
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular crowding effects on the biochemical properties of amyloid β–heme, Aβ–Cu and Aβ–heme–Cu complexes†
Meng
Li
 ab,
Zhenqi
Liu
ac,
Jinsong
Ren
ac and
Xiaogang
Qu
*ac
ab,
Zhenqi
Liu
ac,
Jinsong
Ren
ac and
Xiaogang
Qu
*ac
aLaboratory of Chemical Biology, Division of Biological Inorganic Chemistry, State Key Laboratory of Rare Earth Resource Utilization, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, Jilin 130022, P. R. China. E-mail: xqu@ciac.ac.cn; Fax: +86-431-85262656
bCollege of Pharmaceutical Sciences, Hebei Medical University, Shijiazhuang, 050017, P. R. China
cUniversity of Science and Technology of China, Hefei, Anhui 230026, P. R. China
First published on 2nd July 2020
Abstract
Heme as a cofactor has been proposed to bind with β-amyloid peptide (Aβ) and the formed Aβ–heme complex exhibits enhanced peroxidase-like activity. So far, in vitro studies on the interactions between heme, Cu and Aβ have been exclusively performed in dilute solution. However, the intracellular environment is highly crowded with biomolecules. Therefore, exploring how Aβ–heme–Cu complexes behave under molecular crowding conditions is critical for understanding the mechanism of Aβ neurotoxicity in vivo. Herein, we selected PEG-200 as a crowding agent to mimic the crowded cytoplasmic environment for addressing the contributions of crowded physiological environments to the biochemical properties of Aβ–heme, Aβ–Cu and Aβ–heme–Cu complexes. Surprisingly, experimental studies and theoretical calculations revealed that molecular crowding weakened the stabilization of the Aβ–heme complex and decreased its peroxidase activity. Our data attributed this consequence to the decreased binding affinity of heme to Aβ as a result of the alterations in water activity and Aβ conformation. Our findings highlight the significance of hydration effects on the interaction of Aβ–heme and Aβ–Cu and their peroxidase activities. Molecular crowding inside cells may potentially impose a positive effect on Aβ–Cu but a negative effect on the interaction of Aβ with heme. This indicates that Aβ40–Cu but not Aβ40–heme may play more important roles in the oxidative damage in the etiology of AD. Therefore, this work provides a new clue for understanding the oxidative damage occurring in AD.
Introduction
Alzheimer's disease (AD) is a devastating neurodegenerative disease.1 One of the important pathological features of AD is the formation of senile plaques, which is caused by the aggregation and cerebral deposition of amyloid β-peptide (Aβ). Targeting, preventing and even reversing aggregation of Aβ have been considered to be effective strategies for AD therapy.2–5 Although promising, till now, there is no known cure for AD, due to the complex pathogenesis of AD.6–8Numerous studies indicate that heme plays a critical role in the neurotoxicity of Aβ, which can bind with Aβ to form an Aβ–heme complex with enhanced peroxidase-like activity.7–9 The complex can catalyze the oxidation of neurotransmitters by H2O2 and induce an abnormality of heme metabolism, leading to enhanced neural damage.7,9 Moreover, metal ions as cofactors are also involved in Aβ aggregate deposition and neurotoxicity. It has been reported that Cu2+ accumulated in amyloid plaques is capable of exacerbating the progression of amyloid pathology in AD.10–12 On the other hand, upon binding to Aβ, the formed Aβ–metal complexes can promote oxygen activation to produce toxic reactive oxygen species (ROS) under physiological conditions.13–16 The produced ROS then in turn result in enhanced aggregation propensity of Aβ especially Aβ oligomerization,17 which is thought to be another contributing factor to Aβ-related neuronal dysfunction, cellular apoptosis and further amyloid plaque formation.17–19 More importantly, it has been reported that the amount of toxic ROS produced by Aβ–heme–Cu is obviously higher than that produced by Aβ–heme or Aβ–Cu complexes in dilute solution,20 which may significantly increase the cytotoxicity. Critically, due to the distribution of Aβ peptides both in the cytoplasm and extracellular spaces in the brain,20,21 Aβ would bind to Cu2+ and heme simultaneously in vivo. Therefore, it is highly appropriate to study the combined effects of heme and Cu on Aβ-induced neurotoxicity under physiological conditions.
So far in vitro analysis of the interaction of Aβ with heme and Cu has been exclusively carried out in dilute solution.20,21 However, in contrast to the in vitro solution environments, the cellular environment where Aβ peptides exist is highly crowded since various macromolecules occupy up to 40% of the cytoplasmic volume.22,23 This so-called macromolecule crowding effect makes the behaviors of peptides in a cellular environment quite different from that in dilute solution. It can accelerate protein aggregation to form a compact state due to the reduced available volume.24 In addition, as the water activity decreased, the protein solubility was reduced, resulting in the self-assembly of amyloidogenic proteins.24–26 Given such a profound influence on Aβ aggregation, investigating the effects of molecular crowding on Aβ–heme–Cu interaction is of great significance. In this study, in order to address the contributions of crowded physiological environments to the biochemical properties of the Aβ–heme–Cu complex, we utilized a concentrated solution of PEG-200, one of the commonly used molecular crowding agents, to mimic the crowded cellular environment.27,28 Aβ40, the most abundant Aβ isoform, was chosen as the peptide model for this study.
Materials and methods
Materials
3,3′,5,5′-Tetramethylbenzidine (TMB) was purchased from Damas-beta (Shanghai, China). H2O2 was bought from Beijing Chemicals (Beijing, China). Heme was purchased from Alfa Aesar (Ward Hill, MA). Glycerol, polyethylene glycol with an average molecular weight of 200 (PEG-200) and 1,1,1,3,3,3-hexafluoropropan-2-ol (HFIP) were obtained from Acros Organics. Other chemicals used in this study were purchased from Sigma-Aldrich. All these reagents were used as received without further purification. Deionized water (D.I. water, 18.2 MΩ cm) used for all experiments was obtained from a Milli-Q system (Millipore, Bedford, MA).Aβ preparation
Aβ40 (lot no. ALX-151-003) was obtained from Enzo Life Sciences and prepared according to previous reports.29–31 Briefly, the peptide was dissolved in HFIP at a concentration of 2 mg mL−1 under shaking at 4 °C for 4 hours in a sealed vial for complete dissolution. After that, the peptide was stored at −20 °C. Before experiments, the solvent HFIP was removed by evaporation under a gentle stream of nitrogen and the peptide was dissolved in phosphate buffered saline (PBS, pH 7.0), which was then used immediately.Preparation of heme
A stock solution was prepared by dissolving heme in dimethyl sulfoxide (DMSO) and stored at −20 °C. The concentration of heme was determined spectrophotometrically using a millimolar extinction coefficient of 179 at 404 nm in 40% DMSO.32 The concentration of the stock solution was 32 mM. The heme working solution was freshly prepared with PBS (pH 7.0), which contained 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 2 mM KH2PO4. The final concentration of DMSO was kept lower than 0.8% for all assays.Preparation of different complexes
Aβ40–heme was formed by incubating Aβ40 with heme at a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for 1 h. Similarly, Aβ40–Cu complex was prepared via incubating CuCl2 solution with Aβ40 at a molar ratio of 1:1 for 1 h. The incubation of CuCl2 solution with the preformed Aβ40–heme at a molar ratio of 1:1 (Cu2+:Aβ40) for 1 h can give the Aβ40–heme–Cu complex, while the sample of Aβ40–Cu–heme was obtained by incubating heme with the preformed Aβ40–Cu complex at a molar ratio of 1:1 (heme:Aβ40) for 1 h. All the complexes were prepared in PBS. The final pH value of all the complexes was kept at 7.0.
:1 for 1 h. Similarly, Aβ40–Cu complex was prepared via incubating CuCl2 solution with Aβ40 at a molar ratio of 1:1 for 1 h. The incubation of CuCl2 solution with the preformed Aβ40–heme at a molar ratio of 1:1 (Cu2+:Aβ40) for 1 h can give the Aβ40–heme–Cu complex, while the sample of Aβ40–Cu–heme was obtained by incubating heme with the preformed Aβ40–Cu complex at a molar ratio of 1:1 (heme:Aβ40) for 1 h. All the complexes were prepared in PBS. The final pH value of all the complexes was kept at 7.0.
Electron paramagnetic resonance (EPR) analysis
EPR spectra were acquired with a Bruker A3000 electron paramagnetic resonance spectrometer. The concentration of EPR samples was 0.25 mM, and the experiments were run at 77 K in a liquid nitrogen finger dewar.Fluorescence titrations
Quenching of the intrinsic fluorescence of tyrosine was monitored according to previous reports.30,31 Different concentrations of heme (0–7 μM) were incubated with Aβ40 (3 μM) in various concentrations of glycerol. Then the fluorescence spectra of these samples were collected with the excitation wavelength at 278 nm. The emission intensity at 306 nm was recorded for analysis. To calculate the binding constants, the 1:1 binding stoichiometric equation29,33 was employed:where [P] indicates the concentration of Aβ40 and [L] is the concentration of heme. ΔI represents the difference in fluorescence intensities of the free and complexed Aβ40 peptides. The binding constant: K = 1/Kd.
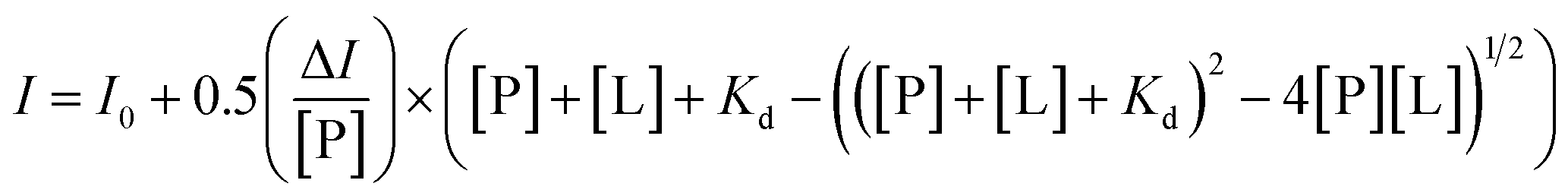
The change in hydration is given by the equation:29,34
| (∂ln(Ks/K0))/∂[Osm] = −Δnw/55.5 |
Peroxidase activities of the different complexes
TMB was utilized as the substrate for evaluating the catalytic abilities of the different Aβ40-related complexes. The measurements were performed by monitoring the absorbance change at 652 nm. To perform this assay, 4 μL TMB (42 mM), 4 μL H2O2 (300 mM) and 4 μL of heme or the formed complexes (80 μM) were successively added into 388 μL PBS (pH 5.0). For the comparison of the different peroxidase activities of these complexes, the absorbance at 652 nm was monitored after 1 h of incubation at 37 °C. Methylene blue (MB) was also utilized to determine the peroxidase activities of these different complexes under neutral pH conditions.8 A PBS solution (200 μL, pH = 7) containing the formed complexes (2 μM), H2O2 (1 mM) and MB (10 μM) was prepared. After 2 h of incubation at 37 °C, the absorbance change of MB at 664 nm was monitored by using a microplate reader (molecular devices, SpectraMax Plus 384).In vitro ROS measurements
The produced ROS by Aβ40 related complexes in the presence of a reducing agent (ascorbate) was detected using Amplex Red/horseradish peroxidase (HRP) assays.35 Different Aβ40 related samples (5 μM) in PBS (pH 7.0) were first mixed with HRP (1 U mL−1) and Amplex Red (50 μM). The formation of resorufin was initiated by the addition of ascorbate (50 μM) in the reaction mixture. The total volume of the sample solution was 400 μL. After incubation for 45 min, the formation of ROS was determined by the measurement of the fluorescence intensity at 590 nm with an excitation wavelength of 540 nm. The interference of PEG-200 on the detection of ROS was eliminated by the measurement of H2O2 with the given concentrations to obtain a calibration curve in the presence of different concentrations of PEG-200. The amount of ROS produced by these complexes was normalized with that produced by Aβ40–heme in dilute solution. Each experiment was performed in triplicate.CD measurements
A Jasco-J810 spectropolarimeter was employed to analyse the secondary conformation of Aβ40. A path length of the cell of 0.1 cm was used. And the spectrum was measured over the wavelength range from 200 nm to 260 nm. The concentration of Aβ40 was 50 μM.Simulated calculation on optimization molecular interactions
All simulations were performed using the Gaussian 09 program. The structure of heme was obtained from the RCSB PDB database and optimized at the B3LYP level of theory in combination with the 6-31G(d,p) basis set. The constructions of Heme–arginine, heme–histidine and histidine–heme–arginine were investigated at the B3LYP/6-31G(d,p) level. All calculations had no imaginary frequency, indicating that the systems were stable. The interaction energies were computed including the correction of the basis set superposition error (BSSE) at the same level of theory.Results and discussion
To clarify the influence of the crowded environment on the interaction of Aβ40 with heme, we monitored the changes in UV-vis spectra of heme. Since Aβ40 bound to heme in a binding stoichiometry of 1:1 in dilute solution at neutral pH,9,20 we also used 1:1 as the molar ratio of heme to Aβ40 in the following experiments. As shown in Fig. 1A, upon addition of Aβ40 to the heme solution, a red shift of the Soret band from about 375 nm to 404 nm and an increase around 530 nm were observed, which indicated the formation of an Aβ40–heme complex.9 It was worth noting that the spectrum of heme in the presence of PEG-200 was different from that of free heme. The free movement and vibration of heme may be limited due to the solution effect, which then resulted in reduced absorbance and broader bands (Fig. 1A).36 Under the crowding conditions, the red shift of the Soret band of heme was not induced by Aβ40 (Fig. 1A). This typical spectral change implied that the binding ability of heme toward Aβ40 was weakened in the crowding environment.
 | ||
| Fig. 1 (A) UV-vis absorption spectra of Aβ40–heme in the presence of PEG-200. The final concentrations of heme and Aβ40 were 20 μM, respectively. (B) Peroxidase activity of Aβ40–heme in the presence of different concentrations of PEG-200. The values were presented as means ± SD of three independent experiments. | ||
Given the significantly reduced interaction between heme and Aβ40 in PEG-200 solution, we then examined the effects of the crowding agent on the peroxidase-like activity of the Aβ40–heme complex via monitoring the catalytic oxidation of TMB by H2O2.37,38 In dilute solution, the catalytic activity of Aβ40–heme was more than three times higher compared to that of free heme (Fig. S1 and S2, ESI†). However, as illustrated in Fig. 1B, the presence of PEG-200 had a dramatic effect on Aβ40–heme peroxidase activity. The higher the concentration of PEG-200 in the sample, the lower the catalytic activity of Aβ40–heme. In fact, the catalytic activity of Aβ40–heme shrunk by about 20 times when the concentration of PEG-200 was up to 40% in PBS (Fig. S2, ESI†). As a very modest catalyst under these conditions, the peroxidase activity of Aβ–heme could not be of significance in vivo, which was consistent with a previous report.39 However, in the control experiments, the catalytic activity of heme increased slightly when examined under crowding conditions (Fig. S1 and S2, ESI†). Interestingly, in the presence of PEG-200 at a higher concentration, the peroxidase activity of Aβ40–heme was even lower than that of heme. These results indicated that the weak interactions may not be the only factor that affected the peroxidase activity of the Aβ40–heme complex under the crowding conditions.
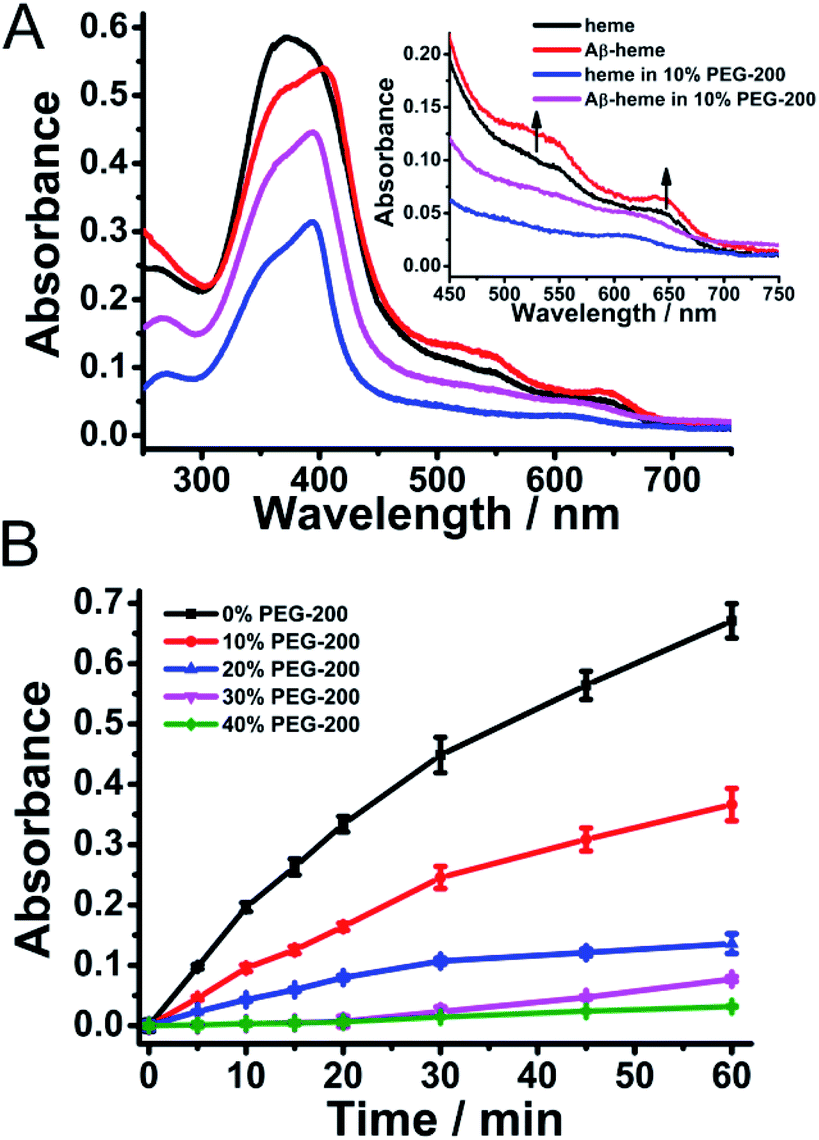
Sequentially, the effect of crowding conditions on the formation of Aβ40–heme–Cu was examined. As shown in Fig. 2A, upon addition of Cu2+ to the preformed Aβ40–heme complex, the characteristic features of Aβ40–heme were still retained, indicating that the addition of Cu2+ did not disrupt the preformed Aβ40–heme complex. Interestingly, if the metal ions were added to Aβ40 before the addition of heme, there were no characteristic features of Aβ40–heme (Fig. S3A, ESI†), indicating that the bound metal ions were able to inhibit the formation of the Aβ40–heme complex. These results were consistent with previous studies reported by Atamna,6 but different from Dey's work.9 The difference may arise from the different experiment conditions. Meanwhile, similar to the behaviour of Aβ40–heme under our experimental conditions, the binding affinity of heme with Aβ40 was too weak to induce the spectral change of heme in Aβ40–heme–Cu in the presence of PEG-200 (Fig. 2A).
 | ||
| Fig. 2 (A) UV-vis absorption spectra of Aβ40–heme–Cu in the presence of PEG-200. The concentrations of heme, Aβ40 and Cu were 20 μM. (B) Peroxidase activity of Aβ40–heme–Cu in the presence of different concentrations of PEG-200. The metal ions were added to Aβ40 simultaneously with the addition of heme or after the Aβ40–heme was formed. The values were presented as means ± SD of three independent experiments. | ||
The binding behaviour of Cu2+ to Aβ was monitored using EPR spectroscopy. As shown in Fig. S4 (ESI†), the EPR spectrum of the Aβ40–Cu complex at pH 7 exhibited a characteristic type 2 Cu2+ signal in the high field region, with g∥ ∼ 2.23 and g⊥ ∼ 2.053. When Cu2+ was added to the preformed Aβ–heme complex, the high field region of the EPR spectrum of the Aβ–heme–Cu complex was identical to that of the EPR spectrum of Aβ–Cu. Furthermore, an identical EPR spectrum was also obtained when heme was added to the preformed Aβ–Cu complex. All these results indicated that the formation of Aβ–Cu was unperturbed by the introduction of heme. Critically, the presence of 10% PEG-200 did not change the binding behavior of Cu2+ to Aβ.
Moreover, the peroxidase activity of the Aβ40–heme–Cu complex which was measured via the oxidation of TMB was comparable with that of the Aβ40–heme complex in solution (Fig. 2B and S5, ESI†). However, under crowding conditions, the peroxidase activity of the Aβ40–heme–Cu complex was obviously inhibited (Fig. 2B and S5, ESI†). It was worth noting that the peroxidase activity of Aβ40–heme–Cu was higher than that of Aβ40–heme in the presence of the same concentration of PEG-200 (Fig. S5, ESI†), which suggested that the peroxidase activity of Aβ40–heme–Cu did not just arise from the interaction between Aβ40 and heme. As a control sample, the Aβ40–Cu complex showed weak peroxidase activity under the same experimental conditions (Fig. S6, ESI†). Interestingly, in the presence of a low concentration of PEG-200, the peroxidase activity of Aβ40–Cu was slightly enhanced (Fig. S6, ESI†), which may be caused by the increased affinity of substrates toward the stable Aβ40–Cu complex in the presence of a low concentration of the crowding agent.40,41 Additionally, the peroxidase activity of the heme treated Aβ40–Cu complex (Aβ40–Cu–heme) was significantly lower than that of the Aβ40–heme–Cu complex, further supporting that heme could not bind to Aβ40 after Aβ40–Cu complex had formed (Fig. S7, ESI†). Critically, to mimic in vivo conditions, the peroxidase activities of these Aβ40 related complexes were also measured at neutral pH. As shown in Fig. S8 (ESI†), similar to the finding under acidic conditions, the catalytic activities of Aβ40–heme, Aβ40–heme–Cu and Aβ40–Cu–heme were significantly inhibited in a neutral pH environment under crowding conditions while the catalytic activity of Aβ40–Cu was slightly enhanced in the presence of a low concentration of PEG-200. That was to say that under crowding conditions, Cu binding to Aβ40 may play a more critical role in contributing to the peroxidase activity of Aβ40–heme–Cu.
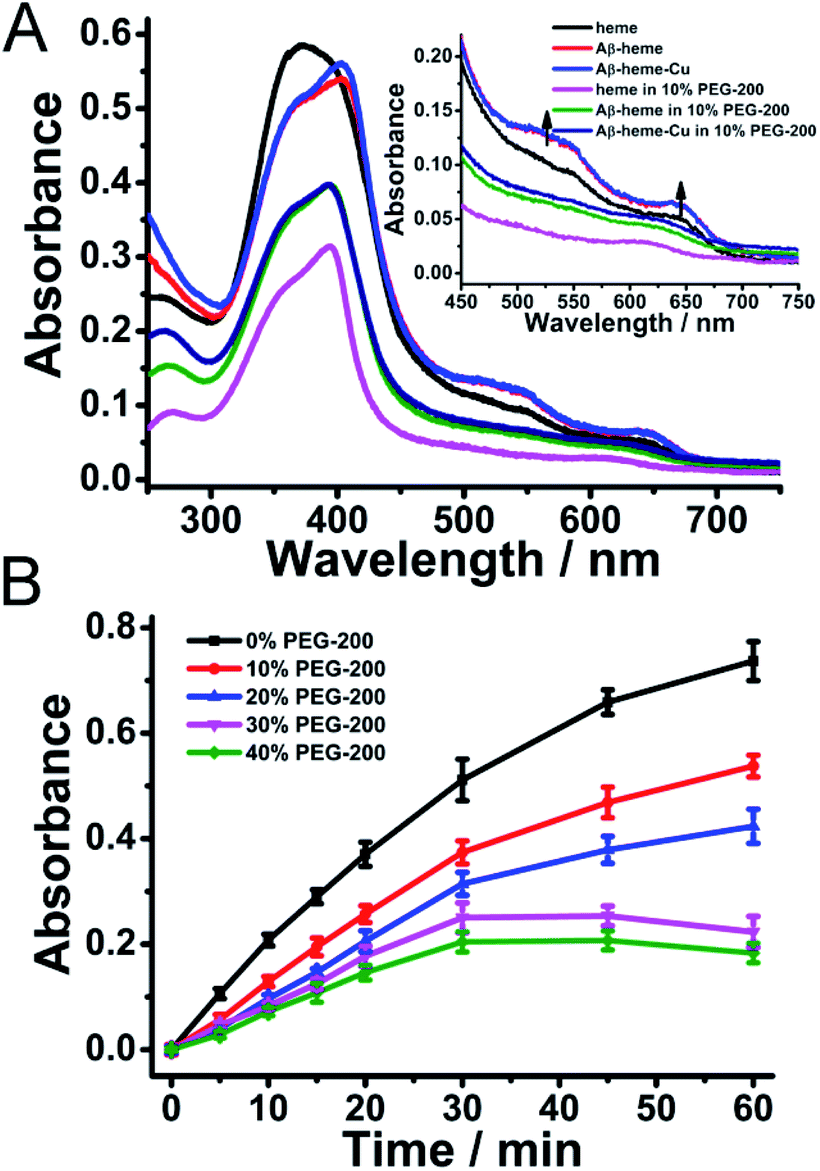
It has been reported that transition metals and heme can bind to Aβ spontaneously to induce the generation of ROS in dilute solution.20 And the ROS was generated by the chemical reduction of O2 by Aβ–Cu+, Aβ–heme (Fe2+), and Aβ–heme (Fe2+)–Cu+ complexes.42,43 Both Aβ–Cu and Aβ–heme can react with O2 in their reduced state, in which O2 can re-oxidize the reduced Cu+ site to the Cu2+ form and the reduced heme site (Fe2+) to the Fe3+ form, respectively, leading to the formation of ROS. In dilute solution, since both the heme and Cu were bound to Aβ, the amount of ROS generated by Aβ–heme–Cu was much more than the individual binder. However, under macromolecular crowding conditions, the binding affinities of Cu and heme with Aβ changed, which would remarkably impact the electron transfer thus affecting the production of ROS. As shown in Fig. 3, upon introduction of PEG-200, the amount of ROS generated by Aβ40–Cu+ was increased to some extent, while the levels of ROS associated with Aβ40–heme (Fe2+)–Cu+ and Aβ40–heme (Fe2+) decreased especially in the presence of higher concentrations of PEG-200. The reduced ROS production and peroxidase activity of Aβ40–heme (Fe2+)–Cu+ were most attributed to the weak interaction between heme and Aβ40 under macromolecular crowding conditions. That was to say that the harmful ROS generated by these Aβ40 related complexes in vivo may be predominantly caused by Aβ40–Cu+ rather than Aβ40–heme (Fe2+). Therefore, Aβ40–Cu but not Aβ40–heme may play more important roles in the oxidative damage in the etiology of AD, which provided a new clue for understanding the oxidative damage occurring in AD.
 | ||
| Fig. 3 Amount of ROS detected for the samples of Aβ40–Cu, Aβ40–heme, and Aβ40–heme–Cu with ascorbate by a HRP/Amplex Red assay in the presence of different concentrations of PEG-200. The values were presented as means ± SD of three independent experiments. | ||
As we reported previously,29 when Cu2+ binds to Aβ, dehydration occurred, which was accompanied by the release of water from the system. The apparent binding constants of Cu2+ with Aβ were significantly increased in a crowding environment, where the water activity was perturbed, leading to more stable Aβ–Cu2+ aggregates,29 which may facilitate the peroxidase activity of the Aβ–Cu2+ complex and enhance the amount of ROS produced by Aβ–Cu+ under macromolecular crowding conditions, while the reduced stabilization of the Aβ–heme complex decreased the generation of ROS associated with Aβ–heme (Fe2+)–Cu+ and Aβ–heme (Fe2+).
To illustrate the mechanism responsible for the reduced stabilization of the Aβ40–heme complex under molecular crowding conditions, we performed spectroscopic titration to analyze the interaction between heme and Aβ40 peptide with or without PEG-200. The binding constants for heme at different concentrations of PEG-200 (from 0–30%) which were determined by using nonlinear least-squares fit of the data from fluorescence titrations were 3.9 × 105 M−1, 2.6 × 105 M−1, 2.0 × 105 M−1 and 1.2 × 105 M−1, respectively (Fig. S9, ESI†). This reduced binding affinity well explained the decreased stabilization of the complex and suppressed peroxidase activity in PEG solution.
Furthermore, it has been widely reported that His13 and His14 residues in the Aβ peptide can both bind with heme on the proximal side.9 After the Aβ–heme complex was formed, the Arg5 residue on the distal side of Aβ can form a H-bond with the axial water ligand, which then facilitates the O–O bond cleavage, hence making the Aβ–heme complex a peroxidase.9 The water molecule plays a critical role in mediating the catalytic activity of Aβ–heme. However, molecular crowding can alter the water activity, leading to a hydration change.27,28 In order to show the roles of water molecules in the activity of Aβ40–heme and how hydration changes occurred upon heme binding to Aβ40, we modulated the water activity of the solution in the presence of an osmotic agent, glycerol. In the absence of osmolyte, the apparent binding constants were 3.9 ×105 for heme. Intriguingly, the apparent Aβ40 binding constants for heme were significantly decreased (Fig. 4A) in the presence of glycerol that perturbed the water activity, which were consistent with the behaviours in PEG-200. The apparent binding constants for heme binding to Aβ40 in the presence of glycerol at several different osmolalities were measured. As indicated in Fig. 4B, the apparent binding constants decreased with the increasing concentration of the osmolyte, showing that decreasing water activity weakened the binding of heme to Aβ40.
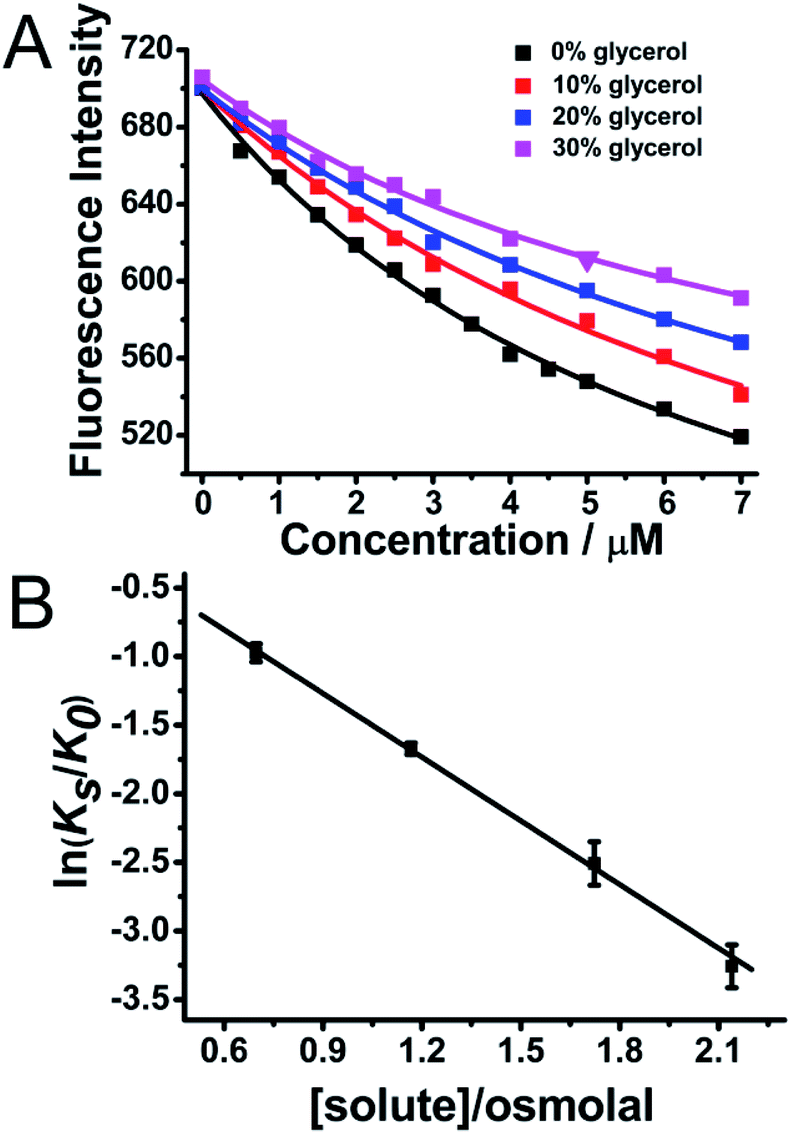 | ||
| Fig. 4 (A) Fluorescence titration of Aβ40 (3 μM) with various concentrations of heme at different concentrations of glycerol. The excitation wavelength was 278 nm and the emission intensity at 306 nm was used for analysis. 1:1 binding model was used to fit the fluorescence titration data. (B) Relationship between the binding constant and the osmolyte concentration. ln(Ks/K0) the change in binding free energy was plotted against solution osmolality. The values were presented as means ± SD of three independent experiments. | ||
The dependence of the equilibrium constant on water activity, as determined by osmotic pressure measurements at 25 °C, is shown in Fig. 4B. From the slopes of the least-squares lines, the amount of water involved in the heme–Aβ40 interaction can be quantified. The negative slopes of the best-fit lines illustrated that Δnw was positive with a value of 87 ± 3, indicating that a substantial number of water molecules were required upon heme binding to Aβ40. Reducing the number of free water molecules in bulk solution was unfavourable for the complex formation. Furthermore, hydration contribution to the thermodynamics of heme binding processes was also quantified. ΔShydration, the entropy change caused by the binding-induced change in the hydration of heme and Aβ40, was calculated44,45 by using the following equation: ΔShydration = 1.3 ± 0.4 cal K−1 mol−1 × Δnw, where 1.3 ± 0.4 cal K−1 mol−1 corresponded to the average difference between the partial molar entropy of water in the bulk state and that of water in the hydration shell of amino acid residues at 298 K, and Δnw was the number of water molecules required from the bulk state upon the binding of heme to Aβ40. The value of ΔShydration was estimated to be 113.4 ± 36.4 cal K−1 mol−1 for heme binding, which was in agreement with a previous report.39 The contribution of hydration entropy to the binding free energy, TΔShydration, was 35.25 ± 10.85 kcal mol−1 (Table 1). The value was five fold larger than the net heme–Aβ40 binding free energy change, ΔGb = −7.35 kcal mol−1, showing that hydration was very important in controlling heme binding to Aβ40.
| Aβ–heme complex | ΔGba [kcal mol−1] | Δnw | TΔShydrationb [kcal mol−1] |
|---|---|---|---|
|
a
G
b = −RTlnK.
b
S
hydration = 1.3 ± 0.4 cal K−1 mol−1 (the average difference between the partial molar entropy of water in the bulk state and water in the hydration shells of amino acid residues) × nw at 298 K.
|
|||
| Aβ–heme | −7.35 | 87 ± 3 | 35.25 ± 10.85 |
To further understand the water activity in the interaction of Aβ40 with heme, the molecular interactions between histidine, arginine and heme in the presence or absence of water were investigated using MD simulations.46,47 As shown in Fig. 5, without water, histidine could bind with heme on the proximal side; meanwhile, the arginine could just form a weak Fe–O bond with the iron center in heme. The introduction of water molecules totally changed the binding behavior of arginine. The guanidinium side chain of arginine provided H-bonds with the axial water ligand bound to the heme iron center, which was in accordance with a previous report.9 This binding behaviour also induced a pull effect by providing a proton required to drive the heterolytic cleavage of the O–O bond, resulting in an enhanced peroxidase-like activity of the Aβ–heme complex in the presence of water. The interaction energy obtained by ab initio calculations (Eint) is summarized in Table S1.† The interaction of histidine and arginine with heme in the absence of water was weaker than that in the presence of water, which further supported that hydration was very important in controlling heme binding to Aβ40.
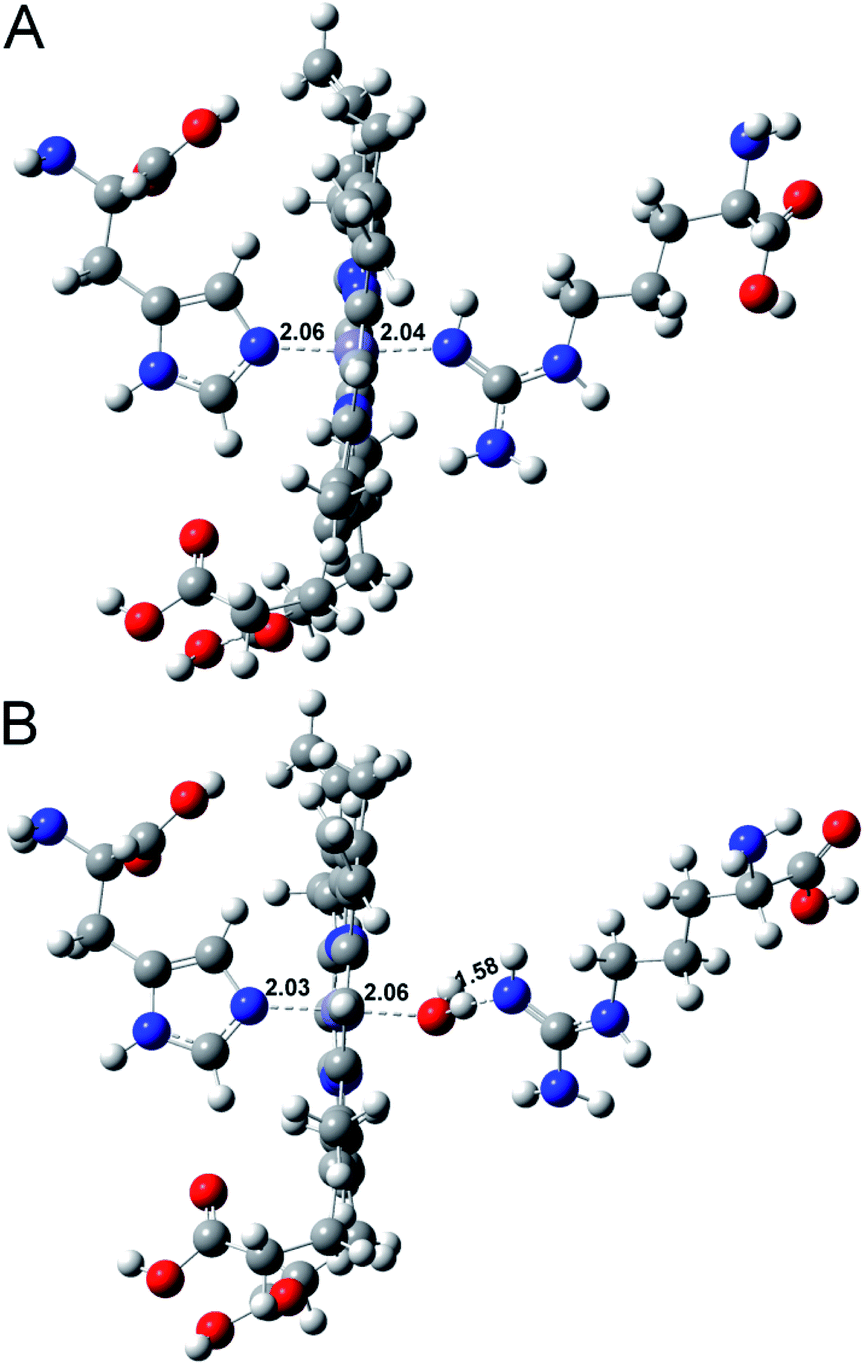 | ||
| Fig. 5 Energy-minimized average interaction models of Arg–heme–His (A) without water or (B) with water. These complexes were shown in a ball-and-stick model. Carbon atoms are indicated in dark gray balls, hydrogen atoms are in light gray, oxygen atoms are in red, and blue balls define nitrogen atoms. Binding bonds are shown by black dashes. | ||
The interactions of heme and Cu with Aβ were also affected by other factors including the structure of Aβ. A large body of research has found that the nucleation step of amyloidogenic protein fibrilization can be accelerated in the crowded environment of PEG.24,48 In accordance with these findings, circular dichroism (CD) spectral analyses (Fig. S10, ESI†) clearly showed that the presence of PEG dramatically promoted amyloid fibril formation of Aβ40. The formed intramolecular or intermolecular β-sheet structure may affect the coordination interaction between heme, Cu and histidine. It has been proposed that Aβ40 aggregates can also bind a full stoichiometric complement of Cu2+ with the same coordination geometry as Aβ40 monomers, resulting in no changes in their secondary structure.49,50 Moreover, Aβ fibrils were currently reported to be formed by in-register stacking of Aβ monomers,49,50 which made the histidine residues at positions 13 and 14 on the Aβ peptide to stack close together in successive peptides to provide a possible Cu2+ coordination site containing four histidine residues. Therefore, the formation of a β-sheet structure under crowding conditions exhibited no effect on Cu binding to Aβ. Furthermore, the linear two-coordinate NHis–Cu+–NHis structure played a critical role in the reduced Cu+–Aβ species induced ROS production.51,52 At this point, in the presence of PEG-200, the stated Aβ aggregate may favor the formation of the toxic Aβ–Cu+ complex via an intermolecular histidine bridge.53 On the other hand, for heme, it was preferred to bind with Aβ monomers rather than Aβ aggregates, leading to the inhibition effect for Aβ fibrilization.7 The formed β-sheet structure would weaken the coordinative interaction of Aβ with heme. These contributions as well as the decrease of water activity could bring additional complexity into the interaction of Cu and heme with Aβ (Scheme 1).
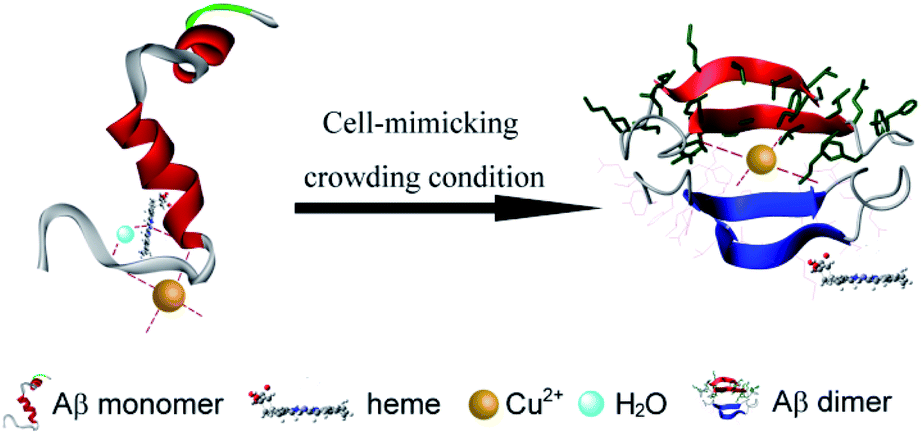 | ||
| Scheme 1 Schematic representation of the interactions of Aβ, Cu2+ and heme under molecular crowding conditions. | ||
Conclusions
In summary, heme as a cofactor has been proposed to interact with Aβ and the formed Aβ–heme complex exhibits peroxidase activity. It is emerging as a promising cytopathology in AD. Together with Cu2+, heme bound to Aβ may cause an enhanced cytotoxicity. Therefore, it is of significant importance to see whether heme still works well in the crowded cellular environment. Herein, we select PEG-200, a commonly used crowding agent, to mimic the crowded cellular environment to address the contributions of crowded physiological environments to the biochemical properties of Aβ–Cu, Aβ–heme and Aβ–heme–Cu complexes. Our present study demonstrates that although molecular crowding cannot disturb the binding of Cu with Aβ, it substantially decreases the stabilization of the Aβ–heme complex, inhibits its peroxidase activity and reduces the ROS levels. The reduced stabilization of the Aβ–heme complex arises from the decreased water activity and the altered Aβ conformation. Our findings highlight the significance of the hydration contributions to the interactions of Aβ–heme and Aβ–Cu and their peroxidase activities. The results suggest that Aβ40–Cu but not Aβ40–heme may play more important roles in the oxidative damage in the etiology of AD. Therefore, our work provides a new clue for understanding the oxidative damage occurring in AD.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21533008, 21820102009, 91856205, 21807024, and 21871249), Key Program of Frontier of Sciences (CAS QYZDJ-SSW-SLH052), Youth Top-notch Talents Supporting (BJ2018007) and Hundred Persons Plan of Hebei Province (E2018050012).Notes and references
- A. Rauk, Chem. Soc. Rev., 2009, 38, 2698 RSC.
- R. Jakob-Roetne and H. Jacobsen, Angew. Chem., Int. Ed., 2009, 48, 3030 CrossRef CAS PubMed.
- L. E. Scott and C. Orvig, Chem. Rev., 2009, 109, 4885 CrossRef CAS PubMed.
- E. Gaggelli, H. Kozlowski, D. Valensin and G. Valensin, Chem. Rev., 2006, 106, 1995 CrossRef CAS PubMed.
- I. W. Hamley, Chem. Rev., 2012, 112, 5147 CrossRef CAS PubMed.
- H. Atamna and W. H. Frey II, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11153 CrossRef CAS PubMed.
- H. Atamna and K. Boyle, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3381 CrossRef CAS PubMed.
- C. Yuan, L. Yi, Z. Yang, Q. Deng, Y. Huang, H. Li and Z. Gao, J. Biol. Inorg Chem., 2012, 17, 197 CrossRef CAS PubMed.
- D. Pramanik and S. G. Dey, J. Am. Chem. Soc., 2011, 133, 81 CrossRef CAS PubMed.
- C. S. Atwood, R. D. Moir, X. D. Huang, R. C. Scarpa, N. M. E. Bacarra, D. M. Romano, M. K. Hartshorn, R. E. Tanzi and A. I. Bush, J. Biol. Chem., 1998, 273, 12817 CrossRef CAS PubMed.
- A. I. Bush, W. H. Pettingell, G. Multhaup, M. D. Paradis, J. P. Vonsattel, J. F. Gusella, K. Beyreuther, C. L. Masters and R. E. Tanzi, Science, 1994, 265, 1464 CrossRef CAS PubMed.
- K. P. Kepp, Chem. Rev., 2012, 112, 5193 CrossRef CAS PubMed.
- J. Dong, C. S. Atwood, V. E. Anderson, S. L. Siedlak, M. A. Smith, G. Perry and P. R. Carey, Biochemistry, 2003, 42, 2768 CrossRef CAS PubMed.
- L. Guilloreau, S. Combalbert, A. Sournia-Saquet, H. Mazarguil and P. Faller, ChemBioChem, 2007, 8, 1317 CrossRef CAS PubMed.
- X. D. Huang, C. S. Atwood, M. A. Hartshorn, G. Multhaup, L. E. Goldstein, R. C. Scarpa, M. P. Cuajungco, D. N. Gray, J. Lim, R. D. Moir, R. E. Tanzi and A. I. Bush, Biochemistry, 1999, 38, 7609 CrossRef CAS PubMed.
- T. Lynch, R. A. Cherny and A. I. Bush, Exp. Gerontol., 2000, 35, 445 CrossRef CAS PubMed.
- C. Guo, L. Sun, X. Chen and D. Zhang, Neural Regener. Res., 2013, 8, 2003 CAS.
- H. Xie, S. Hou, J. Jiang, M. Sekutowicz, J. Kelly and B. J. Bacskai, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7904 CrossRef CAS PubMed.
- C. M. Lee, S. T. Huang, S. H. Huang, H. W. Lin, H. P. Tsai, J. Y. Wu, C. M. Lin and C. T. Chen, Nanomedicine, 2011, 7, 107 CrossRef CAS PubMed.
- D. Pramanik, C. Ghosh and S. G. Dey, J. Am. Chem. Soc., 2011, 133, 15545 CrossRef CAS PubMed.
- D. Pramanik, K. Sengupta, S. Mukherjee, S. G. Dey and A. Dey, J. Am. Chem. Soc., 2012, 134, 12180 CrossRef CAS PubMed.
- S. Muhuri, K. Mimura, D. Miyoshi and N. Sugimoto, J. Am. Chem. Soc., 2009, 131, 9268 CrossRef CAS PubMed.
- L. Stagg, S. Zhang, M. S. Cheung and P. Wittung-Stafshede, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18976 CrossRef CAS PubMed.
- L. A. Munishkina, E. M. Cooper, V. N. Uversky and A. L. Fink, J. Mol. Recognit., 2004, 17, 456 CrossRef CAS PubMed.
- L. A. Munishkina, A. Ahmad, A. L. Fink and V. N. Uversky, Biochemistry, 2008, 47, 8993 CrossRef CAS PubMed.
- M. Bokvist and G. Gröbner, J. Am. Chem. Soc., 2007, 129, 14848 CrossRef CAS PubMed.
- Z. Chen, K. Zheng, Y. Hao and Z. Tan, J. Am. Chem. Soc., 2009, 131, 10430 CrossRef CAS PubMed.
- S. Nakano, H. Karimata, T. Ohmichi, J. Kawakami and N. Sugimoto, J. Am. Chem. Soc., 2004, 126, 14330 CrossRef CAS PubMed.
- H. Yu, J. Ren and X. Qu, ChemBioChem, 2008, 9, 879 CrossRef CAS PubMed.
- J. Geng, M. Li, J. Ren, E. Wang and X. Qu, Angew. Chem., Int. Ed., 2011, 50, 4184 CrossRef CAS PubMed.
- H. Yu, M. Li, G. Liu, J. Geng, J. Wang, J. Ren, C. Zhao and X. Qu, Chem. Sci., 2012, 3, 3145 RSC.
- K. Kuzelová, M. Mrhalová and Z. Hrkal, Biochim. Biophys. Acta, 1997, 1336, 497 CrossRef.
- W. Garzon-Rodriguez, A. K. Yatsimirsky and C. G. Glabe, Bioorg. Med. Chem. Lett., 1999, 9, 2243 CrossRef CAS PubMed.
- X. Qu and J. B. Chaires, J. Am. Chem. Soc., 2001, 123, 1 CrossRef CAS PubMed.
- X. He, H. M. Park, S. J. Hyung, A. S. DeToma, C. Kim, B. T. Ruotolo and M. H. Lim, Dalton Trans., 2012, 41, 6558 RSC.
- L. Liu, J. Lin, Y. Song, C. Yang and Z. Zhu, Chem. Res. Chin. Univ., 2020, 36, 247 CrossRef CAS.
- Y. Song, K. Qu, C. Zhao, J. Ren and X. Qu, Adv. Mater., 2010, 22, 2206 CrossRef CAS PubMed.
- M. Mahajan and S. Bhattacharjya, Angew. Chem., Int. Ed., 2013, 52, 6430 CrossRef CAS PubMed.
- G. Thiabaud, S. Pizzocaro, R. Garcia-Serres, J.-M. Latour, E. Monzani and L. Casella, Angew. Chem., Int. Ed., 2013, 52, 8041 CrossRef CAS PubMed.
- M. Jiang and Z. Guo, J. Am. Chem. Soc., 2007, 129, 730 CrossRef CAS PubMed.
- P. Baumann, M. Spulber, O. Fischer, A. Car and W. Meier, Small, 2017, 13, 1603943 CrossRef PubMed.
- C. Ghosh, D. Pramanik, S. Mukherjee, A. Dey and S. G. Dey, Inorg. Chem., 2013, 52, 362 CrossRef CAS PubMed.
- M. Seal, S. Mukherjee, D. Pramanik, K. Mittra, A. Dey and S. G. Dey, Chem. Commun., 2013, 49, 1091 RSC.
- G. I. Makhatadze and P. L. Privalov, Adv. Protein Chem., 1995, 47, 307 CrossRef CAS PubMed.
- D. N. Dubins, R. Filfil, R. B. Macgregor Jr and T. V. Chalikian, J. Phys. Chem. B, 2000, 104, 390 CrossRef CAS.
- W. Pang, J. Lv, S. Du, J. Wang, J. Wang and Y. Zeng, Mol. Pharm., 2017, 14, 3013 CrossRef CAS PubMed.
- L. Rodríguez-Santiago, J. Alí-Torres, P. Vidossich and M. Sodupe, Phys. Chem. Chem. Phys., 2015, 17, 13582 RSC.
- D. Brambilla, R. Verpillot, B. Le Droumaguet, J. Nicolas, M. Taverna, J. Kona, B. Lettiero, S. H. Hashemi, L. De Kimpe, M. Canovi, M. Gobbi, V. Nicolas, W. Scheper, S. M. Moghimi, I. Tvaroska, P. Couvreur and K. Andrieux, ACS Nano, 2012, 6, 5897 CrossRef CAS PubMed.
- C. J. Sarell, C. D. Syme, S. E. J. Rigby and J. H. Viles, Biochemistry, 2009, 48, 4388 CrossRef CAS PubMed.
- A. T. Petkova, Y. Ishii, J. J. Balbach, O. N. Antzutkin, R. D. Leapman, F. Delaglio and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16742 CrossRef CAS PubMed.
- J. Shearer and V. A. Szalai, J. Am. Chem. Soc., 2008, 130, 17826 CrossRef CAS PubMed.
- R. A. Himes, G. Y. Park, G. S. Siluvai, N. J. Blackburn and K. D. Karlin, Angew. Chem., Int. Ed., 2008, 47, 9084 CrossRef CAS PubMed.
- D. P. Smith, D. G. Smith, C. C. Curtain, J. F. Boas, J. R. Pilbrow, G. D. Ciccotosto, T.-L. Lau, D. J. Tew, K. Perez, J. D. Wade, A. I. Bush, S. C. Drew, F. Separovic, C. L. Masters, R. Cappai and K. J. Barnham, J. Biol. Chem., 2006, 281, 15145 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc01020k |
| This journal is © The Royal Society of Chemistry 2020 |