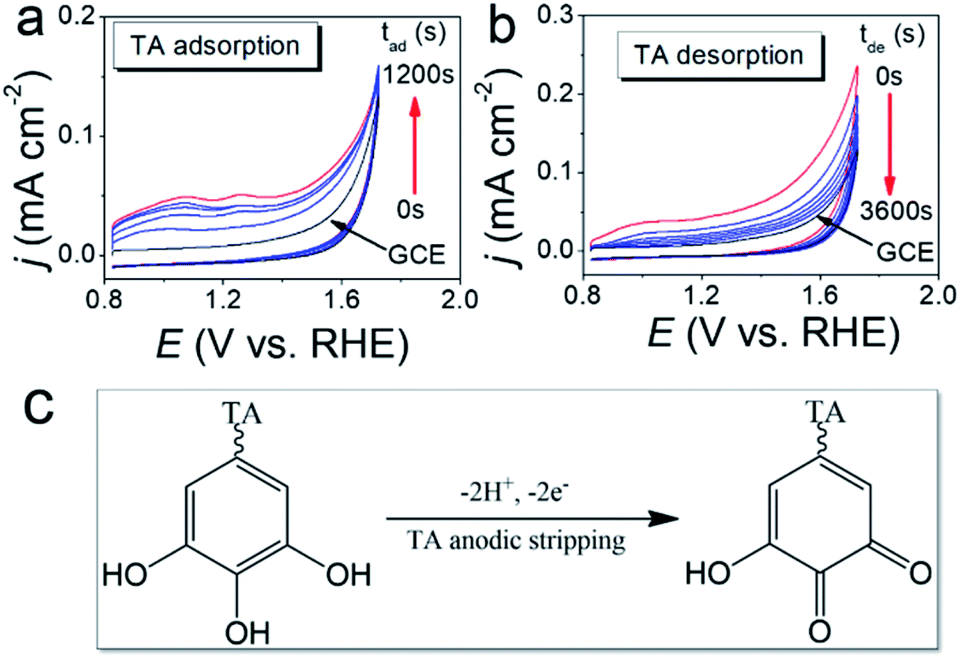
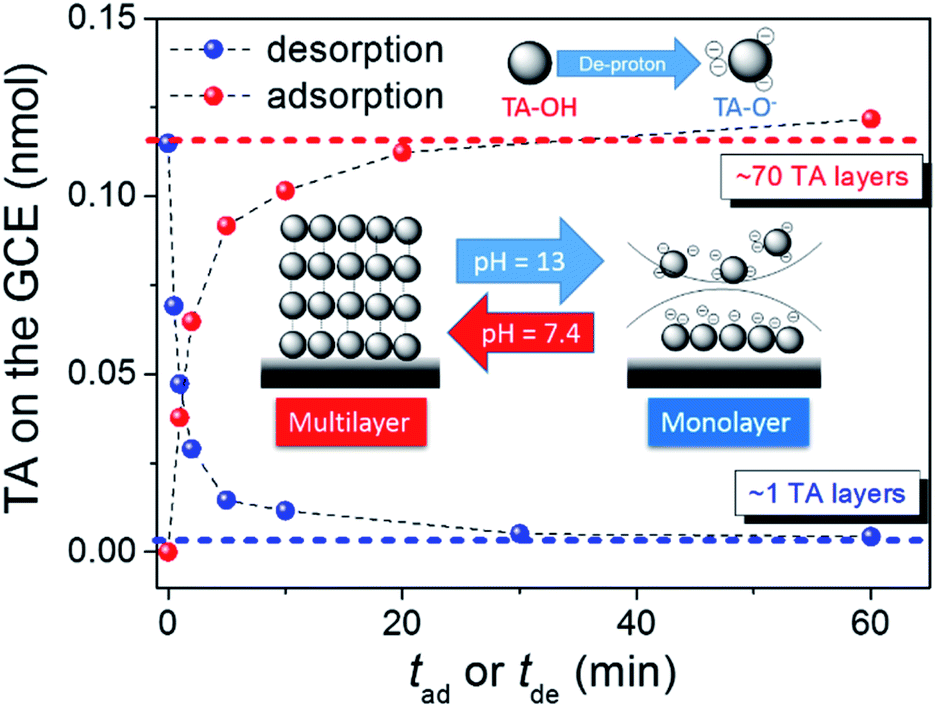
The TA adsorption (TAad) on a GCE is tracked by CVs of GCE immersed in a 20 μM TA aqueous solution (TAaq, pH = 7.4) for various periods (tad) (Fig. 2a). After the adsorptive accumulation of TA on the GCE and rinsing it with deionized water, CV is performed in 1.0 M KOHaq. As shown in Fig. 2a, CVs reveal two broad TA oxidation peaks at peak potentials of (a1) 0.35 V and (a2) 0.15 V vs. RHE assigned for the oxidation of TA to TA–quinone through the 2e−/2H+ process (Fig. 2c). The integrated charges of these peaks were used to determine the amount of TAad. Fig. 3 shows that TAad increased with tad to a limiting plateau where TAad reached a saturated state. An estimated effective radius of 1.28 nm for the TA molecule (Fig. S1†), calculated from the structure and known bond lengths and angles, corresponded to a GCE surface area of 0.07 cm2, occupied per TA. Therefore, the TAad in one monolayer is estimated to be ∼1 × 1012 molecules. Saturated TAad on GCE (s-TAad@GCE) reached ∼70 molecule layers. Interestingly, s-TAad@GCE was re-incubated in 1.0 M KOHaq (pH ∼ 13) for various periods (tde) before the TAad CV measurement (Fig. 2b). TAad sharply decreased from 70 molecule layers to only ∼1 monolayer with an increase in tde (Fig. 3). For TA adsorption on the carbon surface, π–π interactions are the main force for the first TA monolayer (TAi) formation, following the TA multilayer (TAm) formation by attracting more dissolved TA molecules via H-bonding on TAi under neutral conditions. However, the deprotonation of TA–OH to form TA–O− occurs under alkaline conditions (pH > 8), owing to the pKa of TA of ∼7.4, in which the H-bonding between TA molecules break. The electronic repulsion between TA–O− molecules hinders the TAm formation and leaves only π–π interactions between the TAi and carbon surface (Fig. 3). The formation constant between TA and metal ion strongly increases with increasing pH, further enhancing effectively the metal ion capture ability of the TAi-decorated GCE. These results indicate that the stable TA monolayer assists in the formation of ultra-thin and well-dispersed OER electrocatalysts on the GCE, leading to the ultra-low metal loading that characterizes the outstanding OER performance.
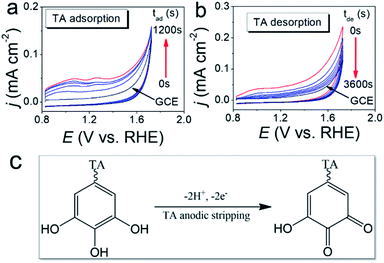 |
| | Fig. 2 (a) CVs of GCE immersed in the 20 μM TA aqueous solution (TAaq, pH = 7.4) for various periods (tad) were recorded in 1.0 M KOHaq with a scan rate of 50 mV s−1. (b) CVs of the s-TAad@GCE (∼70 TA molecule layers) re-incubated in 1.0 M KOHaq (pH ∼ 13) for various periods (tde) were recorded in 1.0 M KOHaq with a scan rate of 50 mV s−1. (c) A 2e−/2H+ process for the oxidation of TA to TA–quinone. | |
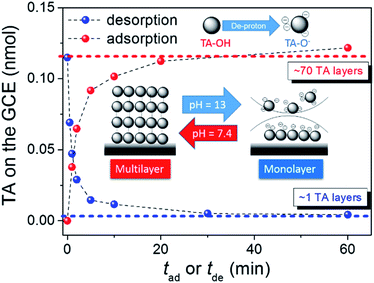 |
| | Fig. 3 The amount of TA adsorption vs. (red solid circle) tad or (blue solid circle) tde. Inset: plot represents the dependence of the TA adsorption onto the carbon surface on the pH of the TA-containing aqueous solution. | |
pH-controllable TA molecule monolayer on the nano-carbon (NC)
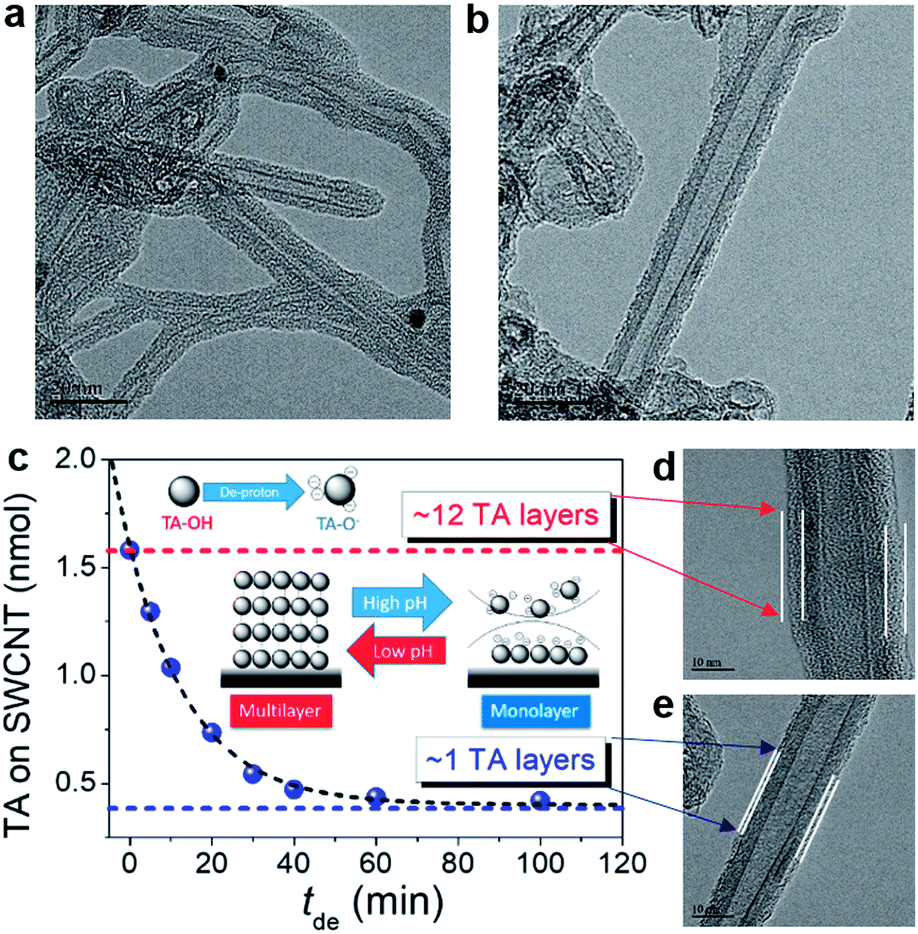
Fig. S2† shows the results of 0.12 wt% TA with 0.12 wt% NCs, including SWCNTs, MWCNTs, GE8, and GE12 (GE8 and GE12 indicate 8 nm and 12 nm GE provided by the manufacturer) well-dispersed in neutral water. Fig. S2a–c† display transmission electron microscopy (TEM) images of well-dispersed TA-modified NCs (TA-NCs). High-resolution TEM (HRTEM) was employed to examine the surface morphology of TA-modified SWCNTs (TA-SWCNTs) (Fig. 4). At higher magnification, a thin layer with ∼4 nm thickness forms around the SWCNT wall, indicating that the SWCNTs are surrounded by TA multi-layers (TAm-SWCNTs). The representative Fourier transform infrared (FT-IR) spectra of SWCNTs and TA-SWCNTs indicate that TA fingerprint vibration peaks between 600 and 1800 cm−1 appear on TA-SWCNTs but not on the SWCNTs (Fig. S3†). Interestingly, TA thickness is reduced to <1 nm after the re-dispersion of TAm-SWCNTs in KOHaq (pH 9–10) for 30 min. This indicates the transfer of TAm-SWCNTs to the TA monolayer on SWCNTs (TAi-SWCNTs), whereby the dispersity of TAi-SWCNTs does not change in pure water. The amount of TA adsorption (TAad) on NCs can be evaluated by cyclic voltammograms (CVs) recorded in KOHaq (Fig. S4†). With TAad on SWCNTs serving as an example, the TAad on the as-prepared TAm-SWCNTs is ∼12 TA molecule layers, which is then reduced to 1–1.5 TA molecule layers after immersion in KOHaq for over 30 min (Fig. 4c). These results demonstrate that pH-tunable TA monolayer formation occurs on NCs (TAi-NCs).
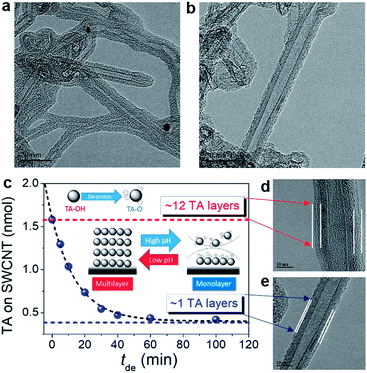 |
| | Fig. 4 HRTEM images of (a) TAm-SWCNTs (from neutral water) and (b) TAi-SWCNTs from the TAm-SWCNTs incubated in KOHaq (pH 9–10) for 30 min. (c) TAad on the TA-SWCNTs vs. tde. Inset: plot represents the dependence of the TA adsorption on the carbon surface on the pH of the TA-containing aqueous solution. (d) and (e) Magnification of the HRTEM images of (a) and (b). | |
Preparation of FeNi3@NC OER electrocatalyst
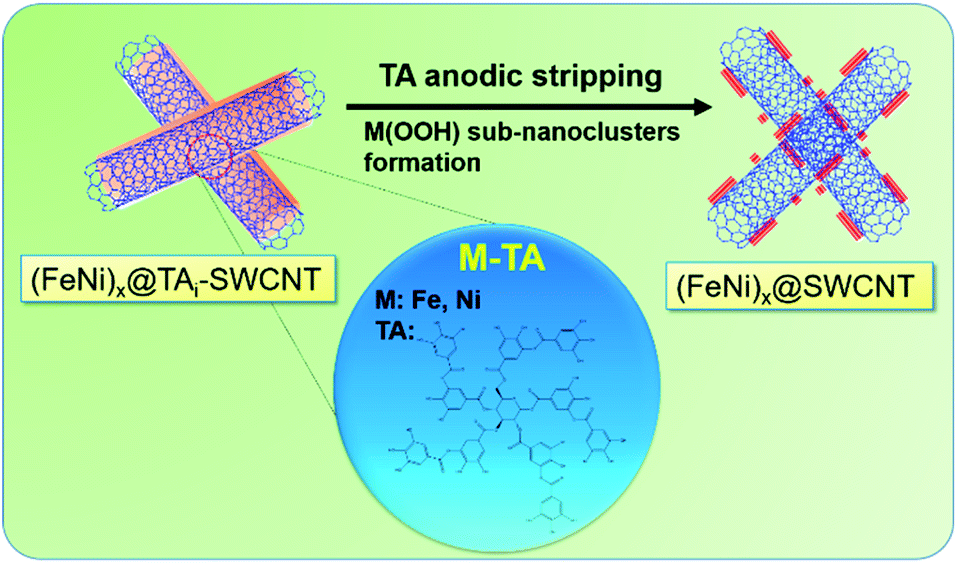
A series of TAm-NCs, including TAm-SWCNTs, TAm-MWCNTs, TAm-GE8, and TAm-GE12, respectively, were incubated in an aqueous solution containing 0.07 mM Fe2+ and 0.21 mM Ni2+ (atomic ratio Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ni 1:3) for 0.5 h. Then, the solution pH was gradually tuned from 7 to 10 in 0.5 h (drastic pH change will cause serious metal oxide precipitation) and pH 10 was maintained for 1 h. TAi-NCs decorated with mixed Fe and Ni (atomic ratio 1:3) (FeNi3@TAi-NCs) were washed and re-dispersed in 1.5 ml deionized water (FeNi3@TAi-NCsaq) as OER electrocatalyst inks. In contrast to TA-SWCNTs, FT-IR spectra of FeNi3@TAi-SWCNTs showed apparent position shifts or intensity changes in the characteristic stretching bands (600–1800 cm−1) due to the interaction between –OH and Fe2+ and Ni2+ ions (Fig. S3†). Furthermore, their electrochemical OER performances were evaluated in a standard three-electrode system in 1.0 M KOHaq (Fig. 5). First, 4 μl of 0.026% FeNi3@TAi-NCsaq was drop-dried on a GCE (catalyst loading 15.1 μg cm−2, details in the Experimental section) serving as a working electrode for the OER CV measurements. After the first CV cycle for the anodic stripping of TAi, FeNi3@TAi-NCs is activated following the FeNi3(OOH)-decorated NCs (FeNi3@NCs); then, the water oxidation for the OER begins at the second CV cycle (Scheme 1). X-ray photoelectron spectroscopy (XPS) was used to investigate the elemental states of FeNi3@TAi-SWCNTs before and after OER activation. In the C 1s spectra of FeNi3@TAi-SWCNTs (Fig. S5a†), the TA characteristic peaks at 287.4, 284.1, and 283.2 eV are attributed to C
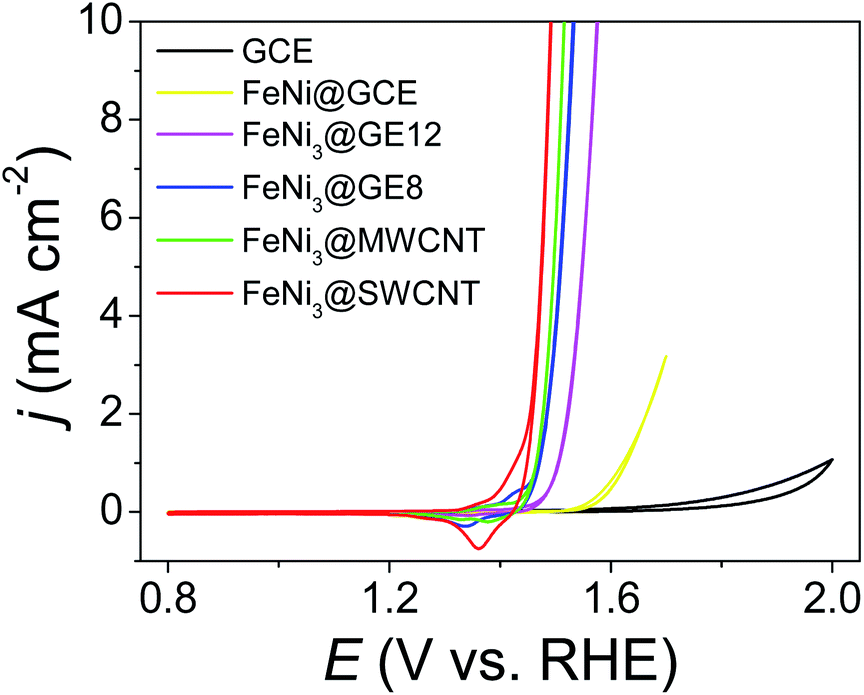
:Ni 1:3) for 0.5 h. Then, the solution pH was gradually tuned from 7 to 10 in 0.5 h (drastic pH change will cause serious metal oxide precipitation) and pH 10 was maintained for 1 h. TAi-NCs decorated with mixed Fe and Ni (atomic ratio 1:3) (FeNi3@TAi-NCs) were washed and re-dispersed in 1.5 ml deionized water (FeNi3@TAi-NCsaq) as OER electrocatalyst inks. In contrast to TA-SWCNTs, FT-IR spectra of FeNi3@TAi-SWCNTs showed apparent position shifts or intensity changes in the characteristic stretching bands (600–1800 cm−1) due to the interaction between –OH and Fe2+ and Ni2+ ions (Fig. S3†). Furthermore, their electrochemical OER performances were evaluated in a standard three-electrode system in 1.0 M KOHaq (Fig. 5). First, 4 μl of 0.026% FeNi3@TAi-NCsaq was drop-dried on a GCE (catalyst loading 15.1 μg cm−2, details in the Experimental section) serving as a working electrode for the OER CV measurements. After the first CV cycle for the anodic stripping of TAi, FeNi3@TAi-NCs is activated following the FeNi3(OOH)-decorated NCs (FeNi3@NCs); then, the water oxidation for the OER begins at the second CV cycle (Scheme 1). X-ray photoelectron spectroscopy (XPS) was used to investigate the elemental states of FeNi3@TAi-SWCNTs before and after OER activation. In the C 1s spectra of FeNi3@TAi-SWCNTs (Fig. S5a†), the TA characteristic peaks at 287.4, 284.1, and 283.2 eV are attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–O, and C–C, respectively.45 After OER activation, the only peak at 284.8 eV assigned to SWCNTs is presented in these spectra. The disappearance of the TA characteristics indicates the dissociation of TA under anodic potential. For the O 1s spectra (Fig. S5b†), the peaks at 533.4 and 532.1 eV of FeNi3@TAi-SWCNTs for the oxygen in hydroxyl and carbonyl of the TA molecule shift to 532.2 and 531.2 eV, respectively, for M–OH after OER activation (FeNi3@SWCNTs). This suggests that M(OOH) forms on NCs during the OER activation. All FeNi3@NCs exhibit outstanding OER performance, improving from that of the FeNi@GCE because of the marked 150–200 mV negative shift in the OER onset potential and sharp increase in the OER current density. Undoubtedly, NCs are good choices for catalyst support. FeNi3@SWCNTs even exhibits superior OER performance to other NCs. In the following, all the results demonstrating SWCNTs to be an adequate catalyst support are discussed.
O, C–O, and C–C, respectively.45 After OER activation, the only peak at 284.8 eV assigned to SWCNTs is presented in these spectra. The disappearance of the TA characteristics indicates the dissociation of TA under anodic potential. For the O 1s spectra (Fig. S5b†), the peaks at 533.4 and 532.1 eV of FeNi3@TAi-SWCNTs for the oxygen in hydroxyl and carbonyl of the TA molecule shift to 532.2 and 531.2 eV, respectively, for M–OH after OER activation (FeNi3@SWCNTs). This suggests that M(OOH) forms on NCs during the OER activation. All FeNi3@NCs exhibit outstanding OER performance, improving from that of the FeNi@GCE because of the marked 150–200 mV negative shift in the OER onset potential and sharp increase in the OER current density. Undoubtedly, NCs are good choices for catalyst support. FeNi3@SWCNTs even exhibits superior OER performance to other NCs. In the following, all the results demonstrating SWCNTs to be an adequate catalyst support are discussed.
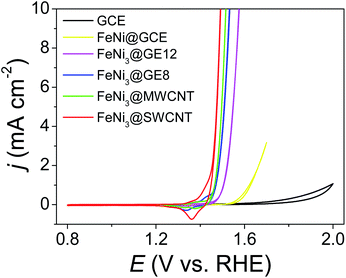 |
| | Fig. 5 CVs of FeNi3@NCs (NCs: SWCNTs, MWCNTs, GE8, and GE12), FeNi(OOH) deposited GCE, and GCE recorded in 1.0 M KOHaq with a scan rate of 5 mV s−1. | |
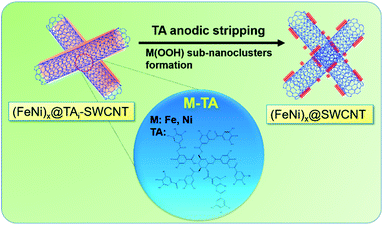 |
| | Scheme 1 (FeNi)x@TAi-SWCNTs is activated to form (FeNi)x@SWCNTs by anodic stripping of TA. | |
OER activity of (FeNi)x@SWCNTs
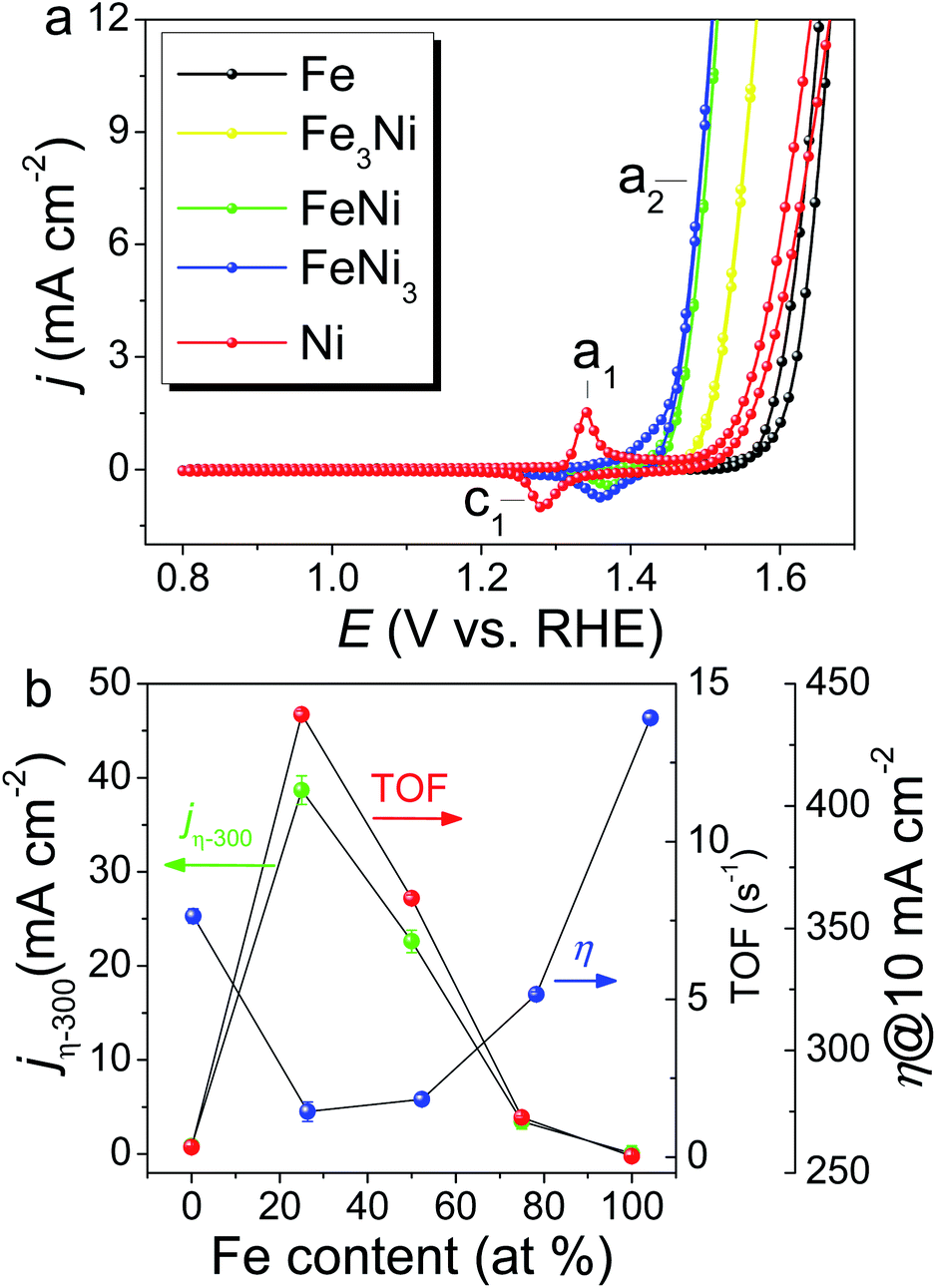
FeNi(OOH) on the SWCNT catalyst ((FeNi)x@SWCNTs) with atomic compositions ranging from 0 to 100 at% Fe was prepared using various stoichiometric Fe/Ni ratios in solution, and the total metal content was kept constant. The metal content of the (FeNi)x@SWCNTs was determined by inductively coupled plasma mass spectroscopy (ICP-MS) as 2.77 wt% on the carbon. Based on a catalyst loading of 15.1 μg cm−2 on the GCE for the OER test, the metal loading was only 0.42 μg cm−2, which is the lowest metal loading record, even three orders of magnitude lower than that of the state-of-the-art OER catalysts.1 Owing to the 1.5 TA molecule layers on the TAi@SWCNTs, one TA molecule can catch ∼2.5 metal ions. In this manner, the final metal loading is minimized to the “pseudo-atomic-scale” on SWCNTs. Additionally, UV-Vis spectra and CVs were used to re-confirm Fe and Ni loading (metal loading) on Fe@SWCNTs and Ni@SWCNTs, respectively. The UV-Vis spectra of TAaq and a mixture of TA and Fe2+ aqueous solution are shown in Fig. S6a.† The absorbance at 550 nm (A550) is dependent on the concentration of the Fe2+–TA complex (Fig. S6b†). The Fe loading on the Fe@SWCNTs was determined by tracking the reduction in A550 after SWCNT adsorption (Fig. S7†). The OER CVs for the (FeNi)x@SWCNT series differ noticeably both in their Ni(OH)2/NiOOH redox characteristics (c1/a1) and OER activities (a2) (Fig. 6a). Owing to the absence of the Ni characteristic UV-Vis signal, the reduction peak, c1, was integrated to determine the Ni content in Ni@SWCNTs.46 The metal loading evaluated from CV is ∼1.2 times that from spectroscopic methods (UV-Vis and ICP-MS). This result is consistent with reports that Ni could exist as γ-NiOOH, in which Ni has an average oxidation state as high as 3.7, and 1.2 electrons are transferred per Ni atom per redox cycle.46 All (FeNi)x@SWCNTs exhibit small Tafel slopes (Fig. S8†). FeNi3@SWCNTs exhibits the lowest Tafel slope of 30.2 mV dec−1, revealing rapid reaction kinetics. Three well-used parameters are defined based on the OER activity: specific current density normalized by the electrochemical surface area (ECSA) according to double-layered capacitances (Cdl) at a constant overpotential (η) of 300 mV (jη−300), η at a specific current density of 10 mA cm−2 (η10), and TOF at a constant η of 300 mV based on the total number of moles of metal (Ni + Fe) determined by ICP-MS. These parameters are plotted as a function of the Fe at% of (FeNi)x@SWCNTs (Fig. 6b). The FeNi3@SWCNTs shows the smallest η10 of 275 mV, which is comparable to the η10 from many outstanding OER electrocatalysts.1 Consistent with earlier reports, a “volcano”-shaped activity trend was demonstrated in jη−300 and TOF, following the Sabatier principle.37 To our surprise, the entire (FeNi)x@SWCNT series exhibits excellent OER catalytic performance. Both TOF and jη−300 surpass the previously reported FeNi OER catalysts (FeNi-OER). The highest OER activity is exhibited by the FeNi3@SWCNTs containing 25% Fe (Fig. 6b), owing to a synergism between Ni and Fe centers.10,47–49 Based on an ultra-low metal loading (0.42 μg cm−2) and high jη−300 = 40 mA cm−2, which is 2–3 times higher than those previously reported for FeNi-OER,1 a remarkably high TOF of 14.03 s−1 is exhibited, which is three orders of magnitude higher than that of FeNi-OER and a 30–70-fold enhancement over FeNi-OER on various carbon supports reported in the literature.1 The mass activity measured at 10 mA cm−2 is 23.8 A mg−1, which is three orders of magnitude higher than those of most of the OER catalysts (1 × 10−3 to 8 × 10−1 A mg−1) (Table S1†). Moreover, the gram-scale production of FeNi3@SWCNTs and long-term stability are demonstrated in Fig. S9.† All the results qualify FeNi3@SWCNTs to be an adequate OER electrocatalyst with both ultra-high activity and good stability.
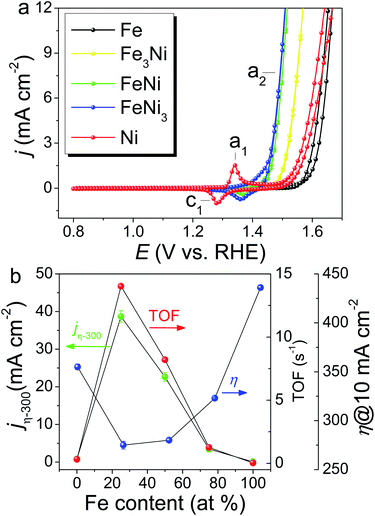 |
| | Fig. 6 (a) OER CVs for the (FeNi)x@SWCNT series recorded in 1.0 M KOH with a scan rate of 10 mV s−1. (b) OER activity of (FeNi)x@SWCNT series, jη−300, η10, and TOF at a constant η of 300 mV based on a metal loading of 0.42 μg cm−2, determined by ICP-MS as a function of Fe at%. | |
Characterization of (FeNi)x@SWCNTs
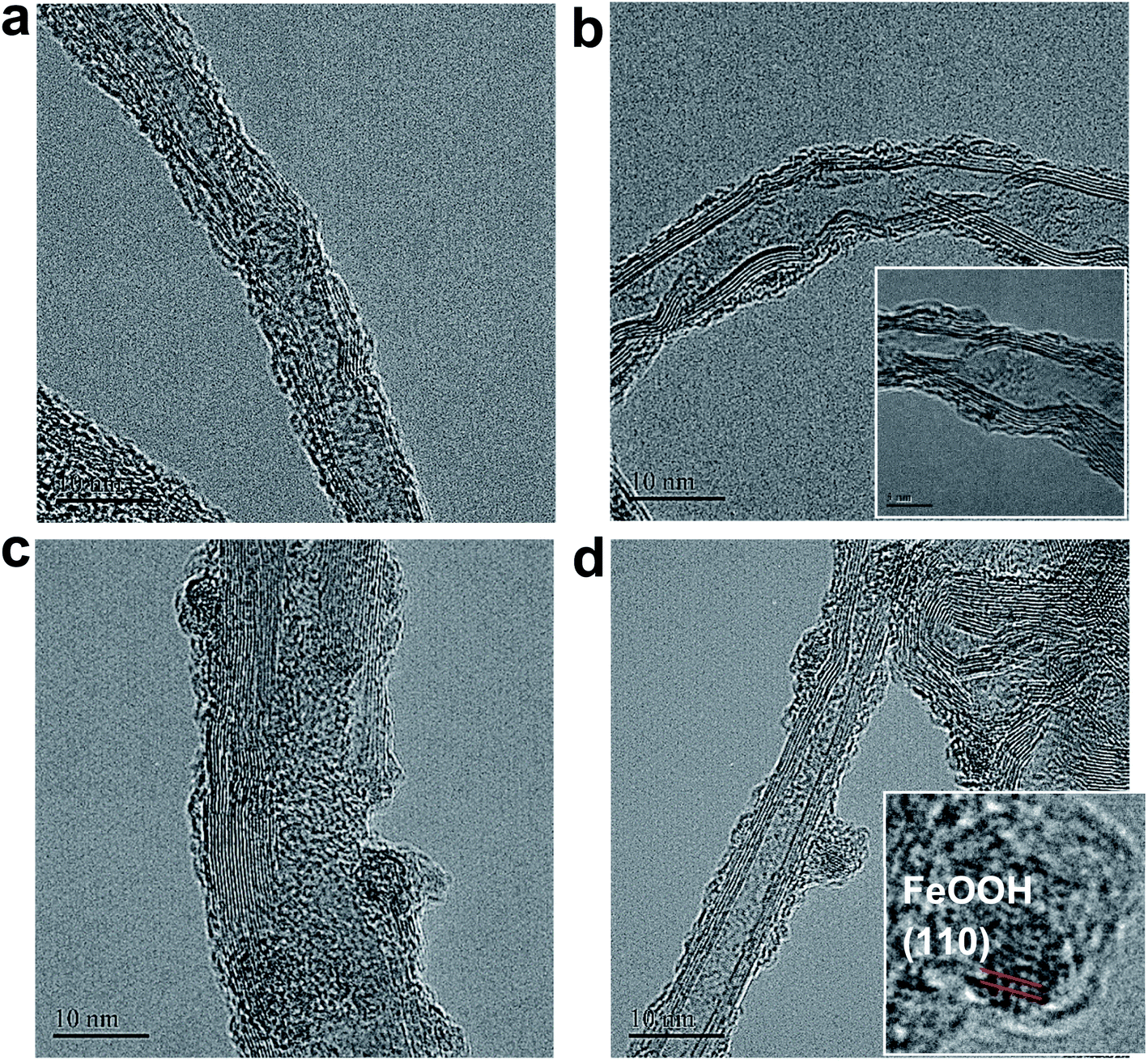
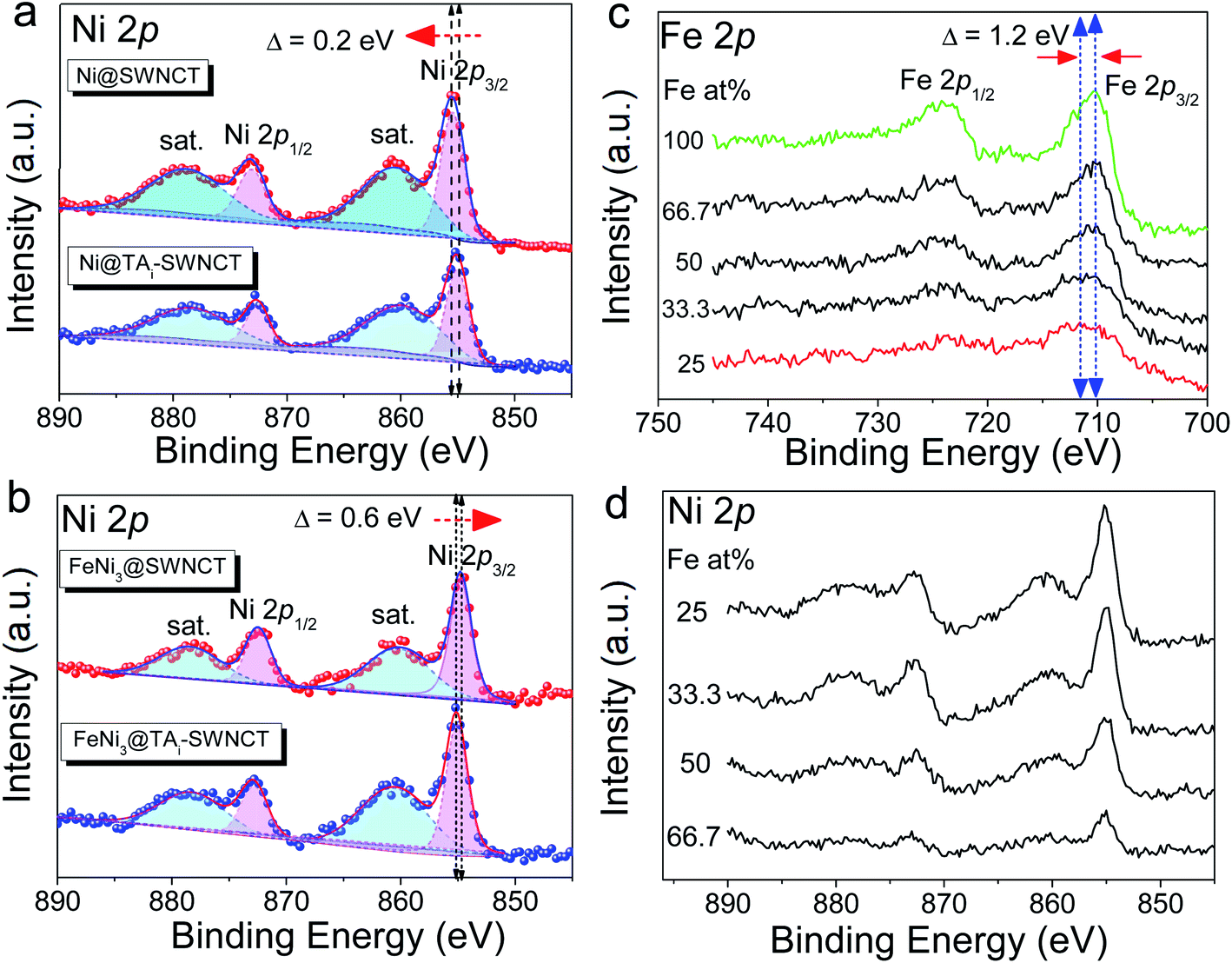
The micro-morphologies of (FeNi)x@SWCNTs were examined by HRTEM. The HRTEM images present two different morphological representations. (FeNi)x@SWCNTs exhibits an amorphous sub-nanometer cluster with a thickness of <1.5 nm, well-dispersed on an SWCNT below 25% Fe (Fig. 7b and S10–S12†). This shows that a homogeneous substitution of <25% Fe cations into NiOOH can maintain the micro-morphology of NiOOH (Fig. 7a) and suggests pseudo-atomic-scale metal loading. Crystalline nanoclusters with a diameter of ∼3 nm appear above 25% Fe (Fig. 7c and d). Lattice fringes with a d-spacing of 0.33 nm shown in the inset of Fig. 7d could be ascribed to the (110) plane of the FeOOH in the nanoclusters, indicating that Fe nucleates as a separate phase above 25% Fe. This could explain why the OER activity did not improve above 25% Fe. In the previous study by Friebel et al.,50 a similar phenomenon as observed based on diverging Fe–O and Ni–O distances by an operando XAS. For the first time, we directly recorded the entire process using micro-morphological images from the atomic-level exposure of active sites in (FeNi)x@SWCNTs. Only the (002) reflections of the carbon support at 24° and a background signal of the Ti sample holder were observed in the X-ray diffraction of (FeNi)x@SWCNTs (Fig. S13†), confirming the amorphous or nanocrystalline characteristics of (FeNi)x@SWCNTs. The surface compositions and chemical states of the (FeNi)x@SWCNTs were determined by XPS. High-resolution Ni 2p spectra of Ni@SWCNTs and FeNi3@SWCNTs (Fig. 8a and b) showed two peaks at 855 and 874.1 eV, corresponding to Ni2+ 2p3/2 and Ni2+ 2p1/2, respectively. For Ni@SWCNTs, the binding energy of Ni2+ 2p3/2 shifted from 855.0 to 856.1 eV after the OER activation energy of Fe 2p3/2 increased from 710 to 712 eV below 25% Fe, indicating a charge transfer from Fe to Ni (Fig. 8c). Above 25% Fe, the binding energy of Fe 2p3/2 returned to that of Fe@SWCNTs, owing to the nucleation of Fe as a separate phase. The Fe phase separation also caused the reduction in the Ni intensity with increase in Fe content (Fig. 8d). The HRTEM images, reinforced by XPS analyses, suggest the model proposed by Friebel et al. in that the active sites in FeNi(OOH) could be Fe cations. The alteration of their electronic properties due to incorporation into γ-NiOOH changes the chemical bonding of these cations with the intermediates involved in the OER, resulting in a lower OER overpotential and, correspondingly, increased OER activity.46
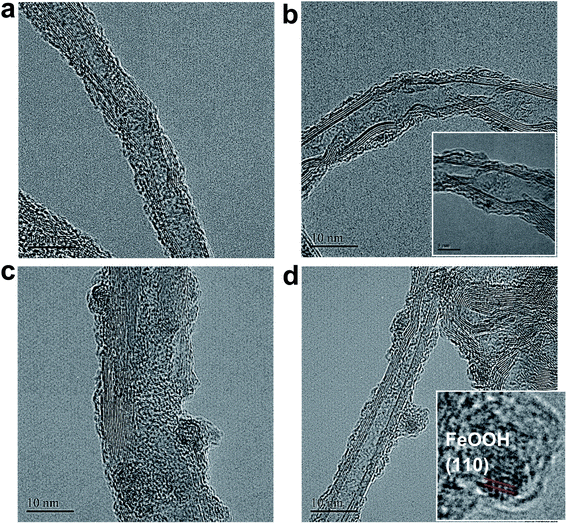 |
| | Fig. 7 HR-TEM images of (a) Ni@SWCNTs, (b) FeNi3@SWCNTs, (c) Fe3Ni@SWCNTs, and (d) Fe@SWCNTs (inset: magnification of (d)). | |
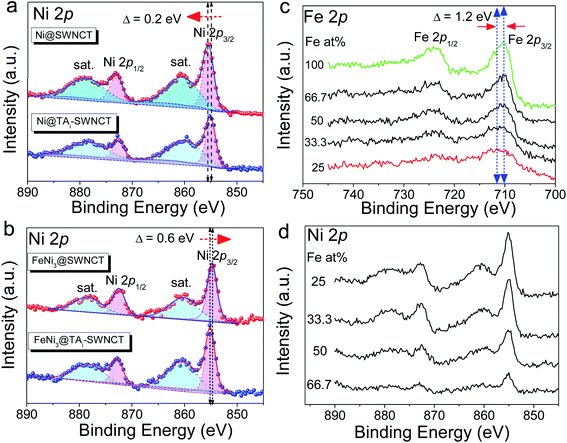 |
| | Fig. 8 High-resolution Ni 2p spectra of (a) Ni@SWCNTs and Ni@TAi-SWCNTs; (b) FeNi3@SWCNTs and FeNi3@TAi-SWCNTs; (c) tracking Fe 2p spectra of (FeNi)x@SWCNTs with Fe at%; (d) tracking Ni 2p spectra of (FeNi)x@SWCNTs with Fe at%. | |
Conclusions
In summary, the bottlenecks—high metal loading and low TOF—of transition-metal-based OER electrocatalysts for large-scale applications have been overcome in this study. Herein, we report on well-dispersed pseudo-atomic-scale M(OOH) nanoclusters on NCs as an earth-abundant OER catalyst with extremely high activity and ultra-low metal loading. As an aside, FeNi3@SWCNT ink loaded in a sprayer can homogeneously spray catalysts onto any conducting substrates for OER application (Fig. S14†). The gram-scale production strategy based on the pH-controllable TA molecule monolayer functionalizes the NC surface. The new strategy makes it possible to fine-tune the chemical and electronic properties of the NC surface and, hence, modulate its catalytic activity. It not only provides a new avenue to engineer efficient, low-cost materials for the OER but also could extend it to the design of other types of catalytic materials.
Experimental
Chemicals
SWCNTs (purity > 90%, ∼3 nm in outer diameter, <20 μm in length, special surface area = 400–600 m2 g−1), MWCNTs (purity > 95%, 20–40 nm in outer diameter, 5–15 μm in length, specific surface area = 40–300 m2 g−1), GE8 (8 nm), and GE12 (12 nm) used in this work were purchased from Uni-onward Corp. Tannic acid (TA), nickel(II) chloride hexahydrate (NiCl2·6H2O), and iron(II) chloride hexahydrate (FeCl2·6H2O) were purchased from Sigma-Aldrich. All chemicals are analytical grade and used as received without further purification.
Preparation of (FeNi)x@TAi-NC OER electrocatalyst inks
The NC (1.8 mg) including SWCNTs, MWCNTs, GE8, and GE12 was added into 1.5 ml of 0.71 mM TA aqueous solution and sonicated (50 W, 20 kHz) for 30 min. A black (concentration 0.12 wt%, pH 6.8), homogeneous ink-like TA-NC suspension was obtained. Then, 0.3 ml TA-NC suspension was mixed with 0.7 ml aqueous solution containing 0.28 mM Fe2+ and Ni2+ (Fe/Ni mole ratio in solution was 1:0, 3:1, 1:1, 1:3, and 0:1, respectively) and stirred for 20 min. Then, the solution pH was gradually tuned from 7 to 10 in 0.5 hours with a 2.0 mM NaOH aqueous solution (drastic pH change will cause serious metal oxide precipitation), and pH 10 was maintained for 1 h. Mixed Fe- and Ni-decorated TAi-NCs, (FeNi)x@TAi-NCs, were washed and re-dispersed in 1.5 ml deionized water as OER electrocatalyst inks ((FeNi)x@TAi-NCsaq).
Characterization
X-ray photoelectron spectroscopy (XPS) and energy-dispersive X-ray spectroscopy (EDS) were used for the surface-element analysis of (FeNi)x@TAi-NCs. The XPS data were acquired using the ULVAC-PHI, PHI5000 VersaProbe/scanning ECSA microprobe. For X-ray diffraction (XRD) measurements (Cu Kα radiation, λ = 1.54 Å), Ti foil (10 × 10 mm) was used as a sample holder for the (FeNi)x@NC OER catalysts. Inductively coupled plasma mass spectrometry (ICP-MS) was used to determine metal loading on the catalysts via an Agilent 7500ce instrument. Prior to the ICP measurement, 20 mg samples were incubated overnight in 2 ml newly prepared aqua regia. Then, plenty of deionized water was added to dilute the aqua regia and carefully boiled to drive HCl away from the solution. After re-adding water and boiling several times, the remaining solution was carefully collected and diluted to a suitable concentration for ICP measurement. Ultraviolet visible (UV-Vis) absorption and cyclic voltammograms (CVs) were used to re-confirm Fe and Ni loading (metal loading) onto Fe@SWCNTs and Ni@SWCNTs, respectively. All UV-Vis spectra were measured using a GBC Cintra 2020 spectrophotometer at 28 °C. In the UV-Vis spectra, the absorbance at 550 nm (A550) is dependent on the concentration of the Fe2+–TA complex. The Fe loading onto Fe@SWCNTs was determined by tracking the reduction in A550 after SWCNT adsorption (ESI†). Owing to the absence of the Ni characteristic UV-Vis signal, the Ni(OH)2/NiOOH redox characteristics (c1/a1) shown in the OER CVs, the reduction peak, c1, was integrated to determine the Ni content in the Ni@SWCNTs. The metal loading evaluated from CV was ∼1.2 times that obtained from spectroscopic methods (UV-Vis and ICP-MS). The micro-morphological analysis of (FeNi)x@NCs was performed with a JEOL JEM-1400 transmission electron microscope (TEM) and a JEOL JEM-2100F field-emission TEM (FE-TEM). The high-resolution TEM (HRTEM) images were obtained with the FE-TEM. Nitrogen adsorption and desorption studies were performed at 77 K on a Micromeritics TriStar 3000 adsorption apparatus. The specific surface area was measured using the Brunauer–Emmett–Teller (BET) method. All UV-Vis spectra were measured using a GBC Cintra 2020 spectrophotometer at room temperature.
Electrochemical experiments
The electrochemical experiments were performed using a CHI 760C potentiostat/galvanostat and a three-electrode electrochemical cell. Hg/HgO and a Pt wire were used as the reference electrode and counter electrode, respectively. In the procedure, 4 μl of 0.026% (FeNi)x@TAi-NCsaq (the metal content of the (FeNi)x@SWCNTs was determined by ICP-MS as 2.77 wt%) was drop-dried on a GCE (0.07 cm2) as a working electrode (metal loading is 0.42 μg cm−2). Then, 5.0 ml of 1.0 M KOH was used as an electrolyte for electrochemical measurements and degassed by bubbling Ar for 5 min before the measurements. The potentials were measured against the reference electrode and converted to the reversible hydrogen electrode (RHE) reference scale using ERHE = EHg/HgO + 0.098 + 0.059pHelectrolyte (for 1.0 M KOH (pH 14), ERHE = EHg/HgO + 0.926). By plotting overpotential η against log(J) from linear scan voltammetry (LSV) curves (or the CV recorded in anodic scanning), Tafel slopes can be obtained. To test the stability of the (FeNi)x@NCs OER electrocatalysts, a galvanostatic measurement at a fixed current density (J) of 10 mA cm−2 was performed. ECSAs were measured by CV at the potential window 0.0–0.15 V versus Hg/HgO, with different scan rates of 20, 40, 60, 80, 100, and 120 mV s−1. By plotting the ΔJ = (Ja − Jc)/2 at 0.08 V versus Hg/HgO against the scan rate, the linear slope that is twice the double-layer capacitance (Cdl) is used to represent the ECSA.
Turnover frequency
The TOF of (FeNi)x@NC OER electrocatalysts was calculated according to the following equation:
where j is the current density at a given overpotential, e.g. η = 300 mV, A is the surface area of the electrode, F is the Faraday constant, and m is the number of moles of the metal on the electrodes. In this study, we assumed that all the metal sites were actively involved in the OER.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Ministry of Science and Technology of the Republic of China, Taiwan.
Notes and references
- J. Kibsgaard and I. Chorkendorff, Nat. Energy, 2019, 4, 430–433 CrossRef.
- J. B. Sambur, T. Y. Chen, E. Choudhary, G. Q. Chen, E. J. Nissen, E. M. Thomas, N. M. Zou and P. Chen, Nature, 2016, 530, 77–80 CrossRef CAS PubMed.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. X. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, Science, 2016, 353, 1011–1014 CrossRef CAS PubMed.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- J. F. Huang and H. Y. Chen, Angew. Chem., Int. Ed., 2012, 51, 1684–1688 CrossRef CAS PubMed.
- J. F. Huang and P. K. Tseng, Chem. Sci., 2018, 9, 6134–6142 RSC.
- C. C. L. McCrory, S. H. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- M. Gong, Y. G. Li, H. L. Wang, Y. Y. Liang, J. Z. Wu, J. G. Zhou, J. Wang, T. Regier, F. Wei and H. J. Dai, J. Am. Chem. Soc., 2013, 135, 8452–8455 CrossRef CAS PubMed.
- W. Ma, R. Z. Ma, C. X. Wang, J. B. Liang, X. H. Liu, K. C. Zhou and T. Sasaki, ACS Nano, 2015, 9, 1977–1984 CrossRef CAS PubMed.
- K. Fan, H. Chen, Y. F. Ji, H. Huang, P. M. Claesson, Q. Daniel, B. Philippe, H. Rensmo, F. S. Li, Y. Luo and L. C. Sun, Nat. Commun., 2016, 7, 9 Search PubMed.
- J. X. Feng, H. Xu, Y. T. Dong, S. H. Ye, Y. X. Tong and G. R. Li, Angew. Chem., Int. Ed., 2016, 55, 3694–3698 CrossRef CAS PubMed.
- B. Zhang, X. L. Zheng, O. Voznyy, R. Comin, M. Bajdich, M. Garcia-Melchor, L. L. Han, J. X. Xu, M. Liu, L. R. Zheng, F. P. G. de Arquer, C. T. Dinh, F. J. Fan, M. J. Yuan, E. Yassitepe, N. Chen, T. Regier, P. F. Liu, Y. H. Li, P. De Luna, A. Janmohamed, H. L. L. Xin, H. G. Yang, A. Vojvodic and E. H. Sargent, Science, 2016, 352, 333–337 CrossRef CAS PubMed.
- J. J. Duan, S. Chen and C. Zhao, Nat. Commun., 2017, 8, 7 CrossRef PubMed.
- K. Fan, Y. F. Ji, H. Y. Zou, J. F. Zhang, B. C. Zhu, H. Chen, Q. Daniel, Y. Luo, J. G. Yu and L. C. Sun, Angew. Chem., Int. Ed., 2017, 56, 3289–3293 CrossRef CAS PubMed.
- Z. H. Xiao, Y. Wang, Y. C. Huang, Z. X. Wei, C. L. Dong, J. M. Ma, S. H. Shen, Y. F. Li and S. Y. Wang, Energy Environ. Sci., 2017, 10, 2563–2569 RSC.
- Y. Xu, W. G. Tu, B. W. Zhang, S. M. Yin, Y. Z. Huang, M. Kraft and R. Xu, Adv. Mater., 2017, 29, 9 Search PubMed.
- C. Q. Dong, T. Y. Kou, H. Gao, Z. Q. Peng and Z. H. Zhang, Adv. Energy Mater., 2018, 8, 9 Search PubMed.
- C. Gu, S. J. Hu, X. S. Zheng, M. R. Gao, Y. R. Zheng, L. Shi, Q. Gao, X. Zheng, W. S. Chu, H. B. Yao, J. F. Zhu and S. H. Yu, Angew. Chem., Int. Ed., 2018, 57, 4020–4024 CrossRef CAS PubMed.
- J. Jiang, F. F. Sun, S. Zhou, W. Hu, H. Zhang, J. C. Dong, Z. Jiang, J. J. Zhao, J. F. Li, W. S. Yan and M. Wang, Nat. Commun., 2018, 9, 12 CrossRef PubMed.
- J. M. V. Nsanzimana, Y. C. Peng, Y. Y. Xu, L. Thia, C. Wang, B. Y. Xia and X. Wang, Adv. Energy Mater., 2018, 8, 7 Search PubMed.
- M. W. Xie, X. L. Xiong, L. Yang, X. F. Shi, A. M. Asiri and X. P. Sun, Chem. Commun., 2018, 54, 2300–2303 RSC.
- F. Yu, H. Q. Zhou, Y. F. Huang, J. Y. Sun, F. Qin, J. M. Bao, W. A. Goddardiii, S. Chen and Z. F. Ren, Nat. Commun., 2018, 9, 9 CrossRef PubMed.
- J. F. Zhang, J. Y. Liu, L. F. Xi, Y. F. Yu, N. Chen, S. H. Sun, W. C. Wang, K. M. Lange and B. Zhang, J. Am. Chem. Soc., 2018, 140, 3876–3879 CrossRef CAS PubMed.
- Z. Cai, L. D. Li, Y. W. Zhang, Z. Yang, J. Yang, Y. J. Guo and L. Guo, Angew. Chem., Int. Ed., 2019, 58, 4189–4194 CrossRef CAS PubMed.
- S. Sultan, M. Ha, D. Y. Kim, J. N. Tiwari, C. W. Myung, A. Meena, T. J. Shin, K. H. Chae and K. S. Kim, Nat. Commun., 2019, 10, 9 CrossRef PubMed.
- L. Yu, Q. Zhu, S. W. Song, B. McElhenny, D. Z. Wang, C. Z. Wu, Z. J. Qin, J. M. Bao, Y. Yu, S. Chen and Z. F. Ren, Nat. Commun., 2019, 10, 10 CrossRef PubMed.
- J. Wang, H. X. Zhong, Z. L. Wang, F. L. Meng and X. B. Zhang, ACS Nano, 2016, 10, 2342–2348 CrossRef CAS PubMed.
- R. Bandyopadhyaya, E. Nativ-Roth, O. Regev and R. Yerushalmi-Rozen, Nano Lett., 2002, 2, 25–28 CrossRef CAS.
- D. H. Lin, N. Liu, K. Yang, L. Z. Zhu, Y. Xu and B. S. Xing, Carbon, 2009, 47, 2875–2882 CrossRef CAS.
- D. H. Lin and B. S. Xing, Environ. Sci. Technol., 2008, 42, 5917–5923 CrossRef CAS PubMed.
- H. Li, C. X. Wei, D. Zhang and B. Pan, Sci. Total Environ., 2019, 655, 807–813 CrossRef CAS PubMed.
- N. Li, D. K. Bediako, R. G. Hadt, D. Hayes, T. J. Kempa, F. von Cube, D. C. Bell, L. X. Chen and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1486–1491 CrossRef CAS PubMed.
- M. Al-Mamun, X. T. Su, H. M. Zhang, H. J. Yin, P. R. Liu, H. G. Yang, D. Wang, Z. Y. Tang, Y. Wang and H. J. Zhao, Small, 2016, 12, 2866–2871 CrossRef CAS PubMed.
- Y. Feng, X. Y. Yu and U. Paik, Sci. Rep., 2016, 6, 8 CrossRef PubMed.
- M. Gorlin, J. F. de Araujo, H. Schmies, D. Bernsmeier, S. Dresp, M. Gliech, Z. Jusys, P. Chernev, R. Kraehnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2017, 139, 2070–2082 CrossRef PubMed.
- X. Y. Lu and C. A. Zhao, Nat. Commun., 2015, 6, 7 Search PubMed.
- Z. Y. Lu, W. W. Xu, W. Zhu, Q. Yang, X. D. Lei, J. F. Liu, Y. P. Li, X. M. Sun and X. Duan, Chem. Commun., 2014, 50, 6479–6482 RSC.
- S. Sultan, M. Ha, D. Y. Kim, J. N. Tiwari, C. W. Myung, A. Meena, T. J. Shin, K. H. Chae and K. S. Kim, Nat. Commun., 2019, 10, 9 CrossRef PubMed.
- H. T. Wang, H. W. Lee, Y. Deng, Z. Y. Lu, P. C. Hsu, Y. Y. Liu, D. C. Lin and Y. Cui, Nat. Commun., 2015, 6, 8 Search PubMed.
- Y. Y. Wang, Y. Q. Zhang, Z. J. Liu, C. Xie, S. Feng, D. D. Liu, M. F. Shao and S. Y. Wang, Angew. Chem., Int. Ed., 2017, 56, 5867–5871 CrossRef CAS PubMed.
- C. Zhang, M. F. Shao, L. Zhou, Z. H. Li, K. M. Xiao and M. Wei, ACS Appl. Mater. Interfaces, 2016, 8, 33697–33703 CrossRef CAS PubMed.
- L. H. Zhuang, L. Ge, Y. S. Yang, M. R. Li, Y. Jia, X. D. Yao and Z. H. Zhu, Adv. Mater., 2017, 29, 7 Search PubMed.
- A. Chilkoti, B. D. Ratner and D. Briggs, Chem. Mater., 1991, 3, 51–61 CrossRef CAS.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS PubMed.
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, J. Am. Chem. Soc., 2012, 134, 17253–17261 CrossRef CAS PubMed.
- K. Fominykh, P. Chernev, I. Zaharieva, J. Sicklinger, G. Stefanic, M. Doblinger, A. Muller, A. Pokharel, S. Bocklein, C. Scheu, T. Bein and D. Fattakhova-Rohlfing, ACS Nano, 2015, 9, 5180–5188 CrossRef CAS PubMed.
- J. Landon, E. Demeter, N. Inoglu, C. Keturakis, I. E. Wachs, R. Vasic, A. I. Frenkel and J. R. Kitchin, ACS Catal., 2012, 2, 1793–1801 CrossRef CAS.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M. J. Cheng, D. Sokaras, T. C. Weng, R. Alonso-Mori, R. C. Davis, J. R. Bargar, J. K. Norskov, A. Nilsson and A. T. Bell, J. Am. Chem. Soc., 2015, 137, 1305–1313 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The area of a single molecule of TA; coverage of TA on GCE (one monolayer); coverage of TA on SWCNTs (one monolayer); pH-controllable TA molecule monolayer on the nano-carbon (NC); UV-Vis of the Fe2+–TA complex determines the Fe loading on the Fe@SWCNTs; Fig. S1–S16; Table S1. See DOI: 10.1039/d0sc01348j |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Wei-Zhe
Xie
* and
Wei-Zhe
Xie