
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen peroxide reduction on single platinum nanoparticles†
Xin
Chang
,
Christopher
Batchelor-McAuley
 and
Richard G.
Compton
*
and
Richard G.
Compton
*
Physical and Theoretical Chemistry Laboratory, Department of Chemistry, University of Oxford, South Parks Road, Oxford, OX1 3QZ, UK. E-mail: Richard.Compton@chem.ox.ac.uk
First published on 8th April 2020
Abstract
Understanding oxygen reduction, key to much of electrochemical energy transformation technology, crucially requires exploration of the role of hydrogen peroxide as a possible intermediate especially on catalysts such as Pt which can bring about the 4e reduction of O2 to water. We reveal that at the single nanoparticle scale the direct platinum catalysed reduction of hydrogen peroxide is found – even at high overpotentials – not to be controlled by the rate mass-transport of the reagents to the interface but by a surface limited process. Further under alkaline (pH 12.3) and near mass-transport free conditions, the single nanoparticle hydrogen peroxide reduction rate goes through a maximum at potentials comparable to the surface deposition of hydrogen (Hupd) with the highest reaction rate occurring when the surface is partially covered in hydrogen.
Introduction
Platinum is a much used electrode material for the four electron reduction of oxygen to water which is essential in maximizing the energy output from fuel cell devices. In particular formation of the two electron product, hydrogen peroxide, leads to significant energy loss. Whilst the net four electron process is often seen with Pt based electrocatalysts, reports suggest that H2O2 is an important intermediate in the reduction reaction and may predominate at high overpotential. It is this behaviour and the underlying mechanism which is of interest in this work.1–3 However experimentally, due to this change in mechanistic behaviour occurring at high overpotentials, the direct study of this process is often at least partially masked by the electrochemical reaction being under mixed kinetic/mass-transport control. Studying reactions at the single nanoparticle scale enables the attainment of high mass-transport conditions.4 For example, for a 25 nm catalytic particle isolated on a surface in aqueous media the associated mass-transport coefficient is of the order 10−2 m s−1. In this work it is evidenced first how the hydrogen underpotential deposition onto individual platinum nanoparticles can be used to give insight into the crystal facets present on the nanoparticle surface and further how the H2O2 reduction reaction can be studied under these high mass-transport conditions, revealing the reaction rate to go through a maximum at ca. −0.5 V vs. Ag/AgCl (pH 12.3).The electrocatalytic activity of individual nanoparticles is conveniently studied via the nano-impact technique.4 Here, an inert microelectrode is submerged into a suspension of the nanomaterial, where the solution also contains the molecular electroactive reagent of study. The nanoparticles held in suspension may randomly collide with the electrode and if a suitably oxidizing or reducing potential is held on the interface, a catalytic reaction may be induced to occur at the single nanoparticle. The resulting change in current gives a direct measure of the single nanoparticle reaction rate and hence the nanomaterial's catalytic activity. In the present case the focus is the H2O2 reduction on platinum nanoparticles. The reduction of H2O2 on palladium nanoparticles in the bulk solution has been reported previously;5 however, this methodology is not suitable for our study, due to platinum being an excellent catalyst for the chemical disproportionation of H2O2. Under alkaline conditions (20 mM KOH) the heterogeneous rate constant for this decomposition reaction has recently been determined to be 3.7 ± 2.1 × 10−5 m s−1.6 Hence, the half-life of a hydrogen peroxide solution in the presence of a 6 pM suspension of 50 nm platinum nanoparticles is ca. 100 s. Consequently, studying the electrochemical response of platinum nanoparticles with both hydrogen peroxide and platinum nanoparticles present in the bulk solution is not experimentally feasible. Herein, this problem is circumvented by producing the reagent, hydrogen peroxide, in situ.
Results and discussion
Production of hydrogen peroxide
Under alkaline conditions gold is a good electrocatalyst for the reduction of oxygen;7 however, on polycrystalline electrodes and under mass-transport limiting conditions the reaction only proceeds as far as the two-electron product hydrogen peroxide.8 At pH 12.3 the formal potential for the oxygen/peroxide redox couple is −0.137 V vs. Ag/AgCl and, as shown in the ESI Section 1,† the voltammetric response of a gold micro-electrode towards the reduction of oxygen (20 mM KOH) is very close to this value with a half-wave potential of −0.147 V. Furthermore, in a solution containing 1.25 mM hydrogen peroxide, no appreciable reduction of the peroxide is observed over the voltammetric range of study. In contrast platinum is capable of both oxidizing (to form oxygen) and reducing (to form water) hydrogen peroxide.9Fig. 1 presents the voltammetric response of a platinum microelectrode in a 1.25 mM solution of hydrogen peroxide. The voltammogram was started at +0.3 V (vs. Ag/AgCl) and subsequently scanned cathodically towards −0.7 V at a scan rate of 100 mV s−1. At this microelectrode the limiting anodic and cathodic currents are comparable in magnitude and on the basis of the known diffusion coefficient for hydrogen peroxide (DH2O2 = 1.42 × 10−9 m2 s−1)10 indicate that at high overpotentials both the oxidation and reduction reactions are essentially two-electron processes involving the formation of oxygen and water respectively. Most notable about the voltammogram present in Fig. 1 is the significant degree of hysteresis exhibited over the course of the voltammetric scan. This hysteresis reflects the sensitivity of the redox process to the underlying oxidation state of the platinum surface.11 The inlay of Fig. 1 depicts the voltammetric response of a platinum surface in the absence of hydrogen peroxide, clearly evidencing the potentials at which hydrogen underpotential deposition and platinum oxide formation occurs. The ability of the platinum metal to catalyse the peroxide redox reaction in both directions is, as noted above, reflected in the fact that the material is an excellent catalyst for the disproportionation reaction.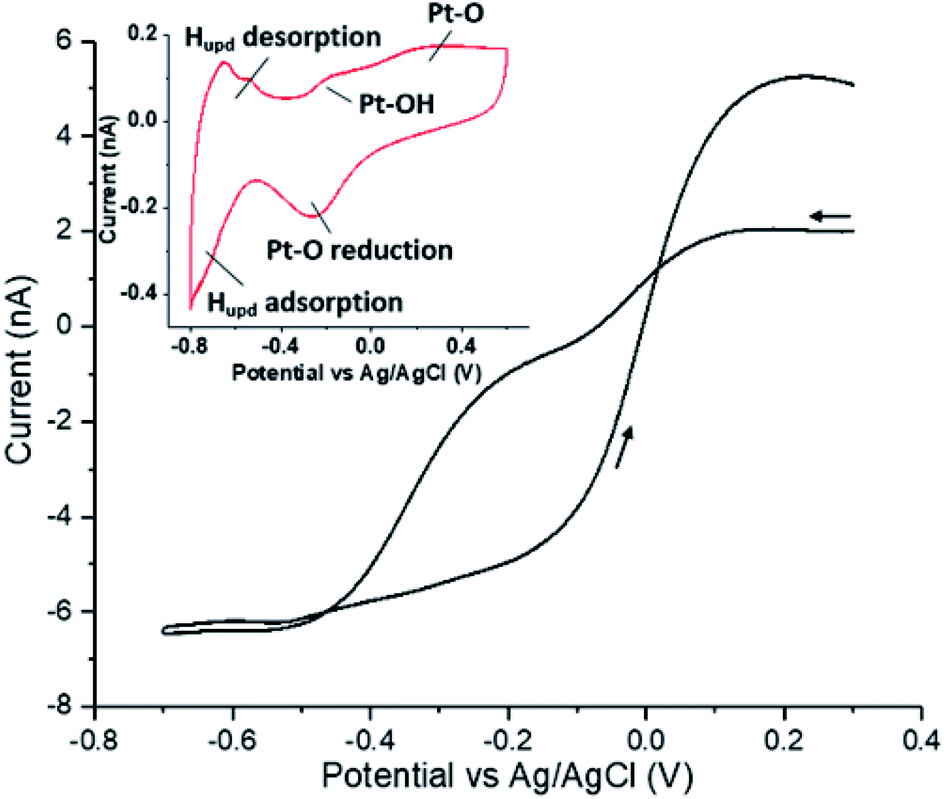 | ||
| Fig. 1 Cyclic voltammogram for hydrogen peroxide disproportionation reaction on a Pt microelectrode in 20 mM KOH + 1.25 mM H2O2 saturated with N2. Inlay: cyclic voltammogram of 20 mM KOH on Pt microelectrode saturated with N2. (CE: graphite, scan rate: 100 mV s−1). | ||
As an alternative approach to having hydrogen peroxide in the bulk solution phase, in this work we utilize the fact that under alkaline conditions and at gold electrode the reduction of oxygen proceeds no further than the production of hydrogen peroxide. This locally produced hydrogen peroxide is used as a reagent and is further catalytically reduced at the impacting nano-particles.
Hydrogen peroxide reduction electrocatalysis
A gold microelectrode (radius = 5 μm) was submerged into an aqueous solution (20 mM KOH) saturated with oxygen (1.24 mM).12 A cathodic potential was applied to the electrode and the resulting chronoamperometric current recorded for a period of 50 s. When the electrode was potentiostated to a potential < −0.2 V vs. Ag/AgCl then a steady-state reductive current of ca. 10 nA was recorded at the microelectrode corresponding to the reduction of the oxygen, local to the electrode, to hydrogen peroxide. Representative chronoamperograms are depicted in the ESI Section 2.† Under these steady-state mass-transport limited reductive conditions the concentration of hydrogen peroxide at the gold interface is equal to the bulk concentration of oxygen scaled by the diffusion coefficients of the reduced and oxidized species. Assuming a diffusion coefficient of 2 × 10−9 m2 s−1 for oxygen, the surface concentration of hydrogen peroxide under these conditions is ca. 1.7 mM.12 Moreover, under steady-state conditions the length scale over which the concentration profile varies is determined by the dimensions and geometry of the electrode; the diffusion layer thickness is approximately equal to the electrode radius and is independent of the molecular diffusion coefficient. Hence, in the present case at a distance of approximately 100 nm away from the electrode the concentration of the hydrogen peroxide will have only decreased by approximately 2% of that at the electrode surface.To the above alkaline and oxygenated aqueous solution, 6 pM of 50 nm platinum nanoparticles were added. ESI Section 3† provides characterization data relating to these platinum nanoparticles evidencing their crystalline and mesoporous structure. In the presence of oxygen, chronoamperograms run at potentials below −0.4 V in this nanoparticle suspension resulted in the observation of small steps in the current superimposed on top of the background signal, where the background current (ca. 10 nA, Fig. S2†) is associated with the reduction of oxygen to hydrogen peroxide at the gold substrate. Fig. 2 presents three such steps in current (black) as recorded at −0.7, −0.6 and −0.5 V vs. Ag/AgCl. These chronoamperometric features are consistent with being due to the arrival of platinum nanoparticles at the gold interface. Notably, these ‘steps’ in current also exhibit a reductive ‘spike’ which occurs at the beginning of the feature. The magnitude of the spikes in the current increases with of the application of higher applied electrode overpotentials, while the magnitude of the steps decreases. In the absence of oxygen these spikes in current are still observable, as evidenced in Fig. 2 which shows representative spikes in current (red) as measured in an alkaline (20 mM KOH) solution degassed with nitrogen and containing 6 pM of the platinum nanoparticles. These spikes in current have been previously observed in acid13 as well as in alkaline solutions14 (see ESI Section 2†) and are associated with the electrochemical deposition of hydrogen onto individual nanoparticles:
| H2O + e− + M ⇌ Pt − H + OH− | (1) |
| HO−2 + H2O + 2e− ⇌ 3OH− | (2) |
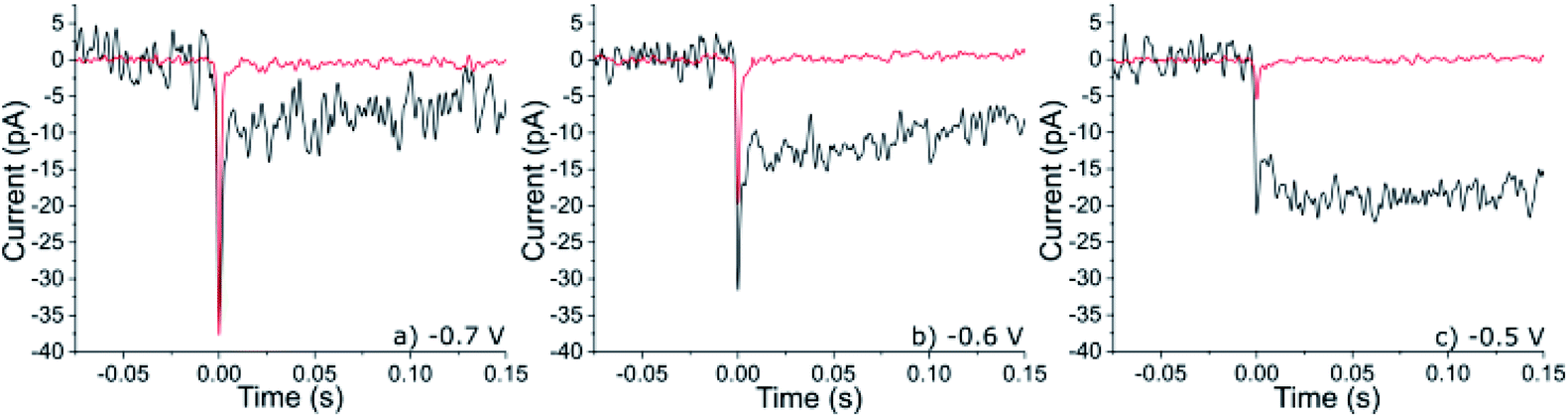 | ||
| Fig. 2 Representative nano-impact events for mesoporous 50 nm platinum nanoparticles impacting a gold microdisc electrode, in the presence (black) and absence (red) of oxygen (sat. 1.24 mM) in the bulk solution phase. The gold electrode has been potentiostated to three different potential of (a) −0.7 V, (b) −0.6 V and (c) −0.5 V (vs. Ag/AgCl). Note to allow direct comparison the background current has been removed and t = 0 has been arbitrarily set. The original full chronoamperometric transients from which this data was taken can be viewed in the ESI Section 2.† | ||
The duration of the steps evidence that once the nanoparticle has arrived at the gold interface it becomes irreversibly adsorbed to the substrate i.e. during the course of the experiment no ‘steps’ down in current have been recorded and the electrode needs to be refreshed by polishing to remove the platinum nanoparticles that have accumulated at the interface, so that the gold electrode only ever supports a sparse array of nanoparticles (<1000, giving a minimum average interparticle separation of 150 nm). Moreover, the measured step size and spike charge are not correlated with the experimental time (Fig S2†). Therefore, each nanoparticle and the resulting current trace can be viewed as independent. As can be seen from Fig. 2 both the magnitude of the spike (surface hydrogen deposition) and the step (catalytic peroxide reduction) in the current profile are potential dependent. No spikes or steps have been observed over studied potentials when the solutions do not contain platinum nanoparticles. For clarity the above discussed chemical processes resulting in the nano-impact events have been schematically depicted in Fig. 3. We also comment that the experimentally observed frequency of nano-impact events in the absence or presence of oxygen is in both cases lower than the theoretically predicted value, this is consistent with previous reports.15,16
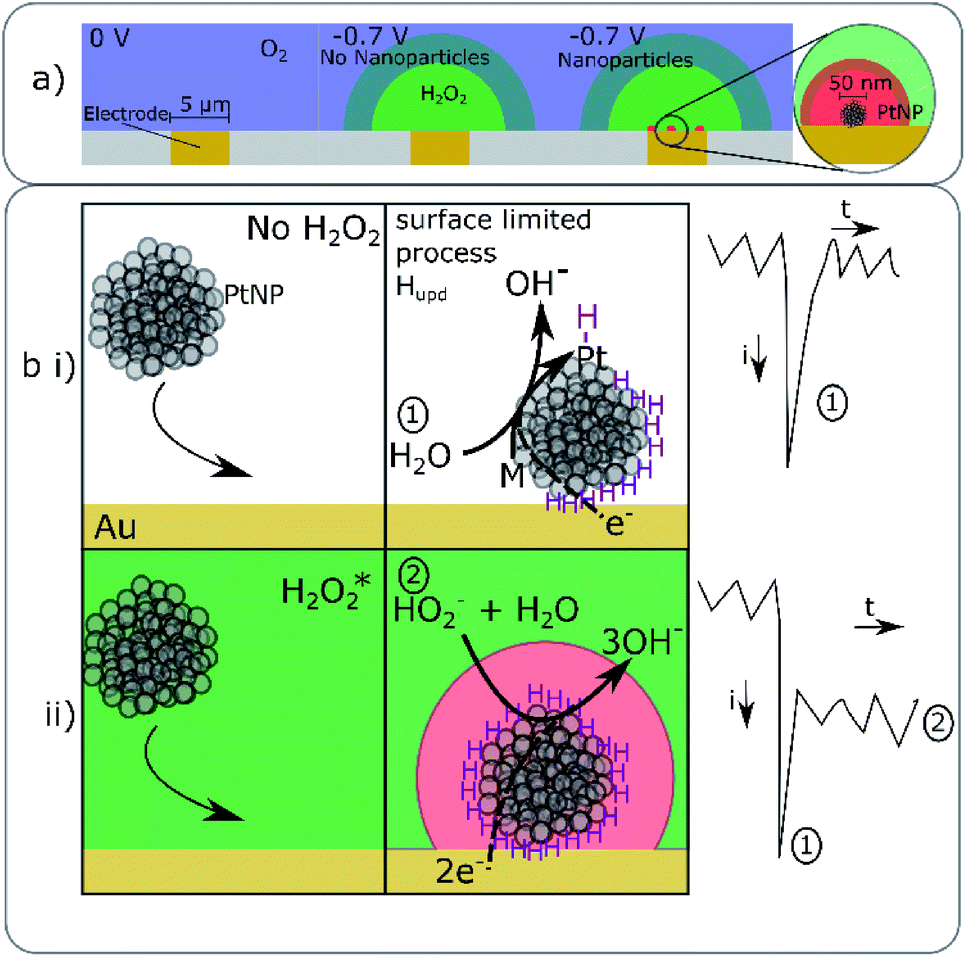 | ||
| Fig. 3 Schematic representation of the chemical processes occurring during the nano-impact reaction. (a) depicts a microelectrode in an oxygenated solution (blue) where at 0 V the oxygen is not reduced at the interface. At an electrode potential of −0.7 V the oxygen is consumed and reduced at the gold interface to hydrogen peroxide (green). Additionally in the presence of the platinum nanoparticles this formed hydrogen peroxide can be further locally reduced to water at the catalytic nano-platinum interface. Where a sparse array of platinum nanoparticles have accumulated at the interface from the solution phase. The depletion of the hydrogen peroxide in the vicinity of the nanoparticle is represented schematically by the red zone surrounding the nanoparticle. (b) visually outlines the processes occurring at the potentiostated (−0.7 V) gold interface as a platinum nanoparticle arrives and the resulting change in the current transient shape. First (b(i)) in the absence of oxygen and hence without hydrogen peroxide being produced at the interface, the nanoparticle arrives and the under potential deposition of hydrogen is induced to occur at the nanoparticle surface resulting in a measured ‘spike’ in current, the process stops when the surface is covered with Hupd. Second, in the presence of oxygen in the bulk solution (b(ii)) hydrogen peroxide (green) is produced locally at the electrode, upon impact of the nanoparticle to the electrode surface the nanoparticle again becomes modified with hydrogen (Hupd) leading to a spike in current as in (a). The nanoparticle is also able to catalyse the further reduction of the hydrogen peroxide to water resulting in a step in current being measured, again the local hydrogen peroxide depletion is represented by the red layer surrounding the nanoparticle. *Note when the platinum nanoparticle is in the vicinity of the electrode (as shown in b(ii)) it will catalyse the disproportionation of the formed hydrogen peroxide, however, for clarity this process has not been schematically represented. | ||
However, as shown in the ESI Section 2,† in the presence of oxygen the impact rate is 3–4 times less than occurs in the absence of oxygen. The physical origin of this difference in impact frequency is presently unknown, although it tentatively indicates that the occurrence of the faradaic reaction serves to alter the mass-transport of the nanoparticles in the vicinity of the electrode. Similar observations have been previously reported in the literature.17–21 Moreover, although their frequency is apparently altered, the magnitude of the measured reductive spikes are unchanged by the presence or absence oxygen (vide infra). Thus indicating that the change in impact frequency is not biasing the system towards the detection of a sub-set of the nanoparticle population.
Fig. 4 presents a summary of the measured single nanoparticle data as recorded as a function of the applied electrode potential. Here the spike charge, as measured in the presence and absence of oxygen, is presented alongside with the measured magnitude of the single nanoparticle catalytic step height. First, at a potential of −0.8 V vs. Ag/AgCl the charge associated with the deposition of hydrogen can, under the assumption of the formation of a monolayer coverage and taking an associated charge density of 210 μC cm−2,22 be used to yield a measure of the electroactive surface area of the nanoparticle. For these 50 nm platinum nanoparticles the electroactive surface area has on the basis of this measurement been determined to be 0.04 ± 0.02 μm2. ESI Section 4† demonstrates that the same surface area is measured when the hydrogen deposition process is studied oxidatively as opposed to reductively; corroborating the assumption that a complete monolayer is being reductively deposited during the experiment when performed at −0.8 V.
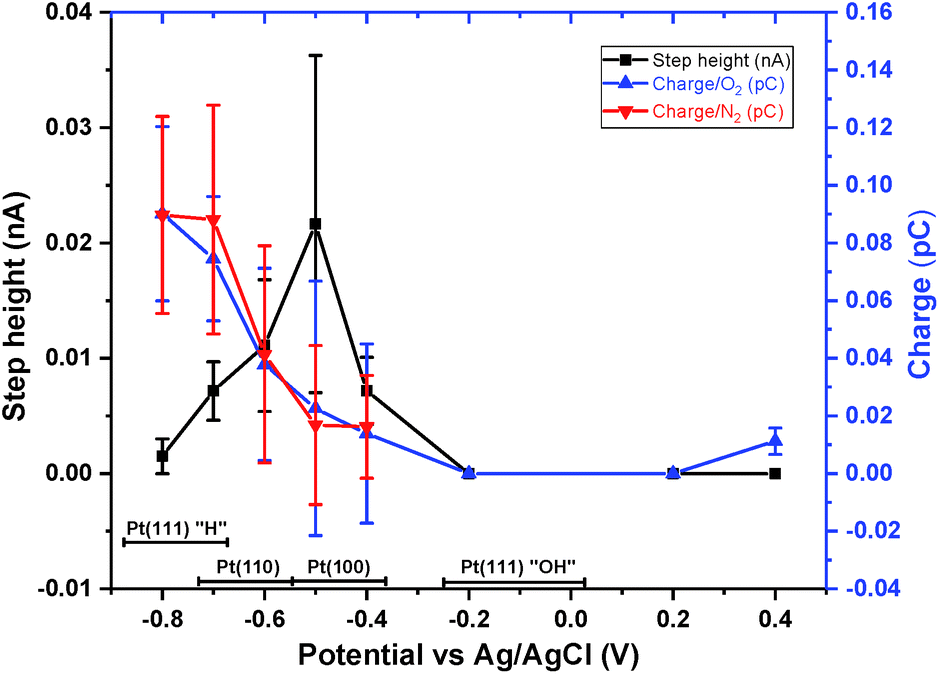 | ||
| Fig. 4 Quantification of the variation in the single platinum particle responses as a function of the applied electrode potential. Black, the measured magnitude of the single particle catalytic current associated with the surface reaction limited reduction of hydrogen peroxide. Blue and red, the magnitude of the spike in current associated with the reductive formation of under potential deposited hydrogen on to the platinum surface, as measured in the presence (blue) and absence of oxygen (red) in the bulk solution phase. Note in the experiment oxygen is reduced via a two-electron process to hydrogen peroxide at the gold electrode substrate. The characteristic potentials for the depositon of hydrogen on to platinum surfaces under alkaline condtions have been indicated on the figure with data taken from ref. 8. | ||
The magnitude of the hydrogen deposition spike is sensitive to the applied electrode potential; moreover, the magnitude of the charge passed is insensitive to the presence or absence of oxygen. Different platinum crystal facets have differing deposition potentials for hydrogen and hence the variation of the magnitude of the hydrogen deposition peak can be used to characterize the dominant crystal faces present on the nanoparticle. The potential ranges associated with the deposition of hydrogen (and concomitantly OH desorption) on to the (100), (110) and (111) surface23 have been indicated on Fig. 4. Under alkaline conditions for the (100) and (110) interfaces the electrochemical signal most likely reflects competitive adsorption/desorption of H/OH. The experimental data shown in Fig. 4 evidences that the nanoparticle interface is dominated by the presence of stepped (110) like surfaces (comprising approximately 60–75%). There is no evidence for any significant contribution from (111) crystal facets, this is most notably indicated by the lack of single nanoparticle electrochemical features in the range of −0.2–0.0 V vs. Ag/AgCl; in this potential region reversible OH adsorption/desorption is expected to occur on the (111) interface. Only at significantly higher potentials (+0.4 V) do spikes of opposite polarity occur; these features are attributed to the onset of platinum oxidation. This relative coverage of the crystal facets is comparable to that found for polycrystalline electrodes which also tend to be dominated by stepped (110) surfaces, and where for polycrystalline platinum approximately 25% of the surface is comprised of (111) and (100) surfaces.24
The current associated with the individual nanoparticle catalyzed reduction of the locally produced hydrogen peroxide varies significantly as a function of the applied potential, reaching a maximum of 21.6 ± 14.6 pA at −0.5 V vs. (Ag/AgCl). For a 50 nm particle the diffusion limited rate associated with the reduction of the hydrogen peroxide can be readily determined to be ca. 100 pA. This experimentally measured maximum (20 pA) reduction rate is significantly below this limit indicating that the catalytic process is under surface reaction control. This conclusion is further corroborated by the fact that, as shown in the ESI Section 5,† the magnitude of the electrocatalytic reaction scales linearly with the nanoparticle surface area. This point is experimentally demonstrated through studying the catalytic process at larger 70 nm particles, the surface area of which is approximately four times greater than that of the 50 nm particles used in the main body of the text. From the data presented in Fig. 4 the heterogeneous rate constant for the reduction of hydrogen peroxide at −0.5 V is determined to be 0.002 ± 0.003 m s−1.
Most notable regarding Fig. 4 is that at potentials more negative than −0.5 V vs. Ag/AgCl the platinum nanoparticle catalyzed reduction of hydrogen peroxide becomes inhibited. This inhibition may reflect the decrease in the surface area available for the catalytic reaction as the surface coverage of deposited hydrogen increases at more negative electrode potentials. This increase in the surface coverage in hydrogen is directly evidenced by the increase in the charge associated with the hydrogen deposition process (spike) as measured on the single nanoparticle scale. However, this conclusion does not necessarily explain why the maximum in the catalytic rate occurs at −0.5 V, not at −0.4 V where the surface coverage of Hupd reaches a minimum (as evidenced in Fig. 4). If the rate determining step were the adsorption of the peroxide in a bridging position then one would anticipate the limiting reaction rate to occur at a potential more positive of ca. −0.4 V, i.e. at a potential where the surface coverage of adsorbed hydrogen was at a minimum. Plausibly the maximum in the hydrogen peroxide reduction rate occurs at potentials close to the potential of zero free charge for the platinum interface.25 Hence, these results evidence the possibility that the rate of the surface reaction is controlled by the interfacial water structure.26 Alternatively, the observed increase in the hydrogen peroxide reduction rate at potentials where the (100) interface is covered with adsorbed hydrogen may indicate that this surface adsorbed species (hydrogen) participates in the reduction reaction. Comparable results have been obtained for the hydrogen oxidation reaction where the catalytic oxidation rate reached a maximum when the platinum surface was only partially covered in hydrogen.27
Conclusions
The potential dependence of the deposition of hydrogen on to individual platinum nanoparticles enables the contribution of different crystal facets to the interfacial structure to be revealed. For the present nanomaterial 60–75% of the interface comprises of stepped Pt(110) like surfaces. The change in the platinum nanoparticle surface functionality is also shown to either directly or indirectly influence the catalytic activity of the particles. Under alkaline (pH 12.3) and near mass-transport free conditions the reduction of hydrogen peroxide reaches a maximum rate at ca. −0.5 V vs. Ag/AgCl with the reaction becoming inhibited at high overpotentials. The maximum in electrocatalytic rate occurs a potential which is consistent with the Pt(100) but not the stepped Pt(110) surface being covered by a near monolayer of adsorbed hydrogen; thus indicating the likely involvement of the adsorbed hydrogen in the reduction rate determining step, and identifying the conditions under which H2O2 formation may be observed in the reduction of oxygen at Pt electrodes.Experimental
Chemicals
Citrate-capped mesoporous 50 nm and 70 nm platinum nanoparticles (PtNPs) were purchased from nanoComposix, San Diego, USA, and detailed analysis of the PtNPs can be seen in Table S1.†28 Potassium hydroxide was purchased from Fisher Scientific, Loughborough, UK, and hydrogen peroxide (30 wt% in H2O) was obtained from Sigma-Aldrich, St. Louis, MO, USA. Oxygen, Nitrogen was supplied by BOC, Surrey, UK. The water used for all experimental solutions was purified by Synergy water purification system, EMD Millipore, with a resistivity of 18.2 MΩ cm at 298 K.Single nanoparticle electrochemistry experiments
All electrochemical experiments were performed with a three-electrode system in a Faraday cage. A gold (radius: 5.0 μm, purchased from ALS, Tokyo, Japan) or a platinum microdisc electrode (radius: 5.0 μm, purchased from Princeton Applied Research, Oak Ridge, TN, USA) was employed as working electrode and polished with alumina slurries in a size sequence of 1.0, 0.3, and 0.05 μm (Buehler, Lake Bluff, IL, USA). The reference electrode was a leakless silver/silver chloride electrode (Ag/AgCl, 3.4 M KCl, eDAQ, E = 0.205 V vs. RHE), and the counter electrode was graphite rod. In all the experiments, the solutions contain 20 mM KOH as supporting electrolyte.29 The solutions containing PtNPs were used for only 20 min each time to avoid aggregation or agglomeration; the successive UV/vis measurements over this period were indistinguishable. All electrochemical measurements were thermostated at 25 ± 0.5 °C, and conducted by a lab built low noise, highly stabilized (1 kHz bandwidth) potentiostat. An NI USB-6289 data acquisition (DAQ, National Instruments, Austin, TX, USA) was connected to a computer interface via an USB. Python 3.5 (Canopy from Enthought, Austin, TX, USA) was employed to control the DAQ. The working electrode was running to virtual ground where a low-noise current amplifier Axopatch 200B (Molecular Devices, San Jose, CA, USA) was used to amplify the current and filtered at an analogue frequency of 10 kHz. The DAQ device oversampled and digitized the incoming analogue signal at a stream rate of 625 kHz, and the digitized signal was subsequently filtered at a bandwidth of 250 Hz. Importantly, the filter and measurement systems used in this work conserve the charge passed.30Conflicts of interest
There are no conflicts to declare.Acknowledgements
X. Chang thanks Natural Sciences and Engineering Research Council of Canada (NSERC) for a Postdoctoral Fellowship.Notes and references
- V. Briega-Martos, E. Herrero and J. M. Feliu, Curr. Opin. Electrochem., 2019, 17, 97–105 CrossRef CAS.
- N. M. Markovic, H. A. Gasteiger and P. N. Ross Jr, J. Phys. Chem., 1995, 99, 3411–3415 CrossRef CAS.
- L. K. Allerston and N. V. Rees, Curr. Opin. Electrochem., 2018, 10, 31–36 CrossRef CAS.
- K. J. Stevenson and K. Tschulik, Curr. Opin. Electrochem., 2017, 6, 38–45 CrossRef CAS.
- J. Y. Park, K. J. Kim, H. Son and S. J. Kwon, Nanomaterials, 2018, 8, 879–887 CrossRef PubMed.
- W. Yu, C. Batchelor-McAuley, X. Chang, N. P. Young and R. G. Compton, Phys. Chem. Chem. Phys., 2019, 21, 20415–20421 RSC.
- P. Quaino, N. B. Luque, R. Nazmutdinov, E. Santos and W. Schmickler, Angew. Chem., Int. Ed., 2012, 51, 12997–13000 CrossRef CAS PubMed.
- Electrocatalysis, ed. J. Lipkowski and P. N. Ross, Wiley, 1998 Search PubMed.
- I. Katsounaros, W. B. Schneider, J. C. Meier, U. Benedikt, P. U. Biedermann, A. A. Auer and K. J. J. Mayrhofer, Phys. Chem. Chem. Phys., 2012, 14, 7384–7391 RSC.
- S. van Stroe-Biezen, F. Everaerts, L. Janssen and R. Tacken, Anal. Chim. Acta, 1993, 273, 553–560 CrossRef CAS.
- I. Katsounaros, W. B. Schneider, J. C. Meier, U. Benedikt, P. U. Biedermann, A. A. Auer and K. J. Mayrhofer, Phys. Chem. Chem. Phys., 2012, 14, 7384–7391 RSC.
- R. Davis, G. Horvath and C. Tobias, Electrochim. Acta, 1967, 12, 287–297 CrossRef CAS.
- P. A. Defnet, C. Han and B. Zhang, Anal. Chem., 2019, 91, 4023–4030 CrossRef CAS PubMed.
- X. Jiao, C. Batchelor-McAuley, N. P. Young and R. G. Compton, Phys. Chem. Chem. Phys., 2018, 20, 23847–23850 RSC.
- X. Xiao, F. F. Fan, J. Zhou and A. J. Bard, J. Am. Chem. Soc., 2008, 130, 16669–16677 CrossRef CAS PubMed.
- S. E. Kleijn, S. C. Lai, T. S. Miller, A. I. Yanson, M. T. Koper and P. R. Unwin, J. Am. Chem. Soc., 2012, 134, 18558–18561 CrossRef CAS PubMed.
- A. N. Patel, A. Martinez-Marrades, V. Brasiliense, D. Koshelev, M. Besbes, R. Kuszelewicz, C. Combellas, G. Tessier and F. Kanoufi, Nano Lett., 2015, 15, 6454–6463 CrossRef CAS PubMed.
- R. Hao, Y. Fan and B. Zhang, J. Am. Chem. Soc., 2017, 139, 12274–12282 CrossRef CAS PubMed.
- W. Ma, H. Ma, J. F. Chen, Y. Y. Peng, Z. Y. Yang, H. F. Wang, Y. L. Ying, H. Tian and Y. T. Long, Chem. Sci., 2017, 8, 1854–1861 RSC.
- S. E. Fosdick, M. J. Anderson, E. G. Nettleton and R. M. Crooks, J. Am. Chem. Soc., 2013, 135, 5994–5997 CrossRef CAS PubMed.
- M. Guix, S. M. Weiz, O. G. Schmidt and M. Medina-Sánchez, Part. Part. Syst. Charact., 2018, 35, 1700382–1700412 CrossRef.
- S. Trasatti and O. Petrii, Pure Appl. Chem., 1991, 63, 711–734 CAS.
- F. J. Vidal-Iglesias, R. M. Arán-Ais, J. Solla-Gullón, E. Herrero and J. M. Feliu, ACS Catal., 2012, 2, 901–910 CrossRef CAS.
- J. Solla-Gullón, P. Rodríguez, E. Herrero, A. Aldaz and J. M. Feliu, Phys. Chem. Chem. Phys., 2008, 10, 1359–1373 RSC.
- N. Garcia-Araez, V. Climent and J. Feliu, J. Phys. Chem. C, 2009, 113, 9290–9304 CrossRef CAS.
- I. Ledezma-Yanez, W. D. Z. Wallace, P. Sebastián-Pascual, V. Climent, J. M. Feliu and M. T. M. Koper, Nat. Energy, 2017, 2, 17031 CrossRef CAS.
- X. Jiao, C. Batchelor-McAuley, C. Lin, E. Kätelhön, E. E. L. Tanner, N. P. Young and R. G. Compton, ACS Catal., 2018, 8, 6192–6202 CrossRef CAS.
- W. Yu, C. Batchelor-McAuley, Y.-C. Wang, S.-Q. Shao, S. M. Fairclough, S. J. Haigh, N. P. Young and R. G. Compton, Nanoscale, 2019, 11, 17791–17799 RSC.
- C. Batchelor-McAuley, K. Ngamchuea and R. G. Compton, J. Electroanal. Chem., 2018, 830, 88–94 CrossRef.
- C. Batchelor-McAuley, J. Ellison, K. Tschulik, P. L. Hurst, R. Boldt and R. G. Compton, Analyst, 2015, 140, 5048–5054 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Cyclic voltammograms of oxygen reduction and hydrogen peroxide reduction on gold electrode, underpotential hydrogen deposition (Hupd) reduction and hydrogen peroxide reduction on platinum nanoparticles, TEM characterization and charge distribution of nano-impact events, comparison of measured impact surface areas distribution via different reactions and quantification of the variation in the single platinum particle responses of different sizes. See DOI: 10.1039/d0sc00379d |
| This journal is © The Royal Society of Chemistry 2020 |