
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of a binary alloy nanoparticle catalyst with an immiscible combination of Rh and Cu assisted by hydrogen spillover on a TiO2 support†
Shinya
Masuda
 a,
Kazuki
Shun
a,
Kohsuke
Mori
*ab,
Yasutaka
Kuwahara
ab and
Hiromi
Yamashita
*ab
a,
Kazuki
Shun
a,
Kohsuke
Mori
*ab,
Yasutaka
Kuwahara
ab and
Hiromi
Yamashita
*ab
aDivision of Materials and Manufacturing Science, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: mori@mat.eng.osaka-u.ac.jp; yamashita@mat.eng.osaka-u.ac.jp; Fax: +81-6-6879-7457; Tel: +81-6-6879-7457
bUnit of Elements Strategy Initiative for Catalysts Batteries (ESICB), Kyoto University, Katsura, Kyoto 615-8520, Japan
First published on 1st April 2020
Abstract
This work demonstrated the use of TiO2 as a promising platform for the synthesis of non-equilibrium RhCu binary alloy nanoparticles (NPs). These metals are regarded as immiscible based on their phase diagram but form NPs with the aid of the significant hydrogen spillover on TiO2 with concurrent proton–electron transfer. The resulting RhCu/TiO2 exhibited 2.6 times higher catalytic activity than Rh/TiO2 during hydrogen production from the hydrolysis of ammonia borane (AB), due to a synergistic effect. Theoretical simulations showed a higher energy value for the adsorption of AB on the RhCu alloy and a lower activation energy for the rate determining N–B bond dissociation by the attack of H2O during AB hydrolysis compared to monometallic Rh. High-angle annular dark-field scanning transmission electron microscopy and energy-dispersive X-ray spectroscopy confirmed the formation of RhCu alloy NPs with a mean diameter of 2.0 nm on the TiO2. H2-temperature programmed reduction and in situ X-ray absorption fine structure analyses at elevated temperature under H2 demonstrated that Rh3+ and Cu2+ precursors were simultaneously reduced only on the TiO2 support. This effect resulted from the improved and limited reducibility of Cu2+ and Rh3+, respectively. The rate of hydrogen spillover of TiO2 is faster as compared to γ-Al2O3 and MgO as evidenced by sequential H2/D2 exchanges during in situ Fourier transform infrared analyses. Density functional theory calculations also showed that the migration of H atoms on TiO2 proceeds with a lower energy barrier than that on Al2O3, and the reduction of Cu2+ species is facilitated by H spillover on the support rather than by direct reduction by H2. These results confirm the vital role of TiO2 in the formation of the alloy and may represent a new strategy for the synthesis of different non-equilibrium solid solution alloys.
Introduction
Over the past decade, metal nanoparticles (NPs) have been studied intensively with regard to a wide range of potential industrial,1,2 biomedical,3–5 electronic,6,7 and catalytic applications.8–11 This is due to the unique properties of NPs, which are different from those of bulk metals, and the small size effects they exhibit.12 In addition, binary alloys, whose nanostructure can consist of solid solution, segregated, or core–shell structures, have been widely researched as advanced catalysts because they show superior catalytic activity and selectivity compared to single metal NPs.12–17 Various geometric and electronic effects can appear as a result of differences in certain characteristics of each metal, such as work functions, ionization potentials, and electronegativity.14,18,19 These factors can, in turn, have significant effects on the properties of binary alloys. Therefore, the formulations of binary alloys are a vital factor in terms of optimizing their performance.The replacement of expensive noble metals with low-cost base metals is also an important consideration that affects the economic aspects of these materials. However, there are several combinations of noble and base metals, such as Au–Co, Ru–Ni,20 and Rh–Cu,21 that are difficult to process into solid solution alloys based on their phase diagram. Thus, even though such combinations have unexplored potential, the catalytic properties of these solid solution alloys have not yet been elucidated. In this regard, Kitagawa et al. have reported the use of a conventional procedure to synthesize solid solution alloys with immiscible combinations of metals using a polyol method.17,21,22 In addition, Humphrey et al. also successfully synthesized solid solution alloys by employing a combination of the polyol method and microwave heating.23,24 These research groups demonstrated that the resulting binary alloys with immiscible combinations show interesting performance and characteristics for specific catalytic reactions. The development of new and facile techniques for the synthesis of such solid solution alloys with immiscible metal combinations remains desirable, even though these NPs are not thermodynamically stable and readily grow to form larger particles.25,26 One means of mitigating this problem is to fix the NPs on supports having relatively high surface areas, such as metal oxides, so as to maintain their small sizes particularly in the field of catalysis. Interestingly, metal NPs are affected by the supports employed during synthesis and will exhibit different properties depending on the support.26–28 Therefore, the selection of an appropriate support for use during the growth of metal NPs is very important, and it is crucial to understand the phenomena that occur on various supports.29
Hydrogen spillover is one such phenomenon that is of interest in heterogeneous catalysis. This process involves the surface migration of dissociated H atoms from noble metal particles to nonmetal supports.30–36 This spillover effect has been widely studied30,34–36 and applied to catalytic systems and the fabrication of hydrogen storage materials.37–43 Several types of hydrogenation have been identified in which substrates adsorbed on supports are hydrogenated by activated H atoms generated on metal catalysts and spilled over to supports.38,42 Hydrogen spillover has also emerged as a promising strategy for high-density hydrogen storage under near-ambient conditions, based on the utilization of carbon-based materials,39,43 metal–organic frameworks,40,44 and zeolites.45 Hydrogen spillover is initiated by the formation of protons (H+) and electrons (e−) from H atoms at metal–support interfaces.30 The protons diffuse to O2− anions to form O–H and H–O–H bonds on the support surface, whereas the electrons simultaneously promote the partial reduction of metals in reducible metal oxides.46,47 In the case of a TiO2 support, Ti4+ ions are partially reduced to Ti3+ by the electrons produced by activated H atoms that migrate through the TiO2 lattice by exchange between Ti3+–Ti4+ pairs.48 Consequently, hydrogen spillover rarely occurs on non-reducible supports, since the concurrent transfer of protons and electrons will not proceed on such materials.
Our group has previously reported that a combination of Ru and Ni (which are essentially immiscible at equilibrium due to the positive enthalpy of formation for their solid solution alloy) will form a solid solution alloy on a TiO2 support when using conventional impregnation and hydrogen reduction procedures.20,49 This highly specific formation of the binary alloy is believed to involve intraparticle hydrogen spillover from the Ru surface to the Ni surface on the TiO2 support. This synthesis can be performed without using a polymeric surfactant, and the strong metal–support interaction obtained from the TiO2 support allows the loading of small size NPs of the alloy.50,51 However, the mechanism by which solid solution alloys are generated from immiscible metals on a TiO2 support and the applicable metal combinations remain unclear.
In the work reported herein, we determined that a combination of Rh and Cu, which are also thermodynamically immiscible according to the associated phase diagram (Fig. S1†),52 forms binary solid solution alloy NPs on a TiO2 support. These NPs exhibit superior catalytic performance compared to monometallic Rh during the hydrolysis of ammonia borane (AB) owing to a significant synergistic effect obtained from the two metals. The present study elucidated the specific mechanism responsible for the alloy formation based on in situ characterization by means of H2 temperature-programmed reduction (TPR), X-ray adsorption fine structure (XAFS) analysis, and Fourier transform infrared (FTIR) spectroscopy. Density functional theory (DFT) calculations were also performed to clarify not only the promotional effect of the RhCu alloy but also the enhanced hydrogen spillover occurring on the TiO2 support.
Experimental
Materials
Rutile TiO2 (JRC-TIO-6) and anatase TiO2 (JRC-TIO-8) were kindly supplied by the Catalysis Society of Japan. MgO was purchased from Wako Pure Chemical Industries, Ltd. Ethanol, RhCl3, CuCl2·2H2O, and ZrO2 were purchased from Nacalai Tesque. γ-Al2O3 was obtained from Strem Chemicals, Inc. Ammonia borane (AB, NH3BH3) was obtained from the Aldrich Chemical Co. All commercially available compounds were used as received. Distilled water was employed as the reaction solvent.Preparation of catalysts
TiO2 (0.5 g) was added to a mixture of distilled water (50 mL) and ethanol (50 mL), followed by the addition of RhCl3 (21.0 mg) and CuCl2·2H2O (17.1 mg). This mixture was stirred at room temperature for 1 h, after which the solvents were evaporated under vacuum. Finally, the sample was reduced with H2 at a heating rate of 5 °C min−1 (20 mL min−1, 350 °C) for 2 h to yield RhCu/TiO2 (Rh 2.0 wt%; Rh![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cu = 1:1). RhCu/ZrO2, RhCu/γ-Al2O3, and RhCu/MgO with the same Rh and Cu loadings were also synthesized according to the same procedure.
:Cu = 1:1). RhCu/ZrO2, RhCu/γ-Al2O3, and RhCu/MgO with the same Rh and Cu loadings were also synthesized according to the same procedure.
Characterization
Transmission electron microscopy (TEM) micrographs were obtained with a field emission (FE) TEM instrument (Hf-2000, Hitachi). Scanning transmission electron microscopy (STEM), elemental mapping and line analysis were performed using a JEOL-ARM 200F instrument equipped with a Kevex EDX detector (JED 2300T) operated at 200 kV. Temperature-programmed reduction with H2 (H2-TPR) was conducted using a BEL-CAT (BEL Japan, Inc.) instrument by heating 30 mg samples at 5 °C min−1 from 50 to 600 °C under a 5.0% H2/Ar flow. In these experiments, as-deposited samples before H2 reduction were employed. Rh K-edge and Cu K-edge in situ X-ray absorption fine structure (XAFS) spectra were recorded in the transmission mode at the 01B1 beamline station in conjunction with a Si (111) monochromator at SPring-8, JASRI, Harima, Japan (proposal numbers 2017A1057 and 2017B1084). In a typical experiment, spectra were acquired while a pellet sample was held in a batch-type in situ XAFS cell. XAFS data were processed using the ATHENA code (Demeter).53 The Fourier transformation (FT) of k3-weighted normalized extended XAFS (EXAFS) data was performed over the range of 3.0 Å < k/Å−1 < 12 Å to obtain the radial structure function.Hydrolysis of AB
In a typical experiment, a quantity of the RhCu/TiO2 catalyst (20 mg) combined with 8 mL water was transferred into a Schlenk-type reaction vessel (30 mL) connected to a gas buret. After the system was purged three times with Ar, 2 mL of a 2 mmol aqueous solution of AB was added to the vessel and allowed to react at 30 °C. The reaction was considered to start at the moment that the AB solution was added to the vessel and the reaction progress was monitored periodically. TOF values were defined as (H2 mol)/((total Rh mol) min).Evaluation of hydrogen spillover using in situ FTIR
In situ Fourier transform infrared (FTIR) spectra were acquired using a JASCO FT/IR-6600 instrument over the range of 4400–400 cm−1. In each case, an approximately 30 mg quantity of the sample (the as-prepared Rh supported catalyst) was pressed into a self-supported wafer (ϕ10 mm). This wafer was subsequently mounted in a specially designed quartz IR cell (Makuhari Rikagaku Garasu Inc.) equipped with a water-cooling condenser and an electric heater and connected to a gas-exchange system. Spectra were acquired using a previously reported procedure.54 The specimen was first reduced at 350 °C under a H2 flow (20 mL min−1) for 2 h. Following this, the sample was heated under N2 at 150 °C for 1 h to remove physisorbed water (after which no further changes in the spectrum were observed) and the baseline spectrum was collected.Spectra were also obtained after the sample was heated under a H2 atmosphere at 50 °C for 30 min, to ensure equilibration. Following this initial heating step, the gas flow was switched to D2 for 10 min and then back to H2 for 10 min. This switching process was repeated again and the resulting three-dimensional changes over time in the peak at 1200 cm−1 (attributed to the δD–O–D stretching vibration) were monitored.
DFT calculations
All density functional theory (DFT) calculations were performed with the DMol3 program in the Materials Studio 17.2 software package.55,56 The generalized gradient approximation (GGA) exchange-correlation functional proposed by Perdew, Burke, and Ernzerhof (PBE) was used to obtain a better understanding of the semiconductor electronic properties of these materials. This function was combined with the double-numerical basis set plus polarization functions (DNP) in conjunction with a cutoff value of 4.0 Å. We adopted the medium level in the DMol3 program for the integration grid. Rh24 and Rh12Cu12 clusters with their (111) facets exposed were selected as models of monometallic and binary clusters, respectively, with the bottom two layers fixed at the corresponding bulk positions and the top layers allowed to relax during geometry optimizations. Adsorption energy values (Ead) were estimated from the differences between the optimized energies of individual substances and the combined cluster model and AB. Ead was defined as Eadsorbate + Ecluster − Eadsorbate/cluster, where Eadsorbate is the total energy of the free adsorbate, Ecluster is the total energy of the bare cluster and Eadsorbate/cluster is the total energy of the adsorbate–cluster system. These activation energy calculations were based on a process previously reported by Peng et al.57Simulations of the reduction of Cu–O groups via H2 spillover were performed using a TiO2(101) plane with atomic thickness and a unit cell of 10.929 × 9.188 × 16.394 Å together with a hexagonal Al2O3(100) plane with atomic thickness and a unit cell of 9.518 × 12.991 × 14.277 Å. In addition, a γ-Al2O3 (100) plane with atomic thickness and a unit cell of 16.826 × 16.136 × 24.318 Å was also used for calculations. Periodic boundary conditions were applied, with some of the top-layer atoms allowed to relax during geometry optimizations and the other layers fixed at the corresponding bulk positions. The slab was separated by a vacuum space with a height of 20 Å and tetrahedral Rh5 clusters were loaded on the surface of each oxide when modeling H2 dissociation.
Results and discussion
Synergistic catalytic effect of RhCu/TiO2
Various Rh, Cu, and RhCu supported catalysts were prepared using a conventional impregnation method. Briefly, aqueous solutions of RhCl3 and CuCl2·2H2O containing ethanol were deposited on various supports, including TiO2 (rutile: R), TiO2 (anatase: A), γ-Al2O3, ZrO2, and MgO. The resulting samples were subsequently reduced under a H2 flow (20 mL min−1) at 350 °C, without applying a prior calcination step.The catalytic performance of each sample was assessed by monitoring hydrogen production during the hydrolysis of AB. AB was used because this compound has emerged as one of the most practical hydrogen storage compounds due to its high stability as a solid under ambient conditions and superior hydrogen storage capacity (19.6 wt%).49,58–60Fig. 1A provides the turnover frequency (TOF) values for various Rh and RhCu supported catalysts, while the time courses of reactivities are summarized in Fig. S2.† The RhCu/TiO2 (R) showed the highest catalytic activity, which was 2.6 times that of a single Rh catalyst, while the other RhCu supported catalysts did not show any significant enhancement as compared to the corresponding Rh catalysts, and the activities of the Cu supported catalysts were negligible (Fig. S2†). The RhCu/TiO2 (R) exhibited the highest promotional alloying effect (Fig. 1B) when the atomic ratio of Rh and Cu was adjusted to 7:3. The volcano-type relationship between activity and composition clearly suggests the formation of a solid solution RhCu alloy structure on the TiO2 and an unusual synergistic effect originating from the combination of Rh and Cu. Transmission electron microscopy (TEM) images of Rh and RhCu NPs supported on TiO2 (R) (Fig. S3†) also indicate that the improved activity of the RhCu was not due to the size of the NPs, since the average diameters of the Rh and RhCu NPs were approximately 1.9 and 2.0 nm, respectively.
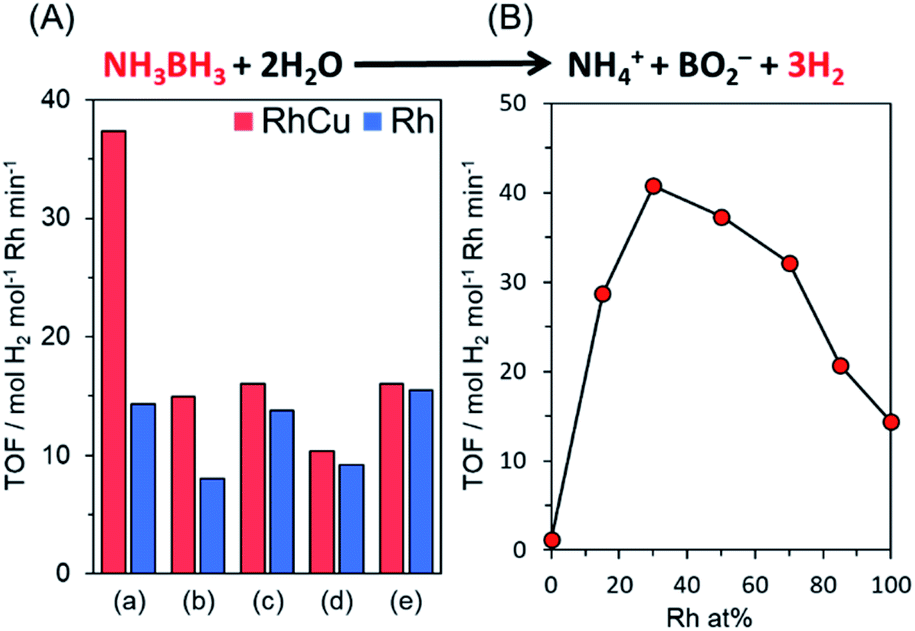 | ||
| Fig. 1 (A) TOF values for hydrogen production from AB hydrolysis over Rh and RhCu supported catalysts on (a) TiO2 (rutile), (b) TiO2 (anatase), (c) γ-Al2O3, (d) ZrO2 and (e) MgO and (B) TOF values obtained using RhCu supported TiO2 (R) catalysts with different Rh:Cu ratios (catalytic conditions: catalyst 20 mg, 10 mL 0.2 M aqueous AB solution, 30 °C). | ||
In addition, when Rh/TiO2 (R) and RhCu/TiO2 (R) specimens were calcined at 500 °C prior to the H2 reduction trials, both specimens showed the same level of activity (Fig. S4†). This effect is attributed to the separation of Rh and Cu ions at high temperatures. From these results, it is evident that a unique RhCu binary alloy was formed on the TiO2 support during simple H2 reduction at 350 °C. These catalysts supported on TiO2 (R) were subsequently characterized, as described below (in which TiO2 (R) is simply referred to as TiO2).
To investigate the alloying effect of Rh and Cu during AB hydrolysis, DFT calculations were performed based on Rh24 and Rh12Cu12 clusters. During these calculations, the level of the highest occupied molecular orbital (HOMO) could be tuned through the coupling of the electronic structure as the cluster composition was varied. When a portion of the Rh24 cluster was replaced with Cu metal, the HOMO level was found to be raised from −4.069 eV (Rh24) to −3.977 eV (Rh12Cu12). This result suggests that the RhCu alloy is theoretically more electron-rich than monometallic Rh, and this finding is supported by Rh 3d X-ray photoelectron spectroscopy (XPS) analysis of the Rh/TiO2 and RhCu/TiO2 (Fig. S5†). In these data, the RhCu/TiO2 XPS peak is shifted to a lower binding energy compared to that generated by the Rh/TiO2. This shift is attributed to a change in the electronic state of Rh due to electron donation from the Cu. Charge transfer from Cu atoms to Rh atoms occurs because of the respective electronegativity values of Rh and Cu (2.28 and 1.90). Note that there is no clear difference in the Cu 2p XPS spectra of Cu/TiO2 and RhCu/TiO2. Cu0 and Cu+ are difficult to distinguish because they appeared at almost the same position in XPS spectra. Therefore, even if electrons are transferred from Cu to Rh, we cannot observe a clear change in the Cu 2p XPS spectra because electron-poor Cu0 appears at an almost similar position to pure Cu0. And, we roughly estimated the ratio of the peak derived from single Rh metal and the RhCu alloy by fitting. The ratio of the peak derived from single Rh metal and the RhCu alloy is estimated to be 42.2% and 57.8%, respectively.
Previous experimental and theoretical studies reported the possible mechanism for AB hydrolysis using metal NPs. Feng et al. observed changes of the Cu state during AB hydrolysis using in situ XAFS, which showed that AB molecules interact with the catalyst firstly and water can approach both the catalyst and AB to attack the N–B bond.61 Peng et al. demonstrated the plausible AB hydrolysis mechanism using DFT calculations.57 Thus, we show the possible mechanism of AB hydrolysis in Fig. 2A, and the details are as follows. In this cycle, the reaction is initiated by the dissociation of the N–B bond in AB, followed by the attack of a H2O molecule to generate BH3(OH)− and NH4+. Subsequently, hydrogen is produced by a second attack of H2O on the BH3(OH)− ion. Finally, two hydrogen molecules are released from BH2(OH)2− accompanied by the formation of BO2−. It should be noted that the final step proceeds readily because the associated energy barrier is lower than those for the other steps. Based on this mechanism, we estimated the activation energy (Ea) values for the first and second steps for Rh24 and Rh12Cu12 clusters (Fig. 2A, Table 1). Ea for the first step was determined to be 24.8 kcal mol−1 on the Rh12Cu12, which is much lower than that on the Rh cluster, for which Ea was 38.5 kcal mol−1. In addition, the Ea values for the second step were lower than those for the first step for both the Rh and RhCu clusters. These results demonstrate that the initial attack by H2O is the rate determining step during this reaction, and is promoted by the presence of Rh12Cu12 clusters having electron-rich Rh species. Calculated models in the rate-limiting step are shown in Fig. S7.† It is well known that a greater adsorption energy value (Ead) for the reaction intermediate on the metal catalyst corresponds to a lower reaction barrier, according to the Brønsted–Evans–Polanyi (BEP) relationship.62 The Ead for adsorption on the Rh12Cu12 cluster was calculated to be 22.1 kcal mol−1, which is 1.6 times the value of 14.1 kcal mol−1 for the Rh24 cluster (Fig. 2B, Table 1). Thus, sites comprising neighboring Rh–Cu pairs with an electronic imbalance evidently act to promote interactions with AB molecules, enhancing the catalytic activity.
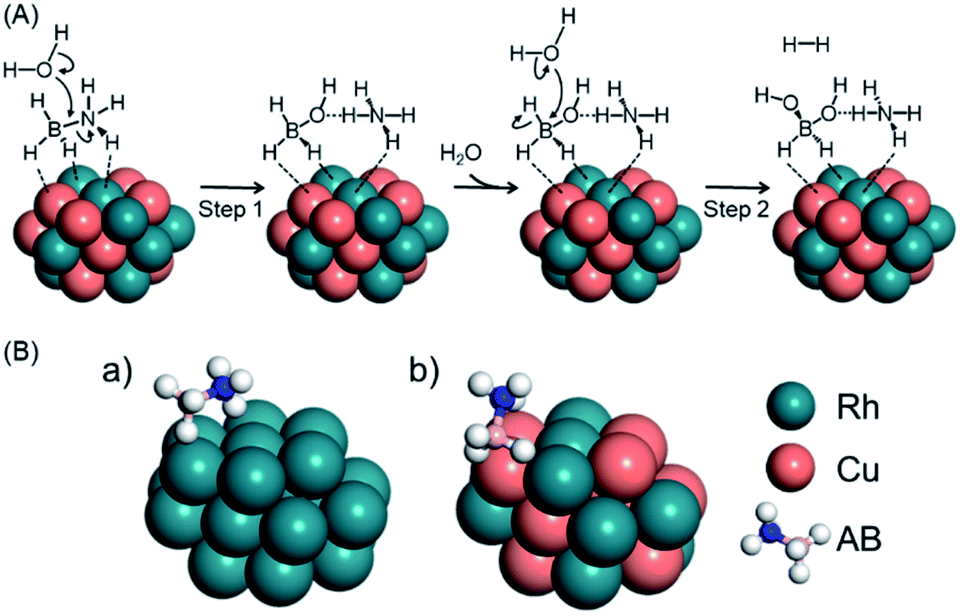 | ||
| Fig. 2 (A) Schematics showing possible processes for AB hydrolysis and (B) the calculated model for the adsorption of AB on (a) Rh24 or (b) Rh12Cu12 clusters with the (111) facet exposed. | ||
| Clusters | (A) Activation Energy | (B) Adsorption Energy of AB/kcal mol−1 | ||
|---|---|---|---|---|
| First attack of H2O (step 1)/kcal mol−1 | Second attack of H2O (step 2)/kcal mol−1 | |||
| a | Rh24 | 38.5 | 10.4 | 14.1 |
| b | Rh12Cu12 | 24.8 | 15.0 | 22.1 |
Characterization of RhCu/TiO2
The formation of a solid solution RhCu alloy on the TiO2 support was confirmed by various characterization experiments. Fig. 3 presents a high-angle annular dark field scanning TEM (HAADF-STEM) image, a bright field STEM (BF-STEM) image, energy dispersive X-ray (EDX) maps, and a line analysis for a RhCu/TiO2 catalyst after reduction at 350 °C under a H2 atmosphere. The average size of the RhCu NPs was estimated to be 2.0 nm based on the STEM image, while the EDX maps confirm the formation of a RhCu alloy, since both Rh and Cu signals appeared in the same region. The EDX line analysis shows both Rh and Cu signals in the same area, demonstrating the presence of a solid solution RhCu alloy. Fig S8† presents EDX line analysis of smaller RhCu NPs at around 2–3 nm. The EDX line analysis showed that this nanostructure was found to be a solid solution structure since the Rh and Cu signal appeared at a similar position. However, the atomic ratio of Rh:Cu is 68:32 in this particle from the X-ray intensity of Rh and Cu, indicating the formation of the Rh-rich RhCu alloy even in smaller particles. Lattice fringes are evident in the BF-STEM image of the RhCu NPs and the corresponding d spacing was estimated to be 0.214 nm in various regions. This value is almost equal to the average of the spacings of the Rh (111) planes (0.219 nm) and the Cu (111) planes (0.207 nm). These findings again confirm that Rh and Cu form a solid solution structure on the TiO2.
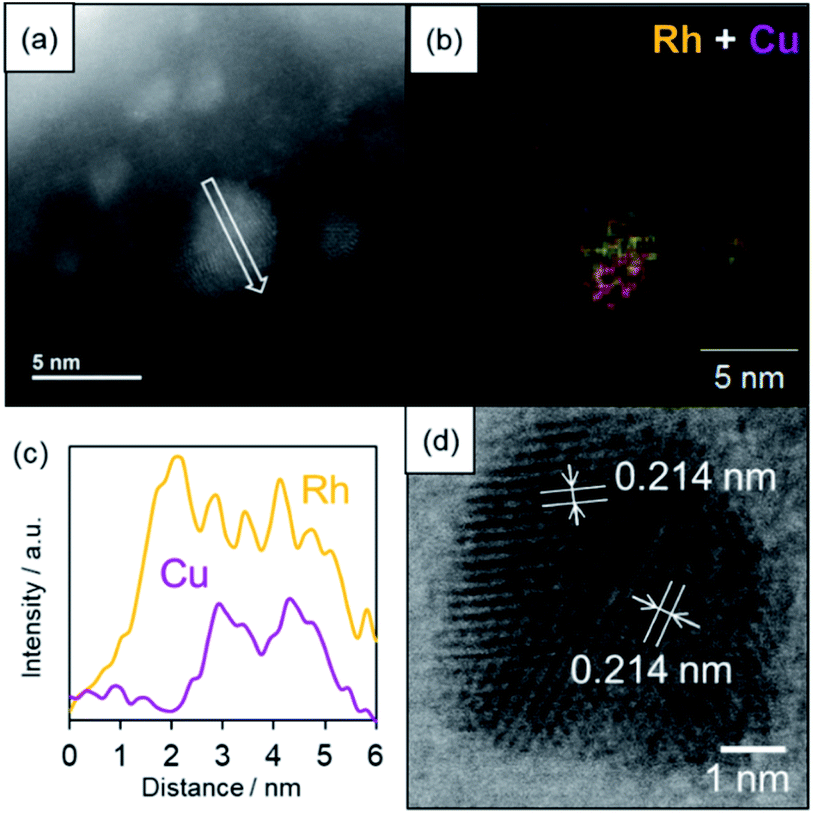 | ||
| Fig. 3 HAADF-STEM and BF-STEM images and EDX analysis of the RhCu/TiO2. (a) A HAADF-STEM image, (b) an EDX map of Rh + Cu in the region indicated in (a), (c) EDX line analysis along the arrow in (a), and (d) a BF-STEM image showing the lattice distances. | ||
The reduction temperatures of the supported metal precursors were determined using H2-TPR (Fig. S9†). Typically, a Rh precursor will be reduced faster than a Cu precursor because the reduction potential of Rh3+ is more positive than that of Cu2+ (Cu2+ + 2e− → Cu:E0 = 0.34 vs. RHE, Rh3+ + 3e− → Rh:E0 = 0.76 vs. RHE) although both ions show a positive reduction potential. The reductions of Rh3+ species in the Rh/γ-Al2O3, Rh/MgO, and Rh/TiO2 were found to occur at around 70, 120, and 150 °C, respectively. In contrast, the reductions of Cu2+ species in the Cu/γ-Al2O3 and Cu/MgO were observed to proceed above 300 °C, while reduction in the Cu/TiO2 occurred at a significantly lower temperature of approximately 240 °C. Interestingly, the RhCu/TiO2 exhibited two-step reduction peaks approximately at 140 and 210 °C. It is considered that the peak at lower temperature (around 140 °C) represents reduction of some of the Rh3+ to Rh0 (Rh nuclei) and the peak at higher temperature (around 210 °C) represents simultaneous reduction of other Rh3+ and Cu2+. This result indicates that the Rh3+ and Cu2+ precursors were reduced simultaneously on the TiO2 support to produce a solid solution alloy. A similar trend was also observed on the TiO2 (A). In contrast, the RhCu/γ-Al2O3 and RhCu/MgO specimens produced two reduction peaks, suggesting the individual reductions of Rh3+ and Cu2+ precursors.
To further elucidate changes in the local structures of the Rh, Cu and RhCu supported catalysts, in situ XAFS analyses were performed during a reduction sequence under H2 at elevated temperatures (Fig. S10–S15†). The Rh K-edge FT-EXAFS spectrum produced by the RhCu/TiO2 (Fig. 4A) exhibits a sharp singlet attributed to Rh–O bonds at approximately 1.9 Å below 200 °C, whereas another peak attributed to metallic Rh bonds appeared at approximately 2.4 Å above 200 °C. This transition in the FT-EXAFS spectra demonstrates the reduction of Rh3+ on the TiO2 support. Fig. 4C summarizes changes in the intensities of the peaks ascribed to Rh–O and Rh–Rh(Cu) bonds during heating of the RhCu/TiO2. The Cu K-edge in situ FT-EXAFS spectra of RhCu/TiO2 and the changes in the intensities of peaks due to Cu–O and Cu–Cu(Rh) bonds during heating are shown in Fig. 4B and D. Similarly, Fig. S10–S15† present the in situ XAFS spectra acquired from Rh, Cu and RhCu supported on TiO2, γ-Al2O3 and MgO and the transition data for all specimens are shown in Fig. S16–S18.† The intersection between the transitions of two peaks was defined as the reduction temperature (TR) for each metal precursor on the various supports, and these values are summarized in Table 2.
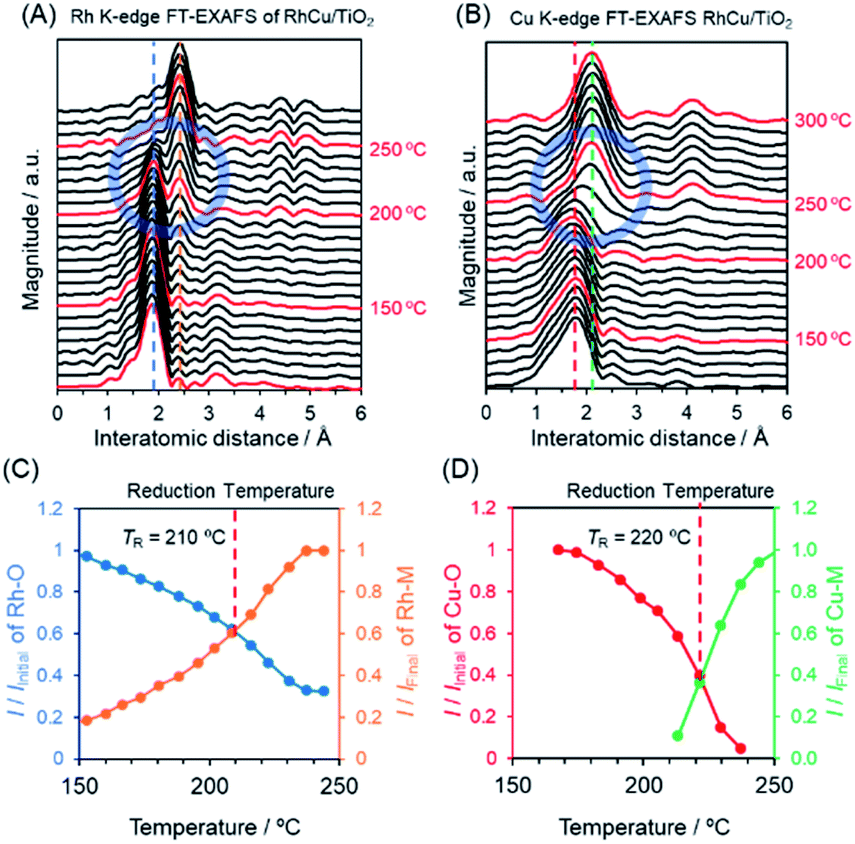 | ||
| Fig. 4 In situ FT-EXAFS spectra of the (A) Rh K-edge of RhCu/TiO2 and (B) Cu K-edge of RhCu/TiO2 acquired during reduction under H2 at elevated temperature. Variations in the intensities of peaks attributed to (C) Rh–O and Rh-M (metal) bonds based on the data in (A) and (D) Cu–O and Cu-M bonds based on the data shown in (B). (C and D) Vertical axis shows relative intensity compared to that of initial data (before H2 reduction) attributed to the Rh–O bond and compared to that of final data (after H2 reduction) attributed to the Rh–Metal(M) bond. | ||
| Rh K-edge | Cu K-edge | |
|---|---|---|
| Rh/TiO2 | 200 °C | |
| Cu/TiO2 | 280 °C | |
| RhCu/TiO2 | 210 °C | 220 °C |
| Rh/γ-Al2O3 | 60 °C | |
| Cu/γ-Al2O3 | 310 °C | |
| RhCu/γ-Al2O3 | 130 °C | 300 °C |
| Rh/MgO | 130 °C | |
| Cu/MgO | 310 °C | |
| RhCu/MgO | 130 °C | 310 °C |
The TR for Rh3+ on the Rh/TiO2 was 200 °C and this value was slightly increased to 210 °C in the case of the RhCu/TiO2. As expected, the TR for Cu2+ on the Cu/TiO2 was drastically decreased (from 280 to 220 °C) after mixing with Rh3+. The TR values for Rh3+ and Cu2+ in the RhCu/TiO2 were intermediate between those corresponding to the single Rh and Cu samples, which suggests that the two precursors were simultaneously reduced on the TiO2 surface. However, the TR values for Cu2+ on the γ-Al2O3 and MgO supports were above 300 °C, and remained high even after mixing with Rh3+. These observations indicate that neither the Rh nor the Cu precursor significantly affects the reduction profile of the other, and that the reductions of these two metals occur individually on the γ-Al2O3 and MgO surfaces. In these cases, segregated monometallic NPs and/or Rhcore–Cushell-type NPs are presumably obtained as opposed to the binary alloy.
It is unlikely that Cu2+ will be reduced as fast as reduction of Rh3+ on the TiO2 support because the reduction potential of Cu is lower than that of Rh. Based on the above results, it is probable that the reductions of Rh3+ and Cu2+ species on the TiO2 support proceed via a unique mechanism compared to the mechanisms on the other supports. A mechanism by which Rh3+ and Cu2+ are reduced on the TiO2 support can be proposed by considering the contribution of hydrogen spillover from the Rh to the Cu.30 Hydrogen spillover is known to promote the reduction of metals on various supports through hydrogen dissociation on a second precious metal.31,32 During the initial stage of the reduction, it is thought that pre-reduced Rh nuclei dissociate hydrogen molecules, after which the hydrogen is transferred to the Cu2+ precursor by spillover such that the ions are reduced to Cu0. Metal oxide supports with reversible reducibility, such as TiO2, also tend to promote spillover ability.33
The hydrogen spillover characteristics of the TiO2, γ-Al2O3, and MgO supports were compared by acquiring in situ FTIR spectra during sequential H2 and D2 gas flows over Rh supported catalysts. If rapid hydrogen spillover occurred on a support, we would expect to observe rapid exchange between gas phase hydrogen and support protons via the spillover process.54 Initially, physisorbed water was removed from the pre-reduced samples over the span of 1 h, after which no further changes in the spectrum were observed. Subsequently, the sample was placed under a H2 atmosphere and heated at 50 °C, and then allowed to equilibrate. Following this, the gas flow was switched to D2 instead of H2 and then back to H2. This gas exchange procedure was repeated, and the 3D changes over time in the 1200 cm−1 peak (attributed to the δD–O–D stretching vibration) are shown in Fig. 5A–C and S19.†Fig. 5C displays the normalized intensity of the 1200 cm−1 peak for Rh supported on TiO2, γ-Al2O3, and MgO supports. After switching from H2 to D2, the Rh/MgO required more than 150 s to saturate, while the Rh/γ-Al2O3 saturated at approximately 150 s. It should be noted that the increase in the rise in the peak intensity during trials with the Rh/TiO2 was complete within only 60 s. The process required 30 s for each observation to integrate the data and another several seconds to replace the gases after switching from H2 to D2. Thus, the rate of peak saturation for the Rh/TiO2 was too fast and almost fell below the resolution limitation (30 s) of the in situ FT-IR system after the D2 gas reached the catalyst. Based on the H-D exchange kinetics, we can conclude that rates of hydrogen spillover on the Rh supported TiO2 specimen were much higher than thsoe for Rh on non-reducible supports such as γ-Al2O3 and MgO.
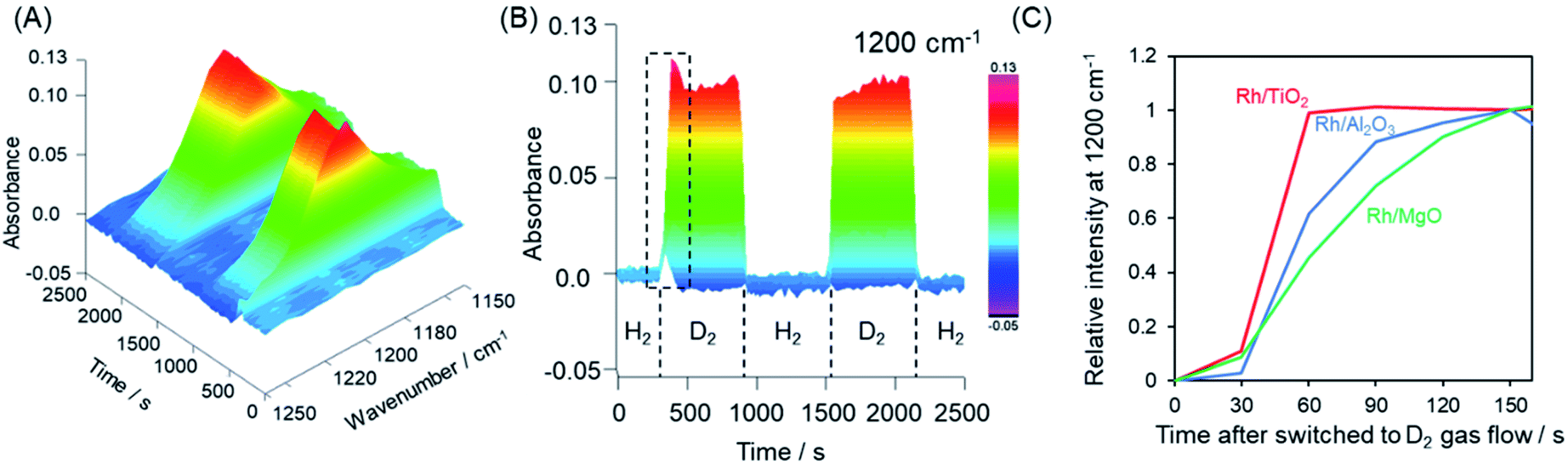 | ||
| Fig. 5 (A) 3D and (B) 2D time courses of the changes in the intensity of the peak at 1200 cm−1 attributed to the δD–O–D stretching vibration as obtained from in situ FTIR spectra acquired during the H2 and D2 gas exchange sequence over Rh/TiO2. (C) Normalized peak intensities after D2 gas introduction within the area indicated by dashed lines in (B) for Rh/TiO2, Rh/Al2O3 and Rh/MgO. | ||
Considering these results, we propose a mechanism for the formation of the solid solution RhCu alloy on the TiO2 support (Fig. 6). During these trials, the sample was heated under a H2 atmosphere such that Rh3+ was partially reduced to generate Rh metal nuclei at around 140 °C. The nuclei seem to be formed at the defect site of TiO2 and hardly involved in alloy formation since they are stable. Following this, hydrogen was dissociated on the surfaces of these Rh nuclei to form Rh–H species, accompanied by the reduction of Ti4+ to Ti3+ at the metal–support interfaces. Electrons migrated from Ti3+ ions to neighboring Ti4+ ions, which allowed simultaneous proton transfer to O2− anions attached to adjacent Ti4+. In this manner, the hydrogen rapidly reached both metal ions (Rh3+ and Cu2+) by moving over the TiO2 surface, and these ions were reduced at the same time to form RhCu clusters. These clusters then aggregated to produce the non-equilibrium RhCu alloy. This proposed mechanism is supported by the synergistic effect that evidently promotes the catalytic activity of the RhCu/TiO2 compared to the Rh/TiO2 following reduction at various temperatures (Fig. S20†). The rate of increase in catalytic activity was found to be significantly dependent on the reduction temperature and increased in the following order: 200 °C (1.38) < 250 °C (2.33) < 300 °C (2.55) < 350 °C (2.60). The decrease of TOF over Rh/TiO2 and RhCu/TiO2 catalysts showed an almost similar rate at reduction temperature over 250 °C, indicating that TOF decreases are related to the particle size. Moreover, RhCu/TiO2 is considered to show a promotional alloying effect at reduction temperature over 220 °C, and in fact, this catalyst suddenly showed more than 2.3 higher activity compared to the Rh/TiO2 catalyst at reduction temperature over 250 °C, indicating that the activity improvement is due to the formation of the solid solution alloy. This result is consistent with the data obtained by H2-TPR and in situ XAFS analyses. In addition, the BET surface area of supports was measured to be 100, 338, 185, 11.3 and 2.53 m2 g−1 for TiO2 rutile, TiO2 anatase, γ-Al2O3, ZrO2 and MgO, respectively. This result showed even if γ-Al2O3 which has a higher surface area than TiO2 rutile is used, the corresponding alloy was not formed. Hence, it can be concluded that the participation of spillover is more important than the distribution of Rh and Cu on the support, for solid solution alloy formation with immiscible Rh and Cu combination.
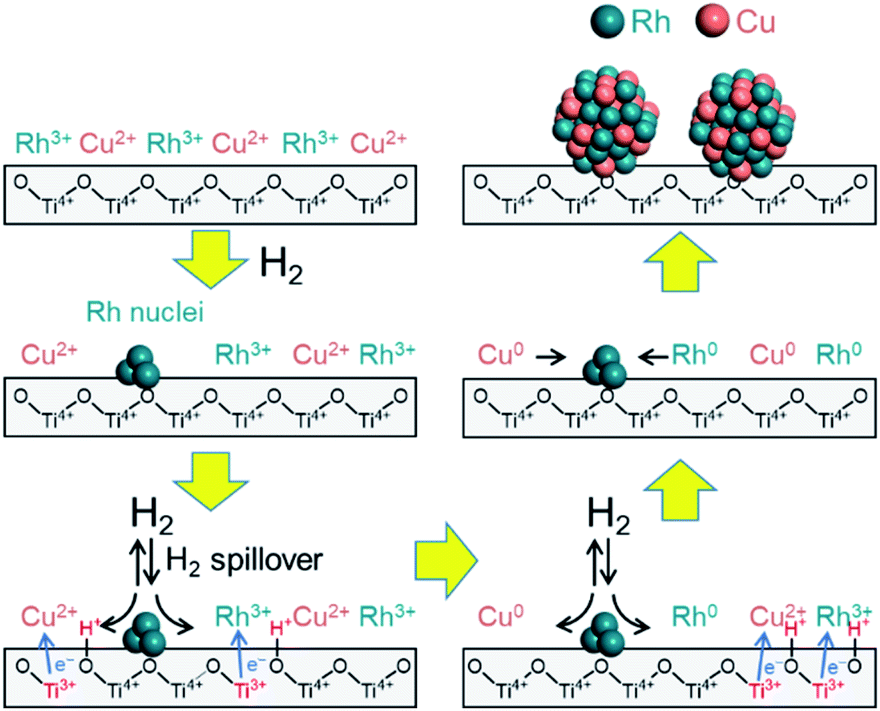 | ||
| Fig. 6 A proposed mechanism for the formation of an immiscible RhCu binary alloy on a TiO2 support assisted by hydrogen spillover. | ||
DFT calculations were also performed to provide further support for this mechanism by which an immiscible alloy is produced based on the spillover effect. The resulting potential energy profiles are summarized in Fig. 7. Rutile TiO2 (101) and hexagonal Al2O3 (100) planes were chosen as models for this theoretical investigation because of their thermodynamic stability, and Rh5 clusters were employed as a model of Rh nuclei. Four representative reaction steps were considered for the reduction of Cu2+ species on the support through the hydrogen spillover from Rh clusters, according to the above proposed reaction mechanism. These steps included the dissociation of H2 on a Rh cluster loaded on the support (step 1), the transfer of a dissociated H atom from a Rh cluster to a neighboring O atom on the support (step 2), the migration of a H atom over the support surface (step 3), and the reduction of Cu2+ by the spilled H atom on the support (step 4).
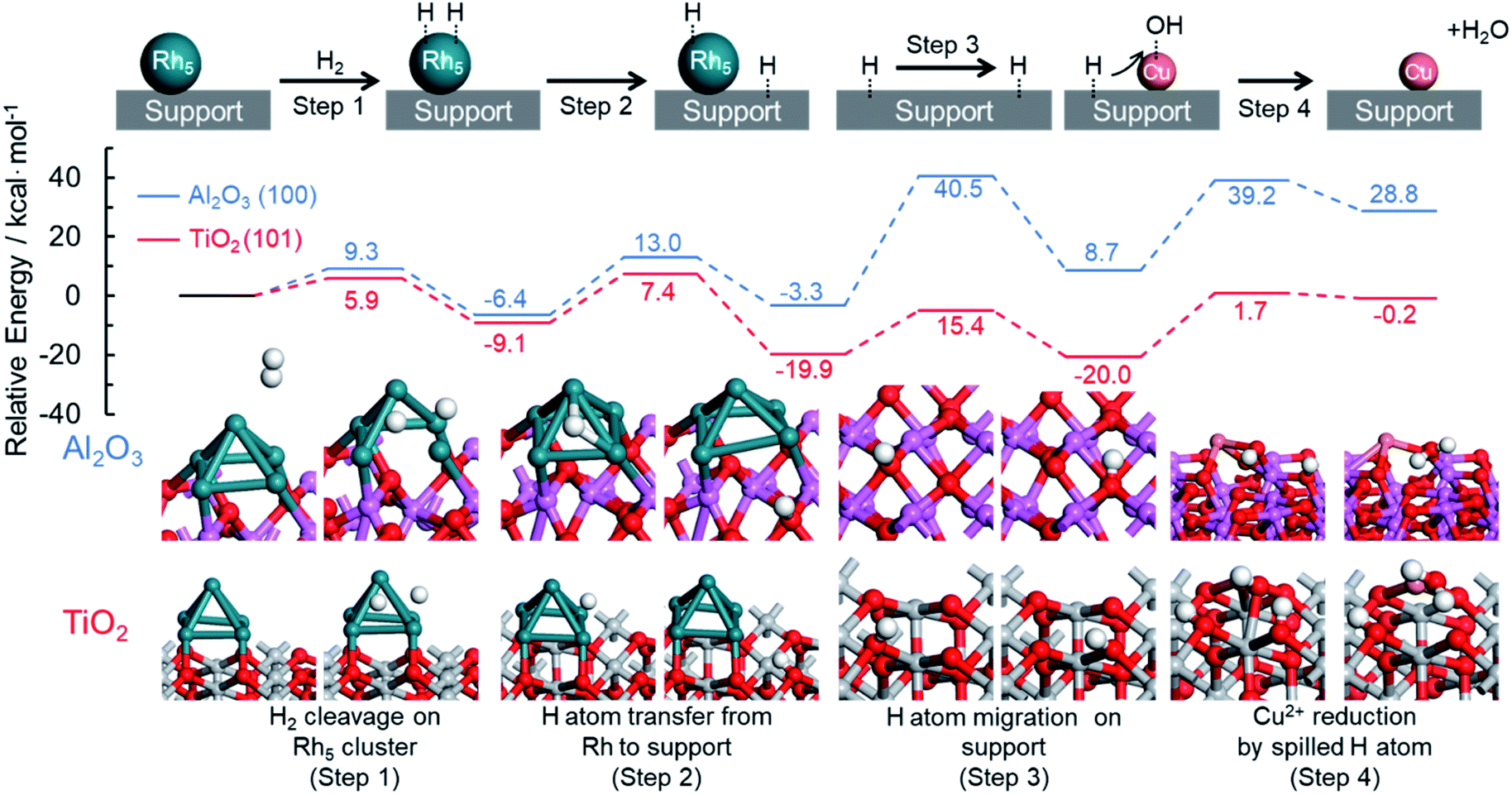 | ||
| Fig. 7 Theoretical pathway for the reduction of Cu species on the TiO2 and Al2O3 supports assisted by hydrogen spillover. | ||
In the case of TiO2 (101), the dissociation of H2 on a Rh5 cluster (step 1) occurs with a barrier of 5.9 kcal mol−1 (Fig. S21†). The Ea associated with H atom transfer from a Rh5 cluster to a neighboring O atom on the support (step 2) was estimated to be 16.5 kcal mol−1 (Fig. S22†). These values are similar to those obtained using Al2O3 (100), for which the Ea values were 9.3 and 19.4 kcal mol−1, respectively. The migration of a H atom over the support surface (step 3) is considered a separate step because of the presence of oxygen sites having different coordination numbers, such as 2-coordinated oxygen (O(2)) or 3-coordinated oxygen (O(3)) sites. The barriers for H atom migration from O(2) to O(2), O(2) to O(3), and O(3) to O(2) were calculated to be 15.0, 37.4, and 12.7 kcal mol−1 on the TiO2 (101), respectively. These results demonstrate that H atom migration over the TiO2(101) preferentially occurs at O(2) sites because TiO2(101) has numerous O(2) sites and because the participation of O(3) is energetically unfavorable. In the case of Al2O3(100), the Ea values were determined to be 29.3, 41.7, and 43.8 kcal mol−1 for the transfer pathways from O(2) to O(3), O(3) to O(2), and O(3) to O(3), respectively (Fig. S23†). Since the number of O(2) sites on Al2O3 is small, H atom migration on this substrate preferentially occurs at the O(3) sites, and even if an O(2) site is adjacent to O(3) sites, there is a relatively high activation energy for migration from O(2) to O(3) sites. These results clearly suggest that H atom transfer on TiO2 is energetically more likely to proceed than that on Al2O3, and hence the rate of hydrogen spillover is faster on the TiO2.
With respect to the reduction of Cu2+ by the spilled H atom on the support (step 4), we calculated the activation energy for the attack of a neighboring H atom on a Cu–OH group on the support, followed by the loss of H2O. The Ea values were estimated to be 21.7 and 30.5 kcal mol−1 on the TiO2(101) and Al2O3(100), respectively (Fig. S24†). Taking into account the overall reactions, the reduction of Cu2+ species on TiO2 proceeds with a barrier of 21.7 kcal mol−1 in the presence of Rh, while that on Al2O3 requires an activation energy of 43.8 kcal mol−1 even in the presence of Rh. The energy of the final state on the Al2O3 appeared to be thermodynamically high since all the steps for reduction of Cu2+ with hydrogen spillover from Rh are described together in Fig. 7. In detail, step 2 and step 3 are thermodynamically unfavorable steps which are related to hydrogen migration to the Al2O3 surface. Moreover, the same calculations were also performed on the γ-Al2O3 model reported by Digne et al., since their structure has been widely accepted for the investigations.63,64 In the theoretical reduction pathway, H atom migration on the support has the highest activation energy (Fig. S27, Table S1†). The obtained activation energy for the hydrogen migration (38.9 kcal mol−1) is almost similar to a value of our result on hexagonal Al2O3 (41.7–43.8 kcal mol−1). In addition, Bokhoven et al. calculated the activation energy of hydrogen spillover on γ-Al2O3 using the model reported by Digne.33 The corresponding value (37.6 kcal mol−1) is the same as our results, indicating the reliability of our calculations. These results produced by the theoretical investigations help to explain the differences in the reduction temperatures for Cu2+ species between the RhCu/TiO2 and RhCu/Al2O3. In preliminary work, the energy changes associated with the direct reduction of Cu2+ by H2 molecules on each support were calculated, giving values of 36.8 and 44.7 kcal mol−1 on the TiO2(101) and Al2O3(100), respectively (Fig. S25†). These values correspond to the reduction of Cu/TiO2 and Cu/Al2O3, respectively. The dissociation energies of gaseous H2 on TiO2(101) and Al2O3(100) without Rh clusters were estimated to be greater than 80 kcal mol−1 (Fig. S26†), and so the reduction of Cu2+ must proceed solely due to spilled H atoms in the presence of Rh. These calculated Ea values and the experimental Cu2+ reduction temperatures on the TiO2 and γ-Al2O3 determined by in situ XAFS are summarized in Table 3. On the TiO2 support, the reduction of Cu2+ species occurs with an Ea of 21.7 kcal mol−1 in the presence of Rh (210 °C) and with an Ea of 36.8 kcal mol−1 in the absence of Rh (280 °C). On the Al2O3 support, these values are 43.8 kcal mol−1 with Rh (300 °C) and 44.7 kcal mol−1 without (310 °C). These correlations between Ea and the experimental reduction temperature are in good agreement with the proposed mechanism for the formation of the solid solution RhCu alloy.
| (A) Activation Energy/kcal mol−1 | (B) Reduction Temperature of Cu2+/°C | ||||
|---|---|---|---|---|---|
| H2 cleavage on the Rh5 cluster | H atom transfer from the Rh5 cluster to the support | H atom migration on the support | Cu2+ reduction by spilled H atoms | ||
| TiO2 (101) | 5.9 | 16.5 | 15.0 | 21.7 | 210 (RhCu/TiO2) |
| Al2O3 (100) | 9.3 | 19.4 | 41.7–43.8 | 30.5 | 300 (RhCu/γ-Al2O3) |
| H2 cleavage on the support | Cu–O reduction by H2 vapor | Reduction temperature/°C | |||
| TiO2 (101) | 82.3 | 36.8 | 280 (Cu/TiO2) | ||
| Al2O3 (100) | 89.3 | 44.7 | 310 (Cu/γ-Al2O3) | ||
The phase diagram for Rh and Cu shows that these metals are essentially immiscible at most compositions and do not form solid solution alloys below 600 °C, because of the positive enthalpy of formation of such alloys. Thus, the synthesis of such alloys is difficult using conventional methods. In addition, γ-Al2O3, MgO, and ZrO2 supports do not afford RhCu alloys under the present synthetic conditions, indicating that the use of TiO2 as a platform for the synthesis of alloy NPs could be a useful means of obtaining solid solution alloy structures with immiscible combinations.
Conclusions
TiO2 has been demonstrated to be an efficient platform for the formation of a RhCu binary alloy with immiscible combination under H2 reduction conditions. A RhCu/TiO2 catalyst synthesized in this manner showed 2.6 times higher activity than Rh/TiO2 during hydrogen production from the hydrolysis of AB. This activity improvement was not obtained when using other supports. Theoretical studies revealed that the adsorption energy of AB was increased on the RhCu binary alloy, whereas the activation energy for dissociation of the N–B bond (the rate-determining step) was decreased compared to the value obtained using monometallic Rh. This change in the energy barrier is attributed to the formation of negatively charged Rh atoms and positively charged Cu atoms in close proximity to one another. The generation of a RhCu binary alloy on the TiO2 support was confirmed by HAADF-STEM and EDX observations. Reduction profiles obtained using H2-TPR and in situ XAFS data suggest the simultaneous reduction of Rh3+ and Cu2+ species on the TiO2 support. The rapid H2/D2 exchange reaction observed by in situ FT-IR spectroscopy suggests that the rate of hydrogen spillover increased on TiO2. From these results, we conclude that Rh3+ ions are first reduced by H2 to generate nuclei, after which the dissociation of hydrogen molecules occurs to form hydride species that act as cocatalysts to reduce neighboring Cu2+ species. This process is assisted by the hydrogen spillover characteristics of the TiO2via a coupled proton–electron transfer mechanism. Theoretical simulations provide evidence for the facilitated migration of H atoms on the TiO2 and show that the spilled H atoms enable the reduction of Cu2+ species with a lower activation energy compared to direct H2 reduction. This study demonstrates a promising approach for the preparation of non-equilibrium solid solution alloys, and we expect that this strategy will be extendable to other combinations of metals that may serve as unique catalysts and/or photocatalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by Grants-in-Aid for Scientific Research (no. A18H020740 and A19H008380) from the Japan Society for the Promotion of Science (JSPS) and MEXT. S.M. thanks JSPS for a Research Fellowship for Young Scientists (no. A19J101530). K.M., Y.K., and H.Y. thank the MEXT program “Elements Strategy Initiative for Catalysts & Batteries (ESICB) (no. JPMXP0112101003)”.Notes and references
- H. M. Chen and R.-S. Liu, J. Phys. Chem. C, 2011, 115, 3513–3527 CrossRef CAS
.
- M. A. El-Sayed, Acc. Chem. Res., 2001, 34, 257–264 CrossRef CAS PubMed
- Y. C. Cao, R. Jin and C. A. Mirkin, Science, 2002, 297, 1536–1540 CrossRef CAS PubMed
- P. V. Kamat, J. Phys. Chem. B, 2002, 106, 7729–7744 CrossRef CAS
- N. L. Rosi, D. A. Giljohann, C. S. Thaxton, A. K. R. Lytton-Jean, M. S. Han and C. A. Mirkin, Science, 2006, 312, 1027–1030 CrossRef CAS PubMed
- S. Nie and S. R. Emory, Science, 1997, 275, 1102–1106 CrossRef CAS PubMed
- P. M. Tessier, O. D. Velev, A. T. Kalambur, J. F. Rabolt, A. M. Lenhoff and E. W. Kaler, J. Am. Chem. Soc., 2000, 122, 9554–9555 CrossRef CAS
- K. Mori, S. Masuda, H. Tanaka, K. Yoshizawa, M. Che and H. Yamashita, Chem. Commun., 2017, 53, 4677–4680 RSC
- M. Haruta, N. Yamada, T. Kobayashi and S. Iijima, J. Catal., 1989, 115, 301–309 CrossRef CAS
- L. N. Lewis, Chem. Rev., 1993, 93, 2693–2730 CrossRef CAS
- A. K. Sinha, S. Seelan, S. Tsubota and M. Haruta, Angew. Chem., Int. Ed., 2004, 116, 1572–1574 CrossRef
- A. K. Singh and Q. Xu, ChemCatChem, 2013, 5, 652–676 CrossRef CAS
- S. Masuda, K. Mori, Y. Futamura and H. Yamashita, ACS Catal., 2018, 8, 2277–2285 CrossRef CAS
- K. Mori, T. Sano, H. Kobayashi and H. Yamashita, J. Am. Chem. Soc., 2018, 140, 8902–8909 CrossRef CAS PubMed
- K. D. Gilroy, A. Ruditskiy, H. C. Peng, D. Qin and Y. Xia, Chem. Rev., 2016, 116, 10414–10472 CrossRef CAS PubMed
- T. Mitsudome, T. Urayama, K. Yamazaki, Y. Maehara, J. Yamasaki, K. Gohara, Z. Maeno, T. Mizugaki, K. Jitsukawa and K. Kaneda, ACS Catal., 2015, 6, 666–670 CrossRef
- K. Kusada, H. Kobayashi, R. Ikeda, Y. Kubota, M. Takata, S. Toh, T. Yamamoto, S. Matsumura, N. Sumi, K. Sato, K. Nagaoka and H. Kitagawa, J. Am. Chem. Soc., 2014, 136, 1864–1871 CrossRef CAS PubMed
- T. Yoshii, K. Nakatsuka, Y. Kuwahara, K. Mori and H. Yamashita, RSC Adv., 2017, 7, 22294–22300 RSC
- M. Miyazaki, S. Furukawa and T. Komatsu, J. Am. Chem. Soc., 2017, 139, 18231–18239 CrossRef CAS PubMed
- S. Masuda, K. Mori, T. Sano, K. Miyawaki, W.-H. Chiang and H. Yamashita, ChemCatChem, 2018, 10, 3526–3531 CrossRef CAS
- T. Komatsu, H. Kobayashi, K. Kusada, Y. Kubota, M. Takata, T. Yamamoto, S. Matsumura, K. Sato, K. Nagaoka and H. Kitagawa, Chem.–Eur. J., 2017, 23, 57–60 CrossRef CAS PubMed
- B. Huang, H. Kobayashi, T. Yamamoto, S. Matsumura, Y. Nishida, K. Sato, K. Nagaoka, S. Kawaguchi, Y. Kubota and H. Kitagawa, J. Am. Chem. Soc., 2017, 139, 4643–4646 CrossRef CAS PubMed
- H. Guo, H. Li, K. Jarvis, H. Wan, P. Kunal, S. G. Dunning, Y. Liu, G. Henkelman and S. M. Humphrey, ACS Catal., 2018, 8, 11386–11397 CrossRef CAS
- S. García, L. Zhang, G. W. Piburn, G. Henkelman and S. M. Humphrey, ACS Nano, 2014, 8, 11512–11521 CrossRef PubMed
- X. Yang, Q. Li, E. Lu, Z. Wang, X. Gong, Z. Yu, Y. Guo, L. Wang, Y. Guo, W. Zhan, J. Zhang and S. Dai, Nat. Commun., 2019, 10, 1611 CrossRef PubMed
- J. Zhang, H. Wang, L. Wang, S. Ali, C. Wang, L. Wang, X. Meng, B. Li, D. S. Su and F. S. Xiao, J. Am. Chem. Soc., 2019, 141, 2975–2983 CrossRef CAS PubMed
- C. Hernandez Mejia, T. W. van Deelen and K. P. de Jong, Nat. Commun., 2018, 9, 4459 CrossRef PubMed
- H. Tang, J. Wei, F. Liu, B. Qiao, X. Pan, L. Li, J. Liu, J. Wang and T. Zhang, J. Am. Chem. Soc., 2016, 138, 56–59 CrossRef CAS PubMed
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed
- R. Prins, Chem. Rev., 2012, 112, 2714–2738 CrossRef CAS PubMed
- D. Xu, W. Li, H. Duan, Q. Ge and H. Xu, Catal. Lett., 2005, 102, 229–235 CrossRef CAS
- G. Jacobs, J. A. Chaney, P. M. Patterson, T. K. Das and B. H. Davis, Appl. Catal., A, 2004, 264, 203–212 CrossRef CAS
- W. Karim, C. Spreafico, A. Kleibert, J. Gobrecht, J. VandeVondele, Y. Ekinci and J. A. van Bokhoven, Nature, 2017, 541, 68–71 CrossRef CAS PubMed
- C. Spreafico, W. Karim, Y. Ekinci, J. A. van Bokhoven and J. VandeVondele, J. Phys. Chem. C, 2017, 121, 17862–17872 CrossRef CAS
- W. C. Conner and J. L. Falconer, Chem. Rev., 1995, 95, 759–788 CrossRef CAS
- V. V. Rozanov and O. V. Krylov, Russ. Chem. Rev., 1997, 66, 107–119 CrossRef
-
S. Ohgoshi, I. Nakamura and Y. wakushima, in Studies in Surface Science and Catalysis, ed. T. lnui, K. Fujimoto, T. Uchijima and M. Masai, Elsevier, 1993, vol. 77, pp. 289–292 Search PubMed
- J. Im, H. Shin, H. Jang, H. Kim and M. Choi, Nat. Commun., 2014, 5, 3370 CrossRef PubMed
- H. Nishihara, P.-X. Hou, L.-X. Li, M. Ito, M. Uchiyama, T. Kaburagi, A. Ikura, J. Katamura, T. Kawarada, K. Mizuuchi and T. Kyotani, J. Phys. Chem. C, 2009, 113, 3189–3196 CrossRef CAS
- K.-S. Lin, A. K. Adhikari, Y.-H. Su, C.-W. Shu and H.-Y. Chan, Adsorption, 2012, 18, 483–491 CrossRef CAS
- Y. Guo, S. Mei, K. Yuan, D.-J. Wang, H.-C. Liu, C.-H. Yan and Y.-W. Zhang, ACS Catal., 2018, 8, 6203–6215 CrossRef CAS
- S. Wang, Z.-J. Zhao, X. Chang, J. Zhao, H. Tian, C. Yang, M. Li, Q. Fu, R. Mu and J. Gong, Angew. Chem., Int. Ed., 2019, 131, 7750–7754 CrossRef
- H. Nishihara and T. Kyotani, Chem. Commun., 2018, 54, 5648–5673 RSC
- G. Zhan and H. C. Zeng, Nat. Commun., 2018, 9, 3778 CrossRef PubMed
- Y. Li and R. T. Yang, J. Phys. Chem. B, 2006, 110, 17175–17181 CrossRef CAS PubMed
- L. Chen, A. C. Cooper, G. P. Pez and H. Cheng, J. Phys. Chem. C, 2008, 112, 1755–1758 CrossRef CAS
- X. Sha, L. Chen, A. C. Cooper, G. P. Pez and H. Cheng, J. Phys. Chem. C, 2009, 113, 11399–11407 CrossRef CAS
- T. Huizinga and R. Prins, J. Phys. Chem. C, 1981, 85, 2156–2158 CrossRef CAS
- K. Mori, K. Miyawaki and H. Yamashita, ACS Catal., 2016, 6, 3128–3135 CrossRef CAS
- S. J. Tauster, S. C. Fung, R. T. K. Baker and J. A. Horsley, Science, 1981, 211, 1121 CrossRef CAS PubMed
- A. Bruix, J. A. Rodriguez, P. J. Ramírez, S. D. Senanayake, J. Evans, J. B. Park, D. Stacchiola, P. Liu, J. Hrbek and F. Illas, J. Am. Chem. Soc., 2012, 134, 8968–8974 CrossRef CAS PubMed
- D. J. Chakrabarti and D. E. Laughlin, J. Phase Equilib., 1982, 2, 511–512 CrossRef
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed
- T. Whittaker, K. B. S. Kumar, C. Peterson, M. N. Pollock, L. C. Grabow and B. D. Chandler, J. Am. Chem. Soc., 2018, 140, 16469–16487 CrossRef CAS PubMed
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS
- C. Y. Peng, L. Kang, S. Cao, Y. Chen, Z. S. Lin and W. F. Fu, Angew. Chem., Int. Ed., 2015, 54, 15725–15729 CrossRef CAS
- K. Fuku, R. Hayashi, S. Takakura, T. Kamegawa, K. Mori and H. Yamashita, Angew. Chem., Int. Ed., 2013, 52, 7446–7450 CrossRef CAS PubMed
- T. B. Marder, Angew. Chem., Int. Ed., 2007, 46, 8116–8118 CrossRef CAS PubMed
- A. Staubitz, A. P. M. Robertson and I. Manners, Chem. Rev., 2010, 110, 4079–4124 CrossRef CAS PubMed
- K. Feng, J. Zhong, B. Zhao, H. Zhang, L. Xu, X. Sun and S.-T. Lee, Angew. Chem., Int. Ed., 2016, 128, 12129–12133 CrossRef
- M. Tang, J. Deng, M. Li, X. Li, H. Li, Z. Chen and Y. Wang, Green Chem., 2016, 18, 6082–6090 RSC
- M. Digne, J. Catal., 2004, 226, 54–68 CrossRef CAS
- J. Gu, J. Wang and J. Leszczynski, ACS Omega, 2018, 3, 1881–1888 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc05612b |
| This journal is © The Royal Society of Chemistry 2020 |