DOI:
10.1039/D0SC01085E
(Edge Article)
Chem. Sci., 2020,
11, 5658-5668
Penta-fluorophenol: a Smiles rearrangement-inspired cysteine-selective fluorescent probe for imaging of human glioblastoma†
Received
24th February 2020
, Accepted 8th May 2020
First published on 11th May 2020
Abstract
Two of the most critical factors for the survival of glioblastoma (GBM) patients are precision diagnosis and the tracking of treatment progress. At the moment, various sophisticated and specific diagnostic procedures are being used, but there are relatively few simple diagnosis methods. This work introduces a sensing probe based on a turn-on type fluorescence response that can measure the cysteine (Cys) level, which is recognized as a new biomarker of GBM, in human-derived cells and within on-site human clinical biopsy samples. The Cys-initiated chemical reactions of the probe cause a significant fluorescence response with high selectivity, high sensitivity, a fast response time, and a two-photon excitable excitation pathway, which allows the imaging of GBM in both mouse models and human tissue samples. The probe can distinguish the GBM cells and disease sites in clinical samples from individual patients. Besides, the probe has no short or long-term toxicity and immune response. The present findings hold promise for application of the probe to a relatively simple and straightforward following of GBM at clinical sites.
1. Introduction
Glioblastoma (GBM) is the most aggressive form of cancer that originates within the central nervous system (CNS).1–3 GBM represents 15% of all primary brain tumors,4 but the cause is unclear, and there is no clear way to prevent it. Generally, the clinical management of GBM, including diagnosis, assessment progress, and therapy effectiveness evaluation, has been mostly reliant on neurological assessment and clinical imaging techniques, such as MRI (magnetic resonance imaging), CT (computed tomography), PET (positron emission tomography) and biopsy tissue sampling.5–8
In the era of precision medicine, the development of new tools for pathological analysis of GBM tissue biopsy samples has become prominent for various reasons, such as (i) the consensus issue of histological diagnosis among pathologists, (ii) the need to improve the diagnostic yield within the limited samples due to the challengeable surgical approach, (iii) the need to improve the predictive values for disease progression, and (iv) the difficulties in performing repeated biopsies. In this vein, useful information on oncogenes, related to the key cellular mechanisms of GBM cells to survive, grow, and proliferate, holds potential as an analytical tool for GBM.9–11 For example, the precise monitoring of GBM genomic heterogeneity and mutational status, related to oncologic cellular mechanisms may well give beneficial information when selecting treatment modalities. Another critical point is about the proposed monitoring of oncogenic metabolism related to glucose, hypoxia, fatty acid, and amino acid in GBM.9,12–14 Among them, we have observed dramatic changes in the amino acid levels of GBM,15 which were recognized in clinical radioisotope imaging: increased levels of glutamate (Glu), glutamine (Gln), and methionine (Met), and a decreased level of aspartate (Asp). Such unusual amino acid levels in GBM could, therefore, be exploited by reprogramming the cellular metabolism and its sub-grouping of GBM, which is dependent on the amino acid level and is important, as a biomarker, for precision diagnosis, therapy, and follow-up treatment.
In this study, we introduce a sensing probe, based on a turn-on type fluorescence response that can estimate the cysteine (Cys) level in human-derived GBM cell lines, xenograft animal models, and human biopsy samples. Fluorescence-based diagnostic tools have been widely used at clinical sites because they are simple-to-operate, highly sensitive, selective, time-saving, economical, user-friendly, and highly applicable for bio-samples.16–21 We have validated a newly developed probe which can isolate GBM cells or tissues from healthy individuals, and examined its potential as a new tool in clinical pathology.
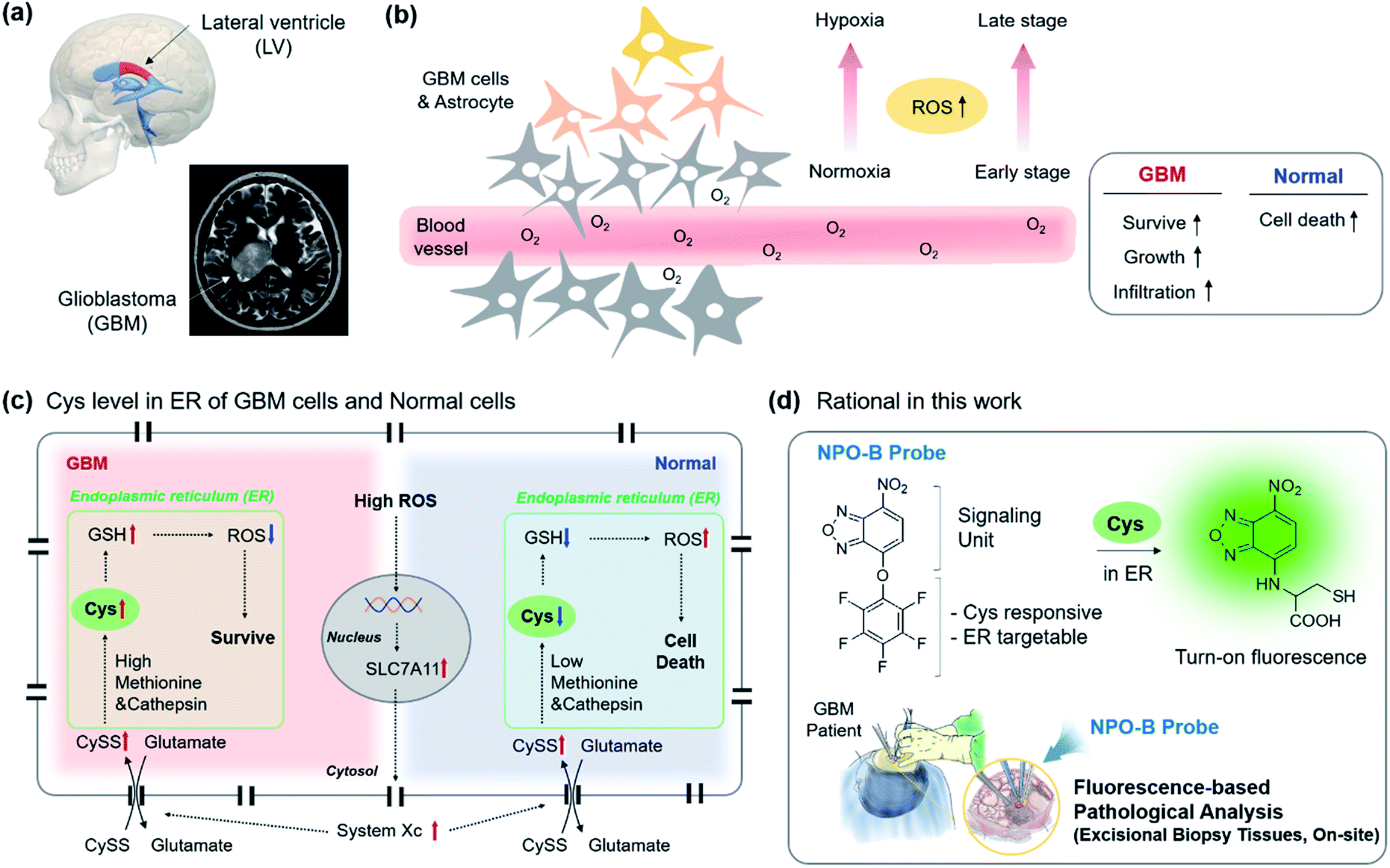
To date, many cases have provided evidence supporting the long-standing hypothesis that the neural stem cell niche that residents in the lateral ventricular (LV)–subventricular zone (SVZ) of the brain is the origin for GBM in humans (Fig. 1a).22 GBM is affected by a high degree of hypoxia due to uncontrolled proliferation and defective vascularization.23 Under hypoxia conditions, healthy cells die via apoptosis due to an increased level of intercellular reactive oxygen species (ROS), while the GBM cells can survive, grow, and infiltrate the organ (Fig. 1b).24–26 The reason is that the increased transcription of the SLC7A11 gene in GBM is induced by the elevated ROS (Fig. 1c).27 The elevated ROS level of the SLC7A11 gene that induces an over-expression of system Xc, a Cys/Glu transporter, is located on the cell membrane, and system Xc leads to the uptake of cystine (CySS) into the cell cytosol.28,29 In the endoplasmic reticulum (ER), the increased CySS is broken into two Cys by the over-expressed cysteine cathepsin, a cysteine protease enzyme that breaks disulfide links.30 The resulting Cys became a precursor in the subsequent glutathione (GSH) synthesis with methionine that is also overexpressed in GBM, and the resulting GSH produced played a vital role in the ROS attenuation.31 As a result, GBM sustains its various characteristics under hypoxia conditions. No reports related to the direct monitoring of the Cys level in clinical GBM samples were disclosed, so we aim to introduce this as a new diagnostic tool to be used for the selective sensing of the up-regulated Cys level in human-derived cells, particularly in ER sub-organelles, and biopsy samples and propose Cys as a new biomarker of GBM within clinical pathology (Fig. 1d).
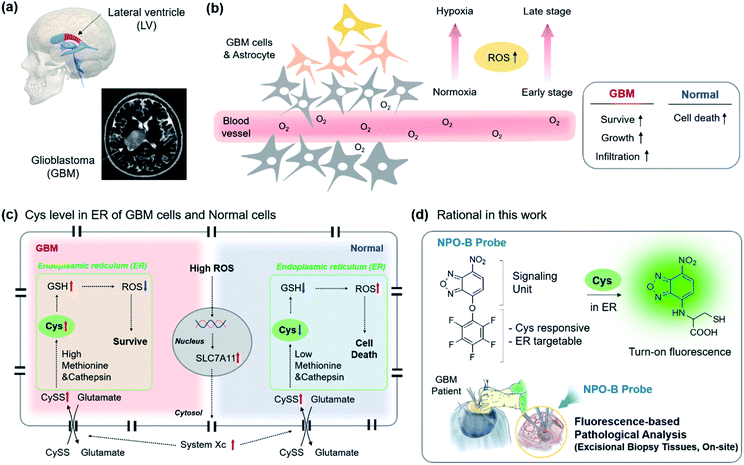 |
| | Fig. 1 A schematic representation of GBM and the Cys-related cellular mechanism, our research rational for GBM identification. (a) 3D illustration and the human MRI image of the GBM site. (b) Schematic drawing of the cells in the GBM site. Reactive-oxygen species (ROS) up-regulation under hypoxia conditions in the distal region from the blood vessel, as well as at the late stage of GBM. Box: ROS effect in the GBM and normal cells. (c) Up-regulated ROS mediated mechanism in the intracellular region. The up-regulation and down-regulation are indicated in red and blue arrows. Green box: endoplasmic reticulum (ER). (d) Rational in this work. The NPO-B probe consisted of a signaling unit (fluorescence) and penta-fluorophenol unit (Cys responsive, ER targetable). The Smiles rearrangement with Cys induced the fluorescence in a turn-on manner. The inset describes the clinical surgery of GBM patients and treatment with the NPO-B probe for pathological analysis. | |
2. Results and discussion
2.1 Rational
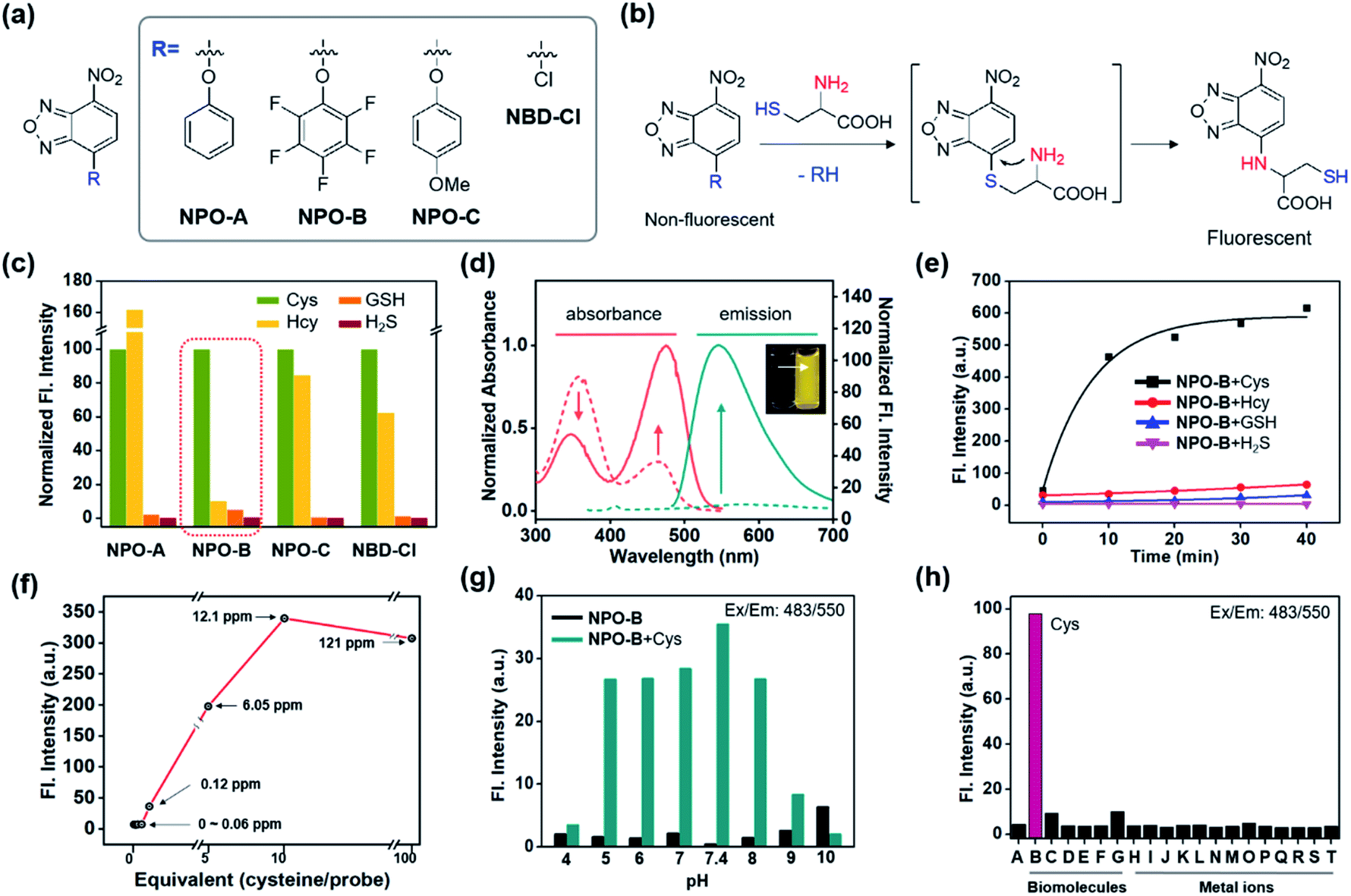
We recently focused on the development of a donor–acceptor (D–A) dipolar type fluorescent platform to monitor biologically essential species, such as enzyme activity, cell organelles, metal ions, and disease biomarkers.32–35 We designed a new molecular sensing platform based on the nitrobenzoxadiazole (NBD) fluorophore, using various aromatic substitutions at the electron-donating site (R): simple phenol (NPO-A), electron-deficient penta-fluorophenol (NPO-B), and electron-rich p-methoxyphenol (NPO-C). As a control probe we used chloro-substituted NBD (NBD-Cl) (Fig. 2a). Interestingly, the addition-substitution of biothiols, such as Cys, homocysteine (Hcy), GSH, and hydrogen sulfide (H2S) occurred at the R-site, and the subsequent Smiles rearrangement (SR),36 with amine-containing biothiols, showed a significant fluorescence response with the formation of D–A type dipolar dye (Fig. 2b). Previously, a similar approach based on the SR reaction for the thiol sensing has been reported,37–40 and in this study, we asserted that the different electron densities at the electron-donating sites could cause different reaction rates toward biothiols. In particular, we hypothesize that the electron-deficient aromatic ring with penta-fluoro substitution (NPO-B) can make a unique hydrophobic macro-environment for amino acid coordination for SR and water molecule coordination, compared to the simple benzene ring in aqueous media. Besides, we hypothesize that the regulation of the level of Cys, which occurs in the ER for the penta-fluorophenol moiety towards ER targeting ability,41,42 makes it possible to trace the Cys level in cells more precisely. With this novel rationale, we prepared the NPO series with the reaction of NBD-Cl and phenol derivatives in the presence of a base (Scheme S1†). The purity of the synthesized compounds was verified by proton/carbon nuclear magnetic resonance (1H/13C NMR) and high-resolution mass spectrometry (HRMS) (data in the ESI†).
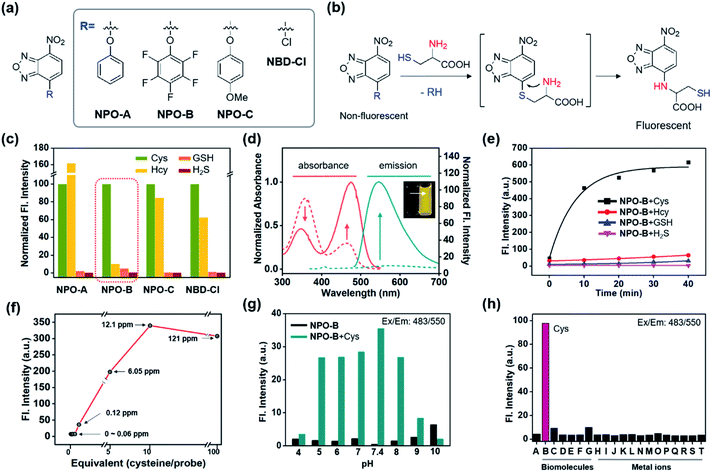 |
| | Fig. 2 Structure, working mechanism, and photophysical property analysis of the NPO probe. (a) Chemical structure of the NPO probes and control compound (NBD-Cl). (b) Working mechanism (Smiles rearrangement) of the NPO probes. (c) Normalized fluorescence intensity (peak height at 550 nm) of NPO probes (10 μM) with biothiols (Cys, Hcy, GSH, and H2S, 50 μM). The emission values were obtained under the excitation at 478 nm. (d) Normalized absorbance and emission spectra changes of NPO-B (10 μM) with Cys (50 μM). The absorption and emission spectra of NPO-B with Cys were measured in DI H2O containing 1% DMSO. (e) Time-dependent fluorescence intensity plot (peak height at 550 nm) of NPO-B (10 μM) with biothiols (50 μM). (f) Fluorescence intensity plot (peak height at 550 nm) of NPO-B (10 μM) with a low concentration of Cys. (g) Fluorescence intensity plot (peak height at 550 nm) of NPO-B (10 μM) with Cys (50 μM) at various pHs (pH 4–10). (h) Fluorescence intensity plot (peak height at 550 nm) of NPO-B (10 μM) with biomolecules and metal ions (50 μM). (A) NPO-B only, (B) L-Cys, (C) DL-Hcy, (D) L-GSH, (E) L-Glu, (F) L-Lys, (G) human serum albumin, (H) PbCl2, (I) MgCl2, (J) FeCl3, (K) Hg(NO3)2, (L) CdCl2, (M) ZnCl2, (N) NiCl2, (O) CoCl2, (P) NaCl, (Q) AuCl3, (R) AgCl, (S) CuCl2, and (T) KCl. The emission spectra were recorded under excitation at the maximum absorption wavelength. All the spectrum and intensity data were recorded in DI H2O for 30 min incubation with the substrate at 37 °C. | |
2.2 Sensing properties of NPO probes
First, the selectivity assay of the NPO series was carried out in the presence of biothiols in aqueous media (deionized water: DI H2O) (Fig. 2c). Interestingly, NPO-B only showed very selective sensing ability toward Cys over the other thiols. The disturbance of Hcy was quite dramatic for NPO-A, NPO-C, and NBD-Cl, indicating that the SR reaction could be affected by the electron density of the leaving groups (R). The NPO-B showed a strong absorbance at 358 nm and 463 nm with negligible emission (Fig. 2d). The insufficient electron-donating ability and free-rotation of aromatic rings at the R-site in aqueous media caused a low emission of NPO itself (Fig. S1†). However, the treatment of Cys in the solution of NPO-B showed changes in absorbance (decrement at 340 nm and increment at 474 nm), with significant emission increments at 550 nm (20-fold) (Fig. 2d). After checking the non-aggregation factor of NPO-B (Fig. S2,† a linear plot depends of concentration 1–100 μM), time-course fluorescence changes for the mixture of the NPO series and biothiols were monitored in DI H2O, under excitation at the maximum absorption wavelength (474 nm) (Fig. 2e and S3–S6†). A significant fluorescence enhancement was observed only in the mixture of NPO-B and Cys, and the signal was almost fully saturated within 10 min. In the concentration-dependent assay, NPO-B showed a high sensitivity under the given conditions, a detection limit of 0.12 ppm, according to the signal-to-noise ratio above 3 (Fig. 2f). The optimal pH range for the sensing of Cys with NPO-B was found to be biological pHs (pH 5–8), including physiological pH (pH 7.4) (Fig. 2g and S7†). In the selectivity assay, NPO-B only showed the fluorescence response towards Cys (“B” in Fig. 2h) among the biomolecules (biothiols, amino acids, and protein) and metal ions. The selectivity of NPO-B was also verified within the Cys oxidation environment. Treatment with air bubbling and hydrogen peroxide (H2O2) reduced the contents of the Cys level, and we observed a decreased emission intensity of NPO-B within these environments (Fig. S8†). The cystine (CySS), which is a reduced form of Cys, showed no emission enhancement. We further investigated the selectivity of NPO-B with thiol substances (cysteamine and ethanethiol), which have a similar chemical structure to Cys (Fig. S8†), and we found a significant emission enhancement only for the Cys. Next, we performed the competitive experiments of NPO-B toward Cys and H2S (Fig. S9†). A similar emission enhancement of NPO-B was observed in the sets of Cys and the mixture of Cys with H2S, not in the set of H2S alone. These results indicate that the reaction of NPO-B is more favorable toward Cys than H2S. The reaction product was analyzed using liquid chromatography mass spectrometry (LC-MS) to understand the sensing mechanism (Fig. S10†). The reaction product of NPO-B and Cys showed 4 main peaks in LC-MS, which corresponded to the SR product (Cys amine-substitute), the disulfide-linked product between the SR product and Cys, each disulfide-linked SR product, and pentafluorophenol side products. High photostability of the reaction mixture (NPO-B with Cys) was observed under strong UV irradiation conditions (Fig. S11a–c,† >70% intensity over 60 min), which is one of the most substantial merits for the fluorescence-based bioimaging. Although absorption and emission intensities of the reaction mixture (NPO-B with Cys) decreased at high temperature after 2 hours, we think that the reaction mixture is stable enough to measure the bio-imaging results due to denaturation of cells and tissue more than at 40 °C (Fig. S11d–h†).
2.3 Quantum chemical calculation
To understand the selectivity of NPO-B toward Cys and the intramolecular substitution reaction of NPO–Cys and NPO–Hcy, we conducted quantum chemical calculations. All calculations were performed using the density functional theory (DFT) method with the APFD functional and 6-31g(d) basis set as implemented in the Gaussian 16 package.43 The detailed calculation procedure is found in the ESI.† As shown in Fig. 2b, the NPO-B reacts with Cys (or Hcy) to form S-bound NPO–Cys (or NPO–Hcy), which further undergoes intramolecular substitution reactions to produce N-bound NPO–Cys (or NPO–Hcy), and the finally produced N-bound NPO–Cys emits strong fluorescence at 550 nm. First, we obtained the optimized structures of NPO–Cys and NPO–Hcy to investigate the stability of S-bound and N-bound isomers of NPO–Cys and NPO–Hcy. N-Bound isomers are found to be more stable and favored than S-bound isomers (Fig. S12†). Next, we further calculated the intrinsic reaction coordinate (IRC) for the intramolecular substitution reaction (i.e., S-bound compounds are converted into N-bound compounds) of NPO–Cys and NPO–Hcy. In fact, the intramolecular substitution reactions of NPO–Cys and NPO–Hcy are accompanied by the proton transfer between NPO–Cys (or NPO–Hcy) and water molecules. Therefore, we calculated the intrinsic reaction coordinate (IRC) for the intramolecular substitution reaction of NPO–Cys with three water molecules and NPO–Hcy with four water molecules. Fig. S13† displays the energies and structures of the S-bound compound, transition state (TS), and N-bound compound along the IRC and the final relaxed N-bound compound. As shown in Fig. S13,† a five-membered ring is formed in the transition state (TS) of NPO–Cys with the simultaneous binding of both N and S atoms of Cys to NPO, whereas a six-membered ring is formed for the TS of NPO–Hcy. In the intramolecular substitution reaction, the TS energy is lower for NPO–Cys (five-membered ring configuration) than for NPO–Hcy (six-membered ring configuration), and additionally, the final N-bound compound is more stable for NPO–Cys than for NPO–Hcy. Overall, the intramolecular substitution reaction is more favored for NPO–Cys than for NPO–Hcy in terms of the TS and the energies of finally produced N-bound compounds. Note that in the IRC calculation, water solvation was not able to be fully calculated for a practical and technical reason, and thus, we used a limited number of water molecules for the IRC for the intramolecular substitution reaction of NPO–Cys and NPO–Hcy. Therefore, the energies of the S-bound compound, TS, and N-bound compound in the IRC might not be directly comparable with the experimental values, but the IRC calculation provides an essential insight into the difference in the intramolecular substitution reaction of NPO–Cys and NPO–Hcy.
2.4 Applications in GBM cell lines
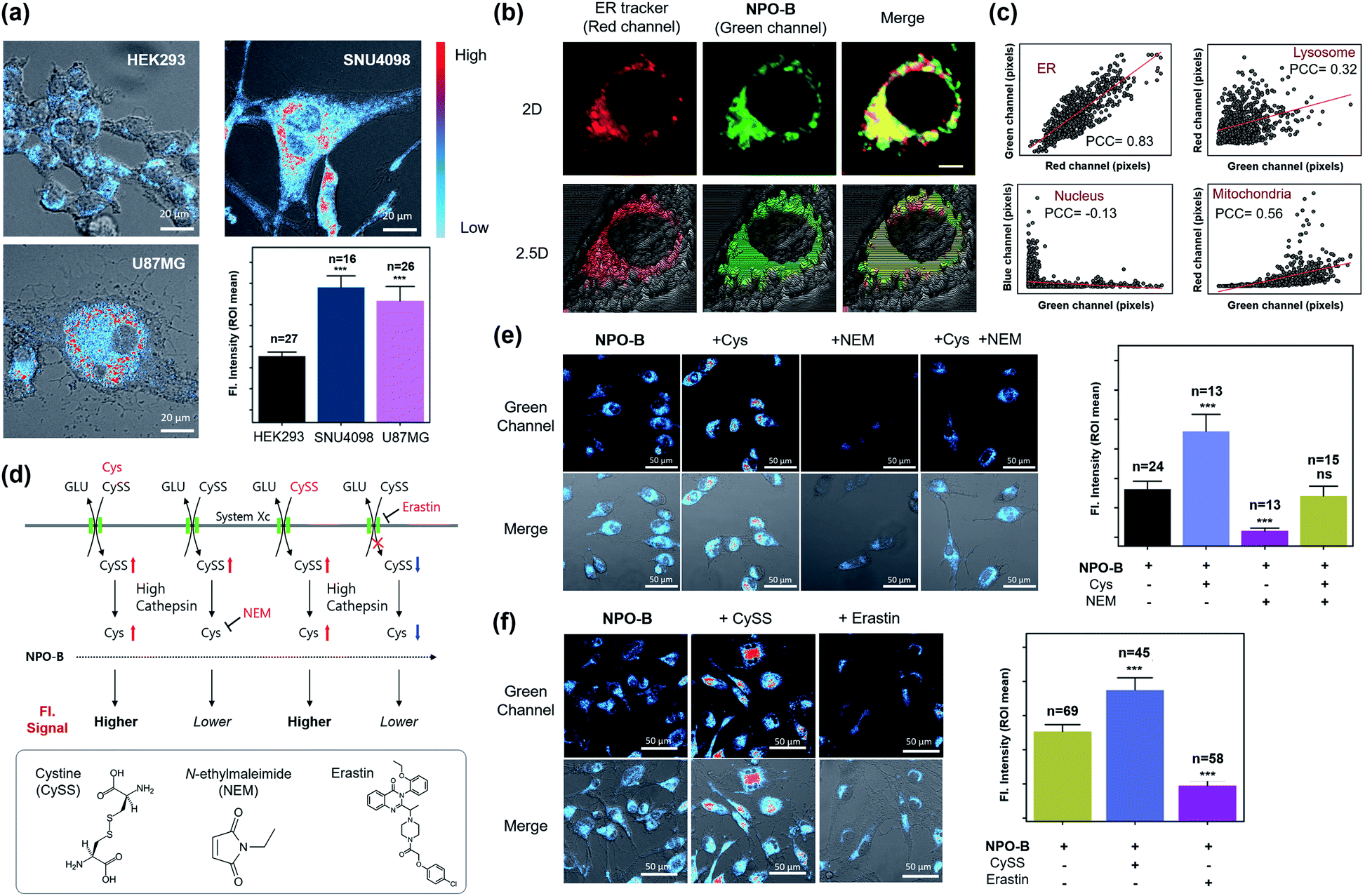
Considering the excellent specificity of NPO-B to trace Cys in vitro, we applied it to the bioimaging of various cell lines using confocal laser scanning microscopy (CLSM). After treatment with NPO-B, strong fluorescence signals were observed in the GBM cell lines, human primary GBM cell line (U87MG), and GBM patient-derived primary cultured cell line (SNU4098). The human ovarian cancer cell line (COV-318) showed a non-significant fluorescence intensity compared with the human embryonic kidney cell line (HEK293), a normal control cell. These data represent that the Cys level is much higher in GBM than that in other cells (Fig. 3a, S14 and S15†). The ER targeting ability of NPO-B was verified by co-staining with ER tracer Red, Pearson correlation coefficient (PCC) value >0.8, in cancer cell lines (Fig. 3b, c and S16†). Additionally, we checked other organelles (lysosome, nucleus, and mitochondria) using each tracker (Fig. S17†). The results showed a higher PCC value in ER than in other organelles, indicating that the NPO-B can track ER with high specificity in U87MG cells (Fig. 3c and S17b†). To understand the Cys-related cellular mechanism, we pre-incubated the cells with external stimuli, Cys, CySS (a precursor of Cys), N-ethyl-maleimide (NEM, biothiol-scavenging reagent),44 and erastin (inhibitor of system Xc)45 (Fig. 3d). As we expected, the cells preincubated with Cys and CySS showed a stronger intensity than the cells incubated with the probe only (Fig. 3e and f). In contrast, cells pretreated with NEM showed little intensity due to the inactivity of Cys (Fig. 3e). After external treatment with NEM, exogenous Cys cells showed a gradual fluorescence enhancement in the cells. The erastin-pretreated cells showed a significantly decreased intensity in the cells due to the blocking of system Xc (Fig. 3f). We additionally observed fluorescence spectra from the vial and the cells (Fig. S18†) and confirmed the spectra well overlapped with each other (Fig. S18e†). We also compared the emission spectra from U87MG (GBM) and HEK293 (normal cells) and found higher emission intensity from the U87MG cells as we expected (Fig. S18f and g†). We do think that these results can prove the reaction of NPO-B with Cys in the cells (Fig. 3 and S18†), and U87MG has a higher concentration of Cys than HEK293. In the cell viability assay, a slightly decreased cell viability of NPO-B at high concentration (50 μM) was observed in the GBM cell lines: SNU4098 (83%) and U87MG (80%), while the other cell lines showed negligible viability changes (Fig. S19†). Also, a slight volume decrease of cytosol for the U87MG cells was observed in the crystal-violet (CV) staining analysis (50 μM NPO-B) (Fig. S20†). We additionally checked the cell viability with pentafluorophenol, a side product of the reaction between NPO-B and Cys, and the result was very similar. This result indicated that more pentafluorophenol was generated in the GBM cell lines, which supported a higher level of Cys in these cell lines (Fig. S19b†). These data prove that the fluorescence signal is generated by the reaction between NPO-B and intercellular Cys, and it is, therefore, possible to determine the level of Cys in the GBM cells.
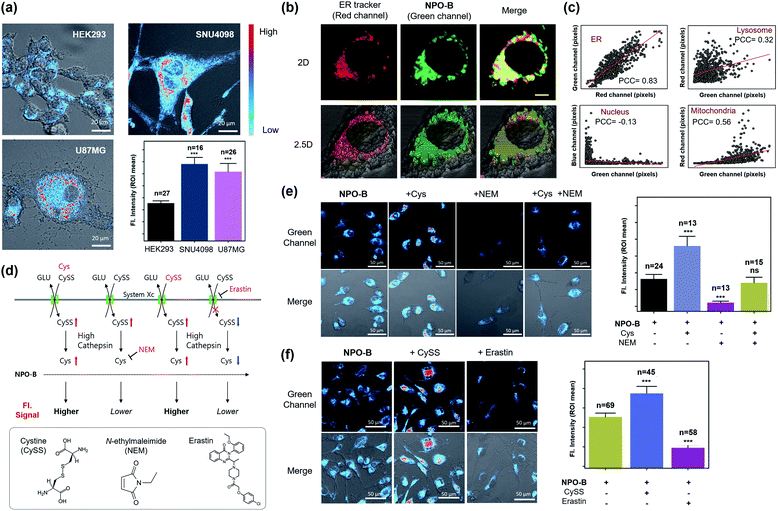 |
| | Fig. 3 Image-based GBM cell line identification study. (a) CLSM images of cell lines treated with NPO-B: HEK293, SNU4098 and U87MG. Cells were incubated with NPO-B (30 μM) for 30 min. Scale bar: 20 μm. The fluorescence intensities represent ROI mean values (ROI was obtained by drawing whole cells based on the DIC image). (b) CLSM images of the U87MG treated ER-Tracker (red) and NPO-B (green). The yellow color represents overwrapped regions of green and red signals. Scale bar: 5 μm. (c) Linear fitting plot to obtain the Pearson correlation coefficient (PCC) in various organelles. Each pixel intensity was collected in whole cells by using the ZEN 2.3 lite program, respectively (see Fig. S17† for CLSM images related PCC of each organelle). (d) Cys-related biological mechanism with CySS, NEM, and erastin. Inset box: chemical structure of CySS, NEM, and erastin. (e) Left: CLSM images of SNU4098 cells treated with NPO-B (30 μM) with Cys (500 μM), NEM (500 μM), and Cys with NEM. Right: Relative fluorescence intensities of each cell (each error bar represents mean ± SD, n = number of cells, ns = non-significant, ***P < 0.001). (f) Left: CLSM images of SNU4098 cells treated with NPO-B (30 μM) with CySS (600 μM) and erastin (20 μM), respectively. Right: Relative fluorescence intensities of each cell (each error bar represents mean ± SD, n = number of cells, ***P < 0.001). All experiments were performed in triplicate and repeated three times independently. An intensity-based pseudo-color was applied in panels (a), (e) and (f) for better visualization. The intensity in cells was measured using the Image-J program by drawing ROI over whole cells based on the DIC image. | |
2.5 Applications in the GBM xenograft model
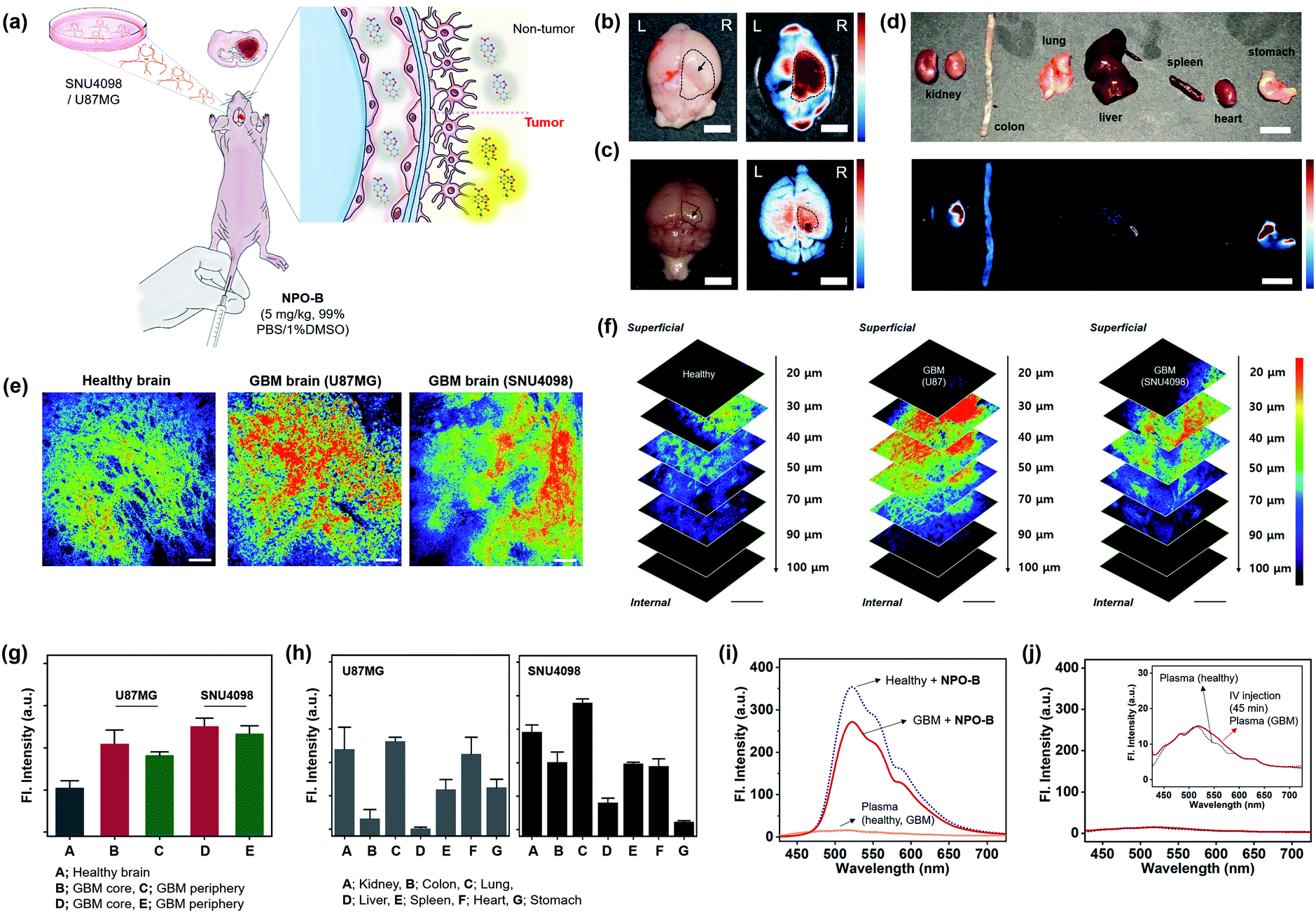
Given that NPO-B has promising Cys sensing ability and biocompatibility with bio-imaging, we demonstrated GBM tissue imaging with NPO-B in the GBM xenograft mouse model. The intracranial mouse model was prepared by unilateral injection of U87MG cells and SNU4098 cells into the brain, using a stereotaxic apparatus (tumor-implantation), and the GBM formation was verified by the tissue analysis of the sacrificed brain (Fig. S21†). We expected that NPO-B could penetrate into the brain by passing through the blood–brain barrier, and give a fluorescence enhancement in the GBM site (Fig. 4a). After intravenous injection of NPO-B (5 mg kg−1) through the tail vein, the organs were sacrificed (45 min circulation) and then analyzed, using the fluorescence tissue imaging system (FTIS) (Fig. 4b–d, 395 nm excitation, 500–550 nm detection channel). A strong fluorescence signal was observed in the GBM cell-implanted site of the mouse brain (U87MG for Fig. 4b and SNU4098 for Fig. 4c). A bio-distribution analysis based on the tissue Images showed that there was no significant fluorescence intensity in the organs, except the U87MG-implanted GBM site of the brain (intensity from the kidney, remaining blood, stomach, and leftover food) (Fig. 4d). In the SNU4098-implanted xenograft mouse, a similar result was observed, but the liver also gave off a signal. The healthy mouse control set didn't show any signal in the brain under the given experimental conditions (Fig. S22†). We also imaged the tissue samples with two-photon microscopy (TPM). TPM based on the nonlinear interaction between light and matter has emerged as an important tool for in vivo and in vitro imaging of tissues due to its various merits including: (i) excitation at near-infrared (NIR) wavelengths in the biological window, which allows for deeper penetration, (ii) focal point excitation for high resolution with 3D image construction, (iii) minimal absorption through highly scattered tissue media, and (iv) suppressed photo-damage and bleaching to the tissues.46,47 In order to monitor the Cys level in each organ using TPM, we sacrificed the mouse organs and then treated them with NPO-B (100 μM) by 60 min incubation at 37 °C in phosphate-buffered saline (PBS) buffer (pH 7.4). As shown in Fig. 4e, strong fluorescence signals were observed in the GBM brain tissues compared to the healthy control set when excited at 900 nm (50 mW laser power at the focal point, detection channel: 458–527 nm). In z-stacked TPM images of each brain sample (20–100 μm from the surface, 10 μm regular intervals) with NPO-B, the signal was the strongest in the zone between 30 μm and 70 μm from the surface, representing the high tissue permeability of NPO-B and its sufficient deep tissue imaging ability under the TPM (Fig. 4f). The TPM intensity analysis result, at the different sites of brain (healthy, GBM core, and GBM periphery), showed that the signal from the GBM model was higher than the healthy control one, and the signal from the core site was higher than that from the periphery site, indicating a higher Cys level in the GBM core site (Fig. 4g). In the TPM images of the other organs with NPO-B, strong fluorescence signals were mainly observed in the kidney, lung, spleen, and the heart for both GBM mouse models, U87MG-implanted and SNU4098-implanted (Fig. 4h, S23 and S24†). To evaluate the differences between the Cys levels in the blood of healthy and GBM mouse models, we treated NPO-B with the extracted plasma, and then checked the fluorescence intensities (Fig. 4i). We observed a slightly lower intensity in the plasma of the GBM model, but it was not significant. In the fluorescence signal analysis of the mouse plasma, after intravenous treatment with NPO-B (5 mg kg−1, 45 min circulation), fluorescence signals were not observed. These data represented that NPO-B seemed to be accumulated in the GBM site and some organs, reacting with Cys, but not a significant reaction with Cys in the blood (Fig. 4j).
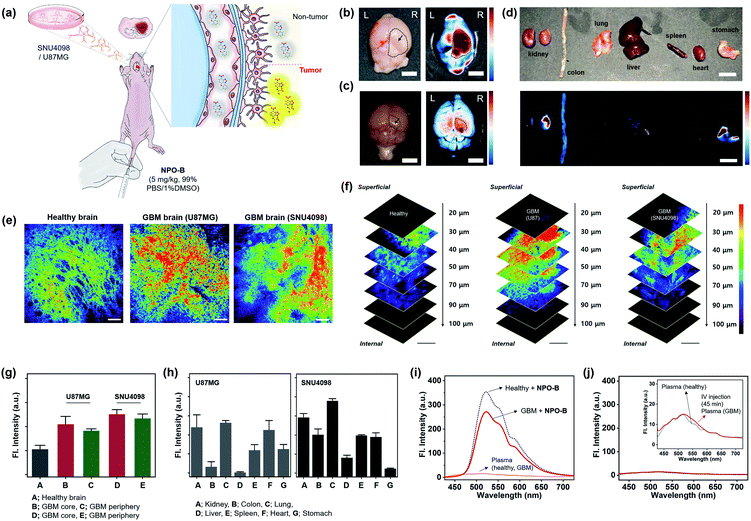 |
| | Fig. 4 GBM identification in the xenograft animal model. (a) Schematic illustration for the application of NPO-B towards the GBM animal model: GBM cell-implanted sites (SNU4098 and U87MG). (b) Ex vivo bright field image (left) and FTIS image (right) of the brain for the U87MG-implanted mouse, after NPO-B injection (i.v.) and 45 min circulation. (c) Ex vivo naked image (left) and FTIS image (right) of the brain for the SNU4098-implanted mouse after NPO-B injection (i.v.) and 45 min circulation. See ESI Fig. S22† for the ex vivo FTIS images of the control group (healthy mouse). (d) Ex vivo bright field image (upper) and FTIS image (bottom) of dissected organs of the U87MG-implanted mouse after NPO-B injection (i.v.) and 45 min circulation. (e) TPM images of the mouse brain after the treatment with NPO-B (100 μM): healthy, U87MG-implanted and SNU4098-implanted. The images were acquired at a middle depth (∼50 μm) of the tissues. (f) TPM images of brain tissue in panel (e) at the indicated depths (0–100 μm). (g) Relative fluorescence intensities of healthy brain tissues and GBM (core, periphery). All fluorescence intensities were analyzed with Image-J; each error bar represents mean ± SEM. (h) TPM intensity plot of the GBM mouse organs (see Fig. S23 and S24† for TPM images). (i) Emission spectra from the isolated mouse plasma (1/3 diluted in PBS buffer) and its NPO-B mixture (10 μM, 30 min incubation at 37 °C). (j) Emission spectra from the isolated mouse plasma (1/3 diluted in PBS buffer) pre-treated with NPO-B (i.v., 45 min circulation). | |
2.6 Applications in GBM human clinical samples
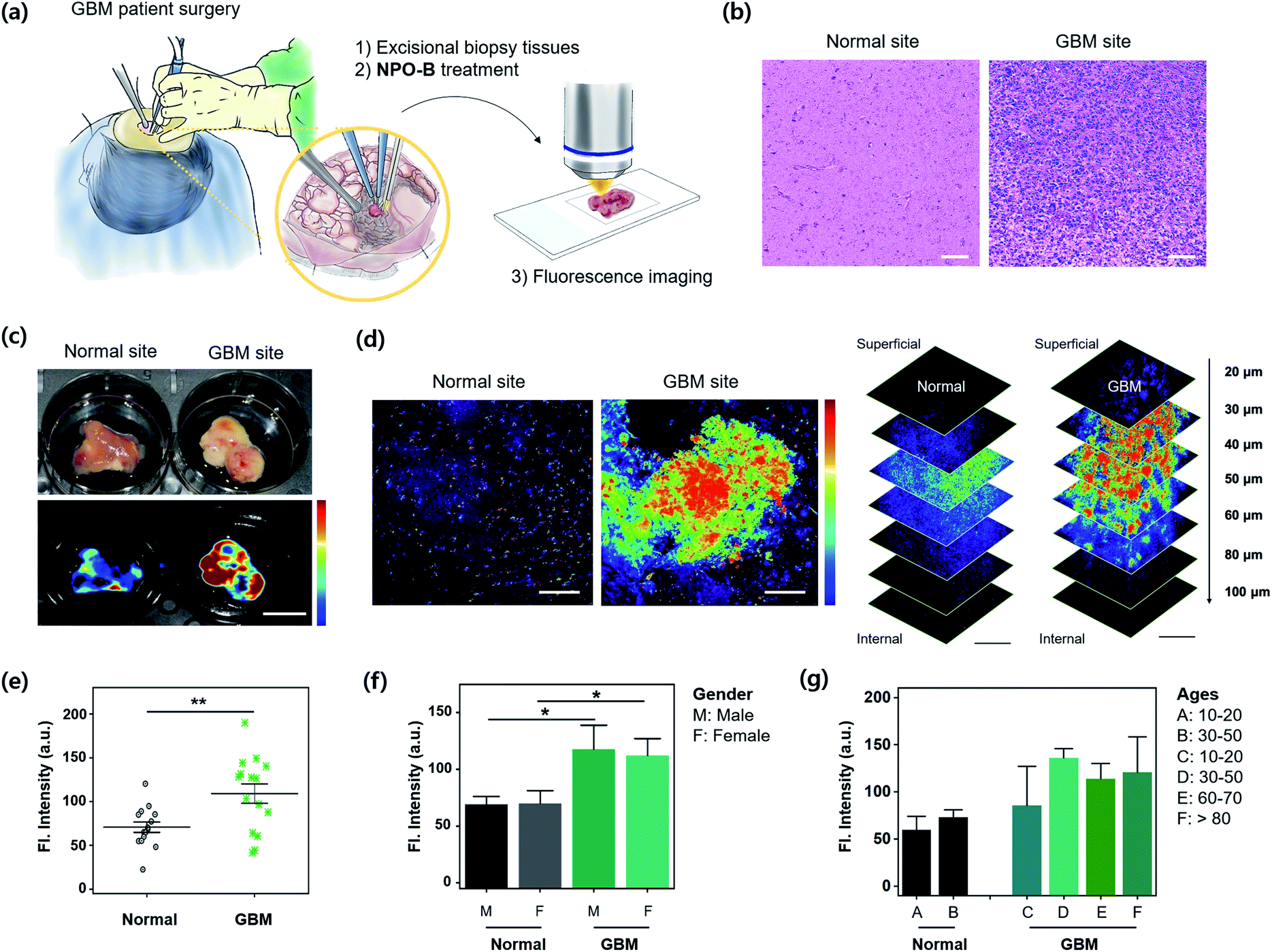
For the practical application of NPO-B, we finally applied NPO-B to GBM human clinical samples. Excisional biopsy brain tissues were collected during a brain surgery and GBM-patient surgery (number of samples: normal = 15, GBM = 15, normal–GBM interface = 5. See the patient information in Table S2†), and then analyzed by FTIS and TPM imaging after treatment with NPO-B (Fig. 5a). First, each site was verified by pathological analysis with H&E staining, and it clearly showed an abnormality for the GBM site (Fig. 5b). Generally, this kind of pathological analysis is essential for disease confirmation, as well as grade decisions, but it is time-consuming (it takes several days for staining) with a complicated procedure, and highly dependent on the pathologist's opinion.48 In this aspect, NPO-B shows strong merits to overcome the drawbacks of the present methods and is offered as a complementary new diagnostic tool. We treated NPO-B with the tissue samples (100 μM) by 30 min incubation at 37 °C in the PBS buffer (pH 7.4) and monitored fluorescence intensities using FTIS (Fig. 5c) and TPM (Fig. 5d, S25 and S26†). Similar to the GBM xenograft mouse results, a significantly much higher fluorescence signal was observed in the GBM tissues than in normal tissues within TPM images (Fig. 5e). The tissue sample of the normal–GBM interface (periphery) also showed higher intensity than the normal tissues (Fig. S26†). We then analyzed the TPM intensities in different genders (male = 14, female = 16, Fig. 5f) and ages (Fig. 5g). As we expected, the GBM group showed higher intensity than the normal group, but the gender-dependent intensity differences were not observed in the normal and GBM groups, respectively. In the age-dependent plotting, the older GBM group gave a higher intensity, and the intensity of the eighty-plus group was approximately twice as high as that of the normal group. The tissue sample of the normal–GBM interface (periphery) also showed higher intensity than that of the normal tissue (Fig. S26†). We confirmed the TPM emission spectra between solutions and biopsy samples via lambda screening of the whole spectrum (Fig. S27†) and found the well-overlapped fluorescence spectra from the confocal dish (Fig. S27a,† NPO-B with Cys) and biopsy samples (Fig. S27b,† NPO-B treated). This result indicated that the fluorescence signal from biopsy samples was generated from the reaction of NPO-B and Cys. These practical application results represented that the level of Cys is significantly higher in the GBM patient, and it can be considered as a new biomarker for the diagnosis and prognosis of GBM.
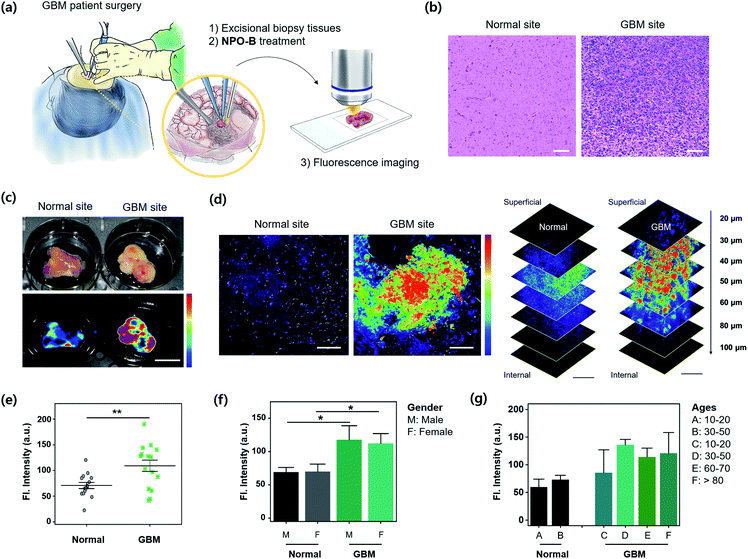 |
| | Fig. 5 Practical application for GBM clinical samples. (a) Experimental scheme for excisional biopsy tissues, isolated from a GBM patient's surgery and treatment with NPO-B. (b) Haematoxylin and eosin (H&E) stained human brain tissue sections from the normal and GBM tissue. Scale bar = 100 μm. (c) Bright field (upper) and the FTIS images (bottom) of freshly isolated brain tissue from the normal and GBM tissue treated with NPO-B [100 μM, 30 min, 37 °C, PBS buffer (pH 7.4)]. (d) TPM images of tissue samples in panel (c). See the experimental conditions in the Method section. Scale bar = 100 μm. The stacked TPM images of tissues acquired following the indicated vertical depths (0–100 μm). Scale bar is 200 μm. See ESI Fig. S25† for additional TPM images of normal and GBM cells (n = 15, each). The fluorescence images were overlaid with pseudo-color (rainbow). (e) Fluorescence intensity plot from TPM images of the brain tissues obtained at the normal (n = 15) and GBM (n = 15) site, each error bar represents mean ± SEM, **P < 0.01. (f and g) Fluorescence intensity plot from TPM images by sex and age. Gender: 8 males, 7 females in the normal group, and 6 males and 9 females in the GBM group. Each error bar represents mean ± SEM vs. normal male, vs. normal female, *P < 0.05. Ages of the group are categorized into six groups (A–F). Each error bar represents mean ± SEM based on TP intensities. | |
2.7
In vivo immune-toxicity analysis
With the promising GBM imaging results, the toxic effects of NPO-B in the liver, renal toxicity, and immuno-toxicity were investigated for its practical application, such as the spraying of NPO-B towards the surgical site of a GBM patient's brain. The toxicity of NPO-B was analyzed in the healthy mouse (C57BL/6J, i.v. injection), and the results were compared with the positive control, the lipopolysaccharide (LPS) treated set.49 As shown in Fig. S28a and b,† neither morphological nor histological change in the liver and kidney was observed in the NPO-B treated set (24 h, 48 h circulation), while the LPS treated group showed significant changes. Besides, no apparent changes in the spleen and lymph nodes were observed. Next, the hematological and immunological parameters, which were related to organ damage and immunotoxicity, were examined. After the administration of NPO-B (5 mg kg−1) and LPS (10 μg/animal), blood was collected from the intra-orbital plexus, and the changes of aspartate aminotransferase (AST) and alanine aminotransferase (ALT),50 which are general markers for hepatic damage, were examined. The NPO-treated group showed no significant values, but the LPS-treated group showed a remarkably increased value (Fig. S28c†). The NPO-B treated set also gave no increment of IL-6, IL-1β, and TNF-α, which are major inflammatory cytokines (Fig. S28d†).51 The enlargement of the spleen and lymph node means an increase of proliferation of the immune cells, and so we compared the immune cell numbers and the stimulation index between both NPO-B and LPS treated groups. As a result, we found that there was no increase of the immune cell numbers and the stimulation index52 in the NPO-B treated group (Fig. S28e†). No CD69 factor change, which is increased by inguinal lymph node cell proliferation, as an early activation marker, was observed in the NPO-B treated set (Fig. S28f†). Together, no effect of NPO-B, related to the population of other types of immune cells, including NK cells, B cells, and macrophages, was observed (Fig. S28g†). NPO-B showed no short-term hepatotoxicity, based on changes in AST and ALT, which suggested that NPO-B is a highly safe contrast agent without any short- or long-term toxicity in the liver and kidney. In the toxicity assay, NPO-B didn't show any toxicity in the liver or kidney, as well as in any immune organs, which proves that NPO-B can be safely applied in a clinical approach, not only for healthy people but also for the patient.
3. Conclusions
Simple methods to monitor the presence or recurrence of brain cancer, particularly GBM, are invaluable tools to improve the outcomes in a patient's treatment. In this study, we have reported a small molecular probe that addresses this challenge by targeting Cys, which is a new biomarker of GBM. We created and characterized a turn-on type fluorescent probe, NPO-B, that allows the tracing of Cys with high selectivity, sensitivity, and biocompatibility. We applied the probe for the first time to monitor the Cys level in human-derived GBM cell lines, a GBM xenograft mouse model, and human clinical biopsy GBM samples. We then systematically analyzed its photophysical properties and provided the sensing mechanism in vitro as well as in cell lines. The NPO-B treated GBM cell lines showed a bright fluorescence under a CLSM, and it also worked in the GBM-site imaging of the xenograft mouse model using FTIS and TPM. Furthermore, we found that the probe could distinguish the GBM site of patients from the healthy control through fluorescence analysis of the TPM tissue samples. Our findings show that NPO-B has promising application for a relatively simple and straightforward screening of GBM from excisional biopsy brain tissues, and this can be used to replace the current complex protocols. The lack of short-/long-term toxicity and immune response results also shows that NPO-B can be fully utilized throughout various clinical applications. In addition to diagnostic applications, the probe alone or its hybrids with nanoparticles have the potential to be applied in image-guided tumor resection, all of which could benefit from a selective fluorescence probe that senses Cys.
4. Methods
4.1 General information vivo immune-toxicity analysis
ESI† is available for the reagents, instruments, and analytical methods in this study.
4.2 Synthesis of the NPO series
The NPO series probes were prepared with the reaction of 4-chloro-7-dinitrobenzofurazan and phenol derivatives in the presence of a base. See details in the ESI (Scheme S1†) and synthetic protocols.
4.3 Cell culture
The Human primary glioblastoma cell line (U87MG), immortalized human cervical cancer cell line (HeLa), human ovarian cancer cell line (COV-318), and human embryonic kidney cell line (HEK293) were obtained from the Korean Cell Line Bank. The cells were cultured in Dulbecco's modified Eagle's medium (Hyclone, US) supplemented with 10% fetal bovine serum (Hyclone) and 1% penicillin–streptomycin (Gibco). The Glioblastoma cell line (SNU4098) was isolated from human GBM tissues (patient-derived clinical sample) and it was obtained from the Korean Cell Line Bank. The SNU4098 cells were cultured in Opti-MEM, supplemented with 5% fetal bovine serum (Gibco™, US) and 1% antibiotic-antimycotic (Gibco™, US). The cell lines were kept in humidified air, containing 5% CO2 at 37 °C.
4.4 Animals
6 week-old female BALB/c nu/nu mice (Taconic, provided by Daehan Biolink Co., Ltd., Eumseong, Rep. of Korea) were housed at an ambient temperature of 23 ± 1 °C and relative humidity of 60 ± 10% under a 12 h light/dark cycle, and were allowed free access to water and food. All of the experiments performed with mice were carried out in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publications no. 80-23) revised in 1996 and protocols approved by the Institutional Animal Care and Use Committee of Kyung Hee University (KHUASP(SE)-19-002).
4.5 Intracranial xenograft (tumor implantation) animal model
U87MG and SNU4098 cell lines were used for the xenograft. Mice were anesthetized with tribromoethanol (312.5 mg kg−1, i.p.) and mounted in a stereotaxic apparatus (myNeuroLab, St. Louis, MO, USA). Each mouse received a unilateral injection of 1.0 × 106 U87MG cells or SNU4098 cells per 5 μL in medium without FBS, according to the following coordinates: anteroposterior: −3.0 mm from bregma; mediolateral: 1.8 mm from bregma; and dorsoventral: −3.0 mm from the skull. The flow for the injection was regulated using an electronic pump at 1 μL min−1 for 5 min followed by 2 min with a needle at the injection site to avoid reflux. Sham operations followed the same procedure for the infusion of the medium without FBS. After surgery, mice were allowed to recover from anesthesia in a temperature-controlled chamber and then placed in individual cages.
4.6
Ex vivo tissue fluorescence imaging
VISQUE® InVivo Elite (Vieworks Co., Ltd., Rep. of Korea) was used as an ex vivo fluorescence tissue imaging system (FTIS). The imaging experiment was carried out in a dark room (wrapped in aluminum foil).
4.6.1 Animal samples.
After intravenous injection of NPO-B, mouse organs (brain, lung, heart, liver, spleen, colon and kidney) were isolated and washed with PBS buffer (3 times). Then, the ex vivo tissue imaging experiment for the tissues was conducted. Brain: lens zoom: 6×, focus: 0.0 cm, iris: F3.6, mode: GFP, LED: blue, type: single-frame, exposure time: 5 s, binning: 1 × 1, intensity min = 170.0, max = 480.0. Other organs: lens zoom: 3×, focus: 1.0 cm, iris: F3.6, mode: GFP, LED: blue, type: single-frame, exposure time: 5 s, binning: 1 × 1, intensity min = 170.0, max = 480.0.
4.6.2 Human samples.
Human tissue samples were washed with PBS buffer (3 times) and treated with NPO-B (100 μM) for 30 min. After incubation, the tissue samples were washed with PBS buffer (3 times) to remove remaining NPO-B. Next, the tissue samples were washed again with PBS buffer (3 times) and treated with 4% PFA to fix for 30 min. The imaging experiment was conducted, and the tissues were washed with PBS buffer (3 times) again before imaging. Conditions: lens zoom: 2.5×, focus: 1.5 cm, iris: F2.5, mode: GFP, LED: blue, type: single-frame, exposure time: 1 s, binning: 1 × 1, intensity min = 1031.0, max = 3218.6.
4.7 Two-photon microscopy (TPM) tissue imaging
The imaging experiment was carried out in a dark room; tissues samples were wrapped in aluminum foil. The scanning two-photon microscope (TPM, Leica, Nussloch, Germany) was equipped with a Titanium Sapphire laser (Chameleon vision, Coherent, USA) and a 25× water immersion objective lens was used for ex vivo imaging of these tissue samples. Two-photon excitation at 900 nm with a laser power of approximately 50 mW at the focal plane provided the best image quality. The fluorescence intensity of TPM images was analyzed using Leica software (Germany) and Image-J (NIH, USA).
4.7.1 Animal samples.
Mice were anesthetized and perfused transcardially with PBS buffer (0.05 M). The mice were dissected to isolate organs: brain, lung, liver, kidney, spleen, colon, heart and stomach and to perform further ex vivo studies. Each organ was washed with PBS buffer (3 times). The mice tissues were placed in dry ice for 5 min. After this, the frozen organs were cut into several pieces using a surgical blade (No. 11, Reather safety razor Co., LTD, Japan). The sliced tissue samples showed an average thickness of about 100 μm, and were transferred into a 24 well plate (SPL Life Science, Rep. of Korea), and washed with PBS buffer (3 times). Next, they were treated with NPO-B (100 μM) and incubated for 1 h at 37 °C in a shaking incubator. After incubation, the tissue samples were washed with PBS buffer (3 times), and treated with 4% PFA for the tissue fixation. Two-photon microscopy (TPM) imaging experiments were conducted with additional washing of tissues with PBS buffer (3 times).
4.7.2 Human samples.
Patient-derived human clinical sample (GBM) assay with TPM used the same method described above.
4.8 Human GBM samples
The human GBM tissue samples were snap-frozen in liquid nitrogen immediately during the surgery and were stored at −80 °C. This study was approved by the Institutional Review Boards (IRB) of Seoul National University Hospital (#H-1404-056-572).
4.9 Immuno-toxicity analysis
C57BL/6J mice were kept in a specific pathogen-free condition in the animal facility at the Seoul National University College of Medicine (Rep. of Korea). Six-to eight-week old male mice weighing 20–25 g were used for the experiments. The animal protocol for the experiments was reviewed and approved by the Ethics Committee of Seoul National University. See more details in the ESI.†
Conflicts of interest
The authors declare the following competing financial interest(s): the authors are listed as inventors on a pending patent application related to technology described in this work.
Acknowledgements
This research was supported by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) of Korea funded by the Ministry of Science & ICT (2018-M3A9H3021707) and by the Medical Research Center Program (2017-R1A5A2014768 and 2019-M3A9H1103783). This research was also supported by the Basic Science Research Program of the NRF of Korea funded by the Ministry of Education (2018-R1A6A1A03025124 and 2018-R1D1A1B07043383). This research was also supported by the NRF of Korea (2017-R1A2B2010948 for J. S. K., 2017-R1E1A1A01074529 for J. K. H., and 2019-R1A6A1A11044070 for S. P.).
Notes and references
- M. Weller, W. Wick, K. Aldape, M. Brada, M. Berger, S. M. Pfister, R. Nishikawa, M. Rosenthal, P. Y. Wen, R. Stupp and G. Reifenberger, Nat. Rev. Dis. Primers, 2015, 1, 15017 CrossRef PubMed.
- K. Aldape, K. M. Brindle, L. Chesler, R. Chopra, A. Gajjar, M. R. Gilbert, N. Gottardo, D. H. Gutmann, D. Hargrave, E. C. Holland, D. T. W. Jones, J. A. Joyce, P. Kearns, M. W. Kieran, I. K. Mellinghoff, M. Merchant, S. M. Pfister, S. M. Pollard, V. Ramaswamy, J. N. Rich, G. W. Robinson, D. H. Rowitch, J. H. Sampson, M. D. Taylor, P. Workman and R. J. Gilbertson, Nat. Rev. Clin. Oncol., 2019, 16, 509–520 CrossRef CAS PubMed.
- D. N. Louis, A. Perry, G. Reifenberger, A. von Deimling, D. Figarella-Branger, W. K. Cavenee, H. Ohgaki, O. D. Wiestler, P. Kleihues and D. W. Ellison, Acta Neuropathol., 2016, 131, 803–820 CrossRef PubMed.
- Q. T. Ostrom, H. Gittleman, G. Truitt, A. Boscia, C. Kruchko and J. S. Barnholtz-Sloan, Neuro-Oncology, 2018, 20, iv1–iv86 CrossRef PubMed.
- G. Shukla, G. S. Alexander, S. Bakas, R. Nikam, K. Talekar, J. D. Palmer and W. Shi, Chin. Clin. Oncol., 2017, 6, 40 CrossRef PubMed.
- K. Yamashita, A. Hiwatashi, O. Togao, K. Kikuchi, R. Hatae, K. Yoshimoto, M. Mizoguchi, S. O. Suzuki, T. Yoshiura and H. Honda, Am. J. Neuroradiol., 2016, 37, 58–65 CrossRef CAS PubMed.
- H.-W. Kao, S.-W. Chiang, H.-W. Chung, F. Y. Tsai and C.-Y. Chen, BioMed Res. Int., 2013, 2013, 14 Search PubMed.
- K. Weigel, Neurosurgery, 2019, 84, E273 CrossRef.
- M. M. Kim, A. Parolia, M. P. Dunphy and S. Venneti, Nat. Rev. Clin. Oncol., 2016, 13, 725–739 CrossRef CAS PubMed.
- D. S. Nørøxe, H. S. Poulsen and U. Lassen, ESMO Open, 2016, 1, e000144 CrossRef PubMed.
- A. Shergalis, A. Bankhead III, U. Luesakul, N. Muangsin and N. Neamati, Pharmacol. Rev., 2018, 70, 412–445 CrossRef CAS PubMed.
- Y. Zhou, L. Yang, X. Zhang, R. Chen, X. Chen, W. Tang and M. Zhang, BioMed Res. Int., 2019, 2019, 6581576 Search PubMed.
- W. X. Mai, L. Gosa, V. W. Daniels, L. Ta, J. E. Tsang, B. Higgins, W. B. Gilmore, N. A. Bayley, M. D. Harati, J. T. Lee, W. H. Yong, H. I. Kornblum, S. J. Bensinger, P. S. Mischel, P. N. Rao, P. M. Clark, T. F. Cloughesy, A. Letai and D. A. Nathanson, Nat. Med., 2017, 23, 1342 CrossRef CAS PubMed.
- Z. Dong and H. Cui, Semin. Cancer Biol., 2019, 57, 45–51 CrossRef CAS PubMed.
- S. Liu, H. Ma, Z. Zhang, L. Lin, G. Yuan, X. Tang, D. Nie, S. Jiang, G. Yang and G. Tang, Theranostics, 2019, 9, 1144–1153 CrossRef CAS PubMed.
- H.-W. Liu, L. Chen, C. Xu, Z. Li, H. Zhang, X.-B. Zhang and W. Tan, Chem. Soc. Rev., 2018, 47, 7140–7180 RSC.
- Y. Tang, D. Lee, J. Wang, G. Li, J. Yu, W. Lin and J. Yoon, Chem. Soc. Rev., 2015, 44, 5003–5015 RSC.
- M. Gao, F. Yu, C. Lv, J. Choo and L. Chen, Chem. Soc. Rev., 2017, 46, 2237–2271 RSC.
- J. Zhang, X. Chai, X.-P. He, H.-J. Kim, J. Yoon and H. Tian, Chem. Soc. Rev., 2019, 48, 683–722 RSC.
- L.-Y. Niu, Y.-Z. Chen, H.-R. Zheng, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, Chem. Soc. Rev., 2015, 44, 6143–6160 RSC.
- J. Chan, S. C. Dodani and C. J. Chang, Nat. Chem., 2012, 4, 973 CrossRef CAS PubMed.
- S. Facchino, M. Abdouh and G. Bernier, Cancers, 2011, 3, 1777–1797 CrossRef CAS PubMed.
- S. Jawhari, M.-H. Ratinaud and M. Verdier, Cell Death Dis., 2016, 7, e2434 CrossRef CAS PubMed.
- E. Singer, J. Judkins, N. Salomonis, L. Matlaf, P. Soteropoulos, S. McAllister and L. Soroceanu, Cell Death Dis., 2015, 6, e1601 CrossRef CAS PubMed.
- A. R. Monteiro, R. Hill, G. J. Pilkington and P. A. Madureira, Cells, 2017, 6, 45 CrossRef PubMed.
- W.-J. Huang, W.-W. Chen and X. Zhang, Oncol. Lett., 2016, 12, 2283–2288 CrossRef CAS PubMed.
- M. D. Polewski, R. F. Reveron-Thornton, G. A. Cherryholmes, G. K. Marinov and K. S. Aboody, Stem Cells Dev., 2017, 26, 1236–1246 CrossRef CAS PubMed.
- M. D. Polewski, R. F. Reveron-Thornton, G. A. Cherryholmes, G. K. Marinov, K. Cassady and K. S. Aboody, Mol. Cancer Res., 2016, 14, 1229–1242 CrossRef CAS PubMed.
- R. J. Bridges, N. R. Natale and S. A. Patel, Br. J. Pharmacol., 2012, 165, 20–34 CrossRef CAS PubMed.
- R. Löser and J. Pietzsch, Front. Chem., 2015, 3, 37 Search PubMed.
- T. A. Ogunrinu and H. Sontheimer, J. Biol. Chem., 2010, 285, 37716–37724 CrossRef CAS PubMed.
- D. Kim, H. Moon, S. H. Baik, S. Singha, Y. W. Jun, T. Wang, K. H. Kim, B. S. Park, J. Jung, I. Mook-Jung and K. H. Ahn, J. Am. Chem. Soc., 2015, 137, 6781–6789 CrossRef CAS PubMed.
- D. Kim, S. H. Baik, S. Kang, S. W. Cho, J. Bae, M.-Y. Cha, M. J. Sailor, I. Mook-Jung and K. H. Ahn, ACS Cent. Sci., 2016, 2, 967–975 CrossRef CAS PubMed.
- Y. Jung, Y. Kim, N. H. Kim, J. Lee, K.-H. Kim, J. Jung, Y. Huh, H.-J. Jang, J. Joo, S. Park and D. Kim, Dyes
Pigm., 2019, 162, 104–111 CrossRef CAS.
- H. Moon, Y. Jung, Y. Kim, B. W. Kim, J. G. Choi, N. H. Kim, M. S. Oh, S. Park, B. M. Kim and D. Kim, Org. Lett., 2019, 21, 3891–3894 CrossRef CAS PubMed.
- C. M. Holden and M. F. Greaney, Chem.–Eur. J., 2017, 23, 8992–9008 CrossRef CAS PubMed.
- M. D. Hammers and M. D. Pluth, Anal. Chem., 2014, 86, 7135–7140 CrossRef CAS PubMed.
- X. Dai, Z.-Y. Wang, Z.-F. Du, J. Cui, J.-Y. Miao and B.-X. Zhao, Anal. Chim. Acta, 2015, 900, 103–110 CrossRef CAS PubMed.
- J. Wang, L. Niu, J. Huang, Z. Yan, X. Zhou and J. Wang, Dyes Pigm., 2018, 158, 151–156 CrossRef CAS.
- P. Wang, Y. Wang, N. Li, J. Huang, Q. Wang and Y. Gu, Sens. Actuators, B, 2017, 245, 297–304 CrossRef CAS.
- A. Goujon, A. Colom, K. Straková, V. Mercier, D. Mahecic, S. Manley, N. Sakai, A. Roux and S. Matile, J. Am. Chem. Soc., 2019, 141, 3380–3384 CrossRef CAS PubMed.
- N. Wagner, M. Stephan, D. Höglinger and A. Nadler, Angew. Chem., Int. Ed., 2018, 57, 13339–13343 CrossRef CAS.
-
G. W. Trucks, M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 (Revision B.01), Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- C. R. Yellaturu, M. Bhanoori, I. Neeli and G. N. Rao, J. Biol. Chem., 2002, 277, 40148–40155 CrossRef CAS PubMed.
- M. Sato, R. Kusumi, S. Hamashima, S. Kobayashi, S. Sasaki, Y. Komiyama, T. Izumikawa, M. Conrad, S. Bannai and H. Sato, Sci. Rep., 2018, 8, 968 CrossRef PubMed.
- W. Denk, J. Strickler and W. Webb, Science, 1990, 248, 73–76 CrossRef CAS PubMed.
- H. M. Kim and B. R. Cho, Chem. Rev., 2015, 115, 5014–5055 CrossRef CAS PubMed.
-
A. Ruiz, O. Sertel, M. Ujaldon, U. Catalyurek, J. Saltz and M. Gurcan, Pathological Image Analysis Using the GPU: Stroma Classification for Neuroblastoma, 2007 IEEE International Conference on Bioinformatics and Biomedicine (BIBM 2007), 2007, pp. 77–88 Search PubMed.
- M. Ogata, S. Yoshida, M. Kamochi, A. Shigematsu and Y. Mizuguchi, Infect. Immun., 1991, 59, 679–683 CrossRef CAS.
- F. Sherer, G. V. Simaeys, J. Kers, Q. Yuan, G. Doumont, M.-A. Laute, C. Peleman, D. Egrise, T. Lahoutte, V. Flamand and S. Goldman, J. Liver, 2015, 4, 1000170 Search PubMed.
- S. Copeland, H. S. Warren, S. F. Lowry, S. E. Calvano and D. Remick, Clin. Diagn. Lab. Immunol., 2005, 12, 60–67 CrossRef CAS PubMed.
- R. I. de Ávila, M. de Sousa Vieira, M. P. N. Gaeti, L. C. Moreira, L. de Brito Rodrigues, G. A. R. de Oliveira, A. C. Batista, D. C. Vinhal, R. Menegatti and M. C. Valadares, Toxicology, 2017, 376, 83–93 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc01085e |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence h,
Veronica Jihyun
Kim
i,
Ji Yeoun
Lee
ij,
Yun Sik
Dho
k,
Yuna
Jung
a,
Junho K.
Hur
ac,
Chan
Park
ab,
Junyang
Jung
ab,
Youngbuhm
Huh
ab,
Ja-Lok
Ku
l,
Sojin
Kim
k,
Tamrin
Chowdhury
k,
Sungnam
Park
h,
Veronica Jihyun
Kim
i,
Ji Yeoun
Lee
ij,
Yun Sik
Dho
k,
Yuna
Jung
a,
Junho K.
Hur
ac,
Chan
Park
ab,
Junyang
Jung
ab,
Youngbuhm
Huh
ab,
Ja-Lok
Ku
l,
Sojin
Kim
k,
Tamrin
Chowdhury
k,
Sungnam
Park