
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA regioselectively 1,1′,3,3′-tetrazincated ferrocene complex displaying core and peripheral reactivity†
Gordon W.
Honeyman
a,
David R.
Armstrong
a,
William
Clegg
 b,
Eva
Hevia‡
a,
Alan R.
Kennedy
a,
Ross
McLellan
a,
Samantha A.
Orr§
a,
John A.
Parkinson
a,
Donna L.
Ramsay
a,
Stuart D.
Robertson
*a,
Stephen
Towie
a and
Robert E.
Mulvey
*a
b,
Eva
Hevia‡
a,
Alan R.
Kennedy
a,
Ross
McLellan
a,
Samantha A.
Orr§
a,
John A.
Parkinson
a,
Donna L.
Ramsay
a,
Stuart D.
Robertson
*a,
Stephen
Towie
a and
Robert E.
Mulvey
*a
aWestCHEM, Department of Pure & Applied Chemistry, University of Strathclyde, Glasgow, G1 1XL, UK. E-mail: r.e.mulvey@strath.ac.uk; stuart.d.robertson@strath.ac.uk
bChemistry, School of Natural and Environmental Sciences, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK
First published on 27th March 2020
Abstract
Regioselective 1,1′,3,3′-tetrazincation [C–H to C–Zn(tBu)] of ferrocene has been achieved by reaction of a fourfold excess of di-t-butylzinc (tBu2Zn) with sodium 2,2,6,6-tetramethylpiperidide (NaTMP) in hexane solution manifested in the trimetallic iron–sodium–zinc complex [Na4(TMP)4Zn4(tBu)4{(C5H3)2Fe}], 1. X-ray crystallographic studies supported by DFT modelling reveal the structure to be an open inverse crown in which two [Na(TMP)Zn(tBu)Na(TMP)Zn(tBu)]2+ cationic units surround a {(C5H3)2Fe}4− tetraanion. Detailed C6D6 NMR studies have assigned the plethora of 1H and 13C chemical shifts of this complex. It exists in a major form in which capping and bridging TMP groups interchange, as well as a minor form that appears to be an intermediate in this complicated exchange phenomenon. Investigation of 1 has uncovered two distinct reactivities. Two of its peripheral t-butyl carbanions formally deprotonate toluene at the lateral methyl group to generate benzyl ligands that replace these carbanions in [Na4(TMP)4Zn4(tBu)2(CH2Ph)2{(C5H3)2Fe}], 2, which retains its tetrazincated ferrocenyl core. Benzyl-Na π-arene interactions are a notable feature of 2. In contrast, reaction with pyridine affords the crystalline product {[Na·4py][Zn(py*)2(tBu)·py]}∞, 3, where py is neutral pyridine (C5H5N) and py* is the anion (4-C5H4N), a rare example of pyridine deprotonated/metallated at the 4-position. This ferrocene-free complex appears to be a product of core reactivity in that the core-positioned ferrocenyl anions of 1, in company with TMP anions, have formally deprotonated the heterocycle.
Introduction
Second generation metallation, where the Brønsted base is a multicomponent compound or mixture, instead of a single organolithium reagent 1 or lithium amide reagent2 as in the first generation types, is now well established.3 In terms of synthetic utility, Knochel's salt-activated4 metal amide reagents have proved most popular, typified by magnesium complexes such as (TMP)MgCl·LiCl5 and zinc complexes such as (TMP)2Zn·2MgCl2·2LiCl,6 though many others have also been applied successfully in deprotonation reactions.7 The vast majority of these reactions have involved monodeprotonations of organic substrates, while di-deprotonative metallations are usually unwanted side reactions, generally indirectly identified through detection of difunctionalised products following electrophilic interception protocols.8 Much less developed are polymetallation reactions, where di- or higher order-deprotonation is a deliberate aim. A special category of metallation called template magnesiation is an exception.9 Here, the degree as well as the regioselectivity of deprotonation are dictated mainly by the preformed structure of the second-generation base. For example, the 24-atom template ring structure of hexameric [{KMg(TMP)2(nBu)}6] can deprotonate six naphthalene molecules with its six Mg-fixed butyl bases, while retaining the [(KNMgN)6] ring in the product [{KMg(TMP)2(2-C10H7)}6].10 The related sodium template [Na4Mg2(TMP)6(nBu)2] with its 12-atom (NaNMgNNaN)2 template ring di-deprotonates para-terphenyl on one terminal Ph ring to generate [Na4Mg2(TMP)6(3,5-para-terphenyl-di-ide)], or four times (twice on each terminal Ph ring) to afford [{Na4Mg2(TMP)6}2(3,3′′,5,5′′-para-terphenyl-tetra-ide)].9aFerrocenes and their functionalized derivatives are important molecules in a remarkably wide variety of fields such as in materials, as pharmaceuticals and as components of chiral and achiral ligands for catalysis.11 Previously, such fourfold deprotonation was accomplished with ferrocene in [Na4Mg4(Ni–Pr2)8{Fe(C5H3)2}], using a sodium magnesium diisopropylamide base,12 while recently a first-generation potassium alkoxy neopentyl complex has also achieved tetradeprotonation though the ferrocenylpotassium product could not be characterised.13 Ferrocene is typically more susceptible to mono- or di-deprotonation (one deprotonation per ring),14 meaning controlled tetradeprotonation would provide access to a potentially useful synthetic intermediate.15 Since zinc is a key component of many synergistic compounds16 and offers more onward functionalization opportunities than magnesium (e.g., through Negishi cross-coupling processes),17 it would be useful to develop a complementary zinc chemistry of this controlled higher order deprotonation. This work sets out on this path, reporting the first reaction exhibiting four C–H to C–Zn exchanges in an open inverse crown product15a,18 the openness of which enables peripheral and core reactivities not observed previously in inverse crown chemistry.
Results and discussion
Synthesis and solid-state studies of tetrametallated complex 1
In earlier work we showed it was possible to effect mono- and di-zincation of ferrocene using the appropriate amount of TMP-zincate reagent TMEDA·Na(μ-TMP)(μ-tBu)ZntBu,19 with crystal structures of TMEDA·Na(μ-TMP)[μ-(C5H4)Fe(C5H5)]ZntBu (A) and [TMEDA·Na(μ-TMP)ZntBu]2(C5H4)2Fe (B) being obtained in each case though NMR studies established that stoichiometric control was not perfect with each solution containing traces of the incorrect stoichiometric product. A hint that higher order zincation might be realised came from isolation of a poorly soluble red powder on treating ferrocene with four equivalents of the base mixture in the absence of Lewis donating TMEDA.20 Different isomers of a putative tetrazincation product were implicated on the sole evidence of a routine 1H NMR spectrum, forming the starting point for the present study. Following many attempts, we have managed to coax red crystals from this reaction (Scheme 1) and analysed them by X-ray crystallography.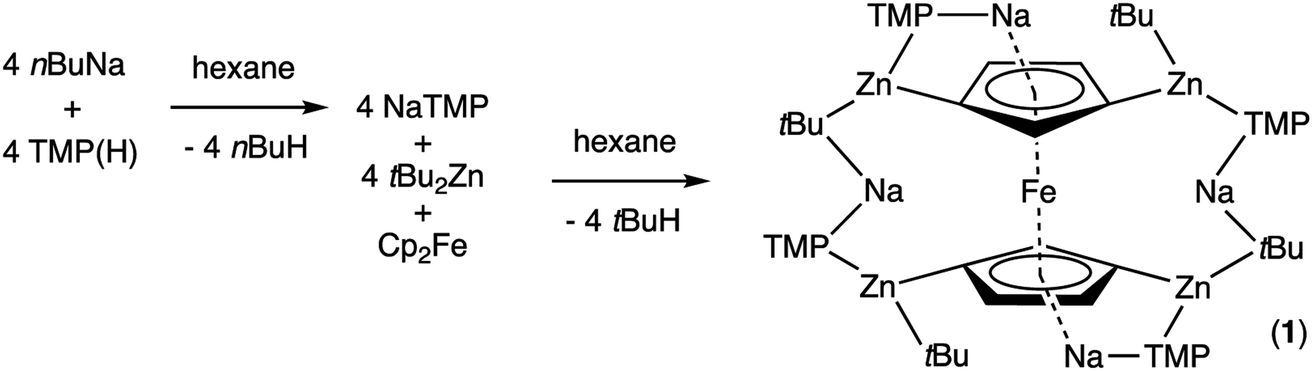 | ||
| Scheme 1 Synthetic protocol for accessing the tetrazincated ferrocene complex. | ||
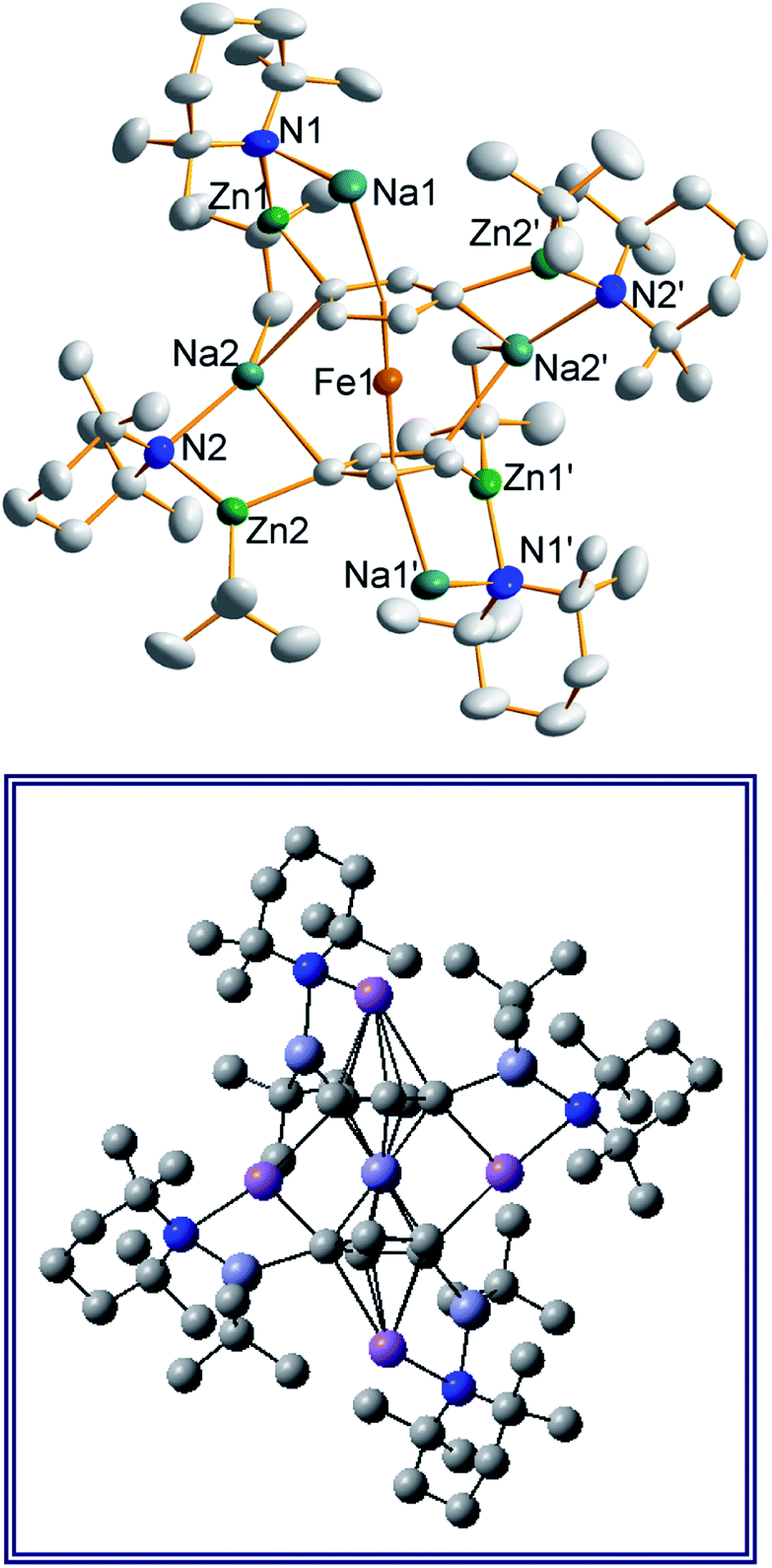
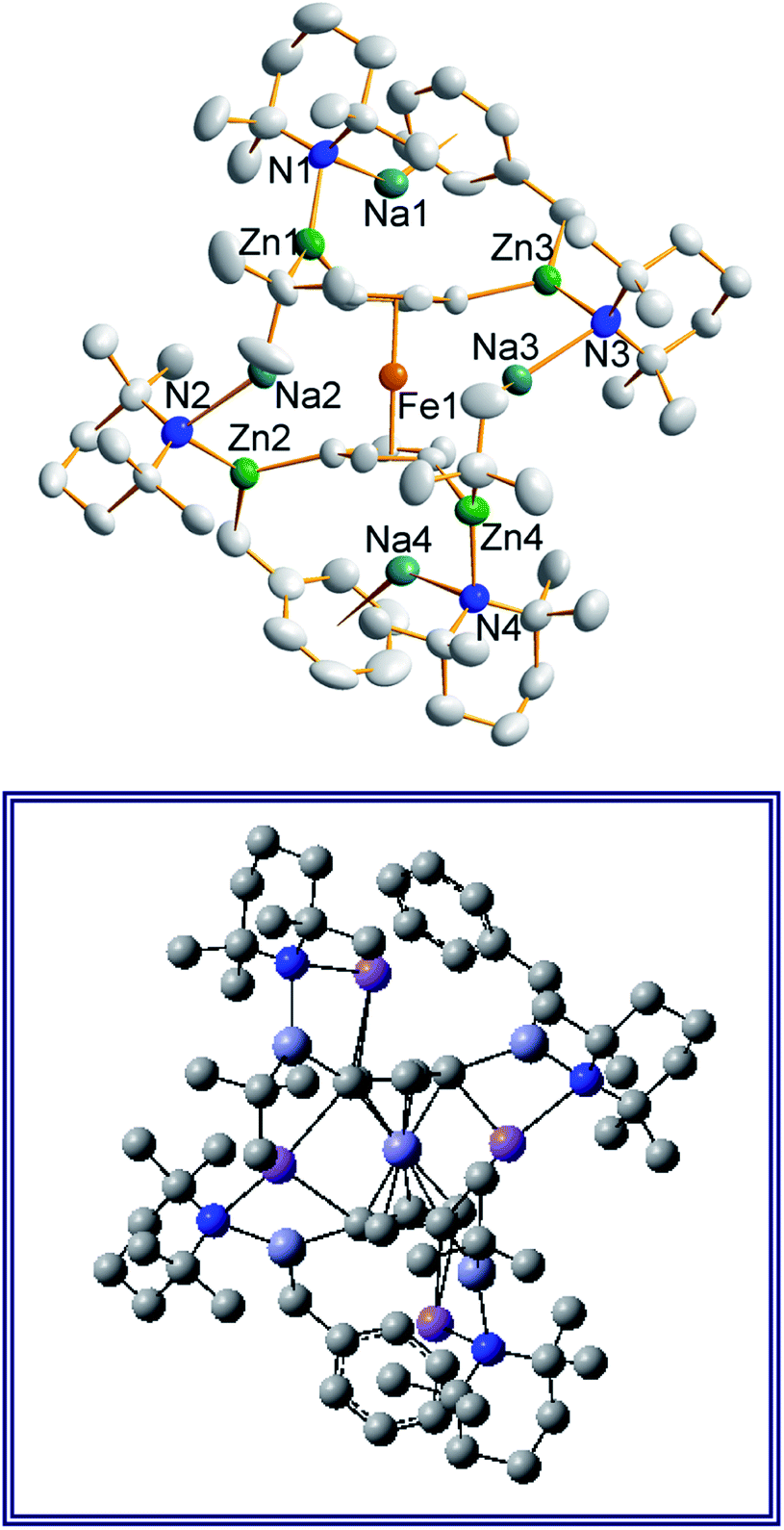
Frustratingly, these crystals and different batches from several reproducible reactions proved highly disordered. There are 3 crystallographically independent molecules in the structure, one of which is completely disordered around a centre of symmetry and one molecule has a disordered tBu group. However, one molecule is well ordered (Fig. 1) and so it is reasonable to conclude that the bulk of the product is the expected tetrazincated ferrocene [Na4(TMP)4Zn4(tBu)4{(C5H3)2Fe}], 1. The four zinc atoms occupy the 1,1′,3,3′ sites on the ferrocene scaffold commensurate with tetradeprotonation having occurred. Two zinc atoms bridge through a TMP ligand to the sodium atoms capping the C5H3 rings, while the other two zinc atoms each bridge through a TMP ligand to the centrally disposed sodium centres positioned either side of the iron. The remaining coordination site on each zinc atom is filled by an alkyl ligand. Consequently, with the two zinc atoms bound to each Cp ring being inequivalent, this can be considered as a planar chiral complex (see NMR section for further discussion). In earlier work the magnesiated ferrocene [Na4Mg4(NiPr2)8{Fe(C5H3)2}] has been interpreted as an inverse crown10,21 comprising a 16-membered [(NaNMgN)4] cationic (4+ charged) ring that hosts a [Fe(C5H3)2}] (4− charged) guest. Though related, 1 has structural features that sets it apart from this magnesiated structure in having a heteroleptic ligand set of alternating amido and alkyl groups and a less complete (or totally open) [{Na(TMP)Zn(alkyl)}4]4+ unit in the cationic moiety surrounding the [(C5H3)2Fe]4− core. A DFT optimisation 1calc (Fig. 1 inset, with enforced C2 symmetry corresponding to the symmetry of the experimental structure) starting from the experimental X-ray established coordinates of 1 predicts the cationic moiety in this structure is totally open having terminal tBu ligands bound to Zn2. This is demonstrated by the closest methyl group of this terminal tBu group to the adjacent sodium which is almost 3 Å away (calculated distance is 2.922 Å) compared with the bridging tBu group of TMEDA·Na(μ-TMP)(μ-tBu)ZntBu which has significantly shorter distances of 2.750(10) Å 22 and 2.818(13)/2.837(15) Å for the tBu groups bridging sodium and zinc in complex 3 (vide infra) and 2.883 Å for the corresponding Na2–CtBu distance in this complex. Previously studied tetramagnesiated metallocenes (Ru and Os examples, besides the aforementioned Fe example) have been analysed via DFT calculations from which it was concluded that two isomeric forms (assigned Z and W for Ci and C2 symmetrical variants respectively) are possible with the Z form (characterized by having a symmetry imposed linear Na⋯Fe⋯Na unit) being the prevalent one.23 In this case the solid-state structure appears to adopt the W form (Fig. 2) as evidenced by the bent Na⋯Fe⋯Na unit. This variation is perhaps unsurprising since the guest molecule (tetradeprotonated ferrocene) is identical but the inverse crown host ring (assuming a long-distance contact between Na and the methyl group of the tBu ligand) surrounding it formally contains 20 atoms as opposed to 16 and so a deformation of this ring in the case of the W isomer is plausible.
 | ||
| Fig. 1 Molecular structure of one independent molecule of the tetrazincated ferrocene [Na4(TMP)4Zn4(tBu)4{(C5H3)2Fe}] (1, top) and DFT optimised structure (1calc, bottom). Ellipsoids are shown at 50% probability and all hydrogen atoms have been removed for clarity. Selected bond lengths (Å) and angles (°) for 1calc: Na1–N1, 2.374; Zn1–N1, 2.046; Zn1–CFc, 2.120; Zn1–CtBu, 2.045; Na2–CtBu, 2.883; Na2–N2, 2.414; Na2, CFc, 2.624, 2.651; Zn2–CFc, 2.085; Zn2–N2, 2.041; Zn2–CtBu, 2.061; Na1–CtBu, 2.922; Na1–N1–Zn1, 92.1; Na1–CFc–Zn1, 83.1; N1–Zn1–CFc, 102.7; N1–Na1–CFc, 80.5; Na2–N2–Zn2, 83.9; Na2–CFc–Zn2, 77.3; N2–Zn2–CFc, 112.6; N2–Na2–CFc, 85.2; CFc–Na2–CFc, 87.0. | ||
 | ||
| Fig. 2 Alternative perspective looking through the Na⋯Na axis, emphasising the “W-isomeric” form of complex 1. | ||
Also revealing is the space-filling model of 1calc (Fig. 3) in showing that Na1 is much less well protected in this arrangement making it more susceptible to onward reactivity than the other sodium environment between the planes of the cyclopentadienyl rings, which is clearly more hidden and thus less available to coordinate potential substrates.
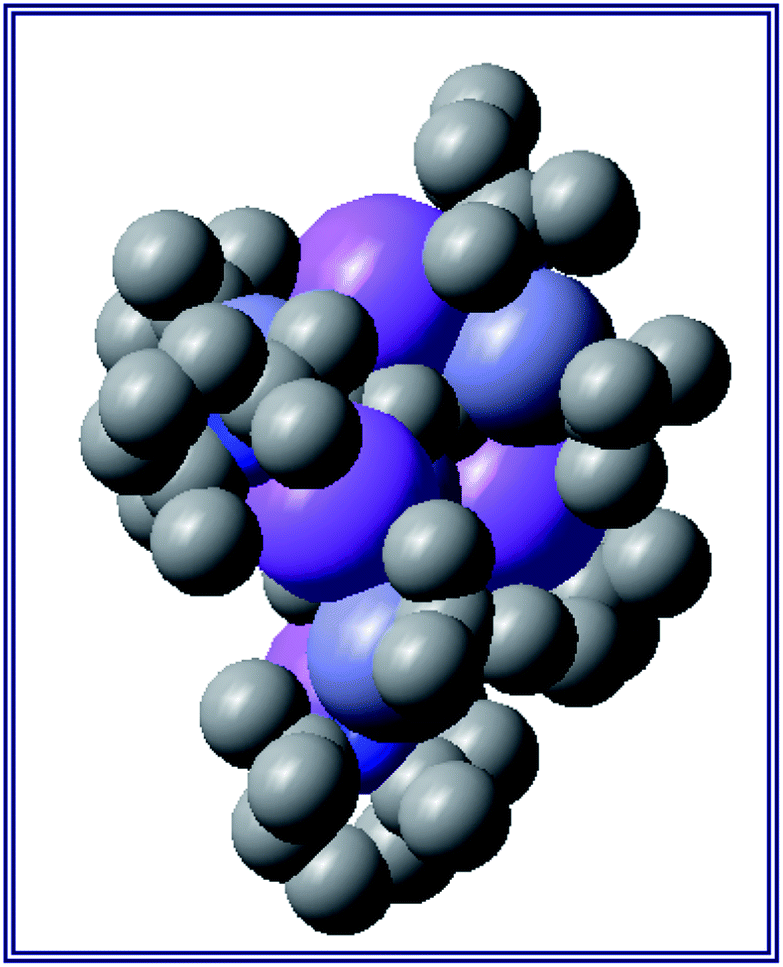 | ||
| Fig. 3 Space-filling model of complex 1calc. | ||
Solution state study of 1
Solution phase NMR analysis of complex 1 solubilized in C6D6 revealed dynamic characteristics not considered in the solid-state study. These characteristics were revealed following extensive and detailed 1H and 13C NMR data analysis using both 2D [1H, 1H] and 2D [1H, 13C] correlation data sets. Full details of the deductive reasoning behind the data interpretation are provided in the ESI (Fig. S1–S10 and Tables S1–S3†). Basic evidence existed for the presence in solution of a minor species alongside the major form. Following data analysis, it was clear that the major form of the complex in solution reflected the crystal structure (W-isomer) form. No definite conclusion could be drawn regarding the structure of the minor species although it can tenuously be assigned to a Z-isomeric intermediate. The distinction of NMR resonances for Cp ring protons arising from major and minor forms of the complex was supported by 2D [1H, 1H] EXSY NMR data for the sample studied at 298 K. This revealed clear evidence of chemical exchange between major and minor forms. Deeper investigation of this phenomenon revealed a further, concerted, intramolecular structural rearrangement of the major form of the complex. We speculate that the minor form could be present for one of two reasons or both. The minor (speculatively ‘open’) form of the complex could be an ‘active’ structure which allows for bond making and breaking to occur at the Cp ring. Alternatively, the minor form of the complex could be an ‘open’ intermediate species, which exists during rearrangement of the major form of the complex (Fig. 4).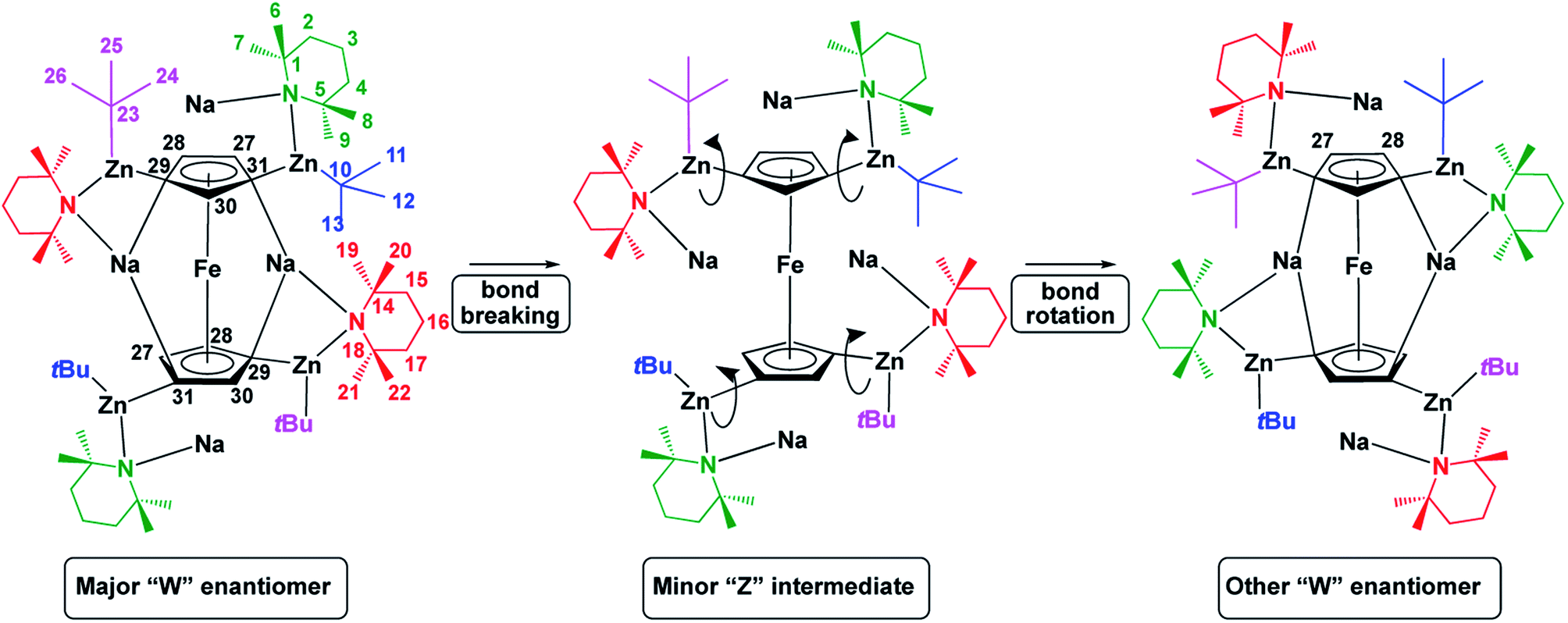 | ||
| Fig. 4 Potential interconversion of major isomer of 1 to its other enantiomer via a minor intermediate, with proton numbering system used in discussion shown for clarity. Colour coding is used to clarify movement of ligands upon rearrangement and not to specify whether a ligand is bridging/capping for example. | ||
The minor species could also be present serving both purposes or neither. It was not possible to define a structure for the minor species but we were intrigued by the observed changes in chemical shifts revealed through analysis of the chemical exchange process. Particularly noteworthy were the large changes in chemical shifts for TMPbMe20 and TMPbMe22 (Δδ1H = 0.788 ppm and 0.612 ppm to lower chemical shift respectively; see Fig. 4 for numbering system) whilst deshielding occurred for TMPcMe6 and TMPcMe7 (Δδ1H = 0.476 ppm and 0.308 ppm to higher chemical shift respectively; note b = bridging and c = capping). These changes suggest significant rearrangement of the structure in which the TMPb rings stack above the Cp rings and the TMPc rings assemble in a planar fashion adjacent to the Cp rings. This is consistent with chemical exchange-related EXSY NMR cross-peaks assigned to the intramolecular rearrangement of the major form of the complex. This reveals that capping and bridging groups interchange. Supporting evidence for this comes via chemical exchange cross-peaks between CpH27 and CpH28 resonances. This is only possible through intramolecular rearrangement of the major form to an identical but mirror image form. We therefore speculate that the minor form of the complex exists as a unique species, present with a relatively long lifetime (and therefore observable by NMR) as a half-way transition point between the oppositely handed major forms of the complex. Such an explanation may help to uncover how the chemistry of this complex is performed in the solution phase. A full analysis of the exchange rate characteristics, based on inspection of the Cp proton resonances, were derived through this interpretation and are reported in the ESI.† Overall, we conclude that an “opening” mechanism exists for this complex in solution in which the TMP and tBu groups relocate into quite different magnetic environments, after which the structure “closes” into either the same handed or the oppositely handed major form of the complex.
Reactivity of complex 1
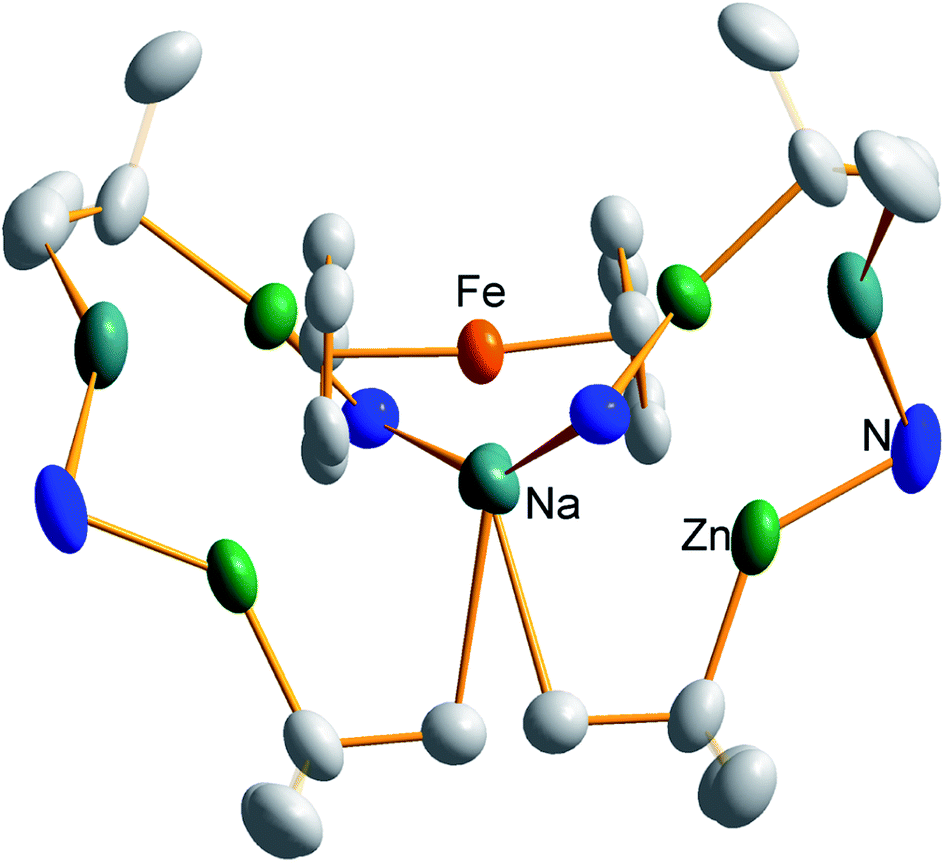
Quenching of complex 1 with D2O resulted in the formation of D4-Fc as evidenced by a GC-MS analysis with the predominant peak occurring at m/z 190.1, (Fig. S11†) consistent with tetradeuterated Fe(C5H3D2)2. On comparing with the predicted mass spectrum (Fig. S12†), it appears that a small amount of tri-deuterated ferrocene is also present due to the presence of small peaks at 189 and 187, which should be absent for pure tetra-deuterated ferrocene. That notwithstanding, it is clear that D4-Fc is the major product. Other peaks observed include C5H3D2 at m/z 67.2 and Fe(C5H3D2) at 123.0.Further investigation of the tetrazincated ferrocenyl complex 1 revealed novel and unexpected onward reactivity in its reactions with aromatic ring compounds. With toluene, 1 reacts through its peripheral alkyl ligands to deprotonate the lateral methyl arm and form the bis-benzyl derivative [Na4(TMP)4Zn4(tBu)2(CH2Ph)2{(C5H3)2Fe}] (complex 2, Scheme 2) in which the tetrasodium tetrazincated ferrocenyl scaffold remains intact. Interestingly, only two of the four tbutyl arms of 1 react, specifically the zinc-bonded terminal ones located above and below the plane of the cyclopentadienyl rings, with the other two (more protected) bridging tbutyl groups remaining unreacted. To the best of our knowledge the transformation of complex 1 into complex 2 is the first example of peripheral reactivity of an inverse crown complex, whereby an anionic component of the cationic {Na4(TMP)4Zn4(tBu)4}4+ unit engages in a deprotonation reaction. Interestingly, however, the location of toluene deprotonation is at the most thermodynamically acidic position24 and is thus in contrast to the synergistically-operative reactivity of TMEDA·Na(μ-TMP)(μ-tBu)ZntBu, which preferentially deprotonates toluene at a ring position via a stepwise TMP deprotonation followed by a t-butyl deprotonation of TMP(H) releasing butane and reforming TMP to generate TMEDA·Na(μ-TMP)(μ-C6H4Me)ZntBu.25 In gross features the structural motif of 2 is similar to that of 1 (Fig. 5, see Table 1 for selected bond lengths and angles). A tetrazincated core is encapsulated by a heteroleptic inverse crown which is now closed due to the bridging nature of the newly formed benzyl anions which, in turn, coordinate the zinc in a σ-fashion through the CH2 anion and the sodium in more of a π-fashion through the aromatic ring.26 This σ–π distinction has been observed in other mixed alkali metal–zinc complexes.26b,27
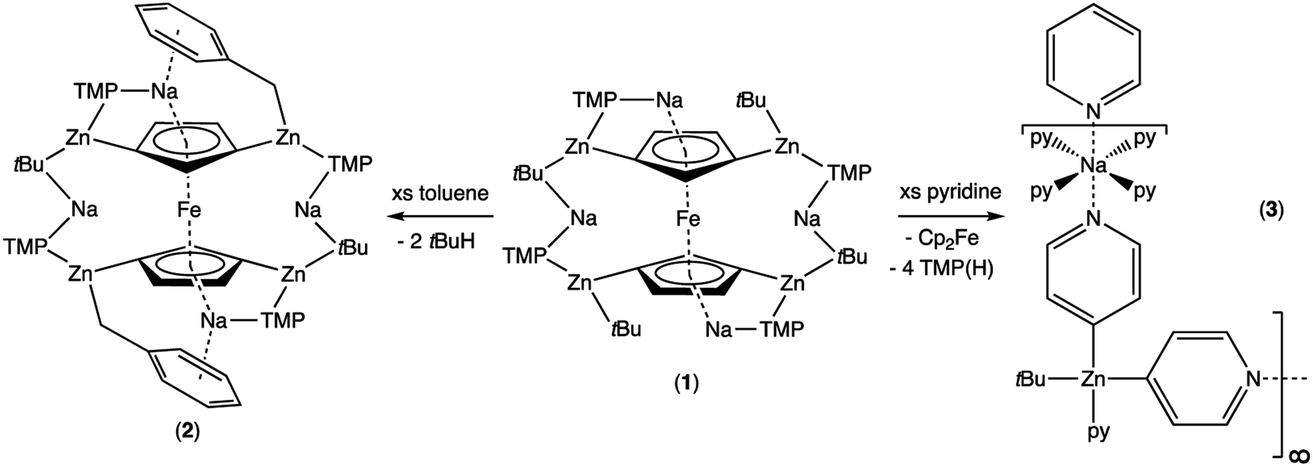 | ||
| Scheme 2 Divergent reactivity of complex 1 with toluene and pyridine. | ||
 | ||
| Fig. 5 Molecular structure of the tetrazincated ferrocene complex [Na4(TMP)4Zn4(tBu)2(CH2Ph)2{(C5H3)2Fe}] (2, top) and DFT optimised structure (2calc, bottom). Ellipsoids are shown at 50% probability and all hydrogen atoms have been removed for clarity. | ||
| M(1) | M(2) | M(3) | M(4) | |
|---|---|---|---|---|
| a Distance to benzyl is to centroid of aromatic ring. b Distance is to closest methyl carbon. c Angle at benzyl is from centroid of aromatic ring. d Angle at ferrocene is from centroid of Cp ring. | ||||
| Zn–CFc | 2.087(10), 2.108 | 2.022(10), 2.038 | 2.016(10), 2.037 | 2.076(10), 2.107 |
| Zn–CBz | — | 2.031(11), 2.057 | 2.049(10), 2.058 | — |
| Zn–CtBu | 2.072(12) | — | — | 2.059(12) |
| Zn–NTMP | 2.017(9), 2.040 | 2.013(9), 2.027 | 2.018(8), 2.027 | 2.020(9), 2.040 |
| Na–NTMP | 2.447(9), 2.467 | 2.386(10), 2.422 | 2.355(10), 2.424 | 2.477(9), 2.463 |
| Na–CFc | 2.456(4)a | 2.707(11), 2.562(12), 2.707, 2.597 | 2.698(10), 2.562(11), 2.705, 2.604 | 2.498(4)a |
| Na–CBz | 2.711(3)a | — | — | 2.686(5)a |
| Na–CtBu | — | 2.847(15)b | 2.812(13)b | — |
| C–Zn–CFc | 120.2(5) | 116.6(5) | 116.8(4) | 116.8(4) |
| C–Zn–NTMP | 134.2(4) | 127.5(4) | 127.8(4) | 135.1(4) |
| CFc–Zn–NTMP | 103.8(4), 103.4 | 115.8(4), 118.0 | 114.8(4), 117.9 | 104.1(4), 103.3 |
| CBz–Na–NTMP | 141.8(2)c | — | — | 139.6(3)c |
| CBz–Na–CFc | 112.2(1)c,d | — | — | 114.5(2)c,d |
| CFc–Na–NTMP | 106.0(3)d | — | — | 105.8(2)d |
| CFc–Na–CFc | — | 86.4(3), 86.5 | 85.9(3), 86.6 | — |
| CFc–Na–NTMP | — | 152.1(3), 84.0(3), 85.4 | 149.1(3), 84.3(3), 85.3 | — |
| Na–NTMP–Zn | 92.6(3), 93.3 | 83.5(3), 81.4 | 84.0(3), 81.5 | 91.0(3), 93.2 |
| Na–CFc–Zn | 84.9(4), 84.8 | 75.4(3), 74.4 | 75.6(3), 74.6 | 84.6(3), 84.6 |
For completeness, we also carried out a DFT optimisation giving 2calc, again starting from the experimental X-ray established coordinates of 2. The ΔE of the reaction of complex 1 with two equivalents of toluene to yield complex 2 and two molar equivalents of isobutane was calculated as being −35.40 kcal mol−1. The 1H NMR spectrum of complex 2 (Fig. S13†) was, as that of complex 1, highly complicated within the aliphatic region. However, the 3.5–4.5 ppm region was similar to that of complex 1, showing two sets of equal integration singlets indicative of a symmetrical 1,1′,3,3′-tetrametallated ferrocene.
Treating complex 1 with neat pyridine resulted in considerably divergent reactivity from that with toluene. The product complex 3 was shown by X-ray crystallography (Fig. 6) to be a chain polymer of formula {[Na·4py][Zn(py*)2(tBu)·py]}∞, where py is solvating pyridine (C5H5N) and py* is an anionic derivative of pyridine formed through deprotonation of the heterocycle at the 4-position (4-C5H4N). The structure can be considered an ate arrangement in that the sodium centre is formally positive (surrounded by six solvating Lewis-donor pyridyl N-atoms in an approximate octahedral arrangement); while the monoanionic zinc moiety adopts a tetrahedral zincate structure consisting of a tbutyl anion, two pyridyl C-anions and a neutral pyridine molecule. The polymer propagates via a pair of pyridyl anions acting as a N,C ditopic linker between sodium (N bound) and zinc (C bound). With respect to sodium, these linking molecules are trans-disposed, effecting a near linear unit to the polymer while the near tetrahedral nature of the zinc [C23–Zn–C28 bond angle = 114.28(18)°] enforces the zig–zagging conformation (Fig. 7). Each polymeric chain propagates via a 21 screw axis acting parallel to the crystallographic c direction. The chains pack into a layered structure. The layers lie parallel to the ab plane and each layer is composed only of Na(py)6 or ZntBu(py)3 units. These alternate in the c direction as shown in Fig. S14.†
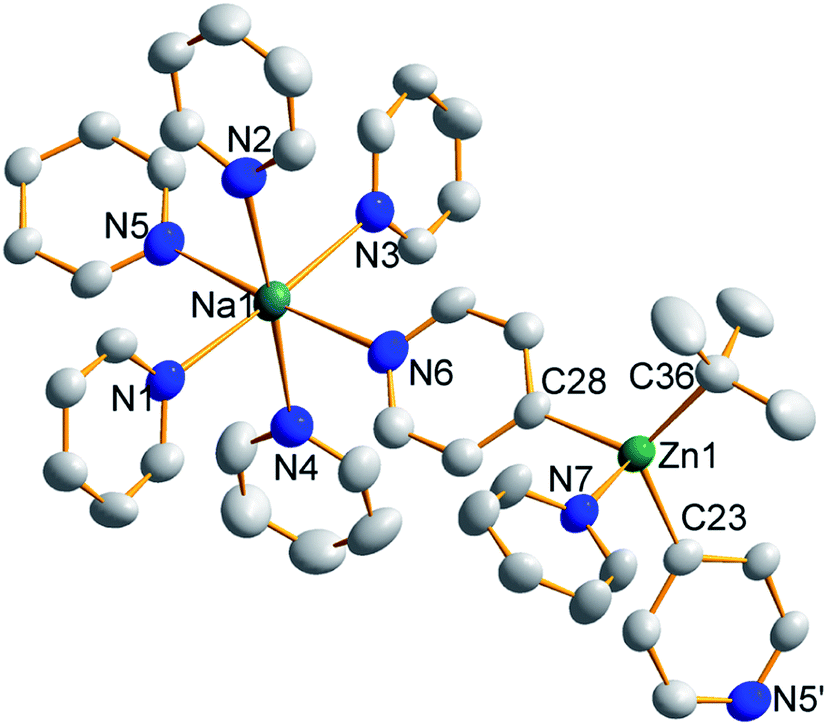 | ||
| Fig. 6 Structure emphasising the asymmetric unit of sodium zincate {[Na·4py][Zn(py*)2(tBu)·py]}∞ (3). Ellipsoids are shown at 50% probability and all hydrogen atoms have been removed for clarity. Selected bond lengths (Å) and angles (°): Na1–N1, 2.512(4); Na1–N2, 2.586(5); Na1–N3, 2.533(5); Na1–N4, 2.600(5); Na1–N5, 2.583(5); Na1–N6, 2.548(4); Zn1–N7, 2.254(3); Zn1–C23, 2.057(5); Zn1–C28, 2.061(4); Zn1–C36, 2.049(4); N1–Na1–N2, 89.52(15); N1–Na1–N3, 176.30(15); N1–Na1–N4, 88.09(15); N1–Na1–N5, 91.30(15); N1–Na1–N6, 93.76(13); N2–Na1–N3, 87.09(15); N2–Na1–N4, 177.04(16); N2–Na1–N5, 88.38(14); N2–Na1–N6, 92.62(14); N3–Na1–N4, 95.25(15); N3–Na1–N5, 87.13(15); N3–Na1–N6, 87.87(14); N4–Na1–N5, 89.93(15); N4–Na1–N6, 89.28(14); N5–Na1–N6, 174.85(15); C23–Zn1–C28, 114.28(18); C23–Zn1–C36, 117.38(19); C23–Zn1–N7, 97.57(18); C28–Zn1–C36, 119.81(18); C28–Zn1–N7, 99.22(17); C36–Zn1–N7, 102.47(14). | ||
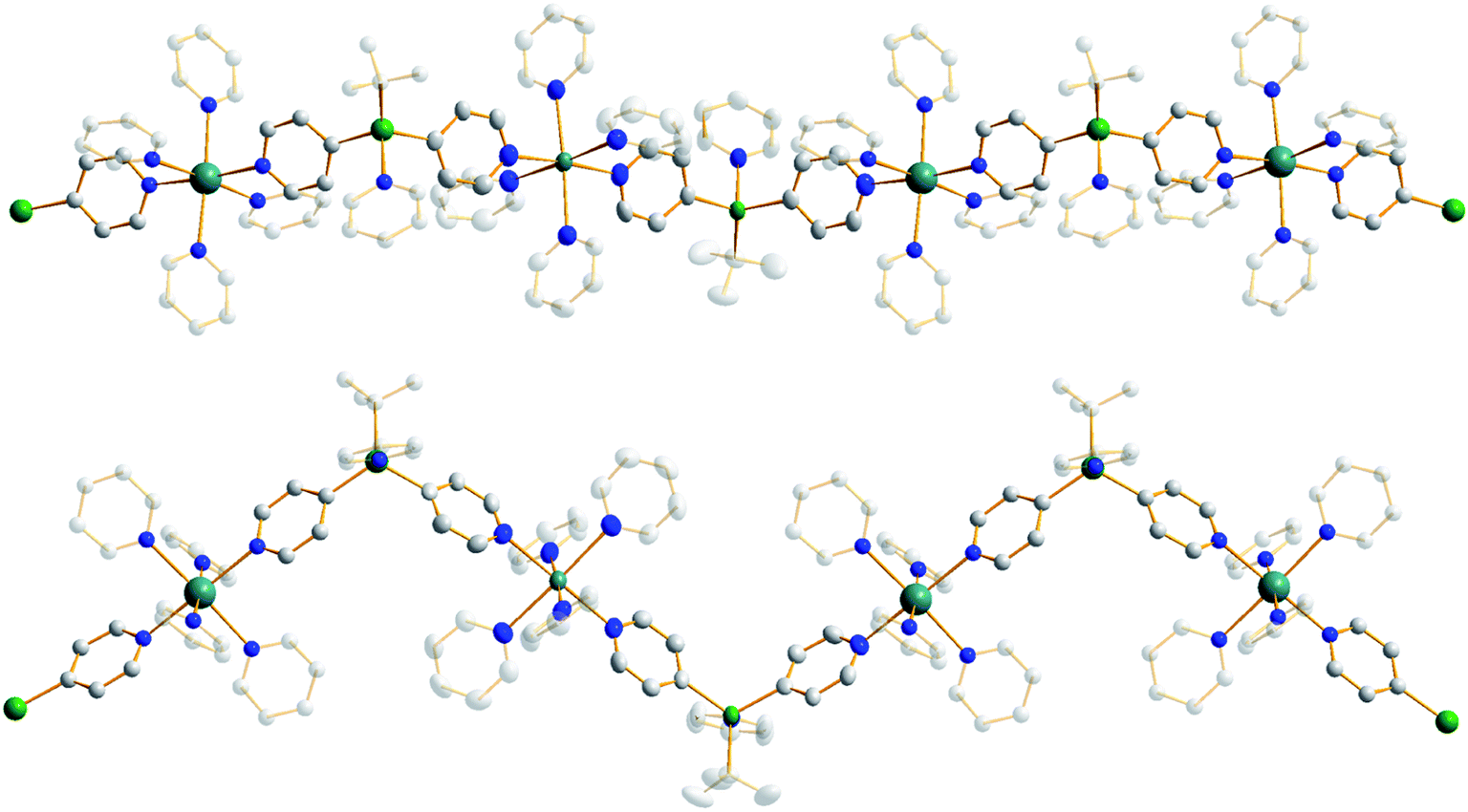 | ||
| Fig. 7 Propagation of {[Na·4py][Zn(py*)2(tBu)·py]}∞ (3) into a polymer shown from the side and above. Ellipsoids are shown at 50% probability, all hydrogen atoms have been removed and all tBu carbon atoms and neutral donor pyridine ligands are highly transparent for clarity. | ||
Reports of characterised 4-metallated pyridine molecules are rare, particularly with respect to either zinc or the s-block. A series of structures have been published utilizing the heavier group 12 metal mercury although these have been prepared via mercury(II) acetate and the aryl boronic acid 28 or by preparing the Grignard reagent via metal–halogen exchange of 4-iodopyridine followed by metathesis with mercuric chloride.29 Metal–halogen exchange is the principal route to metallated pyridines, although direct metallation with bimetallic alkali-metal complexes such as LiCKOR30 or Caubere's base31 is possible albeit metallation is predominantly directed α to the heteroatom.32
This reactivity with pyridine is in complete contrast to the reaction of 1 with toluene in that here, the parent tetrazincated ferrocenyl species exhibits simultaneous core and peripheral reactivity acting as a dual ferrocenyl/TMP base with retention of tbutyl groups in 3 as opposed to acting as a tbutyl base with retention of the TMP and ferrocenyl anions in 2. This was further supported by the presence of TMP(H) and ferrocene in the 1H NMR spectrum of the filtrate.
As the yield of isolated recrystallized complex 3 was low (9%), the reaction was repeated in the less polar solvent hexane with pyridine slowly added until complete dissolution had been reached. Unfortunately, despite several attempts no solid product could be obtained with an orange oil separating from the solution instead. Attempts to recrystallize the product from toluene were also unsuccessful. After isolating the crystals from pyridine solution, the solvent was removed from the remaining solution under vacuum and analysed by 1H NMR spectroscopy but the presence of many overlapping resonances meant it was not possible to determine the presence of more 3 or any other metallation products. To gain more insight the reaction of 1 with pyridine was repeated in situ and then electrophilically quenched with iodine to assess the metallation outcome indirectly. After work-up, the organic products were studied via1H NMR spectroscopy (Fig. 8), with the aromatic region revealing the presence of 4-iodopyridine (as expected) alongside 3-iodopyridine and some pyridine solvent. The ratio of 3-iodopyridine:4-iodopyridine was approximately 57![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :43 suggesting that 3-metallation is slightly more predominant while integration of these resonances versus an internal standard of hexamethylbenzene suggested that the total amount of pyridine metallation was under 20%. That the major 3-metallated product did not preferentially recrystallise perhaps suggests that it is a discrete molecule rather than a polymer (as in 3) and thus is less inclined to precipitate from the highly polar pyridine solution. A literature search identified the report of a 3-zincated pyridyl compound exhibiting an eye-catching infinite stepladder arrangement made by reacting 4-methoxypyridine with the related mixed-metal base PMDETA·K(μ-TMP)(μ-Et)ZnEt. However, in this case the 4-position was blocked by a methoxy group which can be considered as a directing group towards the position ortho to itself (that is the 3-position).33 The Zn–Cpyr distance in this complex [2.062(4) Å] is almost identical to that of 3 [2.061(4) Å].
:43 suggesting that 3-metallation is slightly more predominant while integration of these resonances versus an internal standard of hexamethylbenzene suggested that the total amount of pyridine metallation was under 20%. That the major 3-metallated product did not preferentially recrystallise perhaps suggests that it is a discrete molecule rather than a polymer (as in 3) and thus is less inclined to precipitate from the highly polar pyridine solution. A literature search identified the report of a 3-zincated pyridyl compound exhibiting an eye-catching infinite stepladder arrangement made by reacting 4-methoxypyridine with the related mixed-metal base PMDETA·K(μ-TMP)(μ-Et)ZnEt. However, in this case the 4-position was blocked by a methoxy group which can be considered as a directing group towards the position ortho to itself (that is the 3-position).33 The Zn–Cpyr distance in this complex [2.062(4) Å] is almost identical to that of 3 [2.061(4) Å].
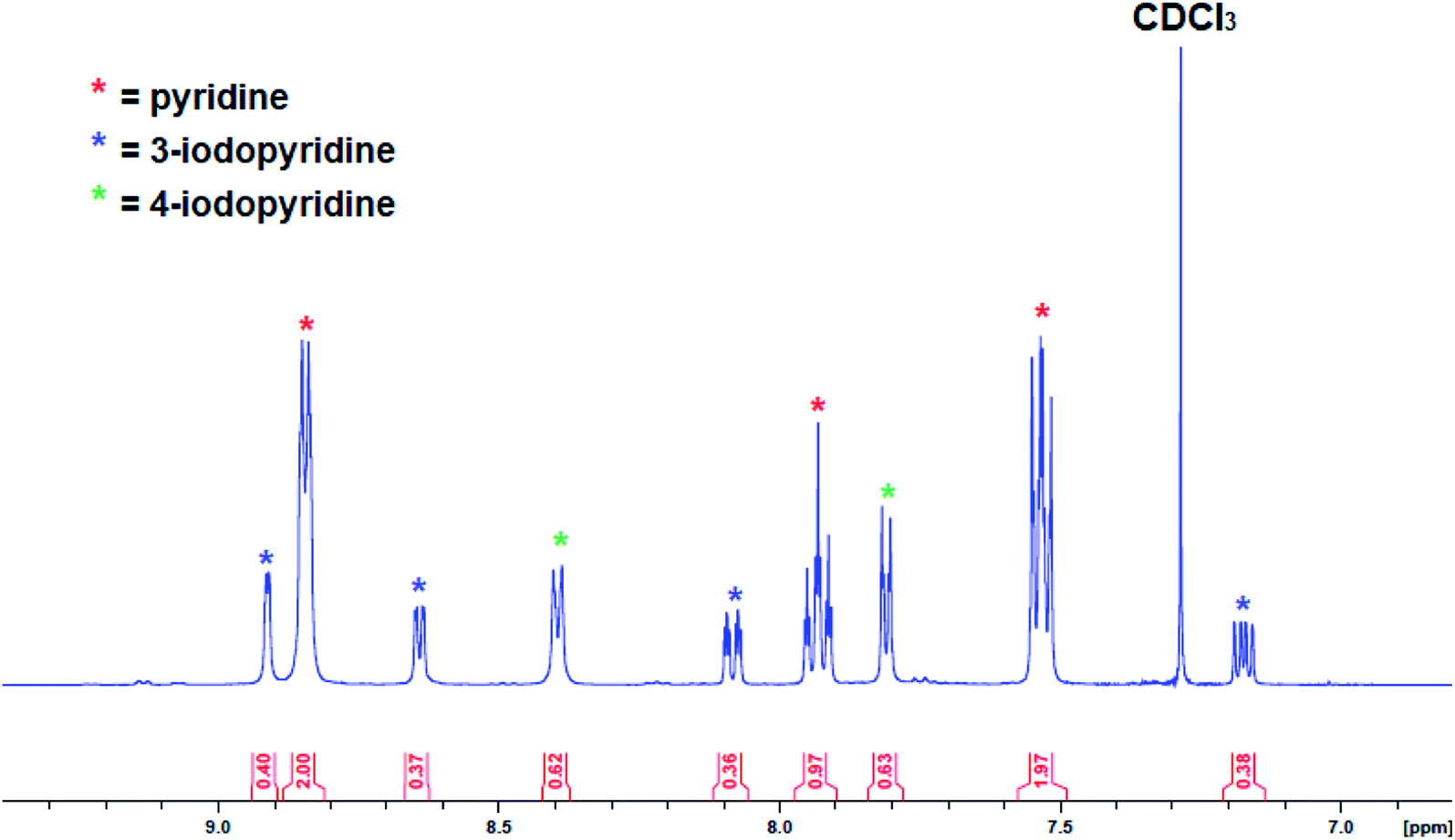 | ||
| Fig. 8 Aromatic region of the 1H NMR spectrum (in CDCl3) of work-up products after reaction of complex 1 with pyridine followed by an iodine quench. | ||
Interestingly, the peripheral reactivity towards toluene appears to be limited solely to the tetrazincated ferrocene complex 1. Dizincated ferrocene complex B is inert to toluene as demonstrated by the molecular structure of the recrystallised product upon refluxing in neat toluene, which is simply B with a toluene molecule of crystallisation trapped within the lattice (Fig. S15†). Likewise, a 1H NMR spectrum of complex B refluxed in pyridine showed no evidence of heterocycle metallation.
Conclusions
Zinc alkyls and homometallic zinc compounds in general are exceptionally poor bases due primarily to insufficient Zn–C bond polarity, but here through the assistance of sodium TMP, a fourfold deprotonation of ferrocene is accomplished. While alkali-metal-mediated zincation (AMMZn) is well established, to our knowledge this is the only example where four C–H to C–Zn(R) transformations have been reported on a single molecule. A single crystal structure determination supported by DFT studies have elucidated a discrete open inverse crown arrangement where two [Na(TMP)Zn(tBu)Na(TMP)Zn(tBu)]2+ cationic units trap a {(C5H3)2Fe}4− tetraanion. Reactions of this polyzincated complex with toluene and pyridine result in different outcomes. The former produces two benzyl anions which replace two of the alkyl substituents in [Na4(TMP)4Zn4(tBu)4{(C5H3)2Fe}] to give [Na4(TMP)4Zn4(tBu)2(CH2Ph)2{(C5H3)2Fe}], which retains the gross inverse crown structure. In contrast, the latter produces {[Na·4py][Zn(py*)2(tBu)·py]}∞, a zig–zag chain polymer possessing both neutral and deprotonated (at the 4-position) pyridine ligands. These initial studies suggest that [Na4(TMP)4Zn4(tBu)4{(C5H3)2Fe}] has broad reactivity scope since it can utilise both peripheral (t-butyl, TMP) and core (ferrocenyl) anions in its reactions. Since methodologies to convert C–Zn bonds to other C–X bonds are well known, such polyzincated molecules may provide access to novel compounds and materials in future work which are of interest to synthetic, macromolecular and supramolecular scientists amongst others.Experimental
General experimental
All reactions and manipulations were conducted under a protective argon atmosphere using either standard Schlenk techniques or an MBraun glove box fitted with a gas purification and recirculation unit. Solvents were dried by heating to reflux over sodium benzophenone ketyl and then distilled under nitrogen prior to use. tBu2Zn was prepared according to literature procedures.22 All other chemicals were obtained from commercial sources and used as received. Elemental analyses were performed at the University of Strathclyde Elemental Analysis Service.X-ray crystallography
Crystallographic data were collected on Bruker APEX2 or Oxford Diffraction instruments using synchrotron, Mo Kα or Cu Kα radiation (λ = 0.6814, 0.71073 and 1.54184 Å respectively). Structures were solved using SHELXS-97 or OLEX2, while refinement was carried out on F2 against all independent reflections by the full matrix least-squares method using the SHELXL-97 or SHELXL-2018/3 program or by the GaussNewton algorithm using OLEX2. All non-hydrogen atoms were refined using anisotropic displacement parameters. Selected crystallographic details and refinement details are provided in Table S4.† CCDC 1972641–1972644 contains the supplementary crystallographic data for this paper.Theoretical calculations
DFT calculations were performed using the Gaussian 03 (ref. 34) package using the B3LYP 35 density functional and the 6-311(d, p)36 basis set. After each geometry optimization a frequency analysis was performed.NMR spectroscopy
NMR data for signal assignment and chemical exchange analysis were acquired on a three r.f. channel Bruker AVANCE II+ NMR spectrometer equipped with a 14.1 T UltraShield™ superconducting magnet housing a 5 mm ∅ BBO-z-ATMA probehead, which was maintained at 298 K. The sample was solubilized in C6D6. Data were acquired and processed under Topspin 3.5 patch level 7 running on a Hewlett-Packard Z420 Workstation equipped with an Intel Xeon CPU E5-1620 v2 processor operating at 3.70 GHz under Windows 7 Professional edition.All data assignments were carried out exclusively within the NMRFAM-SPARKY edition of the NMR data handling software package SPARKY.38
1H NMR (400.13 MHz, NC5D5, 300 K): δ 0.51 [9H, s, tBu], 8.24 [4H, d, 3J(H,H) = 4.8 Hz, −py β H], 8.59 [4H, d, 3J(H,H) = 4.80 Hz, −py α H]. 13C NMR (100.6 MHz, NC5D5, 300 K): δ 0.2 [tBu], 0.7 [tBu], 137.1 [−py β C], 145.5 [−py α C], metallated pyridine carbon could not be resolved.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
W. C. wishes to thank the Science and Technology Funding Council for access to Synchrotron Radiation Source at Daresbury Laboratory.References
- (a) W. N. Setzer and P. v. R. Schleyer, Adv. Organomet. Chem., 1985, 24, 353–451 CrossRef CAS; (b) V. H. Gessner, C. Däschlein and C. Strohmann, Chem.–Eur. J., 2009, 15, 3320–3334 CrossRef CAS; (c) H. J. Reich, Chem. Rev., 2013, 113, 7130–7178 CrossRef; (d) E. Carl and D. Stalke, in Lithium Compounds in Organic Synthesis, ed. R. Luisi and V. Capriati, Wiley-VCH, Weinheim, 2014, pp. 1–32 Search PubMed.
- R. E. Mulvey and S. D. Robertson, Angew. Chem., Int. Ed., 2013, 52, 11470–11487 CrossRef.
- (a) R. E. Mulvey, F. Mongin, M. Uchiyama and Y. Kondo, Angew. Chem., Int. Ed., 2007, 46, 3802–3824 CrossRef; (b) S. D. Robertson, M. Uzelac and R. E. Mulvey, Chem. Rev., 2019, 119, 8332–8405 CrossRef.
- E. Hevia and R. E. Mulvey, Angew. Chem., Int. Ed., 2011, 50, 6448–6450 CrossRef.
- R. Neufeld and D. Stalke, Chem.–Eur. J., 2016, 22, 12624–12626 CrossRef.
- A. Castello-Mico and P. Knochel, Synthesis, 2018, 50, 155–169 CrossRef.
- B. Haag, M. Mosrin, H. Ila, V. Malakhov and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 9794–9824 CrossRef.
- (a) M. Schlosser, J. H. Choi and S. Takagishi, Tetrahedron, 1990, 46, 5633–5648 CrossRef; (b) E. Baston, R. Maggi, K. Friedrich and M. Schlosser, Eur. J. Org. Chem., 2001, 3985–3989 CrossRef; (c) W. Clegg, S. H. Dale, A. M. Drummond, E. Hevia, G. W. Honeyman and R. E. Mulvey, J. Am. Chem. Soc., 2006, 128, 7434–7435 CrossRef; (d) E. Nagaradja, F. Chevallier, T. Roisnel, V. Dorcet, Y. S. Halauko, O. A. Ivashkevich, V. E. Matulis and F. Mongin, Org. Biomol. Chem., 2014, 12, 1475–1487 RSC; (e) M. Y. A. Messaoud, G. Bentabed-Ababsa, M. Hedidi, A. Derdour, F. Chevallier, Y. S. Halauko, O. A. Ivashkevich, V. E. Matulis, L. Picot, V. Thiery, T. Roisnel, V. Dorcet and F. Mongin, Beilstein J. Org. Chem., 2015, 11, 1475–1485 CrossRef; (f) M. Hedidi, W. Erb, F. Lassagne, Y. S. Halauko, O. A. Ivashkevich, V. E. Matulis, T. Roisnel, G. Bentabed-Ababsa and F. Mongin, RSC Adv., 2016, 6, 63185–63189 RSC.
- (a) A. J. Martinez-Martinez, A. R. Kennedy, R. E. Mulvey and C. T. O'Hara, Science, 2014, 346, 834–837 CrossRef; (b) A. J. Martinez-Martinez, S. Justice, B. J. Fleming, A. R. Kennedy, I. D. H. Oswald and C. T. O'Hara, Sci. Adv., 2017, 3, e1700832 CrossRef.
- A. J. Martinez-Martinez, D. R. Armstrong, B. Conway, B. J. Fleming, J. Klett, A. R. Kennedy, R. E. Mulvey, S. D. Robertson and C. T. O'Hara, Chem. Sci., 2014, 5, 771–781 RSC.
- (a) Ferrocenes, Ligands, Materials and Biomolecules, P. Štěpnička, Wiley, Chichester, 2008 Search PubMed; (b) K. Heinze and H. Lang, Organometallics, 2013, 32, 5623–5625 CrossRef; (c) D. Astruc, Eur. J. Inorg. Chem., 2017, 6–29 CrossRef; (d) M. Patra and G. Gasser, Nat. Rev. Chem., 2017, 1, 0066 CrossRef.
- W. Clegg, K. W. Henderson, A. R. Kennedy, R. E. Mulvey, C. T. O'Hara, R. B. Rowlings and D. M. Tooke, Angew. Chem., Int. Ed., 2001, 40, 3902–3905 CrossRef CAS.
- B. Jennewein, S. Kimpel, D. Thalheim and J. Klett, Chem.–Eur. J., 2018, 24, 7605–7609 CrossRef CAS.
- R. A. Benkeser, D. Goggin and G. Schroll, J. Am. Chem. Soc., 1954, 76, 4025–4026 CrossRef CAS.
- For other crystallographically characterized zincated ferrocenyl complexes see: (a) H. R. L. Barley, W. Clegg, S. H. Dale, E. Hevia, G. W. Honeyman, A. R. Kennedy and R. E. Mulvey, Angew. Chem., Int. Ed., 2005, 44, 6018–6021 CrossRef CAS; (b) A. S. Perucha, J. Heilmann-Brohl, M. Bolte, H.-W. Lerner and M. Wagner, Organometallics, 2008, 28, 6170–6177 CrossRef; (c) N. Seidel, K. Jacob, P. Zanello and M. Fontani, J. Organomet. Chem., 2001, 620, 243–248 CrossRef CAS; (d) E. Hevia, A. R. Kennedy and M. D. McCall, Dalton Trans., 2012, 41, 98–103 RSC; (e) W. Clegg, B. Conway, P. Garcia-Alvarez, A. R. Kennedy, J. Klett, R. E. Mulvey and L. Russo, Dalton Trans., 2010, 39, 62–65 RSC.
- (a) M. Uchiyama and C. Wang, Top. Organomet. Chem., 2014, 47, 159–202 CrossRef CAS; (b) A. Hernan-Gomez, E. Herd, M. Uzelac, T. Cadenbach, A. R. Kennedy, I. Borilovic, G. Aromi and E. Hevia, Organometallics, 2015, 34, 2614–2623 CrossRef CAS; (c) A. Hernan-Gomez, S. A. Orr, M. Uzelac, A. R. Kennedy, S. Barroso, X. Jusseau, S. Lemaire, V. Farina and E. Hevia, Angew. Chem., Int. Ed., 2018, 57, 10630–10634 CrossRef CAS PubMed; (d) B.-K. Tao, H. Yang, Y.-Z. Hua and M.-C. Wang, Org. Biomol. Chem., 2019, 17, 4301–4310 RSC; (e) G. Trott, J. A. Garden and C. K. Williams, Chem. Sci., 2019, 10, 4618–4627 RSC.
- (a) S. Sase, M. Jaric, A. Metzger, V. Malakhov and P. Knochel, J. Org. Chem., 2008, 73, 7380–7382 CrossRef CAS; (b) C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS; (c) L. C. McCann, H. N. Hunter, J. A. C. Clyburne and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 7024–7027 CrossRef CAS; (d) D. Haas, J. M. Hammann, R. Greiner and P. Knochel, ACS Catal., 2016, 6, 1540–1552 CrossRef CAS.
- R. E. Mulvey, Chem. Commun., 2001, 1049–1056 RSC.
- W. Clegg, E. Crosbie, S. H. Dale-Black, E. Hevia, G. W. Honeyman, A. R. Kennedy, R. E. Mulvey, D. L. Ramsay and S. D. Robertson, Organometallics, 2015, 34, 2580–2589 CrossRef CAS.
- D. R. Armstrong, W. Clegg, S. H. Dale, D. V. Graham, E. Hevia, L. M. Hogg, G. W. Honeyman, A. R. Kennedy and R. E. Mulvey, Chem. Commun., 2007, 598–600 RSC.
- (a) P. C. Andrikopoulos, D. R. Armstrong, A. R. Kennedy, R. E. Mulvey, C. T. O'Hara and R. B. Rowlings, Eur. J. Inorg. Chem., 2003, 3354–3362 CrossRef CAS; (b) A. R. Kennedy, J. Klett, R. E. Mulvey, S. Newton and D. S. Wright, Chem. Commun., 2008, 308–310 RSC; (c) X.-H. Lu, M.-T. Ma, Y.-M. Yao, Y. Zhang and Q. Shen, Inorg. Chem. Commun., 2010, 13, 1566–1568 CrossRef CAS; (d) A. R. Kennedy, R. E. Mulvey, C. T. O'Hara, G. M. Robertson and S. D. Robertson, Angew. Chem., Int. Ed., 2011, 50, 8375–8378 CrossRef CAS PubMed.
- P. C. Andrikopoulos, D. R. Armstrong, H. R. L. Barley, W. Clegg, S. H. Dale, E. Hevia, G. W. Honeyman, A. R. Kennedy and R. E. Mulvey, J. Am. Chem. Soc., 2005, 127, 6184–6185 CrossRef CAS PubMed.
- P. C. Andrikopoulos, D. R. Armstrong, W. Clegg, C. J. Gilfillan, E. Hevia, A. R. Kennedy, R. E. Mulvey, C. T. O'Hara, J. A. Parkinson and D. M. Tooke, J. Am. Chem. Soc., 2004, 126, 11612–11620 CrossRef CAS PubMed.
- (a) L. Lochmann, H. Jakabův and L. Brandsma, Collect. Czech. Chem. Commun., 1993, 58, 1445–1451 CrossRef CAS; (b) I. A. Topol, G. J. Tawa, R. A. Caldwell, M. A. Eissenstat and S. K. Burt, J. Phys. Chem. A, 2000, 104, 9619–9624 CrossRef CAS.
- (a) D. R. Armstrong, J. Garcia-Alvarez, D. V. Graham, G. W. Honeyman, E. Hevia, A. R. Kennedy and R. E. Mulvey, Chem.–Eur. J., 2009, 15, 3800–3807 CrossRef CAS; (b) W. Clegg, B. Conway, E. Hevia, M. D. McCall, L. Russo and R. E. Mulvey, J. Am. Chem. Soc., 2009, 131, 2375–2384 CrossRef CAS.
- (a) M. G. Davidson, D. Garcia-Vivo, A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Chem.–Eur. J., 2011, 17, 3364–3369 CrossRef CAS PubMed; (b) D. R. Armstrong, M. G. Davidson, D. Garcia-Vivo, A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Inorg. Chem., 2013, 52, 12023–12032 CrossRef CAS PubMed.
- (a) W. Clegg, G. C. Forbes, A. R. Kennedy, R. E. Mulvey and S. T. Liddle, Chem. Commun., 2003, 406–407 RSC; (b) J. A. Garden, A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Dalton Trans., 2011, 40, 11945–11954 RSC.
- D. V. Partyka and T. G. Gray, J. Organomet. Chem., 2009, 694, 213–218 CrossRef CAS.
- T. Mocanu, C. I. Rat, C. Maxim, S. Shova, V. Tudor, C. Silvestru and M. Andruh, CrystEngComm, 2015, 17, 5474–5487 RSC.
- J. Verbeek, A. V. E. George, R. L. P. de Jong and L. Brandsma, J. Chem. Soc., Chem. Commun., 1984, 257–258 RSC.
- P. Gros, Y. Fort and P. Caubere, J. Chem. Soc., Perkin Trans. 1, 1997, 3597–3600 RSC.
- For a review of pyridine metallation chemistry see M. Schlosser and F. Mongin, Chem. Soc. Rev., 2007, 36, 1161–1172 RSC.
- B. Conway, D. V. Graham, E. Hevia, A. R. Kennedy, J. Klett and R. E. Mulvey, Chem. Commun., 2008, 2638–2640 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, V. Tomasi, B. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. B. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03 (Revision C.02), Gaussian Inc., Wallingford, CT, 2004 Search PubMed.
- (a) W. Kohn, A. D. Becke and R. G. Parr, J. Phys. Chem., 1996, 100, 12974–12980 CrossRef CAS; (b) A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- (a) A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS; (b) R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- M. Foroozandeh, R. W. Adams, M. Nilsson and G. A. Morris, J. Am. Chem. Soc., 2014, 136, 11867–11869 CrossRef CAS PubMed.
- W. Lee, M. Tonelli and J. L. Markley, Bioinformatics, 2015, 31, 1325–1327 CrossRef PubMed.
- J. C. Cobas and M. Martin-Pastor, EXSYCalc, 1.0, Mestrelab Research, Santiago de Compostela Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1972641–1972644. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc01612h |
| ‡ Current address: Department für Chemie und Biochemie, Universität Bern, CH3012, Bern, Switzerland. |
| § Current address: School of Chemistry, Monash University, Clayton, Melbourne, VIC 3800, Australia. |
| This journal is © The Royal Society of Chemistry 2020 |