
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chromium-catalyzed cyclopropanation of alkenes with bromoform in the presence of 2,3,5,6-tetramethyl-1,4-bis(trimethylsilyl)-1,4-dihydropyrazine†
Hideaki
Ikeda
 ,
Kohei
Nishi
,
Hayato
Tsurugi
* and
Kazushi
Mashima
*
,
Kohei
Nishi
,
Hayato
Tsurugi
* and
Kazushi
Mashima
*
Department of Chemistry, Graduate School of Engineering Science, Osaka University, Toyonaka, Osaka 560-8531, Japan. E-mail: mashima@chem.es.osaka-u.ac.jp; tsurugi@chem.es.osaka-u.ac.jp
First published on 11th March 2020
Abstract
Chromium-catalyzed cyclopropanation of alkenes with bromoform was achieved to produce the corresponding bromocyclopropanes. In this catalytic cyclopropanation, an organosilicon reductant, 2,3,5,6-tetramethyl-1,4-bis(trimethylsilyl)-1,4-dihydropyrazine (1a), was indispensable for reducing CrCl3(thf)3 to CrCl2(thf)3, as well as for in situ generation of (bromomethylidene)chromium(III) species from (dibromomethyl)chromium(III) species. The (bromomethylidene)chromium(III) species are proposed to react spontaneously with alkenes to give the corresponding bromocyclopropanes. This catalytic cyclopropanation was applied to various olefinic substrates, such as allyl ethers, allyl esters, terminal alkenes, and cyclic alkenes.
Introduction
Cyclopropane is a strained three-membered carbocycle, and a common structural motif in pharmaceutical and biologically active compounds.1 The synthesis of cyclopropanes from easily available starting materials is in high demand, and several stoichiometric synthetic protocols for the C3 ring have been developed: (1) classical reductive cyclization of 1,3-dihalopropanes or β-haloalkenes using metal-based reductants such as lithium and magnesium,2 (2) cyclopropanation of alkenes using haloform (CHX3) and a strong base in phase-transfer conditions to afford geminal dihalocyclopropanes,3 and (3) cyclopropanation of alkenes using nitrogen-, phosphonium-, and sulfur-ylides,4in situ-generated zinc carbenoid from Zn reagents and CH2I2 (Simmons–Smith reaction),5 and in situ-generated chromium carbene species from excess amounts of CrCl2, diamine ligands, and RCHI2.6 In contrast to these stoichiometric reactions, metal-catalyzed cyclopropanation of alkenes using diazomethane and its derivatives is an alternative effective protocol, despite the use of explosive diazomethane derivatives.7 To avoid the use of explosive compounds, the development of metal-catalyzed cyclopropanation reactions using non-explosive reagents was recently explored.8 Uyeda et al. reported that some nickel and cobalt complexes serve as catalysts for Simmons–Smith type reactions of alkenes with less reactive CH2Cl2 and CH2Br2 in the presence of excess zinc powder (Fig. 1a).8f–8i Takai et al. reported that chromium-catalyzed cyclopropanation of alkenes with Me3SiCHI2 proceeds in the presence of catalytic amounts of chromium complex and excess Mn powder as a reducing reagent, from which gem-dichromiomethane complexes (Cr2–SiMe3) are isolated (Fig. 1b),9a and, similarly, Anwander et al. isolated an iodomethyl-bridged dichromium complex by treating CrCl2 with CHI3 as a key intermediate species for cyclopropanation to give iodocyclopropanes.9b | ||
| Fig. 1 Metal-assisted cyclopropanation of alkenes with di- and trihalomethanes; (a) cyclopropanation with excess zinc powder, (b) cyclopropanation with excess or catalytic amounts of chromium, and (c) bromocyclopropanation with catalytic amounts of chromium and organosilicon reductant 1a (This Work). | ||
We have focused our attention on the versatility of a family of organosilicon-based reductants, 1,4-bis(trimethylsilyl)-1,4-dihydropyrazine derivatives and 1,1′-bis(trimethylsilyl)-4,4′-bipyridinylidene, as stoichiometric reagents for reducing not only transition metal complexes10 for reductive C–C bond formation without generating any metal-based waste,10b,10d but also main group halides for producing multiple bonds between main group elements11 and some oxo compounds, such as nitrobenzenes and sulfoxides, in a metal-free fashion to give respectively anilines and thioethers.12c,12d Herein, we report that chromium(III) trichloride with N,N,N′,N′-tetramethylethylenediamine (TMEDA) served as a catalyst for the cyclopropanation of alkenes with bromoform in combination with 2,3,5,6-tetramethyl-1,4-bis(trimethylsilyl)-1,4-dihydropyrazine (1a) to obtain synthetically useful bromocyclopropanes in high yield (Fig. 1c).13
Results and discussion
We then screened conditions by tuning reductants, additives, and supporting ligands to optimize the chromium-catalyzed cyclopropanation of allyl benzyl ether (2a) with bromoform as a model reaction, and the results are summarized in Table 1. When we used a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of CrCl3(thf)3 (5 mol%) and TMEDA (5 mol%) in the presence of 1a (2 equiv.) in 1,2-dimethoxyethane (DME) at 50 °C for 24 h, bromocyclopropane 3a was obtained in 98% yield with high trans (89%) selectivity (entry 1). Cyclopropanation at 25 °C resulted in a slightly lower yield (81%) of 3a with almost the same trans selectivity (entry 1 vs. 2). No cyclopropanation product was obtained when organosilicon compounds 1b–d were used as the reducing reagents (entries 3–5), although 1b–d reduced CrCl3(thf)3 to CrCl2, probably due to coordination of the reduction byproducts, 2,5-dimethylpyrazine (from 1b), pyrazine (from 1c), and 4,4′-bipyridyl (from 1d), to the chromium center, as confirmed by the inhibition of the catalytic reaction when pyrazine was added under the standard conditions. Screening of several multidentate nitrogen-based ligands revealed that TMEDA was the best ligand for this catalytic reaction (entry 1 vs. 12–17; amines, phosphines, and other ligands in ESI†). Notably, no reaction was observed when using typical organic and inorganic reductants, such as tetrakis(dimethylamino)ethylene (TDAE), Zn, and Mn powder (entries 6–8). The coordination of TMEDA to the chromium center was essentially required to produce the catalytic activity: the addition of either ZnCl2 (2 equiv.) or MnCl2 (2 equiv.) to the standard reaction conditions resulted in no reaction (entry 9) or lowered the yield of 3a (entry 10), respectively, due to the removal of TMEDA from the chromium center,9a while under ligand-free conditions, the yield of 3a decreased significantly (entry 11). When isolated CrCl3(tmeda) (5 mol%) was used as the catalyst, the yield of 3a was comparable with that of the in situ CrCl3(thf)3 and TMEDA system (entry 18).
:1 mixture of CrCl3(thf)3 (5 mol%) and TMEDA (5 mol%) in the presence of 1a (2 equiv.) in 1,2-dimethoxyethane (DME) at 50 °C for 24 h, bromocyclopropane 3a was obtained in 98% yield with high trans (89%) selectivity (entry 1). Cyclopropanation at 25 °C resulted in a slightly lower yield (81%) of 3a with almost the same trans selectivity (entry 1 vs. 2). No cyclopropanation product was obtained when organosilicon compounds 1b–d were used as the reducing reagents (entries 3–5), although 1b–d reduced CrCl3(thf)3 to CrCl2, probably due to coordination of the reduction byproducts, 2,5-dimethylpyrazine (from 1b), pyrazine (from 1c), and 4,4′-bipyridyl (from 1d), to the chromium center, as confirmed by the inhibition of the catalytic reaction when pyrazine was added under the standard conditions. Screening of several multidentate nitrogen-based ligands revealed that TMEDA was the best ligand for this catalytic reaction (entry 1 vs. 12–17; amines, phosphines, and other ligands in ESI†). Notably, no reaction was observed when using typical organic and inorganic reductants, such as tetrakis(dimethylamino)ethylene (TDAE), Zn, and Mn powder (entries 6–8). The coordination of TMEDA to the chromium center was essentially required to produce the catalytic activity: the addition of either ZnCl2 (2 equiv.) or MnCl2 (2 equiv.) to the standard reaction conditions resulted in no reaction (entry 9) or lowered the yield of 3a (entry 10), respectively, due to the removal of TMEDA from the chromium center,9a while under ligand-free conditions, the yield of 3a decreased significantly (entry 11). When isolated CrCl3(tmeda) (5 mol%) was used as the catalyst, the yield of 3a was comparable with that of the in situ CrCl3(thf)3 and TMEDA system (entry 18).
|
|
|||
|---|---|---|---|
| Entry | Variation from standard condition | Yield (%)b |
trans:cisb |
| a Reaction condition: 2a (0.1 mmol), bromoform (2 equiv.), CrCl3(thf)3 (5 mol%), ligand (5 mol%), and reductant (above-mentioned amount) in 1,2-dimethoxyethane (DME, 0.10 M) at 50 °C for 24 hours. b Determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as an internal standard. c Isolated yield. TDAE: tetrakis(dimethylamino)ethylene. | |||
| 1 | None | 98 (93)c | 89:11 |
| 2 | 25 °C, 24 h | 81 | 90:10 |
| 3 | 1b (2 equiv.) instead of 1a | 0 | N/A |
| 4 | 1c (2 equiv.) instead of 1a | 0 | N/A |
| 5 | 1d (2 equiv.) instead of 1a | 0 | N/A |
| 6 | TDAE (2 equiv.) instead of 1a | 0 | N/A |
| 7 | Zn (6 equiv.) instead of 1a | 0 | N/A |
| 8 | Mn (6 equiv.) instead of 1a | 0 | N/A |
| 9 | Addition of ZnCl2 (2 equiv.) | 0 | N/A |
| 10 | Addition of MnCl2 (2 equiv.) | 56 | 87:13 |
| 11 | Without TMEDA | 7 | 71:29 |
| 12 | L1 (5 mol%) instead of TMEDA | 97 | 90:10 |
| 13 | L2 (5 mol%) instead of TMEDA | 7 | 57:43 |
| 14 | L3 (5 mol%) instead of TMEDA | 0 | N/A |
| 15 | L4 (5 mol%) instead of TMEDA | 8 | >99:1 |
| 16 | L5 (5 mol%) instead of TMEDA | 0 | N/A |
| 17 | L6 (5 mol%) instead of TMEDA | 0 | N/A |
| 18 | CrCl3(tmeda) (5 mol%) instead of CrCl3(thf)3 and TMEDA | 90 | 88:12 |

|
|||

With the optimized reaction conditions in hand, we analyzed the substrate scope of the alkenes (Table 2). Allyl phenyl ether (2b) was converted to the corresponding bromocyclopropane 3b in 92% yield with high trans selectivity. Other allyl aryl ethers 2c–g with electron-withdrawing and -donating substituents on the phenyl ring were transformed to the corresponding cyclopropanes 3c–g in moderate to high yields, with a cyano group or halogen atoms at the para-position of the aryl ring remaining intact during the cyclopropanation reaction. Reaction of CHBr3 with allyl butyl ether (2h) afforded 3h in 81% yield with a trans:cis ratio of 87:13. The carbonyl group also tolerated the reductive conditions to produce cyclopropanes; benzoyl-substituted alkene 2i was converted to 3i in 75% yield, while allyl carbonate 2j, which is typically used for allylic substitution of nucleophiles, afforded 3j in 60% yield without any decomposition of 2j. Allylamine 2k was also applicable and the corresponding cyclopropylmethylamine 3k was obtained in 64% yield. Simple α-olefins, such as allylbenzene 2l, 5-hexenyl acetate 2m, 1-octene 2n, and vinylcyclohexane 2o, gave the corresponding cyclopropanes 3l–o in good yield. When we applied substrates possessing two olefinic moieties, a terminal and mono-substituted olefinic part was selectively cyclopropanated to give 3p and 3q in moderate yield. Internal alkenes with cis-configuration were also applicable to our catalytic system: cis-1,4-diacetoxy-2-butene (2r) showed a moderate reactivity to give the corresponding cyclopropane 3r in 47% yield, while some cyclic alkenes such as cycloheptene (2s), cyclooctene (2t) and acenaphthylene (2u) were applicable to afford polycyclic compounds 3s, 3t, and 3u in moderate to high yields, though debromination of initially formed bromocyclopropane might be involved for the formation of 3u. Other olefins such as styrene, 1,1-disubstituted alkenes, acyclic internal alkenes, and dienes were not applicable in this cyclopropanation reaction (see ESI† for the limitations of this cyclopropanation).
|
|
|---|
| a Standard reaction condition: 2 (0.4 mmol), bromoform (0.8 mmol, 2 equiv.), CrCl3(thf)3 (0.02 mmol, 5 mol%), TMEDA (5 mol%), and 1a (0.8 mmol, 2 equiv.) in 1,2-dimethoxyethane (DME, 4 mL) at 50 °C for 24 hours. b CrCl3(thf)3/TMEDA: 10 mol%. c NMR yield. Isolated yields after purification by flash column chromatography are noted. |
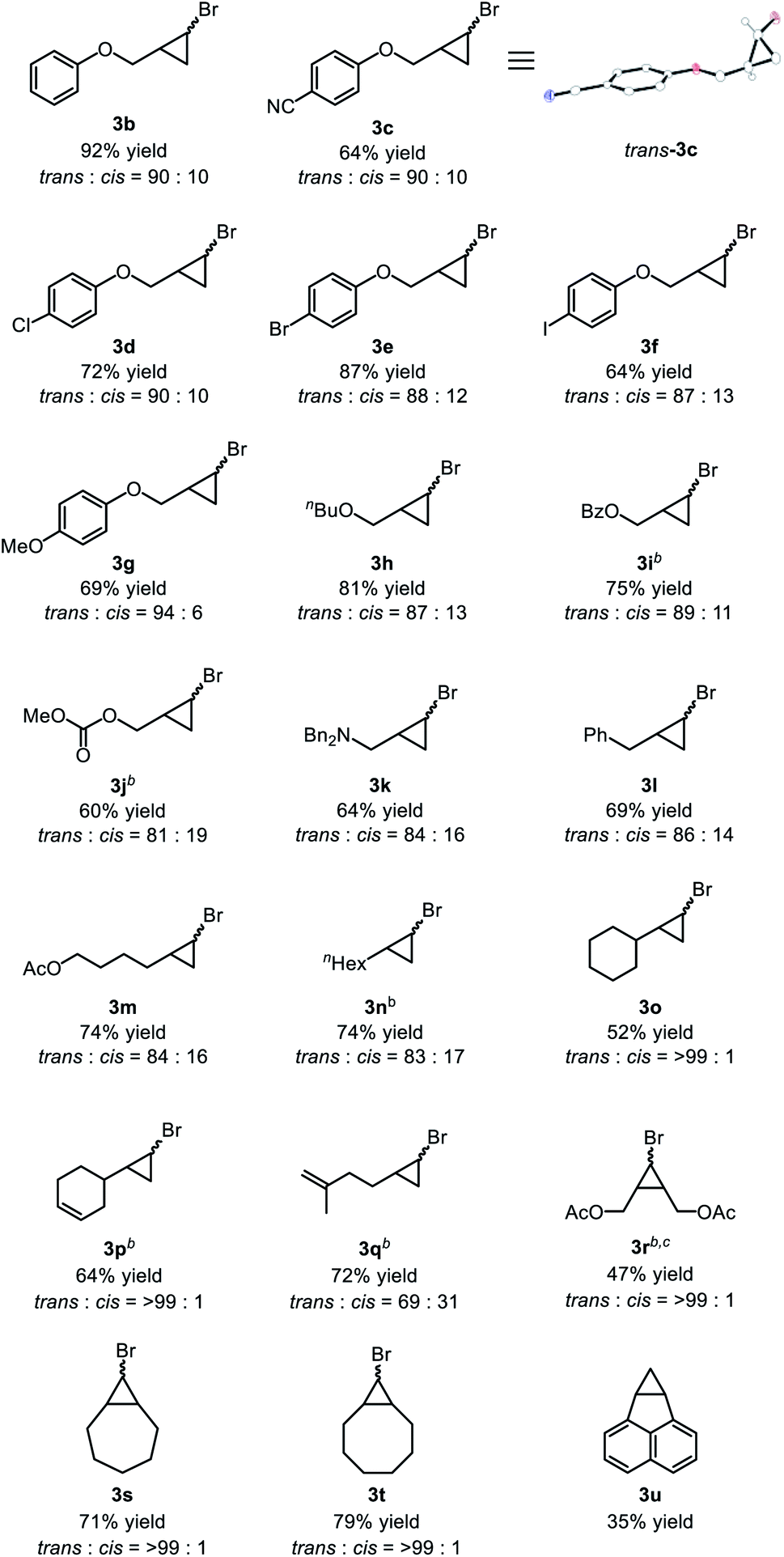
|
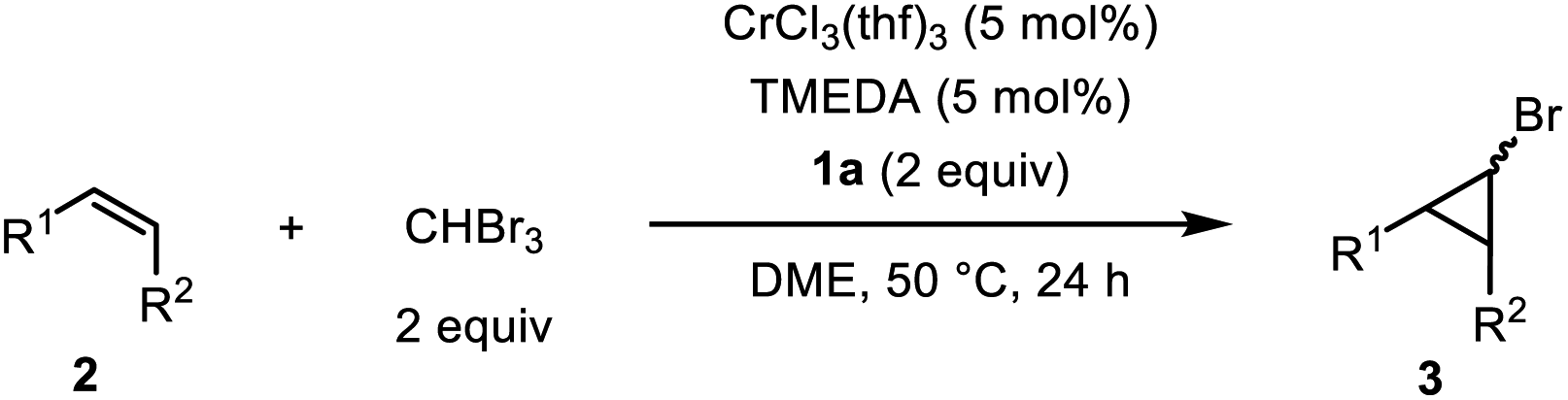
In addition to bromoform, other trihalomethanes were applicable to the catalytic cyclopropanation. It was noteworthy that, in the reactions of 2a with both CHClBr2 and CHCl2Br, the same bromocyclopropane 3a was obtained as the major product in 84% and 66% yield, respectively, along with chlorocyclopropane as a minor product, although it was much easier to cleave the C–Br bond than the C–Cl bond (Scheme 1a).14 Direct synthesis of iodocyclopropane was not accessible under the optimized conditions with CHI3, while cyclopropanation using CHBr3 in the presence of NaI (2 equiv.) produced iodocyclopropane 3a′ instead of 3a in 75% yield (Scheme 1b). When Me3SiCHI2 was used as a C1 source, corresponding silyl-substituted cyclopropane 3a′′ was obtained in quantitative yield (Scheme 1c).
 | ||
| Scheme 1 Cyclopropanation using tri- and dihalomethanes. (a) Reaction with CHClBr2 and CHCl2Br. (b) Reaction with CHBr3 in the presence of NaI. (c) Reaction with Me3SiCHI2. | ||
To elucidate the reaction mechanism, we carried out a kinetic study for the formation of 3a, and the resulting data were analyzed by variable time normalization analysis (see ESI†).15 The overall reaction rate did not change with various concentrations of chromium catalyst (0.004–0.01 M) and alkene 2a (0.08–0.12 M), giving a rate dependence of [Cr]0[2a]0, which is in sharp contrast to the report of Takai et al. who found that chromium-catalyzed cyclopropanation with Me3SiCHI2 obeys first-order dependence on the concentrations of both a chromium carbene complex and 2a, giving a rate dependence of [Cr]1[2a]1.9a Such a difference was further observed in the reaction profile; no induction period was observed under the various reaction conditions.16
Next, to understand how 1a functioned to generate a catalytically active species, we performed some control experiments. Direct activation of CHBr3 by 1a was excluded because no significant rate acceleration was observed when a mixture of CHBr3 and 1a was pre-treated by stirring at 50 °C for 1 hour before adding the chromium catalyst (see ESI†). Although we tried repeatedly to isolate the dichromium species having a bridging bromomethyl group, the target complex could not be isolated and characterized, probably due to the instability of the bromomethyl-bridged dichromium species (see ESI†). In previous reports, however, gem-dichromiomethane complexes (Cr2–X) was isolated as key intermediates prior to generating reactive mononuclear carbene species via disproportionation (Fig. 2). Takai et al. reported the first example of an isolated gem-dichromiomethane complex (Cr2–SiMe3) by introducing a bulky trimethylsilyl-substituent on a carbon atom of diiodomethane, from which silylcyclopropanes were obtained upon treatment with alkenes. The related germanium derivative, Cr2–GeMe3, was also isolated and used for cyclopropanation. Anwander et al. independently observed the formation of a gem-dichromiomethane complex (Cr2–I) from the reaction of CrCl2 and CHI3 at low temperature.
 | ||
| Fig. 2 Proposed reaction pathway for chromium carbene species from isolated dichromium complexes by Takai et al. and Anwander et al. | ||
We next conducted a stoichiometric cyclopropanation reaction of alkene 2a with bromoform in the presence of excess CrCl2 (Scheme 2). The desired cyclopropane 3a was not obtained even at 80 °C (Scheme 2a), although formation of the corresponding cyclopropanes was observed when iodoform and diiodomethane derivatives were used. Moreover, under the catalytic conditions using 1a, the yield of 3a gradually decreased as the catalyst loading was increased from 5 to 100 mol% (Scheme 2b). The lower product yield caused by increasing the amount of the chromium salt suggested that involvement of gem-dichromiomethane species was less likely in our metal-salt free system with 1a compared with other chromium-catalyzed cyclopropanation developed by Takai et al.
 | ||
| Scheme 2 Control experiments. (a) Reaction in the presence of excess amount of CrCl2. (b) Reaction with different catalyst loading. | ||
On the basis of these findings, we propose the reaction mechanism shown in Scheme 3. The initial step is the activation of bromoform by chromium(II) species A to form (dibromomethyl)chromium(III) species B accompanied by the formation of an equimolar amount of chromium(III) trihalide C, which can be reduced by 1a or in situ-generated chromium(I) halide F (vide infra). Species B is dehalogenated by 1a to afford (bromomethylidene)chromium(III) D along with the elimination of Me4-pyrazine and 2 equiv. of Me3SiX (X = Cl, Br), whose reactivity is assumed due to the reductive dehalogenation of vicinal dihaloalkanes by the organosilicon-based reductant 1d leading to the formation of alkenes.12a In addition, the generation of metal carbene species by the dehalogenation of metal carbenoids with zinc powder was proposed for nickel- or cobalt-catalyzed cyclopropanation of alkenes with dibromomethane or dichloromethane (Scheme 3).8b,8h Finally, the reaction of D with alkenes gives 4-membered metallacycle E, whose reductive elimination affords the desired bromocyclopropane together with a low valent nascent chromium(I) species F. The resulting F reacts with chromium(III) trihalide C to regenerate chromium(II) species A through comproportionation. Accordingly, 1a has dual functions to reduce not only a catalyst precursor, CrCl3(tmeda), at the initial step, but also mainly the chromium(III) species B for generating mononuclear chromium carbene species D as a key intermediate.
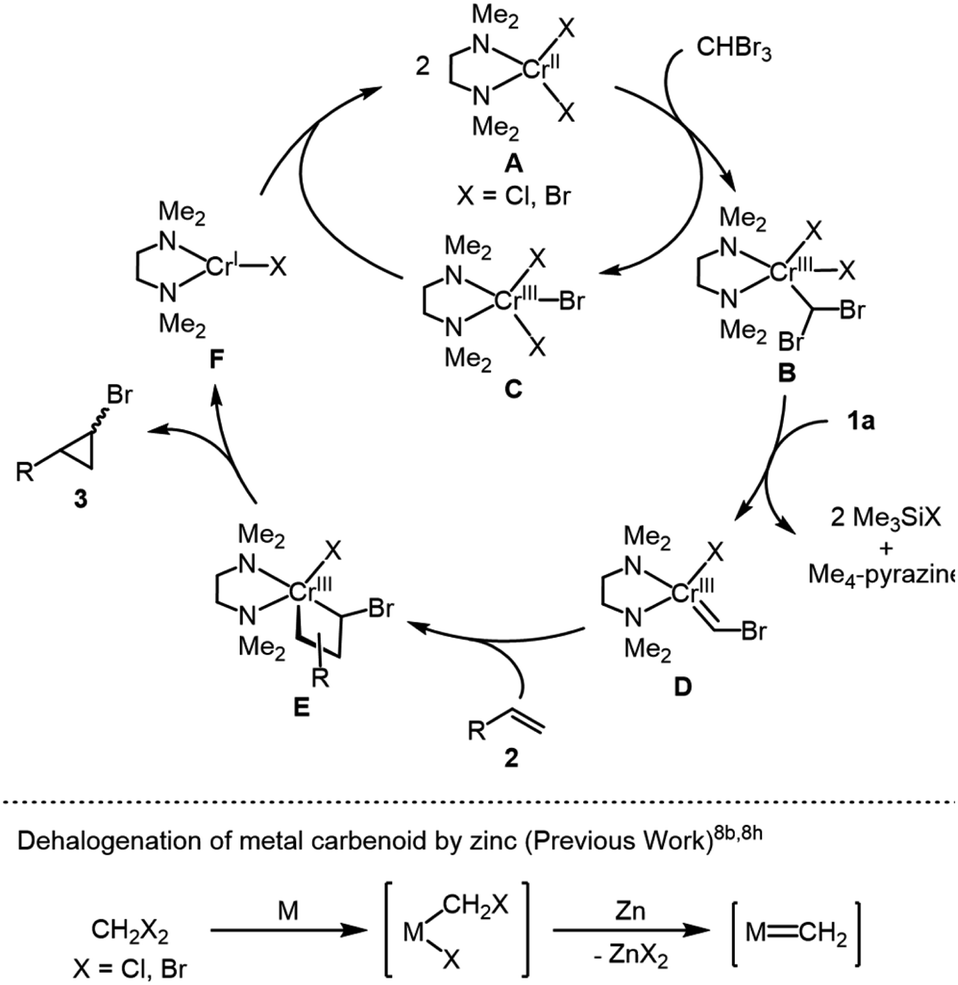 | ||
| Scheme 3 Proposed reaction mechanism. | ||
Conclusions
In summary, we developed chromium-catalyzed bromocyclopropanation of alkenes with bromoform using an organosilicon-based reductant 1a. The desired bromocyclopropanes were obtained in moderate to high yields with good trans selectivity, and the reaction was applicable to allyl ether derivatives, allyl carbonate, allylamine, and simple α-olefins. Control experiments suggested that 1a played an important role in reducing the (dibromomethyl)chromium(III) species to generate mononuclear (bromomethylidene)chromium(III) as a key intermediate. Further exploration to discover the unique metal salt-free reductive transformation of organic compounds is ongoing in our laboratory.Conflicts of interest
The author declares no conflict of interest.Acknowledgements
H. I. thanks the financial support by the JSPS Research Fellow-ships for Young Scientists. This work was supported by JSPS KAKENHI Grant No. JP26708012 (Grant-in-Aid for Young Scientist (A)) to H. T. and JP15H05808 (Precisely Designed Catalysis with Customized Scaffolding) to K. M.Notes and references
- (a) H.-U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151 CrossRef CAS PubMed; (b) H. Lebel, J.-F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977 CrossRef CAS PubMed; (c) H. Pellissier, Tetrahedron, 2008, 64, 7041 CrossRef CAS; (d) D. Y.-K. Chen, R. H. Pouwer and J.-A. Richard, Chem. Soc. Rev., 2012, 41, 4631 RSC; (e) P. Tang and Y. Qin, Synthesis, 2012, 44, 2969 CrossRef CAS; (f) R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845 CrossRef CAS PubMed; (g) C. Ebner and E. M. Carreira, Chem. Rev., 2017, 117, 11651 CrossRef CAS PubMed.
- (a) H. C. Brown and K. A. Keblys, J. Am. Chem. Soc., 1964, 86, 1791 CrossRef CAS; (b) L. Kaplan, J. Am. Chem. Soc., 1967, 89, 1753 CrossRef CAS; (c) J. Barluenga, J. Florez and M. Yus, Synthesis, 1983, 647 CrossRef CAS; (d) W. F. Bailey and Y. Tao, Tetrahedron Lett., 1997, 38, 6157 CrossRef CAS; (e) R. A. Aitken, P. K. G. Hodgson, J. J. Morrison and A. O. Oyewale, J. Chem. Soc., Perkin Trans. 1, 2002, 1, 402 RSC.
- (a) W. V. E. Doering and K. Hoffmann, J. Am. Chem. Soc., 1954, 76, 6162 CrossRef CAS; (b) M. Mąkosza and M. Wawrzyniewicz, Tetrahedron Lett., 1969, 4659 CrossRef; (c) M. Fedoryński, W. Ziólkowska and A. Jończyk, J. Org. Chem., 1993, 58, 6120 CrossRef; (d) X. Creary, Z. Jiang, M. Butchko and K. McLean, Tetrahedron Lett., 1996, 37, 579 CrossRef CAS; (e) M. Lahrech, S. Hacini, J.-L. Parrain and M. Santelli, Tetrahedron Lett., 1997, 38, 3395 CrossRef CAS; (f) M. N. Masuno, D. M. Young, A. C. Hoepker, C. K. Skepper and T. F. Molinski, J. Org. Chem., 2005, 70, 4162 CrossRef CAS PubMed; (g) T. N. Grant and F. G. West, J. Am. Chem. Soc., 2006, 128, 9348 CrossRef CAS PubMed; (h) M. Fedoryński, Chem. Rev., 2003, 103, 1099 CrossRef PubMed.
- (a) P. A. Grieco and R. S. Finkelhor, Tetrahedron Lett., 1972, 13, 3781 CrossRef; (b) H. Fauduet and R. Burgada, Synthesis, 1980, 642 CrossRef CAS; (c) S. Kojima, K. Fujimoto, Y. Shinohara, M. Shimizu and K. Ohkata, Tetrahedron Lett., 2000, 41, 9847 CrossRef CAS; (d) C. D. Papageorgiou, S. V. Ley and M. J. Gaunt, Angew. Chem., Int. Ed., 2003, 42, 828 CrossRef CAS PubMed; (e) X.-M. Deng, P. Cai, S. Ye, X.-L. Sun, W.-W. Liao, K. Li, Y. Tang, Y.-D. Wu and L.-X. Dai, J. Am. Chem. Soc., 2006, 128, 9730 CrossRef CAS PubMed; (f) S. Lakhdar, R. Appel and H. Mayr, Angew. Chem., Int. Ed., 2009, 48, 5034 CrossRef CAS PubMed; (g) N. Kanomata, R. Sakaguchi, K. Sekine, S. Yamashita and H. Tanaka, Adv. Synth. Catal., 2010, 352, 2966 CrossRef CAS; (h) R. Appel, N. Hartmann and H. Mayr, J. Am. Chem. Soc., 2010, 132, 17894 CrossRef CAS PubMed; (i) B.-H. Zhu, R. Zhou, J.-C. Zheng, X.-M. Deng, X.-L. Sun, Q. Shen and Y. Tang, J. Org. Chem., 2010, 75, 3454 CrossRef CAS PubMed; (j) T. Ferrary, E. David, G. Milanole, T. Besset, P. Jubault and X. Pannecoucke, Org. Lett., 2013, 15, 5598 CrossRef CAS PubMed; (k) A.-H. Li, L.-X. Dai and V. K. Aggarwal, Chem. Rev., 1997, 97, 2341 CrossRef CAS PubMed.
- (a) H. E. Simmons and R. D. Smith, J. Am. Chem. Soc., 1959, 81, 4256 CrossRef CAS; (b) J. Furukawa, N. Kawabata and J. Nishimura, Tetrahedron Lett., 1966, 7, 3353 CrossRef; (c) J. Furukawa, N. Kawabata and J. Nishimura, Tetrahedron, 1968, 24, 53 CrossRef CAS; (d) A. B. Charette and N. Wilb, Synlett, 2002, 176 CrossRef CAS; (e) M. Nakamura, A. Hirai and E. Nakamura, J. Am. Chem. Soc., 2003, 125, 2341 CrossRef CAS PubMed; (f) J. Long, Y. Yuan and Y. Shi, J. Am. Chem. Soc., 2003, 125, 13632 CrossRef CAS PubMed; (g) L.-P. B. Beaulieu, J. F. Schneider and A. B. Charette, J. Am. Chem. Soc., 2013, 135, 7819 CrossRef CAS PubMed; (h) S. Taillemaud, N. Diercxsens, A. Gagnon and A. B. Charette, Angew. Chem., Int. Ed., 2015, 54, 14108 CrossRef CAS PubMed; (i) G. Benoit and A. B. Charette, J. Am. Chem. Soc., 2017, 139, 1364 CrossRef CAS PubMed.
- (a) K. Takai, S. Toshikawa, A. Inoue and R. Kokumai, J. Am. Chem. Soc., 2003, 125, 12990 CrossRef CAS PubMed; (b) K. Takai, M. Hirano and S. Toshikawa, Synlett, 2004, 1347 CrossRef CAS; (c) J. M. Concellón, H. Rodríguez-Solla, C. Méjica, E. G. Blanco, S. García-Granda and M. R. Díaz, Org. Lett., 2008, 10, 349 CrossRef PubMed; (d) J. M. Concellón, H. Rodríguez-Solla, C. Méjica, E. G. Blanco, S. García-Granda and M. R. Díaz, J. Org. Chem., 2008, 73, 3828 CrossRef PubMed; (e) M. Murai, C. Mizuta, R. Taniguchi and K. Takai, Org. Lett., 2017, 19, 6104 CrossRef CAS PubMed; (f) M. Murai, R. Taniguchi, C. Mizuta and K. Takai, Org. Lett., 2019, 21, 2668 CrossRef CAS PubMed.
- For selected reviews, see: (a) G. Mass, Chem. Soc. Rev., 2004, 33, 183 RSC; (b) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981 CrossRef CAS PubMed; (c) S. H. Goh, L. E. Closs and G. L. Closs, J. Org. Chem., 1969, 34, 25 CrossRef CAS; (d) D. S. Crumrine, T. J. Haberkamp and D. Suther, J. Org. Chem., 1975, 40, 2274 CrossRef CAS; Ruthenium: (e) H. Nishiyama, Y. Itoh, H. Matsumoto, S.-B. Park and K. Itoh, J. Am. Chem. Soc., 1994, 116, 2223 CrossRef CAS; (f) H. Nishiyama, S.-B. Park, M. Haga, K. Aoki and K. Itoh, Chem. Lett., 1994, 1111 CrossRef CAS; (g) S. Chanthamath, S. Takai, K. Shibatomi and S. Iwasa, Angew. Chem., Int. Ed., 2013, 52, 5818 CrossRef CAS PubMed; Rhodium: (h) W. Hu, D. J. Timmons and M. P. Doyle, Org. Lett., 2002, 4, 901 CrossRef CAS PubMed; (i) D. Marcoux and A. B. Charette, Angew. Chem., Int. Ed., 2008, 47, 10155 CrossRef CAS PubMed; (j) D. Marcoux, S. Azzi and A. B. Charette, J. Am. Chem. Soc., 2009, 131, 6970 CrossRef CAS PubMed; (k) A. Pons, P. Ivashkin, T. Poisson, A. B. Charette, X. Pannecoucke and P. Jubault, Chem.–Eur. J., 2016, 22, 6239 CrossRef CAS PubMed; Cobalt: (l) T. Uchida, B. Saha and T. Katsuki, Tetrahedron Lett., 2001, 42, 2521 CrossRef CAS; (m) Y. Chen, K. B. Fields and X. P. Zhang, J. Am. Chem. Soc., 2004, 126, 14718 CrossRef CAS PubMed; (n) M. P. Doyle, Angew. Chem., Int. Ed., 2009, 48, 850 CrossRef CAS PubMed; Copper: (o) H. Nozaki, S. Moriuti, M. Yamabe and R. Noyori, Tetrahedron Lett., 1966, 1, 59 CrossRef; (p) R. E. Lowenthal, A. Abiko and S. Masamune, Tetrahedron Lett., 1990, 31, 6005 CrossRef CAS; (q) D. A. Evans, K. A. Woerpel, M. M. Hinman and M. M. Faul, J. Am. Chem. Soc., 1991, 113, 726 CrossRef CAS; (r) C. Mazet, V. Kohler and A. Pfaltz, Angew. Chem., Int. Ed., 2005, 44, 4888 CrossRef CAS PubMed.
- (a) H. Kanai and N. Hiraki, Chem. Lett., 1979, 761 CrossRef CAS; (b) H. Kanai, N. Hiraki and S. Iida, Bull. Chem. Soc. Jpn., 1983, 56, 1025 CrossRef CAS; (c) S. Sengmany, E. Léonel, J. P. Paugam and J.-Y. Nédélec, Tetrahedron, 2002, 58, 271 CrossRef CAS; (d) K. Fujii, T. Misaki and T. Sugimura, Chem. Lett., 2014, 43, 634 CrossRef CAS; (e) J. Xu, N. B. Samsuri and H. A. Duong, Chem. Commun., 2016, 52, 3372 RSC; (f) Y.-Y. Zhou and C. Uyeda, Angew. Chem., Int. Ed., 2016, 55, 3171 CrossRef CAS PubMed; (g) S. Pal, Y.-Y. Zhou and C. Uyeda, J. Am. Chem. Soc., 2017, 139, 11686 CrossRef CAS PubMed; (h) J. Werth and C. Uyeda, Angew. Chem., Int. Ed., 2018, 57, 13902 CrossRef CAS PubMed; (i) J. Werth and C. Uyeda, Chem. Sci., 2018, 9, 1604 RSC; (j) C. M. Farley, Y.-Y. Zhou, N. Banka and C. Uyeda, J. Am. Chem. Soc., 2018, 140, 12710 CrossRef CAS PubMed.
- (a) M. Murai, R. Taniguchi, N. Hosokawa, Y. Nishida, H. Mimachi, T. Oshiki and K. Takai, J. Am. Chem. Soc., 2017, 139, 13184 CrossRef CAS PubMed; (b) D. Werner and R. Anwander, J. Am. Chem. Soc., 2018, 140, 14334 CrossRef CAS PubMed.
- (a) T. Saito, H. Nishiyama, H. Tanahashi, K. Kawakita, H. Tsurugi and K. Mashima, J. Am. Chem. Soc., 2014, 136, 5161 CrossRef CAS PubMed; (b) T. Yurino, Y. Ueda, Y. Shimizu, S. Tanaka, H. Nishiyama, H. Tsurugi, K. Sato and K. Mashima, Angew. Chem., Int. Ed., 2015, 54, 14437 CrossRef CAS PubMed; (c) H. Tanahashi, H. Ikeda, H. Tsurugi and K. Mashima, Inorg. Chem., 2016, 55, 1446 CrossRef CAS PubMed; (d) Y. Ueda, N. Tsujimoto, T. Yurino, H. Tsurugi and K. Mashima, Chem. Sci., 2019, 10, 994 RSC; For recent reviews, see: (e) H. Tsurugi and K. Mashima, Chem.–Eur. J., 2019, 25, 913 CrossRef CAS PubMed; (f) H. Tsurugi and K. Mashima, Acc. Chem. Res., 2019, 52, 769 CrossRef CAS PubMed.
- P. K. Majhi, H. Ikeda, T. Sasamori, H. Tsurugi, K. Mashima and N. Tokitoh, Organometallics, 2017, 36, 1224 CrossRef CAS.
- (a) S. Rej, S. Pramanik, H. Tsurugi and K. Mashima, Chem. Commun., 2017, 53, 13157 RSC; (b) S. Pramanik, S. Rej, S. Kando, H. Tsurugi and K. Mashima, J. Org. Chem., 2018, 83, 2409 CrossRef CAS PubMed; (c) A. Bhattacharjee, H. Hosoya, H. Ikeda, K. Nishi, H. Tsurugi and K. Mashima, Chem.–Eur. J., 2018, 24, 11278 CrossRef CAS PubMed; (d) A. Bhattacharjee, H. Hosoya, T. Yurino, H. Tsurugi and K. Mashima, Chem. Lett., 2019, 48, 888 CrossRef CAS.
- (a) G. F. Meils and I. R. Doyle, J. Org. Chem., 1985, 50, 3713 CrossRef; (b) A. Inoue, J. Kondo, H. Shinokubo and K. Oshima, Chem.–Eur. J., 2002, 8, 1730 CrossRef CAS; (c) E. Thomas, A. N. Kasatkin and R. J. Whitby, Tetrahedron Lett., 2006, 47, 9181 CrossRef CAS; For recent reports using bromocyclopropanes for cross-coupling reactions, see: (d) J.-H. Liu, C.-T. Yang, X.-Y. Lu, Z.-Q. Zhang, L. Xu, M. Cui, X. Lu, B. Xiao, Y. Fu and L. Liu, Chem.–Eur. J., 2014, 20, 15334 CrossRef CAS PubMed; (e) P. M. P. Garcia, T. D. Franco, A. Epenoy, R. Scopelliti and X. Hu, ACS Catal., 2016, 6, 258 CrossRef; (f) P. Zhang, C. C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084 CrossRef CAS PubMed; (g) C. P. Johnston, R. T. Smith, S. Allmendinger and D. W. C. MacMillan, Nature, 2016, 536, 322 CrossRef CAS PubMed; (h) C. Le, Y. Liang, R. W. Evans, X. Li and D. W. C. MacMillan, Nature, 2017, 547, 79 CrossRef CAS PubMed.
- K. Takai, K. Nitta and K. Utimoto, J. Am. Chem. Soc., 1986, 108, 7408 CrossRef CAS . Halogen exchange on the methine carbon for (dibromomethyl)chromium(III) intermediate, Cl2Cr(CHBr2), was also observed in CrCl2-mediated carbonyl olefination of benzaldehyde with bromoform: both of β-chlorostyrene and β-bromostyrene were obtained.
- (a) J. Burés, Angew. Chem., Int. Ed., 2016, 55, 2028 CrossRef PubMed; (b) J. Burés, Angew. Chem., Int. Ed., 2016, 55, 16084 CrossRef PubMed.
- In chromium-catalyzed cyclopropanation reported by Takai et al. (ref. 9a), the induction period (ca. 1 hour) was observed due to the slow rate of formation of a reactive chromium carbene species from Cr2–SiMe3via disproportionation when the amount of Cr2–SiMe3 cat. was less than 30 mol%. No induction period in our system indicated that dinuclear chromium intermediate was not included in the catalytic cycle.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1954370. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc00964d |
| This journal is © The Royal Society of Chemistry 2020 |