
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAluminum porphyrins with quaternary ammonium halides as catalysts for copolymerization of cyclohexene oxide and CO2: metal–ligand cooperative catalysis†
Jingyuan
Deng
 a,
Manussada
Ratanasak
b,
Yuma
Sako
c,
Hideki
Tokuda
c,
Chihiro
Maeda
c,
Jun-ya
Hasegawa
*b,
Kyoko
Nozaki
*a and
Tadashi
Ema‡
*c
a,
Manussada
Ratanasak
b,
Yuma
Sako
c,
Hideki
Tokuda
c,
Chihiro
Maeda
c,
Jun-ya
Hasegawa
*b,
Kyoko
Nozaki
*a and
Tadashi
Ema‡
*c
aDepartment of Chemistry and Biotechnology, Graduate School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: nozaki@chembio.t.u-tokyo.ac.jp
bInstitute for Catalysis, Hokkaido University, Kita 21, Nishi 10, Kita-ku, Sapporo, Hokkaido 001-0021, Japan. E-mail: hasegawa@cat.hokudai.ac.jp
cDivision of Applied Chemistry, Graduate School of Natural Science and Technology, Okayama University, Tsushima, Okayama 700-8530, Japan. E-mail: ema@cc.okayama-u.ac.jp
First published on 18th May 2020
Abstract
Bifunctional AlIII porphyrins with quaternary ammonium halides, 2-Cl and 2-Br, worked as excellent catalysts for the copolymerization of cyclohexene oxide (CHO) and CO2 at 120 °C. Turnover frequency (TOF) and turnover number (TON) reached 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 and 55000, respectively, and poly(cyclohexene carbonate) (PCHC) with molecular weight of up to 281000 was obtained with a catalyst loading of 0.001 mol%. In contrast, bifunctional MgII and ZnII counterparts, 3-Cl and 4-Cl, as well as a binary catalyst system, 1-Cl with bis(triphenylphosphine)iminium chloride (PPNCl), showed poor catalytic performances. Kinetic studies revealed that the reaction rate was first-order in [CHO] and [2-Br] and zero-order in [CO2], and the activation parameters were determined: ΔH‡ = 12.4 kcal mol−1, ΔS‡ = −26.1 cal mol−1 K−1, and ΔG‡ = 21.6 kcal mol−1 at 80 °C. Comparative DFT calculations on two model catalysts, AlIII complex 2′ and MgII complex 3′, allowed us to extract key factors in the catalytic behavior of the bifunctional AlIII catalyst. The high polymerization activity and carbonate-linkage selectivity originate from the cooperative actions of the metal center and the quaternary ammonium cation, both of which facilitate the epoxide-ring opening by the carbonate anion to form the carbonate linkage in the key transition state such as TS3b (ΔH‡ = 13.3 kcal mol−1, ΔS‡ = −3.1 cal mol−1 K−1, and ΔG‡ = 14.4 kcal mol−1 at 80 °C).
000 h−1 and 55000, respectively, and poly(cyclohexene carbonate) (PCHC) with molecular weight of up to 281000 was obtained with a catalyst loading of 0.001 mol%. In contrast, bifunctional MgII and ZnII counterparts, 3-Cl and 4-Cl, as well as a binary catalyst system, 1-Cl with bis(triphenylphosphine)iminium chloride (PPNCl), showed poor catalytic performances. Kinetic studies revealed that the reaction rate was first-order in [CHO] and [2-Br] and zero-order in [CO2], and the activation parameters were determined: ΔH‡ = 12.4 kcal mol−1, ΔS‡ = −26.1 cal mol−1 K−1, and ΔG‡ = 21.6 kcal mol−1 at 80 °C. Comparative DFT calculations on two model catalysts, AlIII complex 2′ and MgII complex 3′, allowed us to extract key factors in the catalytic behavior of the bifunctional AlIII catalyst. The high polymerization activity and carbonate-linkage selectivity originate from the cooperative actions of the metal center and the quaternary ammonium cation, both of which facilitate the epoxide-ring opening by the carbonate anion to form the carbonate linkage in the key transition state such as TS3b (ΔH‡ = 13.3 kcal mol−1, ΔS‡ = −3.1 cal mol−1 K−1, and ΔG‡ = 14.4 kcal mol−1 at 80 °C).
Introduction
Conversions of CO2 into value-added products are becoming more and more important from the viewpoint of green and sustainable chemistry.1,2 Among the value-added products, polycarbonates synthesized from epoxides and CO2 have attracted much attention as sustainable and biodegradable materials.2 Since the discovery of the ring-opening copolymerization of epoxides and CO2 by Inoue in 1969,3 a variety of metal complex catalysts have been developed,4–20 such as Co(salen),4–6 Cr(salen),7–9 Zn(β-diiminate),10 and metalloporphyrins.11,12,20 Among them, multinuclear metal complexes4b,6c,9,10b,13–17 and bifunctional monometallic complexes4a,6b,18–20 have exhibited notable catalytic performances. Among several effective metal elements, aluminum is fascinating from the viewpoint of low toxicity, low price, and abundance on the earth.2i,11,20Previously, we have developed bifunctional MgII and ZnII porphyrin catalysts, including 3-Br and 4-Br (Scheme 1), for the synthesis of cyclic carbonates from epoxides and CO2.21 The halide anion, the alkyl group, the linker length, and the number and position of the quaternary ammonium halide group were changed, and high catalytic activities were induced by the cooperative action of the metal center (M) and the halide anion (X−); turnover frequencies (TOFs) and turnover numbers (TONs) reached 12000–46000 h−1 and 103000–310000, respectively. However, these MgII and ZnII complexes showed poor catalytic activity for polycarbonate synthesis.
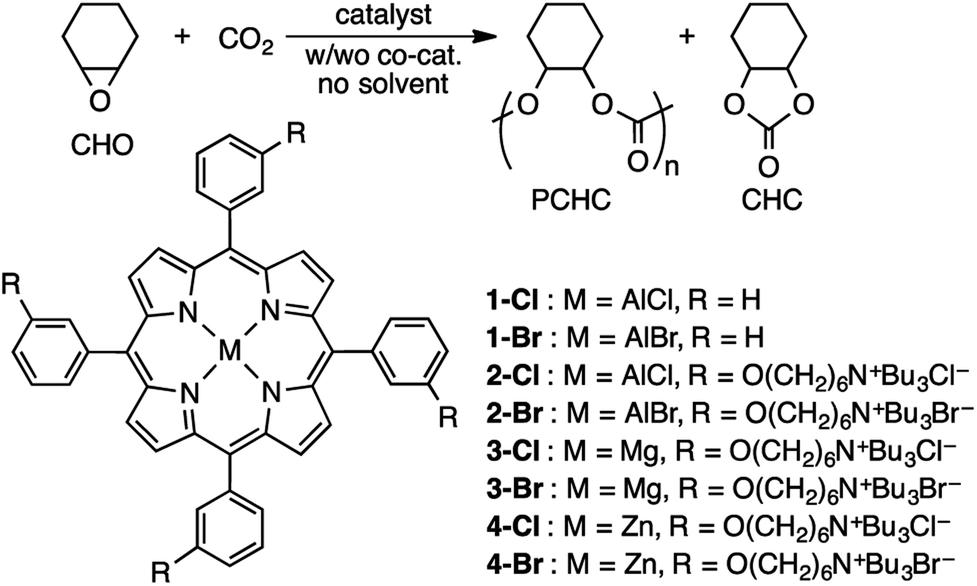 | ||
| Scheme 1 Copolymerization of CHO and CO2. | ||
Here we employed one of the best structures for the synthesis of cyclic carbonates,21 and replaced the central MgII or ZnII ion with the AlIII ion to prepare new bifunctional catalysts with quaternary ammonium halide groups, 2-Cl and 2-Br, for the copolymerization of cyclohexene oxide (CHO) and CO2 (Scheme 1). We used CHO as a monomer despite the low reactivity of the internal epoxide because poly(cyclohexene carbonate) (PCHC) has promising properties in terms of glass transition temperature and tensile strength.22-Cl and 2-Br showed TOFs of 10000 h−1, giving PCHC with molecular weight of up to 281000 with a catalyst loading of 0.001 mol%. DFT calculations suggest that the key transition state in the carbonate-linkage formation step is stabilized by the cooperative effect of (i) the metal activation of the epoxide and (ii) the electrostatic stabilization of the nucleophilic carbonate anion by the quaternary ammonium cation.
Results and discussion
Copolymerization of CHO and CO2
The copolymerization reactions of CHO with CO2 (2 MPa) were conducted at 120 °C in the presence of the catalyst without solvent (Table 1). 2-Cl and 2-Br showed a TOF of 10000 h−1 (entries 1 and 2). To the best of our knowledge, this is one of the highest values for the solvent-free copolymerization of CHO and CO2 among monometallic catalysts reported so far.7,8b,12e1H NMR spectra showed a polycarbonate linkage of >99%; PCHC contained no ether linkages, and a trace amount of cyclohexene carbonate (CHC) was detected. Size-exclusion chromatography (SEC) indicated that PCHC had a bimodal molecular weight distribution with a small polydispersity index (PDI) of 1.02, where the higher-molecular-weight polymers were approximately twice as large as the lower-molecular-weight polymers.13a The molecular weight of PCHC exceeded 67000 in 1 h (entries 1 and 2) and 220000 in 3 h (entries 3 and 4). When the reaction time was extended to 24 h with a catalyst loading of 0.001 mol% (10 ppm), elastic material capped with a rigid shell was formed, and the reaction appeared to stop owing to the high viscosity of the reaction mixture. Nevertheless, PCHC had a molecular weight of 281000 or 263000, corresponding to almost 2000-mers (entries 5 and 6). Although these molecular weights of PCHC are smaller than those calculated by multiplying the TON by the molecular weight of the repeating unit (142), which is due to a chain transfer process,22,23 the values in Table 1 clearly demonstrate the excellent catalytic performances of 2-Cl and 2-Br.
| Entry | Catalyst | S/C | Time (h) | TONb | Polycarbonateb (%) | M n (kg mol−1) | PDIc (Mw/Mn) |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: CHO (2.0 mL, 20 mmol), catalyst (0.0025 mol%), CO2 (2.0 MPa), 120 °C, in a 50 mL autoclave. b TON and the polycarbonate-linkage selectivity were determined by 1H NMR analysis. c Determined by size-exclusion chromatography (SEC) analysis using THF as an eluent and polystyrene as a molecular weight standard. Peaks had bimodal shapes. d CHO (3.1 mL, 31 mmol), catalyst (0.001 mol%). e CHO (3.2 mL, 32 mmol), catalyst (0.001 mol%). f PPNCl (4 equiv. of 1-Cl) was added. | |||||||
| 1 | 2-Cl | 40000 |
1 | 10000 |
>99 | 68/32 | 1.02/1.02 |
| 2 | 2-Br | 40000 |
1 | 10000 |
>99 | 67/31 | 1.02/1.02 |
| 3 | 2-Cl | 40000 |
3 | 19200 |
99 | 232/106 | 1.05/1.07 |
| 4 | 2-Br | 40000 |
3 | 19600 |
99 | 229/101 | 1.04/1.08 |
| 5d | 2-Cl | 100000 |
24 | 45000 |
96 | 281/96 | 1.09/1.27 |
| 6e | 2-Br | 100000 |
24 | 55000 |
97 | 263/108 | 1.09/1.12 |
| 7f | 1-Cl | 40000 |
1 | 1600 | 96 | 3.9/1.6 | 1.03/1.14 |
| 8 | 3-Cl | 40000 |
1 | 27 | 52 | 0.41/0.25 | 1.04/1.02 |
| 9 | 4-Cl | 40000 |
1 | 48 | 63 | 0.44/0.26 | 1.06/1.01 |
Control experiments were done to address the origin of the high catalytic power of bifunctional AlIII catalyst 2-Cl. A binary catalyst system, 1-Cl with bis(triphenylphosphine)iminium chloride (PPNCl), showed a TOF of 1600 h−1, producing a smaller size of PCHC under otherwise the same reaction conditions (entry 7), which demonstrates the importance of the peripheral quaternary ammonium chlorides in 2-Cl. On the other hand, the corresponding bifunctional MgII and ZnII complexes, 3-Cl and 4-Cl, showed TOFs of 27 h−1 and 48 h−1, respectively, and low polycarbonate selectivity (52% and 63%) (entries 8 and 9). Clearly, the identity of the central metal ion has a crucial impact on the catalytic process.24
Kinetic studies
In kinetic measurements, we used diglyme as a solvent to avoid the generation of a highly viscous or rigid polymer product. To monitor the reaction progress, in situ infrared (IR) spectroscopy was employed. Typical examples of the reactions with bifunctional catalyst 2-Br and binary catalyst system 1-Br/PPNBr are shown in Fig. 1. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond stretching signals for PCHC and CHC appear at 1750 cm−1 and 1825 cm−1, respectively. The linearity between the IR signal intensity and the amount of PCHC was verified by calibration. 2-Br was highly selective toward PCHC, and the formation of CHC was not detected at all (Fig. 1a). In contrast, 1-Br/PPNBr was less selective, and the CHC formation was clearly observed (Fig. 1b). CHC was formed only at the early stage of the copolymerization reaction, and the formation of CHC ceased after a certain reaction time. Accordingly, there was an induction period in the PCHC production, which overlapped with the period of the CHC formation. CHC can be formed easily by the backbiting reaction of the carbonate anion with the C atom attached to the Br atom because bromide is a good leaving group, slowing down the PCHC formation at the outset. Once the polycarbonate linkage is formed, the CHC formation is no longer favored because the resulting alkylcarbonate is a poor leaving group.
O bond stretching signals for PCHC and CHC appear at 1750 cm−1 and 1825 cm−1, respectively. The linearity between the IR signal intensity and the amount of PCHC was verified by calibration. 2-Br was highly selective toward PCHC, and the formation of CHC was not detected at all (Fig. 1a). In contrast, 1-Br/PPNBr was less selective, and the CHC formation was clearly observed (Fig. 1b). CHC was formed only at the early stage of the copolymerization reaction, and the formation of CHC ceased after a certain reaction time. Accordingly, there was an induction period in the PCHC production, which overlapped with the period of the CHC formation. CHC can be formed easily by the backbiting reaction of the carbonate anion with the C atom attached to the Br atom because bromide is a good leaving group, slowing down the PCHC formation at the outset. Once the polycarbonate linkage is formed, the CHC formation is no longer favored because the resulting alkylcarbonate is a poor leaving group.
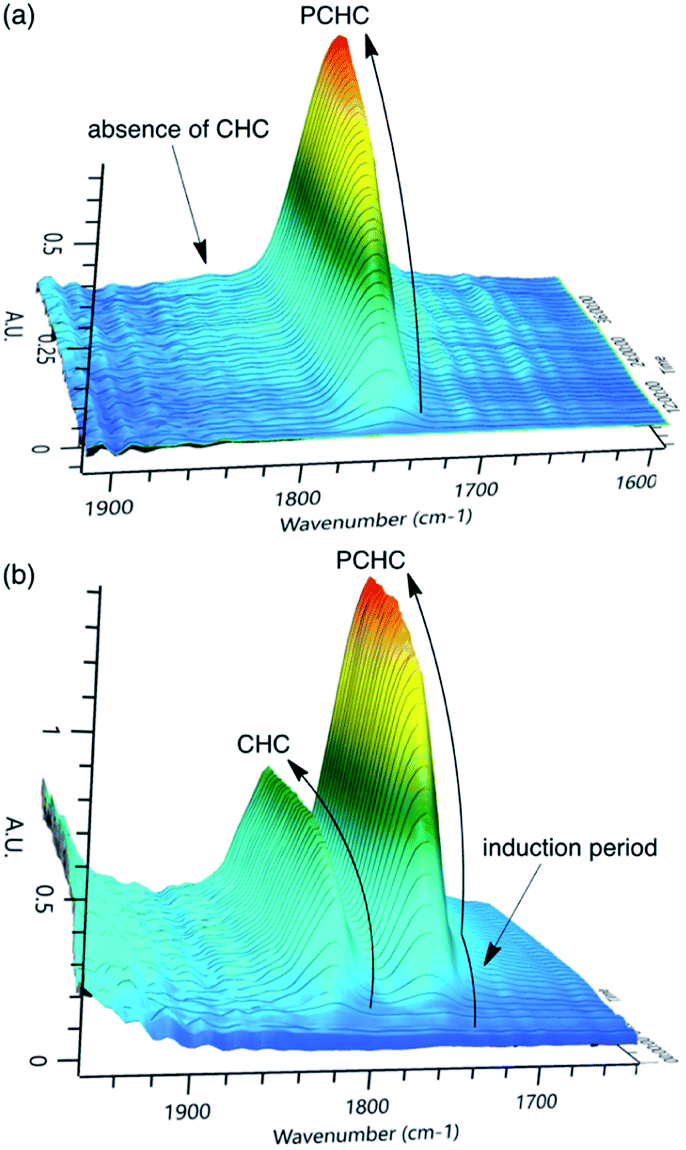 | ||
| Fig. 1 3D stack plots of in situ IR spectra for the copolymerization of CHO and CO2 in diglyme at 80 °C with (a) 2-Br and (b) 1-Br/PPNBr. | ||
Based on the plots of the initial reaction rates at 68 °C vs. different concentrations of either CHO, CO2, or 2-Br (ESI†), the copolymerization reaction was found to be first-order in both [CHO] and [2-Br] and zero-order in [CO2] in the range of the CO2 pressure from 10 to 30 bar: r = k[CHO]1[CO2]0[2-Br]1, where r and k stand for the reaction rate and the rate constant, respectively. This rate law strongly suggests that the epoxide-ring opening is involved in the rate-determining step, where CO2 is not involved.
To determine the activation energies (Ea) and activation parameters (ΔH‡, ΔS‡, and ΔG‡), kinetic experiments were conducted at different temperatures. The initial reaction rates for the PCHC formation with 2-Br are plotted against the reaction temperatures in Fig. 2a, and the Arrhenius plot is shown in Fig. 2b. The activation energy for the PCHC formation was calculated to be 13.2 kcal mol−1. For comparison, the corresponding activation energy with 1-Br/PPNBr was also determined in the same way to be 16.2 kcal mol−1 (ESI†). The Eyring plot for the PCHC formation with 2-Br yielded the following activation parameters: ΔH‡ = 12.4 kcal mol−1, ΔS‡ = −26.1 cal mol−1 K−1, and ΔG‡ = 21.6 kcal mol−1 at 80 °C (ESI†).
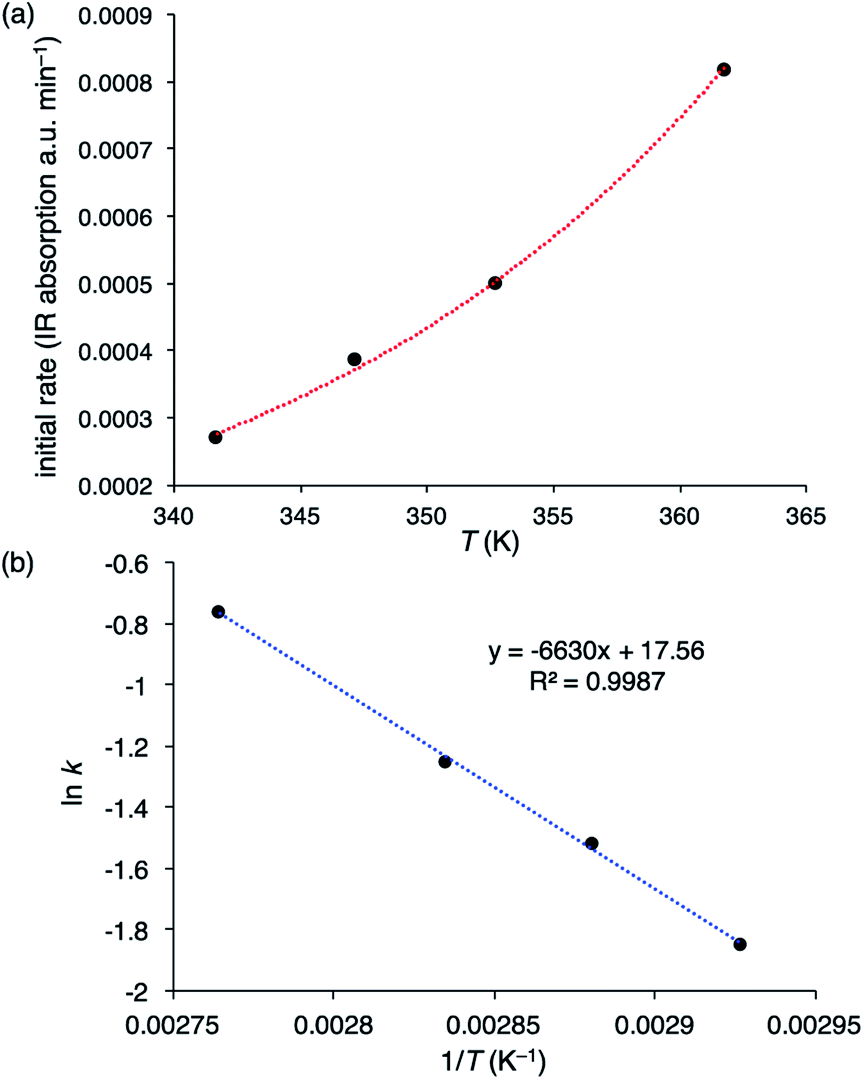 | ||
| Fig. 2 (a) Plot of initial rates of copolymerization vs. temperatures (342, 347, 353, and 362 K) with [2-Br] = 0.055 mM, [CHO] = 1.98 M, and a CO2 pressure of 20 bar in diglyme. (b) Arrhenius plot for the formation of PCHC with 2-Br. | ||
Proposed reaction pathway
Based on the results of the kinetic experiments and the rate law, it can be inferred that one equivalent of CHO and 2-Br are involved in the transition state of the rate-determining step while CO2 is not involved. A plausible reaction pathway is shown in Scheme 2. First, CHO activated by the metal center in reactant complex R is ring-opened by the initiating group (X = Cl or Br) to generate intermediate I1. The resulting alkoxide species subsequently undergoes CO2 insertion to give intermediate I3avia weak CO2 adduct I2, which should be fast because the reaction is zero-order in [CO2]. The carbonate anion will dissociate from the metal center to form an ion pair with the tethered quaternary ammonium cation, allowing for the metal coordination of another CHO (I3b). This carbonate anion is ready for the nucleophilic attack without diffusing into the bulk solution (Scheme 3a). CHO activated by the metal coordination is ring-opened by the carbonate anion, forming the carbonate linkage (I4b). The overall polymerization rate can be substantially improved by accelerating the rate of this ring-opening step. The quaternary ammonium cation in 2 may enhance the nucleophilicity of both the halide anion and the carbonate anion. In addition, the bulky structure of 2 may also prevent the activated CHO from homopolymerization.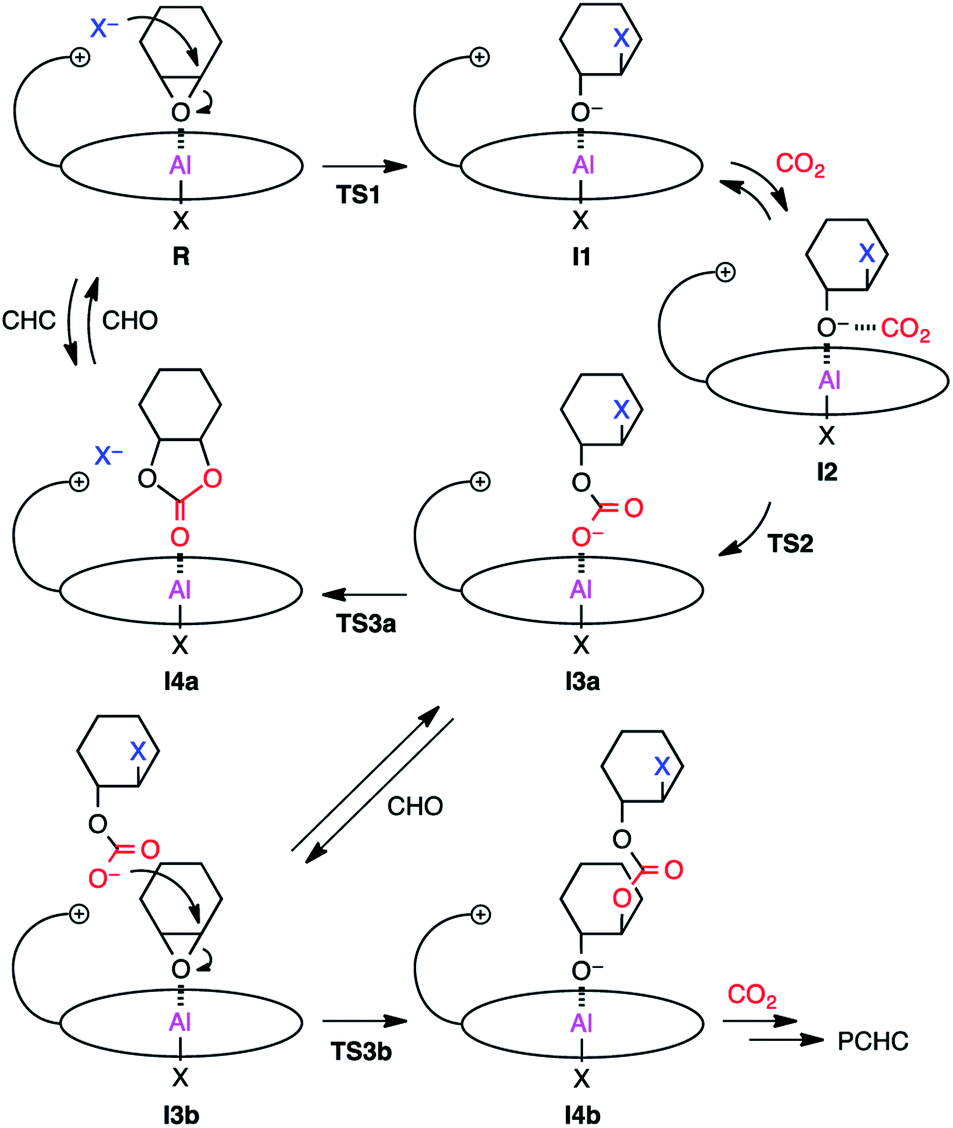 | ||
| Scheme 2 Plausible reaction pathways for the formation of PCHC and CHC with 2-Cl or 2-Br. | ||
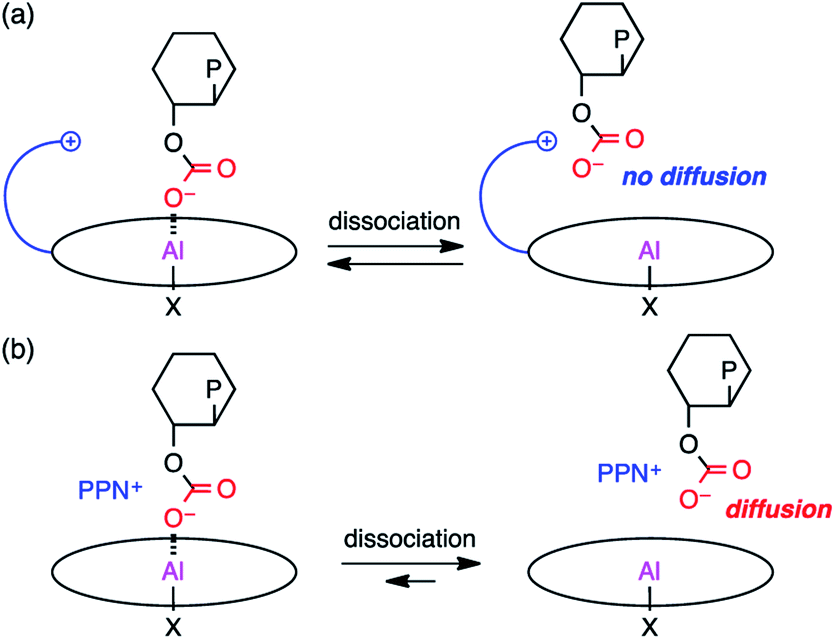 | ||
| Scheme 3 Roles of the quaternary ammonium cation in (a) bifunctional catalyst 2 and (b) binary catalyst system. | ||
On the other hand, a scenario for the binary catalyst system is completely different from that for the bifunctional catalyst. The initiation step will take a longer time because the nucleophilic co-catalyst seldom encounters the catalyst. In addition, the carbonate anion intermediate may dissociate from the metal center to diffuse into the bulk solution, suspending the elongation process (Scheme 3b). The diffused carbonate anion may undergo a backbiting reaction to afford the undesired side product, CHC. Under high dilution conditions, this side reaction is likely to occur, lowering the PCHC selectivity.
DFT calculations
DFT calculations are powerful tools for the understanding of reaction mechanisms of epoxide/CO2 copolymerizations.24,25 Here we performed DFT calculations along the proposed reaction pathway (Scheme 2) using monosubstituted AlIII complex 2′ as a model for 2-Br (Fig. 3). MgII complex 3′ was also employed as a model for 3-Br to pursue the origin of the high catalytic activity of AlIII complex 2-Br. The potential energy profiles for the 2′- and 3′-catalyzed reactions of CHO with CO2 are shown in Fig. 3. The structures of the key transition state TS3b in the 2′- and 3′-catalyzed reactions are shown in Fig. 4, while all of the other transition-state and intermediate structures are given in ESI.†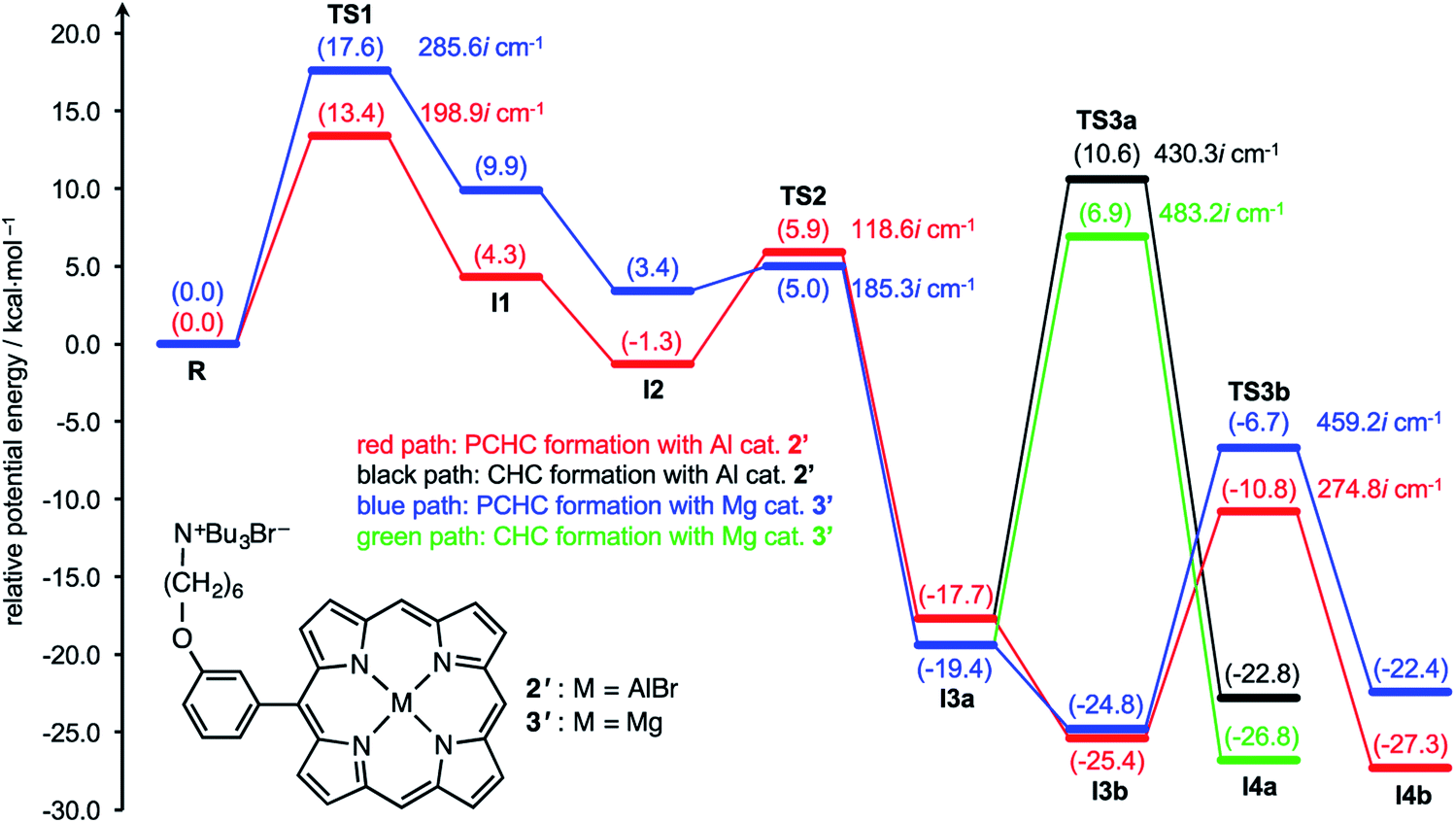 | ||
| Fig. 3 Potential energy profiles for the 2′ and 3′-catalyzed reactions of CHO with CO2. Each transition state and intermediate is designated in the proposed reaction pathway (Scheme 2). Computations were performed at the ωB97XD/6-31G* level with the self-consistent reaction field (SCRF) method (Et2O). The potential energies relative to reactant complex R are given in kcal mol−1. The energies of CO2 and the second CHO are included in the former steps where they do not appear explicitly in the computational model. | ||
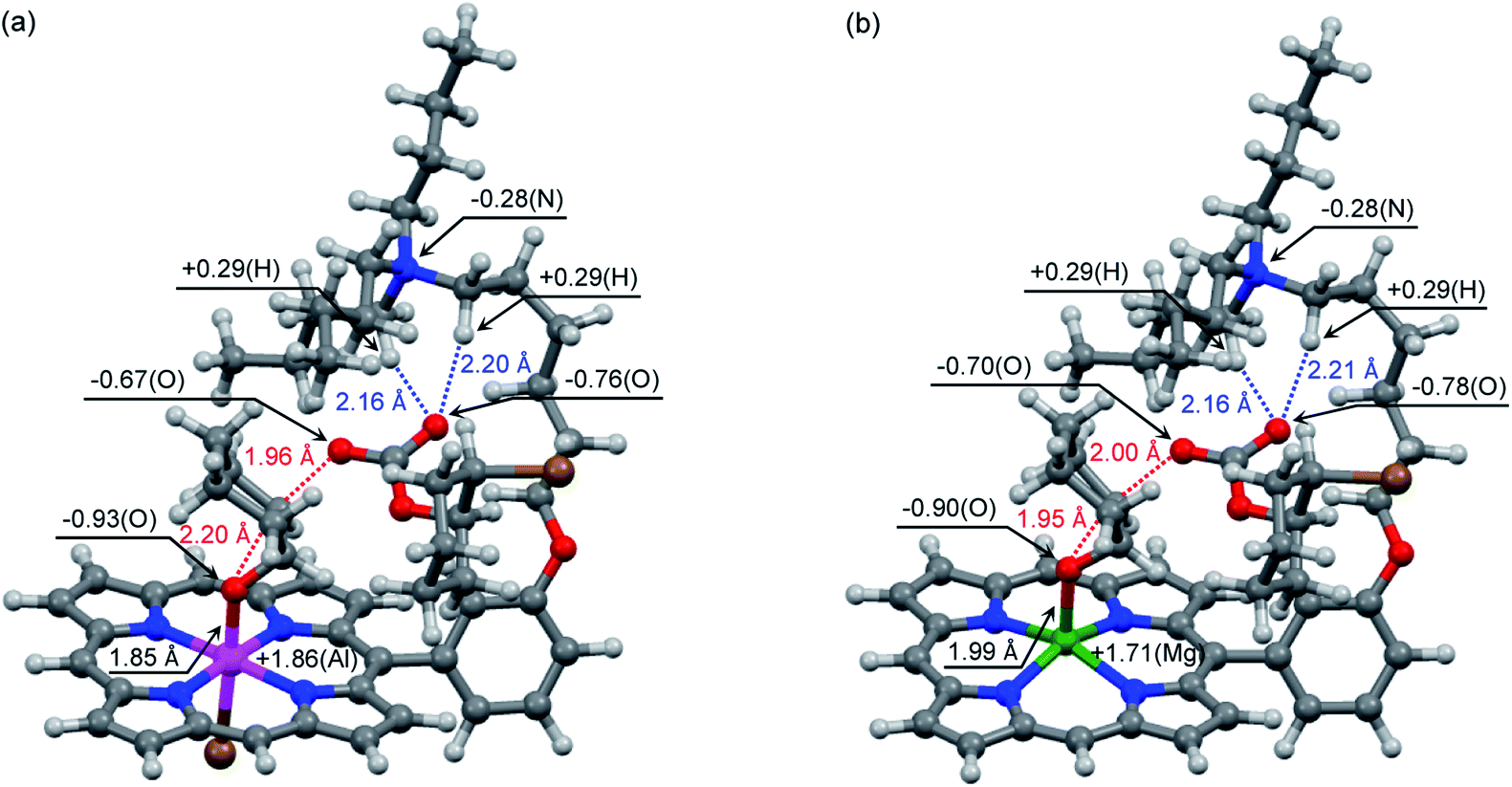 | ||
| Fig. 4 Optimized structures of transition state TS3b in the reaction catalyzed by (a) AlIII complex 2′ and (b) MgII complex 3′. | ||
Fig. 3 clearly indicates that AlIII complex 2′ favors the formation of PCHC over that of CHC; the activation energy for TS3b (Ea = 14.6 kcal mol−1), leading to PCHC, is much smaller than that for TS3a (Ea = 28.3 kcal mol−1), leading to CHC. This is consistent with the experimental results that PCHC was produced highly selectively with 2-Br (Table 1). We consider that the carbonate-linkage formation such as TS3b is the rate-determining step in the production of PCHC because the energy barrier for TS3b (Ea = 14.6 kcal mol−1) is greater than that for TS1 (Ea = 13.4 kcal mol−1) and because the carbonate linkages are formed many times in the chain propagation. CO2 insertion is not the rate-determining step since the energy barrier for TS2 is very small (Ea = 7.2 kcal mol−1), which is consistent with the fact that the reaction rate was independent of the CO2 pressure (ESI†). The calculated activation parameters of AlIII complex 2′ for TS3b were as follows: ΔH‡ = 13.3 kcal mol−1, ΔS‡ = −3.1 cal mol−1 K−1, and ΔG‡ = 14.4 kcal mol−1 at 80 °C. This ΔH‡ value is close to the experimental value for the PCHC formation with 2-Br (ΔH‡ = 12.4 kcal mol−1) although precise comparisons are difficult partly because of the structural differences between 2′ and 2-Br and between the model and real polymeric substrates.
The activation energy for TS3a in the formation of CHC with MgII complex 3′ (Ea = 26.3 kcal mol−1) is much greater than the previously reported value in the 3′-catalyzed reaction of propylene oxide with CO2 (Ea = 17.1 kcal mol−1).21b This is partly because TS3a adopts an unfavorable 1,2-diaxial conformation and because the secondary C atom undergoes the nucleophilic attack in the formation of CHC.25 Interestingly, as compared with MgII complex 3′, AlIII complex 2′ further disfavors the CHC formation (TS3a). This is probably because the stronger Lewis acid (AlIII) reduces the nucleophilicity of the carbonate anion. Likewise, 2′ disfavors the CO2 insertion (TS2) as compared with 3′. In sharp contrast, AlIII complex 2′ effectively stabilizes the oxyanion species generating in the epoxide-ring opening steps (TS1 and TS3b) as compared with MgII complex 3′. These behaviors can be rationalized by the Lewis acidity of the metal ions: AlIII > MgII. Actually, the most significant difference in the key transition state, TS3b, between 2′ and 3′ can be seen around the metal center (Fig. 4); the natural atomic charges on the Al and Mg atoms are +1.86 and +1.71, respectively, and the Al–O and Mg–O distances are 1.85 and 1.99 Å, respectively. Therefore, AlIII complex 2′ can stabilize the negatively charged O atom in TS3b more effectively than MgII complex 3′.
Previously, it has been proposed that the energy difference in the carbonate–epoxide substitution (I3avs.I3b in the present case) is closely related to the chain propagation rate.24 In other words, a better catalyst has a higher affinity for the epoxide so that the carbonate anion can be replaced more easily by the epoxide, which leads to the efficient chain propagation. Indeed, Fig. 3 indicates that the transition from I3a to I3b is thermodynamically more favorable with AlIII complex 2′ than with MgII complex 3′, which accords with the previous proposal.24
In addition to the identity of the metal center, the quaternary ammonium cation is also quite important for the catalysis. Previously, we have reported that the tethered quaternary ammonium cation of bifunctional MgII complex 3′ undergoes conformational changes to stabilize various anionic species generated during the catalysis for the formation of propylene carbonate.21b In this study, we confirmed that all the transition-state and intermediate structures are well stabilized by the tethered quaternary ammonium cation. For example, specific electrostatic interactions between the anionic species and the flexible quaternary ammonium cation can be seen in Fig. 4. In addition, we hypothesized that the tethered quaternary ammonium cation may promote the dissociation of the carbonate anion from the Al atom in I3a to give I3b.24 To investigate this possibility, we compared bifunctional catalyst 2′ with AlIII 5-phenylporphyrin complex, having no quaternary ammonium cation, by calculating the energies for the dissociation of the carbonate anion from the Al atom in I3a (ESI†). As a result, the carbonate anion was found to dissociate more easily by 18 kcal mol−1 in the former case than in the latter case. Furthermore, the tethered quaternary ammonium cation is considered to be useful for the suppression of the diffusion of the growing polymer chain into the bulk solution by means of ion pairing (Scheme 3a). The remaining three quaternary ammonium cations in 2 can contribute to the electrostatic stabilization of the anionic intermediates and transition states. The tethered quaternary ammonium salts in 2 also function as a source of the initiator (X−). Clearly, the quaternary ammonium halide groups in 2 play multiple essential roles in the highly efficient and selective formation of PCHC.
Conclusions
Copolymerization of epoxides and CO2 is one of the most straightforward and promising methods for the synthesis of sustainable polymers, polycarbonates. However, only a limited number of metal elements are known to be active for this fascinating transformation. In this work, bifunctional AlIII porphyrin complexes with quaternary ammonium halide groups, 2-Cl and 2-Br, exhibited excellent catalytic activity and selectivity for the copolymerization of CHO and CO2. TOF and TON reached 10000 h−1 and 55000, respectively, and PCHC with molecular weight of up to 281000 was obtained with a catalyst loading of 0.001 mol%. These excellent catalytic performances of 2-Cl and 2-Br resulted from the elaborately designed structures of the bifunctional catalysts with high thermal stability. Kinetic study with in situ IR spectroscopy revealed that the copolymerization reaction was first-order in [CHO] and [2-Br] and zero-order in [CO2]: r = k[CHO]1[CO2]0[2-Br]1, and the activation parameters were determined by the Arrhenius plot and the Eyring plot: Ea = 13.2 kcal mol−1, ΔH‡ = 12.4 kcal mol−1, ΔS‡ = −26.1 cal mol−1 K−1, and ΔG‡ = 21.6 kcal mol−1 at 80 °C. Comparative DFT calculations on two model catalysts, AlIII complex 2′ and MgII complex 3′, allowed us to extract key factors in the bifunctional AlIII catalyst. Both the AlIII metal center and the quaternary ammonium cations work cooperatively to exert high polymerization activity and carbonate-linkage selectivity. The AlIII metal center effectively activates the epoxide, and the flexible quaternary ammonium cations simultaneously stabilize and guide the carbonate anion in the key transition state, such as TS3b (Ea = 14.6 kcal mol−1, ΔH‡ = 13.3 kcal mol−1, ΔS‡ = −3.1 cal mol−1 K−1, and ΔG‡ = 14.4 kcal mol−1 at 80 °C). In addition, the quaternary ammonium cations in 2-Cl and 2-Br are also considered to prevent the anionic intermediates from diffusing into the bulk solution, which enables the efficient copolymerization reaction even with a low catalyst loading of 0.001 mol%. Molecular catalysis utilizing the cooperative effect has recently attracted much attention,26 and the present work will expand the opportunity of designing further new catalysts in future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant No. JP15H05796, JP15H05805, and JP16H01030 in Precisely Designed Catalysts with Customized Scaffolding. We also thank the Cooperative Research Program of Institute for Catalysis, Hokkaido University (Grant 19A1003). JH thanks MEXT for “Priority Issue on Post-K Computer” (Development of new fundamental technologies for high-efficiency energy creation, conversion/storage and use). We thank Ms. Tomoko Amimoto, the Natural Science Center for Basic Research and Development (N-BARD), Hiroshima University for the measurement of MALDI-TOF mass spectra.Notes and references
- (a) M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510 CrossRef CAS PubMed; (b) Y. Tsuji and T. Fujihara, Chem. Commun., 2012, 48, 9956 RSC; (c) I. Omae, Coord. Chem. Rev., 2012, 256, 1384 CrossRef CAS; (d) L. Zhang and Z. Hou, Chem. Sci., 2013, 4, 3395 RSC; (e) M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709 CrossRef CAS PubMed; (f) C. Maeda, Y. Miyazaki and T. Ema, Catal. Sci. Technol., 2014, 4, 1482 RSC; (g) Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed; (h) G. Fiorani, W. Guo and A. W. Kleij, Green Chem., 2015, 17, 1375 RSC; (i) B. Yu and L.-N. He, ChemSusChem, 2015, 8, 52 CrossRef CAS; (j) Q.-W. Song, Z.-H. Zhou and L.-N. He, Green Chem., 2017, 19, 3707 RSC; (k) S.-S. Yan, Q. Fu, L.-L. Liao, G.-Q. Sun, J.-H. Ye, L. Gong, Y.-Z. Bo-Xue and D.-G. Yu, Coord. Chem. Rev., 2018, 374, 439 CrossRef CAS.
- (a) H. Sugimoto and S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5561 CrossRef CAS; (b) D. J. Darensbourg, R. M. Mackiewicz, A. L. Phelps and D. R. Billodeaux, Acc. Chem. Res., 2004, 37, 836 CrossRef CAS PubMed; (c) D. J. Darensbourg, Chem. Rev., 2007, 107, 2388 CrossRef CAS PubMed; (d) S. Klaus, M. W. Lehenmeier, C. E. Anderson and B. Rieger, Coord. Chem. Rev., 2011, 255, 1460 CrossRef CAS; (e) M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141 RSC; (f) X.-B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462 RSC; (g) D. J. Darensbourg and S. J. Wilson, Green Chem., 2012, 14, 2665 RSC; (h) X.-B. Lu, W.-M. Ren and G.-P. Wu, Acc. Chem. Res., 2012, 45, 1721 CrossRef CAS; (i) N. Ikpo, J. C. Flogeras and F. M. Kerton, Dalton Trans., 2013, 42, 8998 RSC; (j) S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459 RSC; (k) Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354 CrossRef CAS; (l) S. J. Poland and D. J. Darensbourg, Green Chem., 2017, 19, 4990 RSC.
- S. Inoue, H. Koinuma and T. Tsuruta, J. Polym. Sci., Part B: Polym. Lett., 1969, 7, 287 CrossRef CAS.
- (a) K. Nakano, T. Kamada and K. Nozaki, Angew. Chem., Int. Ed., 2006, 45, 7274 CrossRef CAS PubMed; (b) K. Nakano, S. Hashimoto and K. Nozaki, Chem. Sci., 2010, 1, 369 RSC; (c) M. Hatazawa, K. Nakabayashi, S. Ohkoshi and K. Nozaki, Chem.–Eur. J., 2016, 22, 13677 CrossRef CAS PubMed; (d) M. Hatazawa, R. Takahashi, J. Deng, H. Houjou and K. Nozaki, Macromolecules, 2017, 50, 7895 CrossRef CAS.
- (a) Z. Qin, C. M. Thomas, S. Lee and G. W. Coates, Angew. Chem., Int. Ed., 2003, 42, 5484 CrossRef CAS; (b) C. T. Cohen, T. Chu and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 10869 CrossRef CAS PubMed.
- (a) X.-B. Lu, L. Shi, Y.-M. Wang, R. Zhang, Y.-J. Zhang, X.-J. Peng, Z.-C. Zhang and B. Li, J. Am. Chem. Soc., 2006, 128, 1664 CrossRef CAS PubMed; (b) W.-M. Ren, Z.-W. Liu, Y.-Q. Wen, R. Zhang and X.-B. Lu, J. Am. Chem. Soc., 2009, 131, 11509 CrossRef CAS PubMed; (c) Y. Liu, W.-M. Ren, W.-P. Zhang, R.-R. Zhao and X.-B. Lu, Nat. Commun., 2015, 6, 8594 CrossRef CAS PubMed.
- K. Nakano, M. Nakamura and K. Nozaki, Macromolecules, 2009, 42, 6972 CrossRef CAS.
- (a) D. J. Darensbourg and J. C. Yarbrough, J. Am. Chem. Soc., 2002, 124, 6335 CrossRef CAS PubMed; (b) D. J. Darensbourg and R. M. Mackiewicz, J. Am. Chem. Soc., 2005, 127, 14026 CrossRef CAS; (c) D. J. Darensbourg and A. L. Phelps, Inorg. Chem., 2005, 44, 4622 CrossRef CAS PubMed; (d) D. J. Darensbourg and A. I. Moncada, Inorg. Chem., 2008, 47, 10000 CrossRef CAS PubMed; (e) D. J. Darensbourg, A. I. Moncada, W. Choi and J. H. Reibenspies, J. Am. Chem. Soc., 2008, 130, 6523 CrossRef CAS PubMed; (f) D. J. Darensbourg, W.-C. Chung and S. J. Wilson, ACS Catal., 2013, 3, 3050 CrossRef CAS.
- S. I. Vagin, R. Reichardt, S. Klaus and B. Rieger, J. Am. Chem. Soc., 2010, 132, 14367 CrossRef CAS PubMed.
- (a) M. Cheng, D. R. Moore, J. J. Reczek, B. M. Chamberlain, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 8738 CrossRef CAS PubMed; (b) D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2003, 125, 11911 CrossRef CAS PubMed; (c) C. M. Byrne, S. D. Allen, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 11404 CrossRef CAS PubMed; (d) W. C. Ellis, Y. Jung, M. Mulzer, R. Di Girolamo, E. B. Lobkovsky and G. W. Coates, Chem. Sci., 2014, 5, 4004 RSC; (e) F. Auriemma, C. De Rosa, M. R. Di Caprio, R. Di Girolamo, W. C. Ellis and G. W. Coates, Angew. Chem., Int. Ed., 2015, 54, 1215 CrossRef CAS PubMed.
- (a) T. Aida, M. Ishikawa and S. Inoue, Macromolecules, 1986, 19, 8 CrossRef CAS; (b) H. Cao, Y. Qin, C. Zhuo, X. Wang and F. Wang, ACS Catal., 2019, 9, 8669 CrossRef CAS.
- (a) S. Mang, A. I. Cooper, M. E. Colclough, N. Chauhan and A. B. Holmes, Macromolecules, 2000, 33, 303 CrossRef CAS; (b) H. Sugimoto and K. Kuroda, Macromolecules, 2008, 41, 312 CrossRef CAS; (c) C. E. Anderson, S. I. Vagin, W. Xia, H. Jin and B. Rieger, Macromolecules, 2012, 45, 6840 CrossRef CAS; (d) W. Xia, S. I. Vagin and B. Rieger, Chem.–Eur. J., 2014, 20, 15499 CrossRef CAS PubMed; (e) R. M. B. Carrilho, L. D. Dias, R. Rivas, M. M. Pereira, C. Claver and A. M. Masdeu-Bultó, Catalysts, 2017, 7, 210 CrossRef.
- (a) F. Jutz, A. Buchard, M. R. Kember, S. B. Fredriksen and C. K. Williams, J. Am. Chem. Soc., 2011, 133, 17395 CrossRef CAS PubMed; (b) J. A. Garden, P. K. Saini and C. K. Williams, J. Am. Chem. Soc., 2015, 137, 15078 CrossRef CAS PubMed; (c) G. Trott, J. A. Garden and C. K. Williams, Chem. Sci., 2019, 10, 4618 RSC.
- (a) M. W. Lehenmeier, S. Kissling, P. T. Altenbuchner, C. Bruckmeier, P. Deglmann, A.-K. Brym and B. Rieger, Angew. Chem., Int. Ed., 2013, 52, 9821 CrossRef CAS PubMed; (b) S. Kissling, M. W. Lehenmeier, P. T. Altenbuchner, A. Kronast, M. Reiter, P. Deglmann, U. B. Seemann and B. Rieger, Chem. Commun., 2015, 51, 4579 RSC.
- H. Nagae, R. Aoki, S. Akutagawa, J. Kleemann, R. Tagawa, T. Schindler, G. Choi, T. P. Spaniol, H. Tsurugi, J. Okuda and K. Mashima, Angew. Chem., Int. Ed., 2018, 57, 2492 CrossRef CAS PubMed.
- M. Cozzolino, K. Press, M. Mazzeo and M. Lamberti, ChemCatChem, 2016, 8, 455 CrossRef CAS.
- M. Mandal, V. Ramkumar and D. Chakraborty, Polym. Chem., 2019, 10, 3444 RSC.
- (a) E. K. Noh, S. J. Na, S. S, S.-W. Kim and B. Y. Lee, J. Am. Chem. Soc., 2007, 129, 8082 CrossRef CAS; (b) S. S, J. K. Min, J. E. Seong, S. J. Na and B. Y. Lee, Angew. Chem., Int. Ed., 2008, 47, 7306 CrossRef.
- J. Liu, W.-M. Ren, Y. Liu and X.-B. Lu, Macromolecules, 2013, 46, 1343 CrossRef CAS.
- (a) X. Sheng, Y. Wang, Y. Qin, X. Wang and F. Wang, RSC Adv., 2014, 4, 54043 RSC; (b) W. Wu, X. Sheng, Y. Qin, L. Qiao, Y. Miao, X. Wang and F. Wang, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2346 CrossRef CAS; (c) X. Sheng, W. Wu, Y. Qin, X. Wang and F. Wang, Polym. Chem., 2015, 6, 4719 RSC.
- (a) T. Ema, Y. Miyazaki, S. Koyama, Y. Yano and T. Sakai, Chem. Commun., 2012, 48, 4489 RSC; (b) T. Ema, Y. Miyazaki, J. Shimonishi, C. Maeda and J. Hasegawa, J. Am. Chem. Soc., 2014, 136, 15270 CrossRef CAS PubMed; (c) C. Maeda, T. Taniguchi, K. Ogawa and T. Ema, Angew. Chem., Int. Ed., 2015, 54, 134 CrossRef CAS PubMed; (d) C. Maeda, J. Shimonishi, R. Miyazaki, J. Hasegawa and T. Ema, Chem.–Eur. J., 2016, 22, 6556 CrossRef CAS; (e) C. Maeda, S. Sasaki and T. Ema, ChemCatChem, 2017, 9, 946 CrossRef CAS.
- (a) T. Aida and S. Inoue, Acc. Chem. Res., 1996, 29, 39 CrossRef CAS; (b) S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2861 CrossRef CAS; (c) D. J. Darensbourg, Green Chem., 2019, 21, 2214 RSC.
- The MALDI-TOF mass spectrum of PCHC showed a bimodal molecular weight distribution, and peak assignments revealed the structures of the higher- and lower-molecular-weight polymers and a mechanistic aspect involving a chain transfer process (ESI†).
- T. Ohkawara, K. Suzuki, K. Nakano, S. Mori and K. Nozaki, J. Am. Chem. Soc., 2014, 136, 10728 CrossRef CAS PubMed.
- (a) Z. Liu, M. Torrent and K. Morokuma, Organometallics, 2002, 21, 1056 CrossRef CAS; (b) G. A. Luinstra, G. R. Haas, F. Molnar, V. Bernhart, R. Eberhardt and B. Rieger, Chem.–Eur. J., 2005, 11, 6298 CrossRef CAS PubMed; (c) M. W. Lehenmeier, C. Bruckmeier, S. Klaus, J. E. Dengler, P. Deglmann, A.-K. Ott and B. Rieger, Chem.–Eur. J., 2011, 17, 8858 CrossRef CAS PubMed; (d) A. Buchard, F. Jutz, M. R. Kember, A. J. P. White, H. S. Rzepa and C. K. Williams, Macromolecules, 2012, 45, 6781 CrossRef CAS; (e) L. P. Carrodeguas, J. González-Fabra, F. Castro-Gómez, C. Bo and A. W. Kleij, Chem.–Eur. J., 2015, 21, 6115 CrossRef PubMed; (f) J. González-Fabra, F. Castro-Gómez, A. W. Kleij and C. Bo, ChemSusChem, 2017, 10, 1233 CrossRef PubMed.
- (a) J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236 CrossRef CAS PubMed; (b) M. D. Wodrich and X. Hu, Nat. Rev. Chem., 2018, 2, 99 CrossRef CAS; (c) B. D. Nath, K. Takaishi and T. Ema, Catal. Sci. Technol., 2020, 10, 12 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, copolymerization reactions, kinetic experiments, and DFT calculations. See DOI: 10.1039/d0sc01609h |
| ‡ Cooperative Research Fellow, Institute for Catalysis, Hokkaido University, Japan. |
| This journal is © The Royal Society of Chemistry 2020 |