
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTetrazine as a general phototrigger to turn on fluorophores†
Axel
Loredo‡
 a,
Juan
Tang‡
a,
Lushun
Wang‡
a,
Kuan-Lin
Wu
a,
Zane
Peng
b and
Han
Xiao
*abc
a,
Juan
Tang‡
a,
Lushun
Wang‡
a,
Kuan-Lin
Wu
a,
Zane
Peng
b and
Han
Xiao
*abc
aDepartment of Chemistry, Rice University, 6100 Main Street, Houston, Texas 77005, USA. E-mail: han.xiao@rice.edu
bDepartment of Biosciences, Rice University, 6100 Main Street, Houston, Texas 77005, USA
cDepartment of Bioengineering, Rice University, 6100 Main Street, Houston, Texas 77005, USA
First published on 7th April 2020
Abstract
Light-activated fluorescence affords a powerful tool for monitoring subcellular structures and dynamics with enhanced temporal and spatial control of the fluorescence signal. Here, we demonstrate a general and straightforward strategy for using a tetrazine phototrigger to design photoactivatable fluorophores that emit across the visible spectrum. Tetrazine is known to efficiently quench the fluorescence of various fluorophores via a mechanism referred to as through-bond energy transfer. Upon light irradiation, restricted tetrazine moieties undergo a photolysis reaction that generates two nitriles and molecular nitrogen, thus restoring the fluorescence of fluorophores. Significantly, we find that this strategy can be successfully translated and generalized to a wide range of fluorophore scaffolds. Based on these results, we have used this mechanism to design photoactivatable fluorophores targeting cellular organelles and proteins. Compared to widely used phototriggers (e.g., o-nitrobenzyl and nitrophenethyl groups), this study affords a new photoactivation mechanism, in which the quencher is photodecomposed to restore the fluorescence upon light irradiation. Because of the exclusive use of tetrazine as a photoquencher in the design of fluorogenic probes, we anticipate that our current study will significantly facilitate the development of novel photoactivatable fluorophores.
Introduction
Photoactivatable fluorophores, also called turn-on fluorophores,1–7 have numerous advanced biological applications, including detection and release of ions8–11 and metabolites,12–15 monitoring of enzyme activity,16–19 and multiple types of specialized microscopy.7,20–25 Most of these photoactivatable fluorophores are masked by a reactive “cage” group intimately attached to the fluorophore to alter its photophysical properties or to serve as an energy-transfer agent for use in a fluorescence resonance energy transfer (FRET) pair.26 Upon light or chemical treatment, these “caged” fluorophores undergo chemical reactions that release the “cage” groups to regenerate the fluorophores in their active forms.27–35 The timing and site of fluorescence restoration are therefore controlled by the application of the “de-caging” treatment. For two reasons, tetrazine has recently emerged as a very effective quenching group in the design of fluorogenic fluorophores. First, tetrazine absorbs visible light at around 500–550 nm, making it an ideal quencher for a series of fluorophores that are useful for FRET studies.36–39 Second, tetrazine derivatives can undergo an Inverse Electron Demand Diels–Alder (IEDDA) cycloaddition reaction with strained hydrophobic alkenes, such as trans-cyclooctene,40 cyclopropenes,41 or norbornene.42 These bioorthogonal reactions are characterized by fast reaction rates and high selectivity. By taking advantage of this dual functionality, tetrazine has been conjugated via a flexible linker to rhodamine,39 boron dipyrromethene (BODIPY) and Oregon Green37 and then used as a quencher moiety through the FRET mechanism. Upon destruction of the tetrazine core with strained alkenes in the IEDDA reaction, fluorescence enhancement up to 20-fold has been observed for these fluorophores.37To further improve the quenching efficiency, recent studies have used tetrazine to quench fluorophore fluorescence via through-bond energy transfer (TBET),43–49 a mechanism that does not require spectral overlap between donors and acceptors.50 In this context, tetrazine was conjugated to fluorophore donors using a conjugated linker that allows for faster energy transfer through bonds relative to nonradiative decay processes. The resulting “caged” fluorophores exhibited superb fluorescence turn-on upon reaction with trans-cyclooctene,45 allowing for enhanced imaging of cellular components due to reduced background fluorescence.
Despite the potential for broad applications of tetrazine-based fluorogenic fluorophores, large and strained hydrophobic alkenes have been exclusively used as chemical triggers for restoring fluorescence. Compared to decaging using chemical triggers, photo-decaging offers superior spatial and temporal control and is generally considered to be more biocompatible. Tetrazine can undergo photolysis, generating relatively inert, unobtrusive nitriles and molecular nitrogen.51 Gaseous tetrazine is known to absorb visible light that permits the n → π* transition and decomposition with almost unit quantum yield.52 Taking advantage of this photolysis reaction, tetrazine has been used for studies on protein folding by incorporating it into peptides as a phototrigger to modulate peptide stapling/unstapling after illumination with 330–400 nm light.53–57 Here, we describe a new approach for conjugating the tetrazine moiety to fluorophores as a phototrigger for initiating fluorophore photoactivation (Fig. 1).
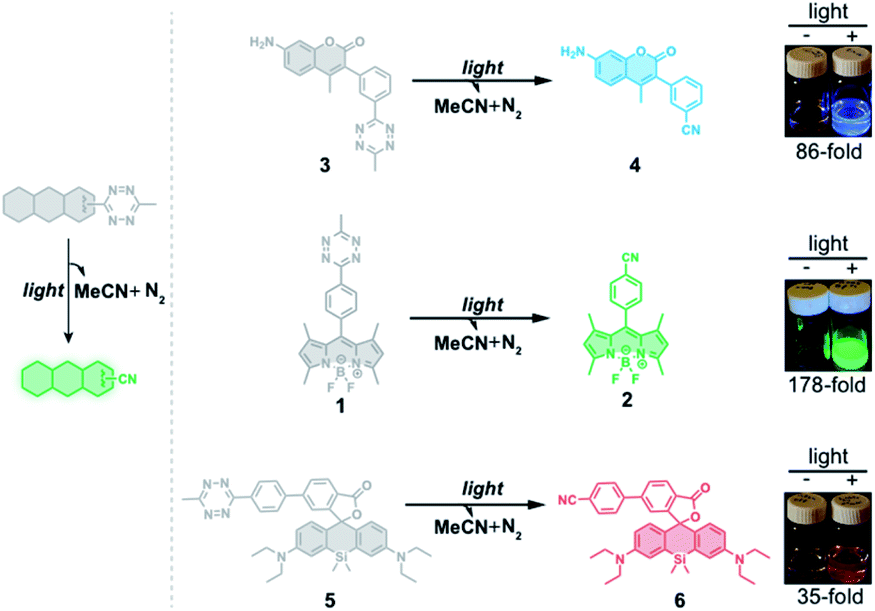 | ||
| Fig. 1 Schematic representation of fluorescence turn-on of tetrazine-fluorophore derivatives using the tetrazine phototrigger. | ||
Results and discussion
Photoactivation of BODIPY fluorophores using the tetrazine phototrigger
To test whether the tetrazine motif can be incorporated into fluorophores to generate photoactivatable fluorophores, we synthesized the methyl-s-tetrazine-BODIPY conjugate (Tz-BODIPY, 1, Fig. 1) as described previously.43 The fluorescence quantum yield for 1 (Φ < 0.01) in acetonitrile is much lower than that for the nitrile counterpart 2 (Φ = 0.60) (Table 1), suggesting the possibility that the photolysis of tetrazine could be used to induce fluorogenic activation of Tz-BODIPY. Indeed, following 20 minute irradiation with 254 nm light (1400 μW cm−2), the fluorescence intensity of Tz-BODIPY increased dramatically. The observed 178-fold fluorescence enhancement at 509 nm is similar to the 340-fold fluorescence increase reported in the literature after chemical triggering with trans-cyclooctene (Fig. 2).43| Compound number | Compound | λ abs (nm) | λ em (nm) | ε × 104 (M−1 cm−1) | Φ MeCN | Fluorescence increased |
|---|---|---|---|---|---|---|
| a Fluorescein in 0.1 M NaOH. b Quinine sulfate in 0.5 M H2SO4 was used as a reference for measuring the quantum yields. c Rhodamine B in water was used as a reference for measuring the quantum yields. d 254 nm light activation (1400 μW cm−2). e Measured after irradiation of compound 5 with 254 nm light. | ||||||
| 1 | Tz-BODIPY | 490 | 509 | 1.9 | 0.004a | 178-Fold |
| 2 | CN-BODIPY | 492 | 509 | 3.6 | 0.60a | — |
| 3 | Tz-Coumarin | 347 | 416 | 1.8 | <0.001b | 86-Fold |
| 4 | CN-Coumarin | 351 | 417 | 1.9 | 0.19b | — |
| 5 | Tz-Si-Rhodamine | 651 | 671 | 2.6 | 0.011c | 35-Fold |
| 6 | CN-Si-Rhodaminee | 652 | 671 | 2.4 | 0.34c | — |
| 7 | Tz-BODIPY-MOR | 495 | 511 | 2.6 | 0.03a | 45-Fold |
| 8 | Tz-BODIPY-Ts | 492 | 510 | 1.8 | 0.02a | 36-Fold |
| 9 | Tz-BODIPY-TPP | 494 | 509 | 3.0 | 0.007a | 97-Fold |
| 10 | Tz-BODIPY-Halo | 491 | 509 | 2.4 | 0.002a | 141-Fold |
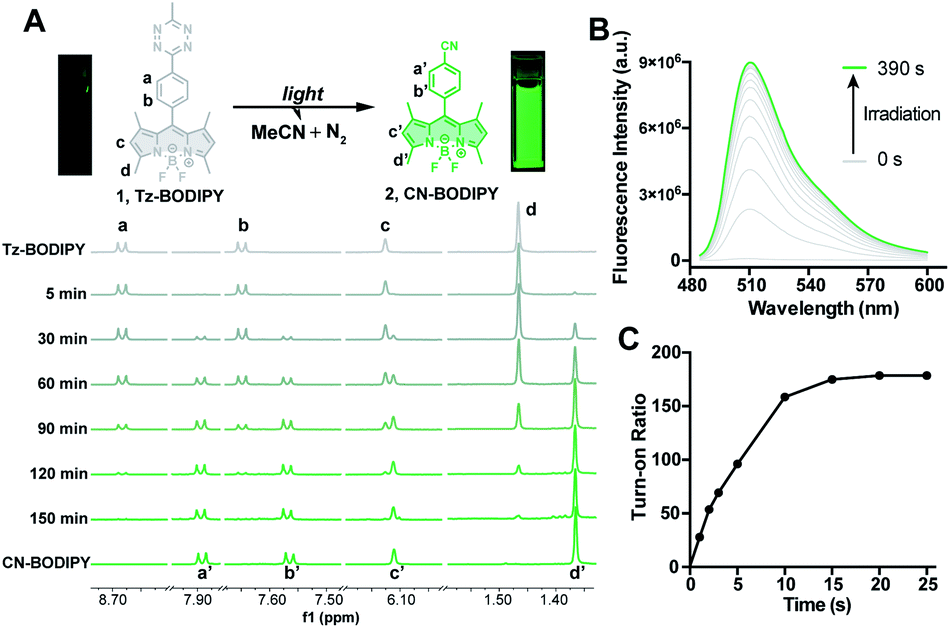 | ||
| Fig. 2 (A) Reaction scheme of light photolysis of Tz-BODIPY 1 and the 1H NMR spectra of Tz-BODIPY 1 obtained at the indicated light irradiation times using a 254 nm handheld UV light. (B) Fluorescent emission spectral change of Tz-BODIPY 1 after light activation. (C) Change of fluorescence intensity at 509 nm during light irradiation using a 254 nm handheld UV light. | ||
The photolysis product was further analyzed by 1H NMR and ESI-MS analysis. Upon light irradiation, the aromatic protons of Tz-BODIPY exhibited an upfield shift. Signals corresponding to phenyl and methyl protons at 8.68, 7.65, 6.12, 3.04, 2.51 and 1.47 ppm gradually disappeared, and new signals appeared at 7.88, 7.56, 6.11, 2.49 and 1.37 ppm (Fig. 2A). These new signals were identical to those observed with synthesized p-cyanophenyl-BODIPY (CN-BODIPY, 2, Fig. 2). ESI-MS analysis of the reaction product yielded an observed mass of 350.16, in agreement with the calculated mass of CN-BODIPY ([M + H+] = 350.15, Fig. S1†). These data, therefore, demonstrate that the photolysis product of Tz-BODIPY is its nitrile counterpart, CN-BODIPY.
Design of photoactivatable fluorophores using the tetrazine phototrigger
To demonstrate that the tetrazine motif can be used as a general phototrigger, methyl-s-tetrazine-coumarin (Tz-Coumarin, 3) and methyl-s-tetrazine-silicon rhodamine (Tz-Si-Rhodamine, 5) were prepared (Fig. 1).45,47 Next, we characterized the spectroscopic and photochemical properties of these fluorophores with the tetrazine phototrigger and their photolysis products using UV-vis and fluorescence spectroscopies (Table 1 and Fig. S2†). As shown in Table 1, the introduction of the tetrazine phototrigger in all the tested fluorophores led to significant reductions in quantum yield. To assess the efficiency of photoactivation for the tetrazine phototrigger, UV-vis and fluorescence spectra were recorded after different irradiation times (Fig. S3†). To our delight, the fluorescence intensity of the Tz-Coumarin and Tz-Si-Rhodamine molecules increased by 86 and 35-fold, respectively, suggesting that the introduction of a tetrazine phototrigger could be a general strategy for preparing photoactivatable fluorophores with diverse structures.Fluorescence imaging using Tz-BODIPY derivatives
Having established the excellent photoactivation properties of the tetrazine phototrigger, we sought to explore the utility of the tetrazine phototrigger for biological imaging. Because of its exceptional turn-on properties, we have used the Tz-BODIPY derivatives in the following study. Ultraviolet light has limited applications as a biological initiator, due to its poor tissue penetration and high phototoxicity. To investigate whether light with longer wavelengths can initiate the tetrazine photolysis reaction, we irradiated Tz-BODIPY with light at 254 nm, 365 nm, and 405 nm, respectively. Gratifyingly, significant fluorescence enhancement was observed upon irradiation at 405 nm, a wavelength that is commonly available in commercial microscopes (Fig. S4†). This wavelength was therefore used for activation in the biological imaging study described below. For this study, it was also important to examine the cellular uptake and intracellular photoactivation of Tz-BODIPY derivatives. A431 cells were incubated with Tz-BODIPY, and intracellular photoactivation was monitored by confocal laser scanning microscopy. As shown in Fig. 3A, while negligible fluorescence was observed before photoactivation of Tz-BODIPY, a 14-fold increase in intracellular green fluorescence was observed after irradiation with 405 nm laser light, suggesting not only good photoactivation but also good cell permeability of Tz-BODIPY. However, Tz-Coumarin and Tz-Si-Rhodamine did not exhibit high fluorescence turn-on in A431 cells due to the high autofluorescence and the possible background fluorescence from the open spiroester form before the irradiation process (Fig. S5†).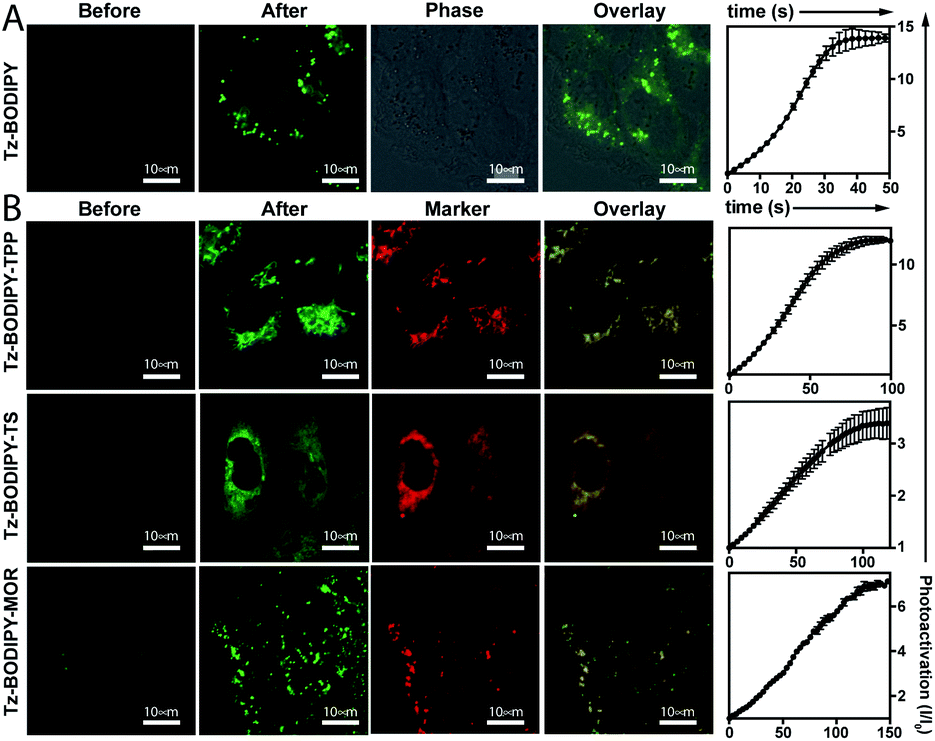 | ||
| Fig. 3 (A) Photoactivation of Tz-BODIPY derivatives in A431 cells using 405 nm laser activation. Confocal images were obtained before and after 405 nm light activation of Tz-BODIPY derivatives. Scale bar = 10 μm. (B) Photoactivation of organelle-targeting Tz-BODIPY probes in A431 cells. A431 cells were incubated with Tz-BODIPY-TPP 9, Tz-BODIPY-MOR 7, or Tz-BODIPY-Ts 8, and the corresponding commercial organelle-targeting dye, followed by photoactivation using a 405 nm laser. Commercial MitoView™ 633, ER-Tracker™ Blue-White DPX, and LysoView™ 633 were used as markers for mitochondria, endoplasmic reticulum, and lysosomes, respectively. Scale bar = 10 μm. | ||
Synthesis of organelle-targeting Tz-BODIPYs and their applications in fluorescence imaging
The combination of subcellular targeting and photodecaging has provided a platform for precise control of site-specific molecular release.58–61 Accordingly, we explored the possibility of combining a tetrazine phototrigger with subcellular targeting moieties. To stably conjugate organelle-targeting moieties to Tz-BODIPY without affecting the its photochemical properties, we developed a robust synthetic approach for functionalizing the BODIPY molecule (Fig. 4).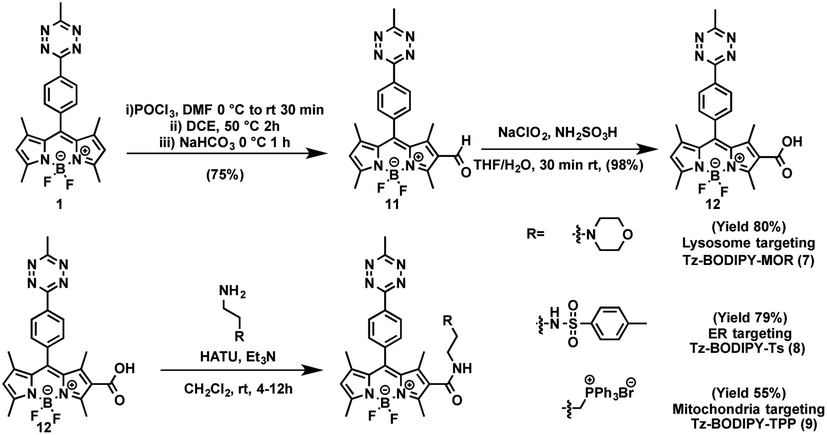 | ||
| Fig. 4 Synthetic route to organelle-targeting Tz-BODIPYs. | ||
In brief, an aldehyde moiety was first introduced at position 2 of BODIPY using the Vilsmeier–Haack reaction,62 followed by a Pinnick oxidation to yield carboxylic acid.63 The resulting carboxyl-functionalized BODIPY was obtained in good yield and coupled to amine-bearing organelle-targeting groups using N-[(dimethylamino)-1H-1,2,3-triazo[4,5-b]pyridin-1-ylmethylene]-N-methylmethanaminium hexafluorophosphate-N-oxide (HATU)-mediated amide coupling to yield organelle-targeting Tz-BODIPYs. Using this protocol, we have prepared Tz-BODIPY with the following organelle-directing groups: phenyl sulfonamide (Tz-BODIPY-Ts, endoplasmic reticulum targeting, Fig. 4),64 morpholine (Tz-BODIPY-MOR, lysosome targeting, Fig. 4),65 and triphenylphosphonium (Tz-BODIPY-TPP, mitochondria targeting, Fig. 4).66 To our delight, all three organelle-targeting Tz-BODIPYs were stable in the absence of light (Fig. S6†) and exhibited significant fluorescence enhancement after light irradiation in acetonitrile (Table 1, Fig. S7†). In order to evaluate the localization of these organelle-targeting Tz-BODIPY derivatives, confocal laser scanning microscopy was carried out on A431 cells. Commercial MitoView™ 633, ER-Tracker™ Blue-White DPX, and LysoView™ 633 were used as markers for mitochondria, endoplasmic reticulum, and lysosomes, respectively. All three BODIPY compounds had negligible fluorescence prior to irradiation with the 405 nm laser, but exhibited distinct increases in fluorescence signals following tetrazine triggering. Turn-on ratios of 12, 3.5 and 7 were observed for Tz-BODIPY-TPP 9, Tz-BODIPY-MOR 7, and Tz-BODIPY-Ts 8, respectively (Fig. 3B).
Most importantly, good co-localization was observed between the green fluorescence of the BODIPY compounds and the red fluorescence of the corresponding organelle-targeting commercial dyes (Fig. 3B). This is reflected by the high Pearson's correlation coefficients found for Tz-BODIPY-Ts (γ = 0.85), Tz-BODIPY-MOR (γ = 0.84), and Tz-BODIPY-TPP (γ = 0.75). These data demonstrate that the photoactivability of Tz-BODIPYs can be productively combined with the specificity of subcellular targeting molecules for organelle imaging.
Super-resolution imaging using tetrazine-caged BODIPY fluorophores
Photoactivatable fluorescent probes should find useful applications in super-resolution imaging (e.g., Photoactivated Localization Microscopy, PALM; Stochastic Optical Reconstruction Microscopy, STORM),67,68 a technology that allows imaging beyond the diffraction limit.21,22,69–72 Thus, we explored the possibility of using Tz-BODIPY for PALM imaging. To site-specifically label proteins of interest, we used HaloTag labeling technology,73–75 which is based on covalent bond formation between HaloTag-fused proteins and a synthetic HaloTag ligand. The HaloTag tetrazine-BODIPY conjugate (Fig. 5A) was synthesized following a strategy similar to that used for the organelle-targeting probes. The fluorescence quantum yield of the compound (Φ = 0.002, Table 1) was in the range of that of the other synthesized Tz-BODIPY dyes. After 40 minutes of 254 nm light irradiation using a handheld UV lamp, a 141-fold fluorescence enhancement was observed (Table 1 and Fig. S7†), while no significant fluorescence increase was detected in the absence of light (Fig. S8†). Next, CHO-K1 cells expressing histone 2B (H2B)-HaloTag were incubated with Tz-BODIPY-Halo 10 for 30 min and then incubated in fresh culture medium for another 30 min. Intracellular photoactivation was initially monitored via confocal laser scanning microscopy. As shown in Fig. S9,† an 8-fold increase in intracellular fluorescence was observed after 405 nm laser irradiation. Co-incubation of compound 10 with the commercial nuclear dye, DRAQ5, confirmed a distinct nuclear localization pattern for the histone conjugate. Subsequently, we utilized Tz-BODIPY-Halo 10 for PALM imaging of histone-2B in CHO-K1 cells by activating Tz-BODIPY-Halo with the 405 nm laser contained in the microscope (Fig. S9†). By capturing sets of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–30000 imaging frames, we reconstructed the super-resolution image of H2B localization (Fig. 5B). Compared to conventional widefield fluorescence imaging, the reconstructed PALM imaging exhibits much better resolution along with high molecular accuracy (ca. 40.8 nm) (Fig. 5C). It is worth mentioning that this PALM imaging can be performed under physiological conditions without the need for cytotoxic reducing buffers to induce the fluorescence on–off transition cycles. Taken together, our data demonstrate that the tetrazine-caged BODIPY fluorophores developed in this study are compatible with existing protein labeling technologies and can be used for super-resolution imaging of living cells.
000–30000 imaging frames, we reconstructed the super-resolution image of H2B localization (Fig. 5B). Compared to conventional widefield fluorescence imaging, the reconstructed PALM imaging exhibits much better resolution along with high molecular accuracy (ca. 40.8 nm) (Fig. 5C). It is worth mentioning that this PALM imaging can be performed under physiological conditions without the need for cytotoxic reducing buffers to induce the fluorescence on–off transition cycles. Taken together, our data demonstrate that the tetrazine-caged BODIPY fluorophores developed in this study are compatible with existing protein labeling technologies and can be used for super-resolution imaging of living cells.
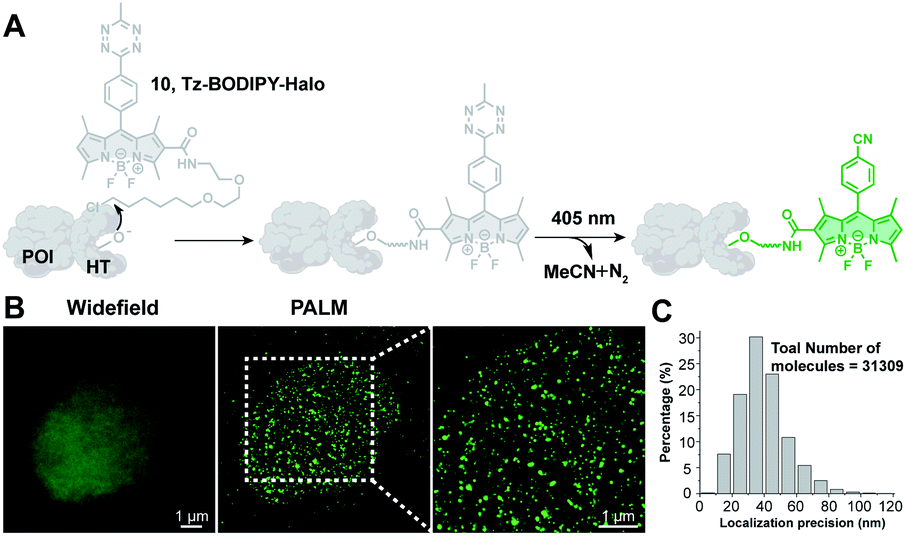 | ||
| Fig. 5 PALM imaging of histone proteins using the Tz-BODIPY-Halo probe. (A) Site-specific labeling of the protein of interest (POI) with the Tz-BODIPY-Halo probe for fluorescence imaging. (B) Widefield image and corresponding PALM images of H2B labeled with H2B-HaloTag in CHO–K1 cells. (C) Histogram plot of the localization accuracy of PALM images in (B). Scale bar = 1 μm. | ||
Conclusions
In summary, we have designed and characterized a unique class of photoactivatable tetrazine-based probes in which the tetrazine moiety serves as both a fluorescence quencher and a phototrigger. The utility of these turn-on probes is further demonstrated by our success with organelle imaging and super-resolution imaging under physiological conditions that do not require the use of a cytotoxic imaging cocktail. We are currently exploring the use of a longer wavelength two-photon laser to dissociate the tetrazine moiety, as this process has been reported for tetrazine by measuring the evolution of HCN after irradiation at 492 and 532 nm.76,77In contrast to the widely used phototriggers (e.g., o-nitrobenzyl and nitrophenethyl groups) altering the electron structure of fluorescent molecules upon light irradiation,78–80 this study affords a new photoactivation mechanism, in which the TBET quencher is photodecomposed to restore the fluorescence. Besides serving as a TBET quencher, the tetrazine moiety has also been exclusively used as a FRET acceptor to design FRET-based fluorogenic probes. Thus, we believe that this work will open up the possibility of using tetrazine as a photo-trigger to activate other FRET-based fluorogenic probes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Cancer Prevention and Research Institute of Texas (CPRIT, RR170014), the Robert A. Welch Foundation (C-1970), the Hamill Innovation Award (Hamill Foundation), the John S. Dunn Foundation Collaborative Research Award (Gulf Coast Consortia) and the NIH (R35-GM133706). H. X. is a Cancer Prevention & Research Institute of Texas (CPRIT) Scholar in Cancer Research.Notes and references
- G. A. Krafft, W. R. Sutton and R. T. Cummings, J. Am. Chem. Soc., 1988, 110, 301–303 CrossRef CAS.
- Y. Zhao, Q. Zheng, K. Dakin, K. Xu, M. L. Martinez and W.-H. Li, J. Am. Chem. Soc., 2004, 126, 4653–4663 CrossRef CAS PubMed.
- T. Kobayashi, Y. Urano, M. Kamiya, T. Ueno, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2007, 129, 6696–6697 CrossRef CAS PubMed.
- J. Fölling, V. Belov, R. Kunetsky, R. Medda, A. Schönle, A. Egner, C. Eggeling, M. Bossi and S. W. Hell, Angew. Chem., Int. Ed., 2007, 46, 6266–6270 CrossRef PubMed.
- G. T. Dempsey, M. Bates, W. E. Kowtoniuk, D. R. Liu, R. Y. Tsien and X. Zhuang, J. Am. Chem. Soc., 2009, 131, 18192–18193 CrossRef CAS PubMed.
- J. C. Vaughan, G. T. Dempsey, E. Sun and X. Zhuang, J. Am. Chem. Soc., 2013, 135, 1197–1200 CrossRef CAS PubMed.
- J. Tang, M. A. Robichaux, K.-L. Wu, J. Pei, N. T. Nguyen, Y. Zhou, T. G. Wensel and H. Xiao, J. Am. Chem. Soc., 2019, 141, 14699–14706 CrossRef CAS PubMed.
- P. Du and S. J. Lippard, Inorg. Chem., 2010, 49, 10753–10755 CrossRef CAS PubMed.
- C.-H. Lee, H.-J. Yoon, J.-S. Shim and W.-D. Jang, Chem.–Eur. J., 2012, 18, 4513–4516 CrossRef CAS PubMed.
- H. K. Agarwal, R. Janicek, S.-H. Chi, J. W. Perry, E. Niggli and G. C. R. Ellis-Davies, J. Am. Chem. Soc., 2016, 138, 3687–3693 CrossRef CAS PubMed.
- C. Deo, S.-H. Sheu, J. Seo, D. E. Clapham and L. D. Lavis, J. Am. Chem. Soc., 2019, 141, 13734–13738 CrossRef CAS PubMed.
- S. Resa, A. Orte, D. Miguel, J. M. Paredes, V. Puente-Muñoz, R. Salto, M. D. Giron, M. J. Ruedas-Rama, J. M. Cuerva, J. M. Alvarez-Pez and L. Crovetto, Chem.–Eur. J., 2015, 21, 14772–14779 CrossRef CAS PubMed.
- Y. Yue, F. Huo, P. Ning, Y. Zhang, J. Chao, X. Meng and C. Yin, J. Am. Chem. Soc., 2017, 139, 3181–3185 CrossRef CAS PubMed.
- R. R. Nani, A. P. Gorka, T. Nagaya, H. Kobayashi and M. J. Schnermann, Angew. Chem., Int. Ed., 2015, 54, 13635–13638 CrossRef CAS PubMed.
- R. R. Nani, A. P. Gorka, T. Nagaya, T. Yamamoto, J. Ivanic, H. Kobayashi and M. J. Schnermann, ACS Cent. Sci., 2017, 3, 329–337 CrossRef CAS PubMed.
- H. Li, Q. Yao, F. Xu, N. Xu, R. Duan, S. Long, J. Fan, J. Du, J. Wang and X. Peng, Biomaterials, 2018, 179, 1–14 CrossRef CAS PubMed.
- Y. Takaoka, H. Tsutsumi, N. Kasagi, E. Nakata and I. Hamachi, J. Am. Chem. Soc., 2006, 128, 3273–3280 CrossRef CAS PubMed.
- H. Ai, K. L. Hazelwood, M. W. Davidson and R. E. Campbell, Nat. Methods, 2008, 5, 401–403 CrossRef CAS PubMed.
- D. Cheng, Y. Pan, L. Wang, Z. Zeng, L. Yuan, X. Zhang and Y.-T. Chang, J. Am. Chem. Soc., 2017, 139, 285–292 CrossRef CAS PubMed.
- L. D. Lavis, T.-Y. Chao and R. T. Raines, ACS Chem. Biol., 2006, 1, 252–260 CrossRef CAS PubMed.
- M. Bates, B. Huang, G. T. Dempsey and X. Zhuang, Science, 2007, 317, 1749–1753 CrossRef CAS PubMed.
- S.-H. Shim, C. Xia, G. Zhong, H. P. Babcock, J. C. Vaughan, B. Huang, X. Wang, C. Xu, G.-Q. Bi and X. Zhuang, Proc. Natl. Acad. Sci., 2012, 109, 13978–13983 CrossRef CAS PubMed.
- J. B. Grimm, B. P. English, H. Choi, A. K. Muthusamy, B. P. Mehl, P. Dong, T. A. Brown, J. Lippincott-Schwartz, Z. Liu, T. Lionnet and L. D. Lavis, Nat. Methods, 2016, 13, 985–988 CrossRef CAS PubMed.
- C. S. Wijesooriya, J. A. Peterson, P. Shrestha, E. J. Gehrmann, A. H. Winter and E. A. Smith, Angew. Chem., Int. Ed., 2018, 57, 12685–12689 CrossRef CAS PubMed.
- F. M. Jradi and L. D. Lavis, ACS Chem. Biol., 2019, 14, 1077–1090 CrossRef CAS PubMed.
- L. Yuan, W. Lin, K. Zheng and S. Zhu, Acc. Chem. Res., 2013, 46, 1462–1473 CrossRef CAS PubMed.
- G. Zheng, Y.-M. Guo and W.-H. Li, J. Am. Chem. Soc., 2007, 129, 10616–10617 CrossRef CAS PubMed.
- V. N. Belov, C. A. Wurm, V. P. Boyarskiy, S. Jakobs and S. W. Hell, Angew. Chem., Int. Ed., 2010, 49, 3520–3523 CrossRef CAS PubMed.
- J. A. Peterson, C. Wijesooriya, E. J. Gehrmann, K. M. Mahoney, P. P. Goswami, T. R. Albright, A. Syed, A. S. Dutton, E. A. Smith and A. H. Winter, J. Am. Chem. Soc., 2018, 140, 7343–7346 CrossRef CAS PubMed.
- P. P. Goswami, A. Syed, C. L. Beck, T. R. Albright, K. M. Mahoney, R. Unash, E. A. Smith and A. H. Winter, J. Am. Chem. Soc., 2015, 137, 3783–3786 CrossRef CAS PubMed.
- Y. Zhou, K. Chu, H. Zhen, Y. Fang and C. Yao, Spectrochim. Acta, Part A, 2013, 106, 197–202 CrossRef CAS PubMed.
- Y. Yu, B. Czepukojc, C. Jacob, Y. Jiang, M. Zeller, C. Brückner and J.-L. Zhang, Org. Biomol. Chem., 2013, 11, 4613–4621 RSC.
- L. Yuan, L. Wang, B. K. Agrawalla, S.-J. Park, H. Zhu, B. Sivaraman, J. Peng, Q.-H. Xu and Y.-T. Chang, J. Am. Chem. Soc., 2015, 137, 5930–5938 CrossRef CAS PubMed.
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef PubMed.
- B. Roubinet, M. Bischoff, S. Nizamov, S. Yan, C. Geisler, S. Stoldt, G. Y. Mitronova, V. N. Belov, M. L. Bossi and S. W. Hell, J. Org. Chem., 2018, 83, 6466–6476 CrossRef CAS PubMed.
- R. J. Blizzard, D. R. Backus, W. Brown, C. G. Bazewicz, Y. Li and R. A. Mehl, J. Am. Chem. Soc., 2015, 137, 10044–10047 CrossRef CAS PubMed.
- N. K. Devaraj, S. Hilderbrand, R. Upadhyay, R. Mazitschek and R. Weissleder, Angew. Chem., Int. Ed., 2010, 49, 2869–2872 CrossRef CAS PubMed.
- K. Lang, L. Davis, J. Torres-Kolbus, C. Chou, A. Deiters and J. W. Chin, Nat. Chem., 2012, 4, 298–304 CrossRef CAS PubMed.
- B. L. Oliveira, Z. Guo, O. Boutureira, A. Guerreiro, G. Jiménez-Osés and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2016, 55, 14683–14687 CrossRef CAS PubMed.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS PubMed.
- D. M. Patterson, L. A. Nazarova, B. Xie, D. N. Kamber and J. A. Prescher, J. Am. Chem. Soc., 2012, 134, 18638–18643 CrossRef CAS PubMed.
- J. Sauer, G. R. Pabst, U. Holland, H.-S. Kim and S. Loebbecke, Eur. J. Org. Chem., 2001, 2001, 697–706 CrossRef.
- J. C. T. Carlson, L. G. Meimetis, S. A. Hilderbrand and R. Weissleder, Angew. Chem., Int. Ed., 2013, 52, 6917–6920 CrossRef CAS PubMed.
- H. Wu, J. Yang, J. Šečkutė and N. K. Devaraj, Angew. Chem., Int. Ed., 2014, 53, 5805–5809 CrossRef CAS PubMed.
- L. G. Meimetis, J. C. T. Carlson, R. J. Giedt, R. H. Kohler and R. Weissleder, Angew. Chem., Int. Ed., 2014, 53, 7531–7534 CrossRef CAS PubMed.
- G. Knorr, E. Kozma, A. Herner, E. A. Lemke and P. Kele, Chem.–Eur. J., 2016, 22, 8972–8979 CrossRef CAS PubMed.
- E. Kozma, G. E. Girona, G. Paci, E. A. Lemke and P. Kele, Chem. Commun., 2017, 53, 6696–6699 RSC.
- A. Wieczorek, P. Werther, J. Euchner and R. Wombacher, Chem. Sci., 2017, 8, 1506–1510 RSC.
- Y. Lee, W. Cho, J. Sung, E. Kim and S. B. Park, J. Am. Chem. Soc., 2018, 140, 974–983 CrossRef CAS PubMed.
- Y.-J. Gong, X.-B. Zhang, C.-C. Zhang, A.-L. Luo, T. Fu, W. Tan, G.-L. Shen and R.-Q. Yu, Anal. Chem., 2012, 84, 10777–10784 CrossRef CAS PubMed.
- R. M. Hochstrasser and D. S. King, J. Am. Chem. Soc., 1975, 97, 4760–4762 CrossRef CAS.
- J. H. Meyling, R. P. Van Der Werf and D. A. Wiersma, Chem. Phys. Lett., 1974, 28, 364–372 CrossRef CAS.
- M. J. Tucker, J. R. Courter, J. Chen, O. Atasoylu, A. B. Smith and R. M. Hochstrasser, Angew. Chem., Int. Ed., 2010, 49, 3612–3616 CrossRef CAS PubMed.
- M. J. Tucker, M. Abdo, J. R. Courter, J. Chen, A. B. Smith and R. M. Hochstrasser, J. Photochem. Photobiol., A, 2012, 234, 156–163 CrossRef CAS PubMed.
- M. Abdo, S. P. Brown, J. R. Courter, M. J. Tucker, R. M. Hochstrasser and A. B. Smith, Org. Lett., 2012, 14, 3518–3521 CrossRef CAS PubMed.
- J. R. Courter, M. Abdo, S. P. Brown, M. J. Tucker, R. M. Hochstrasser and A. B. Smith, J. Org. Chem., 2014, 79, 759–768 CrossRef CAS PubMed.
- S. P. Brown and A. B. Smith, J. Am. Chem. Soc., 2015, 137, 4034–4037 CrossRef CAS PubMed.
- S. Chalmers, S. T. Caldwell, C. Quin, T. A. Prime, A. M. James, A. G. Cairns, M. P. Murphy, J. G. McCarron and R. C. Hartley, J. Am. Chem. Soc., 2012, 134, 758–761 CrossRef CAS PubMed.
- A. Nadler, D. A. Yushchenko, R. Müller, F. Stein, S. Feng, C. Mulle, M. Carta and C. Schultz, Nat. Commun., 2015, 6, 1–10 Search PubMed.
- N. Wagner, M. Stephan, D. Höglinger and A. Nadler, Angew. Chem., Int. Ed., 2018, 57, 13339–13343 CrossRef CAS PubMed.
- D. Kand, L. Pizarro, I. Angel, A. Avni, D. Friedmann-Morvinski and R. Weinstain, Angew. Chem., Int. Ed., 2019, 58, 4659–4663 CrossRef CAS PubMed.
- S. Zhu, J. Bi, G. Vegesna, J. Zhang, F.-T. Luo, L. Valenzano and H. Liu, RSC Adv., 2013, 3, 4793–4800 RSC.
- H. Geng, C. M. Hill, S. Zhu, H. Liu, L. Huang and S. Pan, RSC Adv., 2013, 3, 2306–2312 RSC.
- H. Xiao, P. Li, X. Hu, X. Shi, W. Zhang and B. Tang, Chem. Sci., 2016, 7, 6153–6159 RSC.
- M. Gao, Q. Hu, G. Feng, B. Z. Tang and B. Liu, J. Mater. Chem. B, 2014, 2, 3438–3442 RSC.
- C. W. T. Leung, Y. Hong, S. Chen, E. Zhao, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2013, 135, 62–65 CrossRef CAS PubMed.
- S. Adhikari, J. Moscatelli, E. M. Smith, C. Banerjee and E. M. Puchner, Nat. Commun., 2019, 10, 1–12 CrossRef CAS PubMed.
- G. Beliu, A. J. Kurz, A. C. Kuhlemann, L. Behringer-Pliess, M. Meub, N. Wolf, J. Seibel, Z.-D. Shi, M. Schnermann, J. B. Grimm, L. D. Lavis, S. Doose and M. Sauer, Commun. Biol., 2019, 2, 1–13 CrossRef CAS PubMed.
- G. Y. Mitronova, S. Polyakova, C. A. Wurm, K. Kolmakov, T. Wolfram, D. N. H. Meineke, V. N. Belov, M. John and S. W. Hell, Eur. J. Org. Chem., 2015, 2015, 337–349 CrossRef CAS.
- P. Sengupta, S. B. van Engelenburg and J. Lippincott-Schwartz, Chem. Rev., 2014, 114, 3189–3202 CrossRef CAS PubMed.
- B. Roubinet, M. Weber, H. Shojaei, M. Bates, M. L. Bossi, V. N. Belov, M. Irie and S. W. Hell, J. Am. Chem. Soc., 2017, 139, 6611–6620 CrossRef CAS PubMed.
- M. K. Lee, P. Rai, J. Williams, R. J. Twieg and W. E. Moerner, J. Am. Chem. Soc., 2014, 136, 14003–14006 CrossRef CAS PubMed.
- G. V. Los, L. P. Encell, M. G. McDougall, D. D. Hartzell, N. Karassina, C. Zimprich, M. G. Wood, R. Learish, R. F. Ohana, M. Urh, D. Simpson, J. Mendez, K. Zimmerman, P. Otto, G. Vidugiris, J. Zhu, A. Darzins, D. H. Klaubert, R. F. Bulleit and K. V. Wood, ACS Chem. Biol., 2008, 3, 373–382 CrossRef CAS PubMed.
- Y. Liu, K. Miao, N. P. Dunham, H. Liu, M. Fares, A. K. Boal, X. Li and X. Zhang, Biochemistry, 2017, 56, 1585–1595 CrossRef CAS PubMed.
- Y. Liu, K. Miao, Y. Li, M. Fares, S. Chen and X. Zhang, Biochemistry, 2018, 57, 4663–4674 CrossRef CAS PubMed.
- D. Coulter, D. Dows, H. Reisler and C. Wittig, Chem. Phys., 1978, 32, 429–435 CrossRef CAS.
- J. H. Glownia and S. J. Riley, Chem. Phys. Lett., 1980, 71, 429–435 CrossRef CAS.
- W. Li and G. Zheng, Photochem. Photobiol. Sci., 2012, 11, 460–471 RSC.
- F. M. Raymo, Phys. Chem. Chem. Phys., 2013, 15, 14840–14850 RSC.
- L. Kowalik and J. K. Chen, Nat. Chem. Biol., 2017, 13, 587–598 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc01009j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |