
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrafast amidation of esters using lithium amides under aerobic ambient temperature conditions in sustainable solvents†‡
Michael
Fairley
a,
Leonie J.
Bole
b,
Florian F.
Mulks
 ab,
Laura
Main
a,
Alan R.
Kennedy
a,
Charles T.
O'Hara
a,
Joaquín
García-Alvarez
*c and
Eva
Hevia
*ab
ab,
Laura
Main
a,
Alan R.
Kennedy
a,
Charles T.
O'Hara
a,
Joaquín
García-Alvarez
*c and
Eva
Hevia
*ab
aDepartment of Pure and Applied Chemistry, University of Strathclyde Glasgow, G1 1XL, UK
bDepartment für Chemie und Biochemie, Universität Bern, CH3012, Bern, Switzerland. E-mail: eva.hevia@dcb.unibe.ch
cLaboratorio de Compuestos Organometálicos y Catálisis (Unidad Asociada al CSIC), Centro de Innovación en Química Avanzada (ORFEO-CINQA), Departamento de Química Orgánica e Inorgánica (IUQOEM), Facultad de Química, Universidad de Oviedo, E-33071, Oviedo, Spain
First published on 30th April 2020
Abstract
Lithium amides constitute one of the most commonly used classes of reagents in synthetic chemistry. However, despite having many applications, their use is handicapped by the requirement of low temperatures, in order to control their reactivity, as well as the need for dry organic solvents and protective inert atmosphere protocols to prevent their fast decomposition. Advancing the development of air- and moisture-compatible polar organometallic chemistry, the chemoselective and ultrafast amidation of esters mediated by lithium amides is reported. Establishing a novel sustainable access to carboxamides, this has been accomplished via direct C–O bond cleavage of a range of esters using glycerol or 2-MeTHF as a solvent, in air. High yields and good selectivity are observed while operating at ambient temperature, without the need for transition-metal mediation, and the protocol extends to transamidation processes. Pre-coordination of the organic substrate to the reactive lithium amide as a key step in the amidation processes has been assessed, enabling the structural elucidation of the coordination adduct [{Li(NPh2)(O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CPh(NMe2))}2] (8) when toluene is employed as a solvent. No evidence for formation of a complex of this type has been found when using donor THF as a solvent. Structural and spectroscopic insights into the constitution of selected lithium amides in 2-MeTHF are provided that support the involvement of small kinetically activated aggregates that can react rapidly with the organic substrates, favouring the C–O bond cleavage/C–N bond formation processes over competing hydrolysis/degradation of the lithium amides by moisture or air.
CPh(NMe2))}2] (8) when toluene is employed as a solvent. No evidence for formation of a complex of this type has been found when using donor THF as a solvent. Structural and spectroscopic insights into the constitution of selected lithium amides in 2-MeTHF are provided that support the involvement of small kinetically activated aggregates that can react rapidly with the organic substrates, favouring the C–O bond cleavage/C–N bond formation processes over competing hydrolysis/degradation of the lithium amides by moisture or air.
Introduction
Amide forming reactions are some of the most commonly found reactions in pharmaceutical processes.1 Due to this prevalence and their pivotal role in synthetic processes,2 the investigation of a sustainable method for their formation has been identified by the ACS Green Chemistry Institute as a key research area for the future of pharmaceuticals.3 There is a large variety of elaborate amide bond forming reactions.4,5 While, conceptually, the simplest way of preparing amides should be the direct condensation of carboxylic acids and amines, this amidation process requires extremely harsh reaction conditions (T > 100 °C) in order to avoid the formation of unreactive carboxylate ammonium salts.4b,m–o Thus several alternative approaches have been developed for the construction of amide bonds, including the pre-activation of a carboxylic acid partner to promote the coupling with the relevant amine which allows for the use of milder reaction conditions.5 Examples of this approach include the use of more reactive acyl chlorides, carboxylic acid anhydrides, or other highly activated esters/amides, replacing the OH group with a better leaving group.5 This strategy has been successfully employed for peptide synthesis,6 but it suffers from low atom economy.5,7 Transition-metal catalysed transformations have also been reported to facilitate amidation processes, although in many occasions the use of volatile organic solvents, which are regarded as volatile organic compounds (VOCs) under a protective inert atmosphere (N2 or Ar) is required.8Breaking new ground in this evolving field, Szostak has reported an effective procedure using the utility amide LiHMDS (HMDS = 1,1,1,3,3,3-hexamethyldisilazide) to mediate the amidation of esters and amides, at room temperature, by simply activating the amine component. However, reactions need to be carried out in toluene under inert atmosphere conditions, while using excess lithium amide and the relevant amine (Scheme 1).4j,k,9
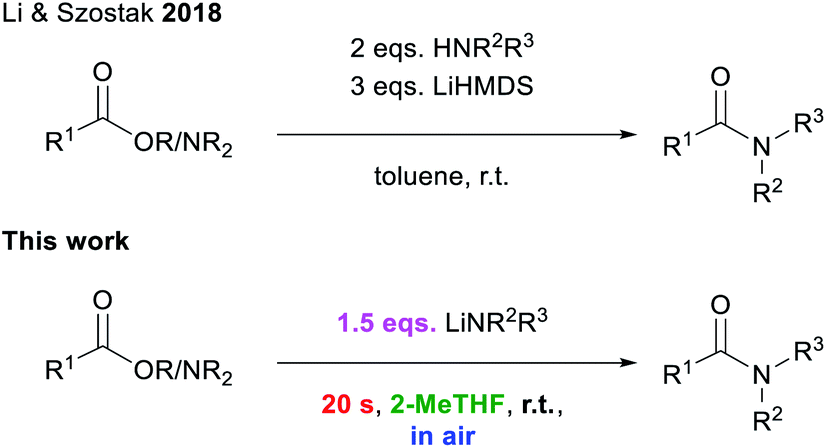 | ||
| Scheme 1 State of the art for transition-metal free amidation procedures.4j,k | ||
In the effort towards more sustainable, safer chemical transformations, our group has been one of the pioneers in using organolithium reagents in non-conventional renewable solvents such as glycerol (Gly) and Deep Eutectic Solvents (DESs), which combine the non-toxic ammonium salt choline chloride (ChCl, as a hydrogen-bond acceptor) with glycerol or water (as a sustainable hydrogen bond-donor).10 Under these conditions not only can higher yields and greater chemoselectivites be accomplished than those obtained using toxic organic solvents, but reactions can also be performed at ambient temperature, in the presence of moisture and air, a trio of conditions normally incompatible with polar organometallic chemistry.10,11 Thus, we have applied fruitfully this sustainable approach for the chemoselective and ultrafast alkylation and arylation of ketones,11a imines11b and nitriles11c as well as the anionic polymerisation of different styrenes.11d,12 Taking a step forward towards developing sustainable, mechanistically well-supported aerobic polar organometallic chemistry, here we report our findings on the mechanism and application of lithium-mediated amidation and transamidation reactions of ethyl esters and an N-Boc-substituted benzamide in air using the biomass derived solvents glycerol and 2-methyltetrahydrofuran (2-MeTHF, Scheme 1).13,14
Solvents are estimated to make up 56% to 85% of the mass involved in pharmaceutical processes.15 Therefore, solvent choice is critical when considering the development of more environmentally benign processes. Biomass-derived solvents such as glycerol (Gly) and 2-MeTHF have emerged as greener alternatives to volatile organic compounds (VOCs) in organic synthesis.16 Thus, for example, 2-MeTHF can be renewably produced from furfural without the use of petrochemicals and shares many of the same properties that make THF a widely used solvent in organic synthesis.14,17 Furthermore, its immiscibility with water enables liquid extractions without the need for toxic classical organic solvents for extracting reaction products.
Results and discussion
Bearing all these precedents in mind, we started our studies on the sustainable formation of aromatic amides by selecting, as a model process, the reaction of ethyl benzoate 1a with lithium N-methylanilide (2a), at room temperature and in the presence of air (Table 1).|
|
|||
|---|---|---|---|
| Entry | Solvent | LiNMePhb [eq.] | Yieldc [%] |
| a Reactions performed in air at ambient temperature using 1 g of solvent and 1 mmol of ester. Reactions stirred for 20 s, then quenched with sat. Rochelle's salt soln. (5 mL). b Lithium amide was added as a 1 M soln. in 2-MeTHF. c Isolated yields are given. d Lithium amide 2a was added as a 0.2 M soln. in THF. e Reactions carried out at 53 °C due to viscosity issues. | |||
| 1 | THFd | 2 | 93 |
| 2 | 2-MeTHF | 3 | 80 |
| 3 | 2 | 81 | |
| 4 | 1.5 | 80 | |
| 5 | 1 | 78 | |
| 6 | 1ChCl/2Gly | 3 | 83 |
| 7 | 1ChCl/2EG | 3 | 59 |
| 8 | 1ChCl/2H2O | 3 | 81 |
| 9 | 1LiCl/3Glye | 3 | 79 |
| 10 | H2O | 3 | 36 |
| 11 | Gly | 3 | 85 |
| 12 | 1.5 | 79 | |
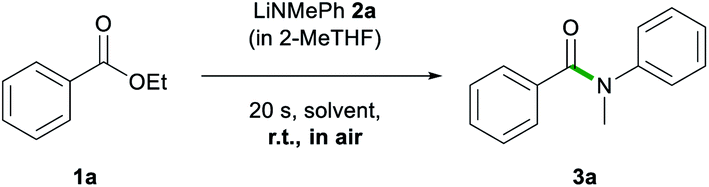
As a control experiment, we first employed THF as the reaction solvent at room temperature, using two equivalents of 2a, which furnished N-methyl-N-phenylbenzamide 3a in excellent yield (93%, Table 1) in as little as 20 s while working under aerobic conditions. We next compared these results by replacing THF with a more sustainable reaction medium, which included biomass-derived 2-MeTHF (entries 2–5) and glycerol (entry 11) as well as several DES combinations (entries 6–9) and water (entry 10). As 2a is a solid, in all cases it was employed as a 1 M solution in 2-MeTHF. Using neat 2-MeTHF as a reaction medium, at room temperature, in air allowed for the isolation of 3a in high yields (80%) upon adding 1.5 equivalents of the lithium amide.18 Switching to the eutectic mixture 1ChCl/2Gly, similar yields were obtained although a three molar excess of 2a was required (83%, entry 6). Good to excellent yields were also obtained employing eutectic mixtures containing other H-bond donors, including ethylene glycol (EG) and water (59% and 81%, entries 7 and 8, respectively). Comparable yields were also obtained when ChCl in the glycerol-based DES was replaced by lithium chloride (79%, entry 9), although, due to the greater viscosity of this mixture, heating was required to ensure adequate stirring. Moving to water as a polar medium, the reaction still proceeded, albeit, with a significantly reduced yield (36%, entry 10) presumably due to an increased rate of hydrolysis of the lithium amide. Interestingly, and as we have previously observed in the addition of organolithium reagents to aromatic nitriles,11c glycerol gave equal or better yields than the eutectic mixtures in the amidation of esters, enabling similar conversions to 3a to those observed for 2-MeTHF when using just 1.5 equivalents of 2a.
Encouraged by these initial findings that show for the first time the potential of lithium amides to enable ester amidations in the presence of air and moisture, which are generally antagonists of these reagents, we next explored the kinetic stability of LiNMePh in Gly and 2-MeTHF.
These two sustainable solvent systems were chosen on the basis of their similar performances when using just 1.5 equivalents of 2a (Table 1). For these studies the order of addition of reagents was reversed; thus, 2a (1.5 eq. of a 1 M solution in 2-MeTHF) was added to the relevant solvent system and allowed to stir in air for a set period of time before introducing ethyl benzoate 1a (Table 2, see ESI for details‡). Remarkably, while only traces of 3a were observed after 30 seconds when Gly was employed (entry 2, Table 2),19 in 2-MeTHF, after 1 minute, 3a forms in a 70% yield (entry 3) which is comparable to that observed when the order of addition of reagents is reversed (80%, entry 4, Table 1). Highlighting the impressive kinetic stability of 2a in 2-MeTHF against decomposition in the presence of air and moisture, it was only after 10 minutes that formation of 3a was almost completely suppressed (13% yield, entry 6).
|
|
|||
|---|---|---|---|
| Entry | Solvent | Time | Yield [%] |
| a Reactions performed in air at ambient temperature using 1 g of solvent and 1 mmol of 1a. Lithium amide 2a was added as a 1 M soln. in 2-MeTHF and stirred for the specified time before addition of 1a. Reactions stirred for another 20 s, then quenched with sat. Rochelle's salt soln. (5 mL). Isolated yields are given. | |||
| 1 | Gly | 10 s | 32 |
| 2 | 30 s | 3 | |
| 3 | 2-MeTHF | 1 min | 70 |
| 4 | 2 min | 62 | |
| 5 | 5 min | 42 | |
| 6 | 10 min | 13 | |
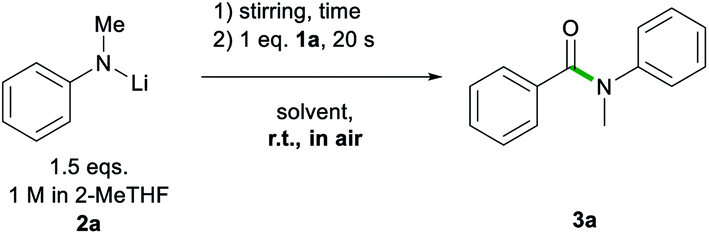
Another advantage of 2-MeTHF over Gly was witnessed on dispensing 2a as a solid rather than as a 2-MeTHF solution (Scheme 2). While the reaction using 2-MeTHF ran smoothly, with reagents quickly mixing into 2-MeTHF to give 3a in 82% yield with no significant difference to that when adding the lithium amide as a solution (entry 4, Table 1), when using neat Gly, partial decomposition of 2a occurred, affording 3a in a diminished 47% yield. Furthermore, the high solubility of 2a in 2-MeTHF allowed the reaction to be scaled up to a 10 mmol scale using just 6 mL of a 2.5 M solution of 2a, yielding 3a in an 89% yield (see ESI for details‡).
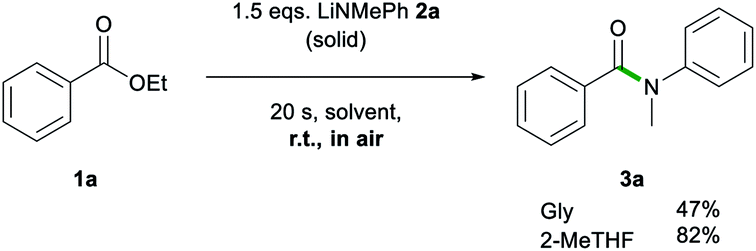 | ||
| Scheme 2 Addition of solid LiNMePh 2a to a solution of ethyl benzoate 1a in Gly and 2-MeTHF. | ||
Building on these results, along with the fact that 2-MeTHF can also be used as the extraction solvent for the isolation of 3a, we chose this solvent to carry out our remaining studies.
Firstly, the feasibility of the in situ formation of the lithium amides through addition of n-butyllithium to the reaction mixture containing ester 1a and the relevant amine was assessed (Scheme 3), encouraged by previous work by Pace that has shown the suitability of 2-MeTHF for the preparation and manipulation of lithium amides.13c,13d
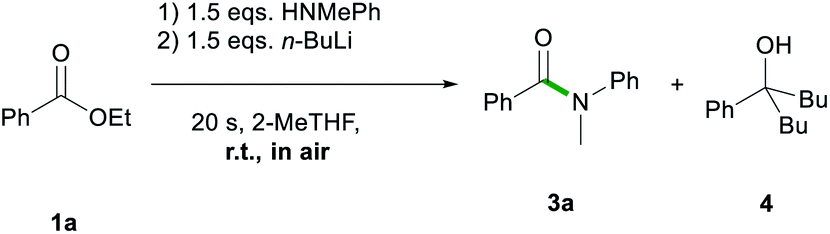 | ||
| Scheme 3 In situ formation of 2a and 2f and their subsequent reactions with 1a in 2-MeTHF. | ||
While full conversion of starting material 1a was observed, the amidation reaction did not take place selectively; thus, 3a was obtained in a 74% yield along with 26% of the double addition product 5-phenylnonan-5-ol (4) (Scheme 3).
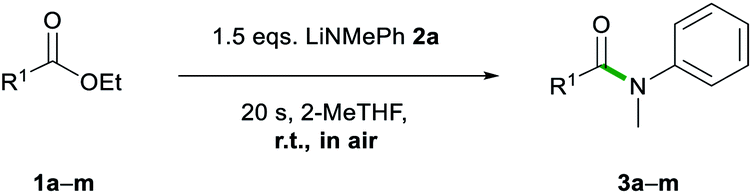
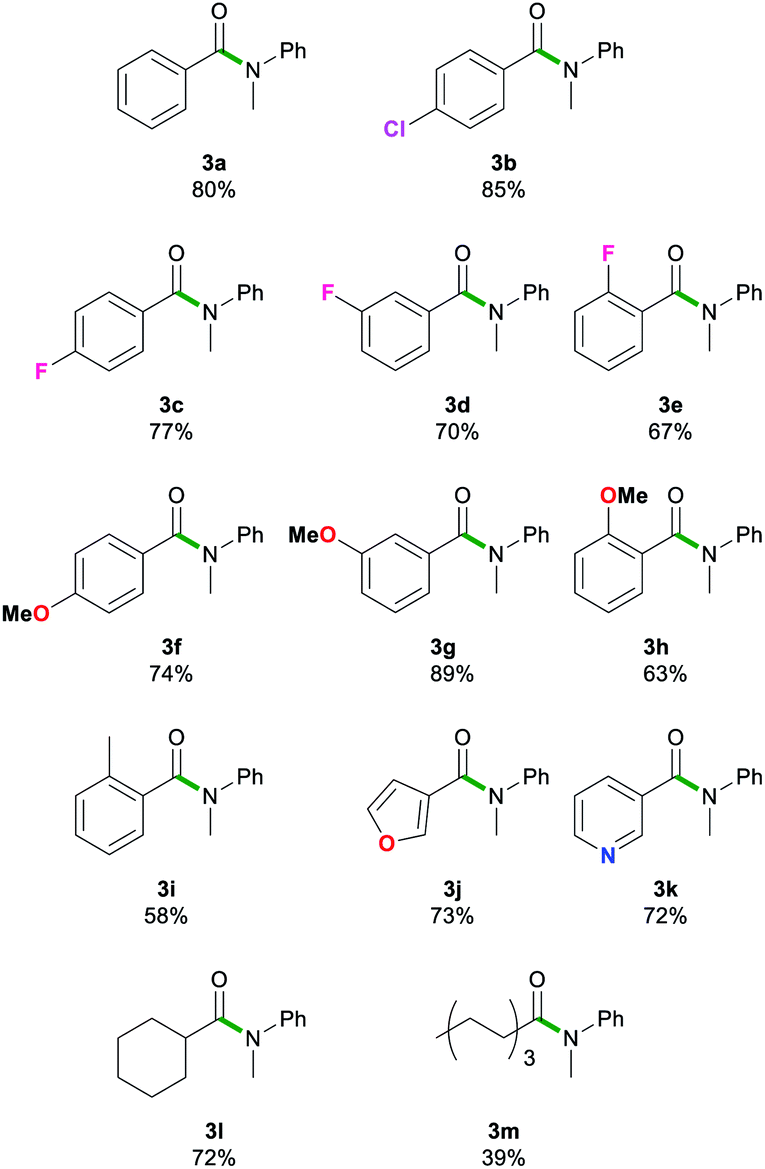
Next, we explored the scope of this air- and moisture-compatible methodology by reacting 2a with assorted esters 1a–m bearing different functional groups. In all cases reactions were carried out using 2-MeTHF in air at room temperature using a small excess of 2a (1.5 equivalents) and quenching the reaction crudes after just 20 seconds, affording amides 3a–3m (Table 3).
| a Reactions performed in air at ambient temperature using 1 g of solvent and 1 mmol of ester. Reactions stirred for 20 s, then quenched with sat. Rochelle's salt soln. (5 mL). Lithium amide 2a was added as a 1 M soln. in 2-MeTHF. Isolated yields are given. |
|---|
|
|
Reactions involving halogen pendant aromatic esters furnished good yields (67–83%) of 3b–e, demonstrating a tolerance to mildly electron withdrawing groups. A slight decrease in yield is apparent when an ortho-substituent, even as small as fluorine, is introduced. When a methoxy group is present in the para and meta positions, good yields are found in 3f and 3g (74–89%). Again, ortho-substituents lead to a decreased yield (63%) for 3h. These results suggest that sterics may have a more prominent role than electronic effects in this type of transformation. The ester substrate scope was also extended successfully to heterocyclic substituents. The 3-furyl group in 3j and 3-pyridyl group in 3k gave high yields (73 and 72%). Aliphatic esters (39–72%, 3l–m) showed no deprotonation of the acidic α-C–H bond. The scope was limited to less bulky esters. Di-ortho-substituted ethyl o,o-dimethylbenzoate and the quaternary carbon centre adjacent to the carbonyl functionality in ethyl pivalate proved to be too sterically encumbered to allow amidation. Esters bearing stronger acidic protons such as ethyl 4-hydroxybenzoate (ethyl paraben) and ethyl 4-aminobenzoate (benzocaine) were incompatible with this method. In all failed reactions, the starting esters were recovered almost quantitatively, likely after hydrolysis during workup. It should also be noted that the reaction of 2a with phenylbenzoate furnished 3a in comparable yields to those found using 1a. These results align well with Szostak's studies using LiHMDS in toluene that show the compatibility of his approach with several alcohol-derived esters.4k
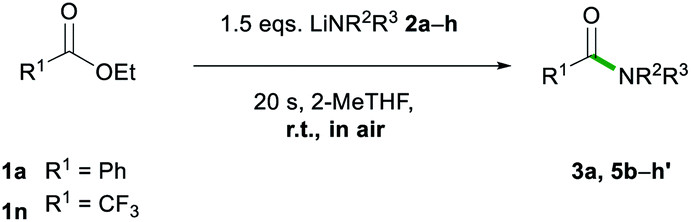
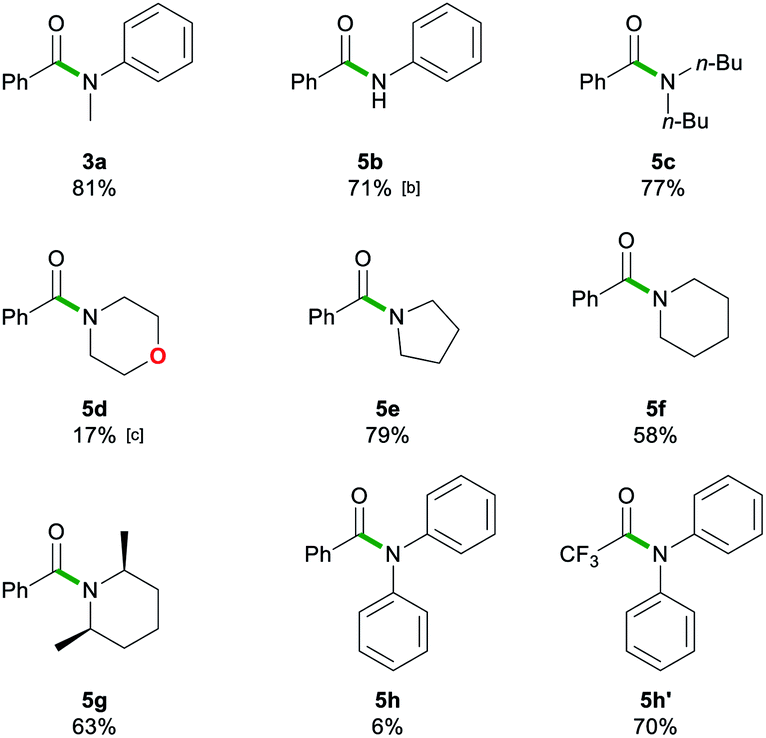
After assessing the range of ester functionalities that could be tolerated by the system, the variation of lithium amides that could be successfully employed was also examined (Table 4).
| a Reactions performed in air at ambient temperature using 1 g of solvent and 1 mmol of ester 1a or 1n. Reactions stirred for 20 s, then quenched with sat. Rochelle's salt soln. (5 mL). Lithium amides 2a–h were added as a 1 M soln. in 2-MeTHF. Isolated yields are given. b 3 eq. of lithium anilide 2b solution. c 0.08 M lithium morpholide 2d solution. Isolated yields are given. |
|---|
|
|
The aromatic primary lithium anilide 2b was found to achieve a good yield of 5b (71%) with a higher amide load of 3 equivalents, whereas when using 1.5 equivalents a modest 36% yield was observed. Interestingly, if the reaction is carried out under identical conditions (1.5 eq. of 2b, 20 s, RT) but under strict inert atmosphere conditions using dry 2-MeTHF, 5b is obtained in a superior 59% yield. These findings suggest that the lower yields observed for LiNHPh are not only a consequence of its reduced basicity in comparison to 2a but also due to its partial degradation under the conditions of the study.
A range of secondary aliphatic amides were found to give moderate to good yields (58–79%, 5c–f) and even in the case of cis-2,6-dimethylpiperidide the steric bulk seems to have had little effect on the yield (63%, 5g). However, when using bulkier 2,2,6,6-tetramethylpiperidide the amidation reaction was completely suppressed. Ethyl trifluoroacetate 1n was found to be a better electronic match with less basic lithium diphenylamide and produced a better yield of 5h′ (70%), while 5h was only afforded in a poor 6% yield. Using lithium morpholide led to the formation of 5d in poor yields (17%), which is attributed to the limited solubility of this amide in 2-MeTHF, so the reaction had to be carried out under much more dilute conditions (see the ESI for details‡). Interestingly, it should be noted that the synthesis of 5d has recently been reported via transamidation of an N-Boc activated amide and free morpholine in acetonitrile at room temperature.4l
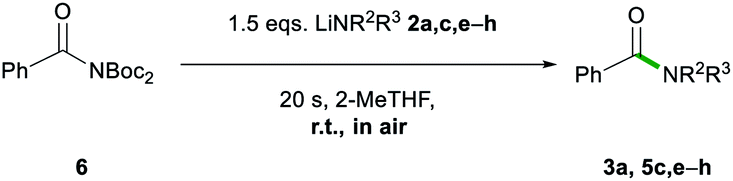
While esters are easier to access as starting materials, the transamidation of amides is of particular importance in biochemical and pharmaceutical applications.4a The examples 3a, 5c, and 5e–5h have, therefore, also been synthesised by transamidation from 6 (Table 5). They afforded fair to good yields (53–77%) which match those observed with the ethyl ester 1a. Interestingly, in the case of lithium diphenylamide, the use of the stabilized N-Boc2 leaving group in 6 circumvented the reactivity problems observed earlier, furnishing 5h in a 72% yield, whereas upon starting from ester 1a, 5h was obtained in a poor 6% yield (vide supra).
| a Reactions performed in air at ambient temperature using 1 g of solvent and 1 mmol of benzamide 6. Reactions stirred for 20 s, then quenched with sat. Rochelle's salt soln. (5 mL). Lithium amides 2a, c, and e–h were added as a 1 M soln. in 2-MeTHF. Isolated yields are given. |
|---|
|
|
Motivated by the fast reactivity and air- and moisture-resistance of the lithium amide reagents in 2-MeTHF, our focus next turned to the characterization of a selection of these reactive intermediates to give insights into the possible mechanism/s operating in these reactions. Previous work by Yoon has shown that KOtBu can mediate ester amidations in technical grade THF in air.20 These reactions seem to operate via a radical mechanism where the presence of oxygen and moisture is crucial for the formation of a reactive acyl radical derived from the ester which, in turn, can react with the amine. Accordingly, when the reaction is performed using an anhydrous solvent under argon the amidation process is completely suppressed. In contrast, we found that carrying out the reaction of ester 1a and LiNMePh (2a) under strict inert atmospheric conditions and stringently dry 2-MeTHF resulted in an 85% yield (almost identical to that when performing the same reaction in air with wet solvent, 81%, Table 1, entry 4). Thus, it seems very unlikely that a related radical mechanism could be in operation here.
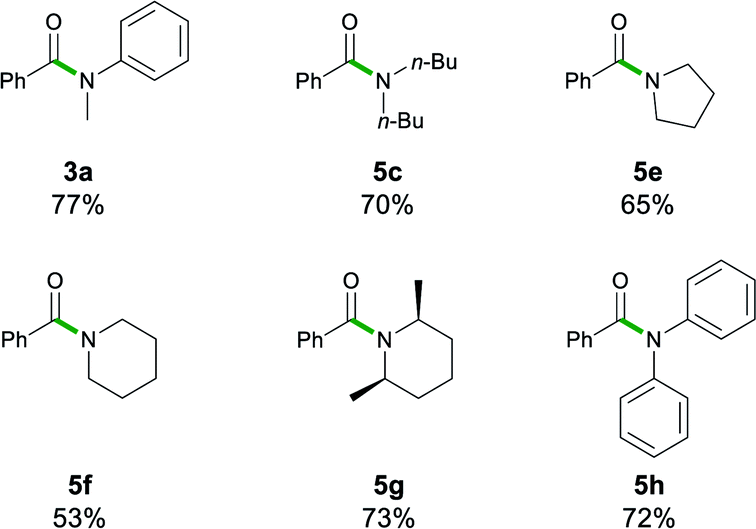
Most recently, insightful DFT studies on LiHMDS mediated transamidation reactions by Hong and Szostak have proposed that these processes take place via the formation of a mixed co-complex between the relevant lithium anilide and LiHMDS (which is present in excess in the reaction media, Scheme 1),4j to which the tertiary amide organic substrate coordinates. This pre-coordination facilitates an intramolecular nucleophilic attack which is the rate determining step of the reaction.4k To investigate if similar substrate pre-coordination could also be in operation in our studies we carried out 1H DOSY NMR studies of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixtures of lithium diphenyl amide 2h with the N,N-dimethylbenzamide 7 in D8-THF and D8-toluene solutions (see Fig. 1 and ESI for details‡).
:1 mixtures of lithium diphenyl amide 2h with the N,N-dimethylbenzamide 7 in D8-THF and D8-toluene solutions (see Fig. 1 and ESI for details‡).
Since the amidation and transamidation reactions presented in Tables 1–5 are very fast, we chose the less nucleophilic lithium amide 2h partnered with tertiary N,N-dimethylbenzamide 7, which lacking an activated leaving group could maximise the likelihood of detecting a possible coordination adduct prior to the nucleophilic addition step. Interestingly, using deuterated THF, a donor solvent with similar capabilities to those of 2-MeTHF, no interaction between 2h and 7 is observed in solution, as is evidenced by the two distinct diffusion coefficients determined for both compounds by DOSY NMR (D = 7.638 × 10−10 m2 s−1 and 1.263 × 10−9 m2 s−1 for 2h and 7, respectively—see Fig. 1A), which are consistent with the presence in solution of free N,N-dimethylbenzamide (MWdet = 153 g mol−1, MWdiff = −3%) and the formation of monomeric trisolvated [Li(NPh2)(THF)3] (MWdet = 363 g mol−1, MWdiff = 8%) (see ESI for details‡). Contrastingly, when 2h and 7 are combined in toluene, a solvent with a significantly lower donor ability,21 coordination complex [{Li(NPh2)(OCPh(NMe2))}2] (8) is obtained as a crystalline solid in 40% yield. X-ray crystallographic studies established the dimeric constitution of 8 (Fig. 2) comprising a planar Li2N2 ring (sum of endocyclic angles: 360°). The Li–N distance in 8 [1.993(2) Å] is noticeably shorter than those found for the related dimeric TMEDA-solvate of 2a [{LiNPh2}(TMEDA)}2] (TMEDA = N,N′-tetramethylethylendiamine) [mean Li–N, 2.139 Å],22 being closer to that reported for monomeric [Li(NPh2)(THF)3] [1.960(2) Å] by Williard.23 Tertiary amide 7 coordinates to Li via its oxygen atom forming a relatively short bond [Li–O, 1.827(2) Å] consistent with the high Lewis basicity of the amide oxygen atoms.24,25 Interestingly, this Li–O bond is retained in toluene solution as revealed by 1H DOSY NMR experiments which show that both {NPh2} and {OCPh(NMe2)} fragments diffuse together in solution as part of the same molecular entity (D = 6.52 × 10−10 m2 s−1, see Fig. 1B and ESI for details‡). While complexes of this type have been predicted in computational studies,4k8 represents to the best of our knowledge the first example known of a coordination adduct of a lithium amide and a tertiary aromatic amide to be structurally characterised.26
 | ||
| Fig. 1 Assessing solvent effects for the co-complexation reaction of LiNPh2 (2h) and PhC(O)NMe2 (7) in (A) D8-THF and (B) D8-toluene using 1H DOSY NMR experiments (see the ESI for details‡). | ||
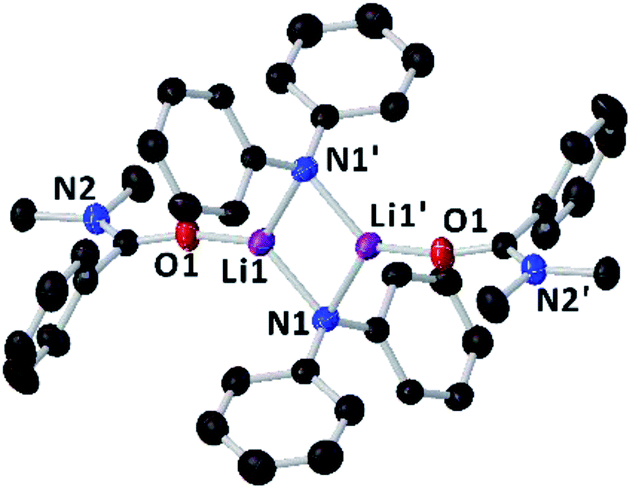 | ||
| Fig. 2 Molecular structure of [{LiNPh2}(OCPh(NMe2)}2] (8) crystallised from toluene. Displacement ellipsoids are drawn at 50% probability and hydrogen atoms are omitted for clarity. | ||
Collectively, these findings suggest that while in the non-Lewis donor solvent toluene, transamidation reactions may take place via the initial formation of a pre-coordination adduct similar to 8, which brings the organic substrate in close proximity to the lithium amide, facilitating nucleophilic addition to its CO bond, this type of activation seems unlikely when using donor solvents like THF (or 2-MeTHF). Alternatively, a plausible explanation of the fast reactivities observed in these solvents could be the formation of kinetically activated smaller aggregates of the lithium amide (vide infra) which could then preferentially undergo transamidation (or ester amidation) reactions over competing degradation processes in the presence of air and/or moisture.
To further investigate solvent effects in these reactions and the possible constitution of the reactive lithium amides in 2-MeTHF, we next prepared and isolated as crystalline solids the 2-MeTHF solvates of lithium N-anilide 2b, N-diphenylamide 2h and 2,2′-bipyridylamide 2i.27 The compounds were prepared by reacting the relevant amine with an equimolar amount of nBuLi, affording white solids that could then be recrystallised from 2-MeTHF/hexane solvent mixtures, furnishing [{Li(NHPh)}2(2-MeTHF)4] (2b-S4), [{Li(NPh2)}2(2-MeTHF)3] (2h-S3) and [{Li(Npy2)}2(2-MeTHF)2] (2i-S2) (py = 2-pyridyl) in 21, 54 and 66% yields, respectively, and whose structures were established by X-ray crystallography (Fig. 3).
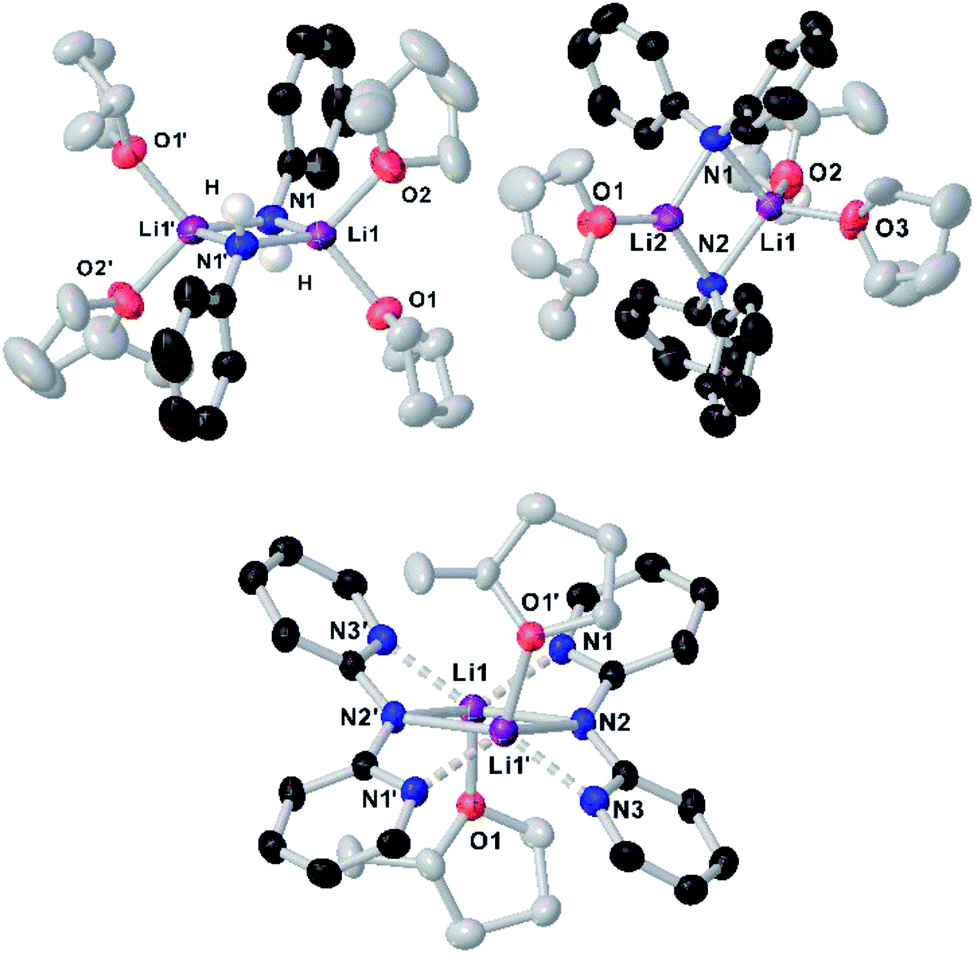 | ||
| Fig. 3 Molecular structures of dimeric (left to right) lithium anilide 2b-S4, lithium diphenylamide 2h-S3 and lithium 2,2′-bipyridylamide 2i-S2 crystallised from 2-MeTHF. Displacement ellipsoids are drawn at 50% probability and hydrogen atoms are omitted for clarity (except those on the nitrogen atoms in 2b-S4). | ||
While all three compounds exhibit dimeric structures in the solid state, displaying a planar Li2N2 core similar to that described for 8, different degrees of solvation are observed. In every case the Me groups of the 2-MeTHF solvent ligands point away from the Li2N2 planes. Thus, for anilide 2b-S4, the symmetrically equivalent Li atoms are tetracoordinated, being solvated by two 2-MeTHF molecules, while upon increasing the steric hindrance in the amide group in diphenylamide 2h-S3, a rare trisolvated dimer is observed with two different Li atoms, one tricoordinate and the other tetracoordinate (Fig. 3). As far as we can ascertain, the only precedent of a structurally authenticated lithium amide solvated by 2-MeTHF is lithium N-methylanilide, which also exhibits a dimeric arrangement, equivalent to 2b-S4 with each lithium coordinated to two molecules of the ethereal solvent.23
Seminal studies by Jackman28 and Williard23 on the constitution of lithium amides in ethereal solvents have proposed a progression of solvation from dimeric Li2A2S2 (A = amide, S = solvent) to LiAS3 when increasing the steric hindrance on the amide ligand and the donor solvent. A closer look into the structures of 2b-S4, 2h-S3 and 2i-S2 shows that along with the steric demands of the amide, electronic effects can also play a role in the degree of solvation. Thus, in centrosymmetric 2i-S2, multidentate 2,2′-bipyridylamide ligands adopt a syn–syn conformation,29 with two N(amido) bridges connecting the Li centers, with each ligand forming two additional dative N(pyridyl)–Li interactions. In contrast with diphenylamide derivative 2h-S3, here the higher hapticity of the amide group favours the formation of a disolvated dimer, with only one molecule of 2-MeTHF coordinated per Li atom. Interestingly 2i failed to undergo transamidation when reacted with 1a, which can be attributed to a certain extent to its reduced nucleophilicity as a consequence of partial delocalization within the dipyridylamide scaffold.
Previous seminal structural studies by Mulvey30,31 and Stalke32 have established the structural diversity of lithium anilide complexes solvated by THF spanning from hexameric [{Li(NHPh)}6(THF)8] (which can be envisaged as a partially dis-assembled ladder motif)31 to a dimeric arrangement similar to 2b-S4 where each lithium binds to two molecules of THF, precluding further association.32 Contrastingly, the trisolvated dimeric structure lithium diphenylamide in 2-MeTHF (Li2A2S3) (vide supra) differs notably to that of the same amide in THF which displays a monomeric LiAS3 motif.23 These differences in aggregation illustrate the subtle effects that small variations in the steric hindrance of the solvent can have on the constitution of the lithium amide.33
In order to get a better understanding of the constitution of these lithium amides in solution, we then carried out detailed 1H DOSY NMR studies of LiNMePh (2a), LiNHPh (2b) and LiNPh2 (2h) in deuterated THF and in non-deuterated 2-MeTHF solutions (Table 6 and see ESI for details‡). It should be noted that for D8-THF the external calibration curve (ECC) method developed by Stalke was employed.34,35 Since no ECC has been developed so far for the estimation of molecular weights of small molecules by DOSY NMR using 2-MeTHF as a solvent, for this solvent we used internal calibration curves (ICCs) using 1,2,3,4-tetraphenylnaphthalene (TPhN), 1-phenylnaphthalene (1-PhN) and TMS as internal reference standards.36
Focusing first on 2-MeTHF, the solvent employed for the reactivity studies (vide supra), the dimeric tetrasolvated Li2A2S4 and trisolvated Li2A2S3 structures found in the solid state for 2a, 2b and 2h, respectively, do not seem to be retained in solution (MWdiff 57, 38 and 35%, respectively, Table 6 and ESI‡). A much better fit is found instead considering the presence of trisolvated LiAS3 monomers (MWdiff 2, −9 and −4%, respectively). Mirroring this trend, a preference for the formation of smaller monomeric aggregates in deuterated THF solutions is also observed for the three lithium amides (see Table 6).37 These findings are consistent with previous work by Collum assessing the constitution of LiNPh2 (2h) in THF/toluene solutions which proposed a dimer/monomer equilibrium in solution. At high concentrations of THF only the monomeric version of 2h is observed, although the degree of THF solvation could not be established.38
|
|
||||
|---|---|---|---|---|
| D 8 -THF | MWdet [g mol−1] | V MWdiff [%] | IV MWdiff [%] | III MWdiff [%] |
| a D8-THF: molecular weights derived from 1H DOSY-ECC-MW determinations at 15 nM concentrations in 0.5 mL D8-THF against tetramethylsilane (TMS) as a reference standard.27a 2-MeTHF: molecular weights derived from 1H DOSY-ICC-MW determinations at 0.2 M concentrations in 0.5 mL 2-MeTHF with 1,2,3,4-tetraphenylnaphthalene (TPhN), 1-phenylnaphthalene (1-PhN) and TMS as internal reference standards.29 See the ESI for full details. | ||||
| LiN(Me)Ph (2a) | 335 | −2 | −23 | 54 |
| LiN(H)Ph (2b) | 331 | −5 | −27 | 47 |
| LiNPh2 (2h) | 393 | <1 | −19 | 87 |
| 2-MeTHF | MWdet [g mol−1] | MWdiff [%] | MWdiff [%] | MWdiff [%] |
|---|---|---|---|---|
| LiN(Me)Ph (2a) | 363 | 2 | −21 | 57 |
| LiN(H)Ph (2b) | 393 | −9 | −31 | 38 |
| LiNPh2 (2h) | 451 | −4 | −23 | 54 |
Collectively, these investigations show that while in the solid state some subtle variations in the degree of aggregation and solvation of the lithium amides using either 2-MeTHF or THF as a donor are observed, nearly identical constitutions are detected in solution when using both of these ethereal solvents, favoring trisolvated LiAS3 monomers. Considering the close interplay of aggregation and solvent effects with reactivity patterns in lithium amide chemistry,39 a plausible rationale for the fast amidation (and transamidation) reactions observed in our study could be the formation of kinetically activated aggregates of the lithium amides in 2-MeTHF solution. Monomer formation should lead to more powerful nucleophilic lithium amides that can react faster with the unsaturated organic substrate (ester or amide) to add across its CO bond, favoring addition over competing degradation by oxygen or moisture.
Conclusions
To conclude, pushing forward the development of more sustainable and air- and moisture-compatible organolithium chemistry, here we introduce renewable and polar 2-MeTHF as a reaction medium to access a wide range of synthetically relevant carboxamides via ester amidation or amide transamidation using lithium amides as metal precursors. Reactions take place at room temperature without the need for external additives in just 20 seconds. 2-MeTHF seems to play a key role in these reactions, ensuring full solubilization of the lithium reagent and favouring the formation of small kinetically activated aggregates that can react rapidly with the organic substrate before decomposition reactions in air or moisture can compete.Experimental
General procedure for (trans)amidation of esters and amides
Additions were performed in air at room temperature in an open Schlenk flask (25 mL) and 1 g of solvent. Lithium amide 2 (1.5 mmol, 1.5 eq.) was added to a stirring solution (960 rpm) of ester 1/amide 6 (1 mmol). After 20 s the reaction was quenched by the addition of sat. Rochelle's salt sol. (sodium potassium tartrate tetrahydrate, 5 mL) before being extracted into 2-MeTHF (3 × 10 mL). Extracts were combined, dried over MgSO4 and concentrated under vacuum.Crude products were purified by silica column chromatography and eluted using hexane:EtOAc (10:1 – 2:1 gradient). Products were identified by GCMS and 1H NMR spectroscopy. Yields were obtained by 1H NMR spectroscopy by integration against a ferrocene internal standard.
General procedure for the synthesis of lithium amides
n-BuLi (19 mL, 30 mmol) was added dropwise to a stirring solution of amine (30 mmol) in hexane (60 mL) and left to stir for 1 h. The resultant suspension was filtered and washed with hexane (3 × 10 mL) before being dried under vacuum. The white solid product obtained was stored in an argon filled glovebox and analyzed by 1H and 7Li NMR spectroscopy (see ESI† for details).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC (EP/S020837/1) and the University of Strathclyde for their generous sponsorship of this research as well as Dr Ross McLellan (University of Strathclyde) and Pasquale Mastropierro (Universität Bern) for their assistance in the structural studies. We are indebted to the MINECO of Spain (Projects CTQ2014-51912-REDC and CTQ2016-75986-P), the Gobierno del Principado de Asturias (Project GRUPIN14-006) and the Fundación BBVA for the award of a “Beca Leonardo a Investigadores y Creadores Culturales 2017” to JGA. We gratefully acknowledge the Leverhulme Trust (research grant, RPG-2016-281 to CTOH). The X-ray crystal structure determination service unit of the Department of Chemistry and Biochemistry of the University of Bern is acknowledged for measuring, solving, refining and summarizing the structure of compound 8. The Synergy diffractometer was partially funded by the Swiss National Science Foundation (SNF) within the R'Equip programme (project number 206021_177033). The data used within this publication can be accessed at https://doi.org/10.15129/fd008fa8-3adc-489c-b9f6-98dfeb98300c.Notes and references
- D. G. Brown and J. Bostrom, J. Med. Chem., 2016, 59, 4443 CrossRef CAS.
- (a) J. M. Humphrey and A. R. Chamberlin, Chem. Rev., 1997, 97, 2243 CrossRef CAS PubMed; (b) J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337 RSC; (c) T. J. Deming, Prog. Polym. Sci., 2007, 32, 858 CrossRef CAS; (d) J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 20, 140 CrossRef CAS.
- (a) D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. J. L. Leazer, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411 RSC; (b) M. C. Bryan, P. J. Dunn, D. Entwistle, F. Gallou, S. G. Koenig, J. D. Hayler, M. R. Hickey, S. Hughes, M. E. Kopach, G. Moine, P. Richardson, F. Roschangar, A. Steven and F. J. Weiberth, Green Chem., 2018, 20, 5082 RSC.
- (a) K. Hollanders, B. Maes and S. Ballet, Synthesis, 2019, 51, 2261 CrossRef CAS; (b) R. M. de Figueiredo, J. S. Suppo and J. M. Campagne, Chem. Rev., 2016, 116, 12029 CrossRef CAS; (c) V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471 CrossRef CAS; (d) A. Ojeda-Porras and D. Gamba-Sanchez, J. Org. Chem., 2016, 81, 11548 CrossRef CAS PubMed; (e) J. D. Muñoz, J. Alcazar, A. de la Hoz, A. Diaz-Ortiz and S. A. A. de Diego, Green Chem., 2012, 14, 1335 RSC; (f) L. Zhu, L. Le, M. Yan, C. T. Au, R. Qiu and N. Kambe, J. Org. Chem., 2019, 84, 5635 CrossRef CAS PubMed; (g) Y. L. Zheng and S. G. Newman, ACS Catal., 2019, 9, 4426 CrossRef CAS; (h) C. L. Allen and J. M. Williams, Chem. Soc. Rev., 2011, 40, 3405 RSC; (i) X. Wang, Nat. Catal., 2019, 2, 98 CrossRef; (j) G. Li and M. Szostak, Nat. Commun., 2018, 9, 4165 CrossRef; (k) G. Li, C. L. Ji, X. Hong and M. Szostak, J. Am. Chem. Soc., 2019, 141, 11161 CrossRef CAS PubMed; (l) M. M. Rahman, G. Li and M. Szostak, J. Org. Chem., 2019, 84, 12091 CrossRef CAS PubMed; (m) L. Gooßen, D. M. Ohlmann and P. L. Lange, Synthesis, 2009, 160 CrossRef; (n) L. Perreux, A. Loupy and F. Volatron, Tetrahedron, 2002, 58, 2155 CrossRef CAS; (o) B. S. Jursic and Z. Zdravkovdki, Synth. Commun., 1993, 23, 2761 CrossRef CAS.
- For selected references see: (a) J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 2, 140 CrossRef; (b) J. W. Bode, Curr. Opin. Drug Discovery Dev., 2006, 9, 765 CAS; (c) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606 RSC; (d) C. E. Garrett, X. Jiang, K. Prasad and O. Repič, Tetrahedron Lett., 2002, 43, 4161 CrossRef CAS; (e) C. Affouard, R. D. Crockett, K. Diker, R. P. Farrell, G. Gorins, J. R. Huckins and S. Caille, Org. Process Res. Dev., 2015, 19, 476 CrossRef CAS; (f) A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557 CrossRef CAS PubMed; (g) P. H. Huy and C. Mbouhom, Chem. Sci., 2019, 10, 7399 RSC.
- (a) S. Chandrudu, P. Simerska and I. Toth, Molecules, 2013, 18, 4373 CrossRef CAS PubMed and references cited therein. (b) N. Sewald and H.-D. Jakubke, Peptides: Chemistry and Biology, Wiley-VCH, Weinheim, 2015 Search PubMed.
- For other short reviews in amide synthesis see: (a) V. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471 CrossRef CAS; (b) J. W. Bode, Top. Organomet. Chem., 2013, 44, 13 CrossRef CAS.
- For selected references see: (a) A. Correa and R. Martin, J. Am. Chem. Soc., 2014, 136, 7253 CrossRef CAS PubMed; (b) H. Wang, G. Tang and X. Li, Angew. Chem., Int. Ed., 2015, 54, 13049 CrossRef CAS PubMed; (c) C. Allen and J. M. Williams, Chem. Soc. Rev., 2011, 40, 3405 RSC; (d) C. W. Cheung, J. A. Ma and X. Hu, J. Am. Chem. Soc., 2018, 140, 6789 CrossRef CAS PubMed; (e) T. Ben Halima, J. Masson-Makdissi and S. G. Newman, Angew. Chem., Int. Ed., 2018, 57, 12925 CrossRef CAS PubMed; (f) A. Ojeda-Porras and D. Gamba-Sanchez, J. Org. Chem., 2016, 81, 11548 CrossRef CAS PubMed.
- G. Li and M. Szostak, Chem. Rec., 2019, 19, 1 CrossRef CAS.
- For recent reviews and concept articles dealing with the use of polar organometallic compounds in unconventional reaction media, see: (a) J. García-Álvarez, E. Hevia and V. Capriati, Eur. J. Org. Chem., 2015, 6779 CrossRef; (b) J. García-Álvarez, E. Hevia and V. Capriati, Chem. - Eur. J., 2018, 24, 14854 CrossRef PubMed.
- (a) C. Vidal, J. Garcia-Alvarez, A. Hernan-Gomez, A. R. Kennedy and E. Hevia, Angew. Chem., Int. Ed., 2014, 53, 5969 ( Angew. Chem. , 2014 , 126 , 6079 ) CrossRef CAS PubMed; (b) C. Vidal, J. Garcia-Alvarez, A. Hernan-Gomez, A. R. Kennedy and E. Hevia, Angew. Chem., Int. Ed., 2016, 55, 16145 ( Angew. Chem. , 2016 , 128 , 16379 ) CrossRef CAS PubMed; (c) M. J. Rodriguez-Alvarez, J. Garcia-Alvarez, M. Uzelac, M. Fairley, C. T. O'Hara and E. Hevia, Chem.–Eur. J., 2018, 24, 1720–1725 CrossRef CAS PubMed; (d) A. Sánchez-Condado, G. A. Carriedo, A. Presa Soto, M. J. Rodríguez-Álvarez, J. García-Álvarez and E. Hevia, ChemSusChem, 2019, 12, 3134 CrossRef PubMed.
- Contemporary to our investigations, Capriati and co-workers have also reported the utilization of organolithium and organomagnesium reagents in water or DESs: (a) V. Mallardo, R. Rizzi, F. C. Sassone, R. Mansueto, F. M. Perna, A. Salomone and V. Capriati, Chem. Commun., 2014, 50, 8655 RSC; (b) F. C. Sassone, F. M. Perna, A. Salomone, S. Florio and V. Capriati, Chem. Commun., 2015, 51, 9459 RSC; (c) L. Cicco, S. Sblendorio, R. Mansueto, F. M. Perna, A. Salomone, S. Florio and V. Capriati, Chem. Sci., 2016, 7, 1192 RSC; (d) G. Dilauro, M. Dell'Aera, P. Vitale, V. Capriati and F. M. Perna, Angew. Chem., Int. Ed., 2017, 56, 10200 ( Angew. Chem. , 2017 , 129 , 10334 ) CrossRef CAS PubMed; (e) G. Dilauro, A. F. Quivelli, P. Vitale, V. Capriati and F. M. Perna, Angew. Chem., Int. Ed., 2019, 58, 1799 ( Angew. Chem. , 2019 , 131 , 1813 ) CrossRef CAS PubMed; (f) C. Prandi, S. Ghinato, G. Dilauro, F. M. Perna, V. Capriati and M. Blangetti, Chem. Commun., 2019, 55, 7741 RSC.
- For some examples of the use of 2-MeTHF as an alternative and sustainable reaction medium in s-block chemistry see: (a) V. Pace, L. Castoldi, P. Hoyos, J. V. Sinisterra, M. Pregnolato and J.-M. Sánchez-Montero, Tetrahedron, 2011, 67, 2670 CrossRef CAS; (b) A. Kadam, M. Nguyen, M. Kopach, P. Richardson, F. Gallou, Z. K. Wan and W. Zhang, Green Chem., 2013, 15, 1880 RSC; (c) V. Pace, A. R. Alcántara and W. Holzer, Green Chem., 2011, 13, 1986 RSC; (d) V. Pace, L. Castoldi, A. R. Alcántara and W. Holzer, Green Chem., 2012, 14, 1859 RSC; (e) C. Carbone, P. O'Brien and G. Hilmersson, J. Am. Chem. Soc., 2010, 132, 15445 CrossRef PubMed; (f) G. Parisi, L. Degennaro, C. Carlucci, M. de Candia, P. Mastrorilli, A. Roller, W. Holzer, C. D. Altomare, V. Pace and R. Luisi, Org. Biomol. Chem., 2017, 15, 5000 RSC; (g) K. de la Vega-Hernández, R. Senatore, M. Miele, E. Urban, W. Holzer and V. Pace, Org. Biomol. Chem., 2019, 17, 1970 RSC; (h) V. Pace, K. de la Vega-Hernández, E. Urban and T. Langer, Org. Lett., 2016, 18, 2750 CrossRef CAS.
- For recent reviews on the applications of 2-MeTHF in organometallic synthesis see: (a) V. Pace, P. Hoyos, L. Castoldi, P. Domínguez de María and A. R. Alcántara, ChemSusChem, 2012, 5, 1369 CrossRef CAS; (b) S. Monticelli, L. Castoldi, I. Murgia, R. Senatore, E. Mazzeo, J. Wackerlig, E. Urban, T. Langer and V. Pace, Monatsh. Chem., 2017, 148, 37 CrossRef CAS PubMed; (c) D. F. Aycock, Org. Process Res. Dev., 2007, 11, 156 CrossRef CAS; (d) V. Pace, Aust. J. Chem., 2012, 65, 301 CrossRef CAS.
- D. J. C. Constable, C. Jimenez-Gonzalez and R. K. Henderson, Org. Process Res. Dev., 2007, 11, 133 CrossRef CAS.
- (a) Y. Gu and F. Jérôme, Green Chem., 2010, 12, 1127 RSC; (b) A. E. Díaz-Álvarez, J. Francos, B. Lastra-Barreira, P. Crochet and V. Cadierno, Chem. Commun., 2011, 47, 6208 RSC; (c) F. Chahdoura, I. Favier and M. Gómez, Chem.–Eur. J., 2014, 20, 10884 CrossRef CAS PubMed; (d) S. Santoro, F. Ferlin, L. Luciani, L. Ackermann and L. Vaccaro, Green Chem., 2017, 19, 1601 RSC.
- (a) O. Sahu, Ann. Agric. Sci., 2018, 16, 389 Search PubMed; (b) Y. Tachibana, S. Kimura and K. Kasuya, Sci. Rep., 2015, 5, 8249 CrossRef; (c) K. Dalvand, J. Rubin, S. Gunukula, M. C. Wheeler and G. Hunt, Biomass Bioenergy, 2018, 115, 56 CrossRef; (d) H. E. Hoydonckx, W. M. Van Rhijn, W. Van Rhijn, D. E. De Vos and P. A. Jacobs, Furfural and Derivatives, Ullmann's Encyclopedia of Industrial Chemistry, 2007, DOI:10.1002/14356007.a14356012_14356119.pub14356002.
- It is noteworthy that 2-MeTHF enabled the use of 1 M solutions of the amides due to significantly better solubility as compared to THF (2a could only be used in a 0.2 M solution in THF).
- The poor stability of 2a in Gly contrasts with that previously reported by us for PhLi (ref. 11c) for the addition to benzonitrile. This difference can be attributed to the addition of phenyllithium solution (in di-n-butyl ether) occurring “on glycerol” as opposed to the present case with the addition of lithium N-methylanilide (in 2-MeTHF) occurring in the bulk solvent.
- B. R. Kim, H. G. Lee, S. B. Kang, G. H. Sung, J. J. Kim, J. K. Park, S. G. Lee and Y. J. Yoon, Synthesis, 2012, 44, 42 CrossRef CAS.
- For selected examples of toluene coordinating as a donor to lithium complexes via π-electrostatic interactions see: (a) H. Niu, R. J. Mangan, A. V. Protchenko, N. Phillips, W. Unkrig, C. Friedmann, E. L. Kolychev, R. Tirfoin, J. Hicks and S. Aldridge, Dalton Trans., 2018, 22, 7445 RSC; (b) K. Samigullin, I. Georg, M. Bolte, H. W. Lerner and M. Wagner, Chem.–Eur. J., 2016, 22, 3478 CrossRef CAS.
- For other examples of structural variations of LiNPh2 see: A. R. Kennedy, J. Klett, C. T. O'Hara, R. E. Muvey and G. M. Robertson, Eur. J. Inorg. Chem., 2009, 5029 CrossRef CAS.
- C. Su, J. Guang and P. G. Williard, J. Org. Chem., 2014, 79, 1032 CrossRef CAS PubMed.
- For examples of a neutral aromatic tertiary organic amide acting as an oxygen donor to other metals see: (a) A. Keys, T. J. Barbarich, S. G. Bott and A. R. Barron, J. Chem. Soc., Dalton Trans., 2000, 577 RSC; (b) K. C. Mullane, T. Cheisson, E. Nakamaru-Ogiso, B. C. Manor, P. J. Carroll and E. J. Schelter, Chem.–Eur. J., 2017, 24, 826 CrossRef; (c) W. Clegg, S. H Dale, E. Hevia, G. W. Honeyman and R. E. Mulvey, Angew. Chem., Int. Ed., 2006, 45, 2371 Search PubMed.
- H. Karlsen, P. Kolsaker, C. Romming and E. Uggerud, J. Chem. Soc., Perkin Trans. 1, 2002, 2, 404 RSC.
- It should be noted that Williard has reported a closely related structure of an adduct of LiN(Me)Ph with two equivalents of N,N-diethylpropionamide; see ref. 23.
- For critical reviews on the relevance of lithium amides in synthesis see: (a) R. E. Mulvey and S. D. Robertson, Angew. Chem., Int. Ed., 2013, 52, 11470 ( Angew. Chem. , 2013 , 125 , 11682 ) CrossRef CAS; (b) V. Capriati, F. M. Perna and A. Salomone, Dalton Trans., 2014, 43, 14204 RSC.
- L. M. Jackman and L. M. Scarmoutzos, J. Am. Chem. Soc., 1987, 109, 5348 CrossRef CAS.
- (a) D. R. Armstrong, E. V. Brouillet, A. R. Kennedy, J. A. Garden, M. Granitzka, R. E. Mulvey and J. J. Trivett, Dalton Trans., 2014, 43, 14409 RSC; (b) D. R. Armstrong, J. A. Garden, A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Angew. Chem., Int. Ed., 2013, 52, 7190 CrossRef CAS.
- W. Clegg, L. Horsburgh, F. M. Mackenzie and R. E. Mulvey, J. Chem. Soc., Chem. Commun., 1995, 2011 RSC.
- For another example of donor-induced partial deaggregation of the polymeric structure of lithium anilide see: W. Clegg, S. T. Liddle, R. E. Mulvey and A. Robertson, Chem. Commun., 2000, 223 RSC.
- R. von Bülow, H. Gornitzka, T. Kottke and D. Stalke, Chem. Commun., 1996, 1639 RSC.
- Stalke recently employed a combination of QTAIM and DFT methods to explain gradually modified aggregation and hence reactivity of lithium amides; see: F. Engelhardt, C. Maaß, D. M. Andrada, R. Herbst-Irmer and D. Stalke, Chem. Sci., 2018, 9, 3111 RSC.
- (a) B. Neufeld and D. Stalke, Chem. Sci., 2015, 6, 3354 RSC; (b) S. Bachmann, B. Gernert and D. Stalke, Chem. Commun., 2016, 52, 12861 RSC; (c) S. Bachmann, R. Neufeld, M. Dzemski and D. Stalke, Chem.–Eur. J., 2016, 22, 8462 CrossRef CAS PubMed.
- For an insightful study on the aggregation of lithium diisopropylamide in hydrocarbon solvents using DOSY NMR see: R. Neufeld, M. John and D. Stalke, Angew. Chem., Int. Ed., 2015, 54, 6994 CrossRef CAS PubMed.
- (a) D. Li, I. Keresztes, R. Hopson and P. G. Williard, Acc. Chem. Res., 2008, 42, 270 CrossRef PubMed; (b) G. Kagan, W. Li, R. Hopson and P. G. Williard, Org. Lett., 2009, 11, 4818 CrossRef CAS PubMed.
- As detailed in the ESI‡ for 2b, DOSY NMR studies also showed a good correlation for its MWdet as the LiA2S2 dimer in d8-THF or 2-MeTHF. However, considering the large excess of Lewis donor present in solution and the X-ray crystallographic studies that show the ability of Li to coordinate to at least two molecules of 2-MeTHF (or THF) furnishing Li2A2S4 dimers, we consider this scenario much less likely to occur.
- J. S. Depue and D. B. Collum, J. Am. Chem. Soc., 1988, 110, 5518 CrossRef CAS.
- For selected references see: (a) Y. Ma, C. Hoepker, L. Gupta, M. F. Faggin and D. B. Collum, J. Am. Chem. Soc., 2010, 132, 15610 CrossRef CAS; (b) K. A. Mack and D. B. Collum, J. Am. Chem. Soc., 2018, 140, 4877 CrossRef CAS PubMed; (c) Y. Zhou, J. Jermaks, I. Keresztes, S. N. MacMillan and D. B. Collum, J. Am. Chem. Soc., 2019, 141, 5444 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor Robert (Rab) Mulvey on the occasion of his 60th birthday, a visionary organolithium chemist and an inspiring mentor to Joaquin Garcia-Alvarez, Charlie O'Hara and Eva Hevia. |
| ‡ Electronic supplementary information (ESI) available: Full experimental details and copies of NMR spectra. CCDC 1973293–1973295 and 1987696. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc01349h |
| This journal is © The Royal Society of Chemistry 2020 |