
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCompleting the triad: synthesis and full characterization of homoleptic and heteroleptic carbonyl and nitrosyl complexes of the group VI metals†‡
Jan
Bohnenberger
 a,
Manuel
Schmitt
a,
Wolfram
Feuerstein
b,
Ivo
Krummenacher
c,
Burkhard
Butschke
a,
Jakub
Czajka
d,
Przemysław J.
Malinowski
d,
Frank
Breher
b and
Ingo
Krossing
*a
a,
Manuel
Schmitt
a,
Wolfram
Feuerstein
b,
Ivo
Krummenacher
c,
Burkhard
Butschke
a,
Jakub
Czajka
d,
Przemysław J.
Malinowski
d,
Frank
Breher
b and
Ingo
Krossing
*a
aInstitut für Anorganische und Analytische Chemie, Freiburger Materialforschungszentrum (FMF), Universität Freiburg, Albertstr. 21, 79104 Freiburg, Germany. E-mail: krossing@uni-freiburg.de
bKarlsruhe Institute of Technology (KIT), Division Molecular Chemistry, Institute of Inorganic Chemistry, Engesserstr. 15, 76131 Karlsruhe, Germany
cInstitut für Anorganische Chemie II, Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
dCentre of New Technologies, University of Warsaw, Banacha 2c, 02-089 Warsaw, Poland
First published on 6th March 2020
Abstract
Oxidation of M(CO)6 (M = Cr, Mo, W) with the synergistic oxidative system Ag[WCA]/0.5 I2 yields the fully characterized metalloradical salts [M(CO)6]+˙[WCA]− (weakly coordinating anion WCA = [F-{Al(ORF)3}2]−, RF = C(CF3)3). The new metalloradical cations with M = Mo and W showcase a similar structural fluxionality as the previously reported [Cr(CO)6]+˙. Their reactivity increases from M = Cr < Mo < W and their syntheses allow for in-depth insights into the properties of the group 6 carbonyl triad. Furthermore, the reaction of NO+[WCA]− with neutral carbonyl complexes M(CO)6 gives access to the heteroleptic carbonyl/nitrosyl cations [M(CO)5(NO)]+ as salts of the WCA [Al(ORF)4]−, the first complete transition metal triad of their kind.
Introduction
Although personal and professional preferences do definitively differ, having a ‘chemical playground’ at hand and being able to explore fundamentally new compounds is one of the great appeals of chemical research. Especially so, when decade- or century-long questions and problems can be answered or solved. The peculiar compound family of carbonyl complexes is part of this century-long research: even about 130 years after the discovery of Ni(CO)4,1 new ‘milestones’ of homoleptic carbonyl complexes are still to be revealed today (Fig. 1).2 And after lying dormant for well over a decade, the synthesis of novel homoleptic transition metal carbonyl complexes has risen anew.From a historical point of view, it is not surprising that homoleptic carbonyl cations were the last family member to be discovered: ready availability of strong and innocent reductants as well as the strong π-back-bonding from electron-rich metal centers to the carbonyl ligands yield (relatively) robust carbonyl anions. In the case of [Fe(CO)4]2−, the synthesis even can be carried out in aqueous solutions.13,14 The cationic counterparts, however, feature weak(er) metal–CO bonds due to a generally poor π-back-bond caused by their electron deficiency and cationic charge,§15 resulting in (super-)electrophilic complexes. Both aspects, the weakly bound CO ligands as well as the reactivity towards most Lewis-bases, lead to an additional challenge for the syntheses of carbonyl cations. The strict absence of Lewis-basic functional groups or lone-pairs in starting materials or solvents and the use of weakly coordinating anions (WCAs) is crucial for a successful synthesis. The most promising approach in the past decades, which yielded the majority of the homoleptic carbonyl cations known today3–12 (Fig. 1), was the use of superacidic media such as SbF5, HF–SbF5 or HSO3F.4 However, the limiting factor was and still is the quality of the [SbF6]−/[Sb2F11]− anions as a WCA: if the carbonyl cation is electron deficient (e.g. does not fulfill the 18-electron rule) and is coordinatively unsaturated, the lone pair orbitals of the fluorine atoms can still be Lewis-basic enough to bind to the metal center. Especially for the group 6 carbonyl cations this is the case, as the tendency to an increased coordination number 7 for Mo and W led to a coordination polymer or fluorine-bridged adducts with the [SbF6]− anion with the respective metals in the oxidation state of +II (Fig. 2).16 Apparently, despite many synthesis attempts, the [SbF6]−/[Sb2F11]− system is not suitable for the synthesis of truly homoleptic group VI carbonyl cations.
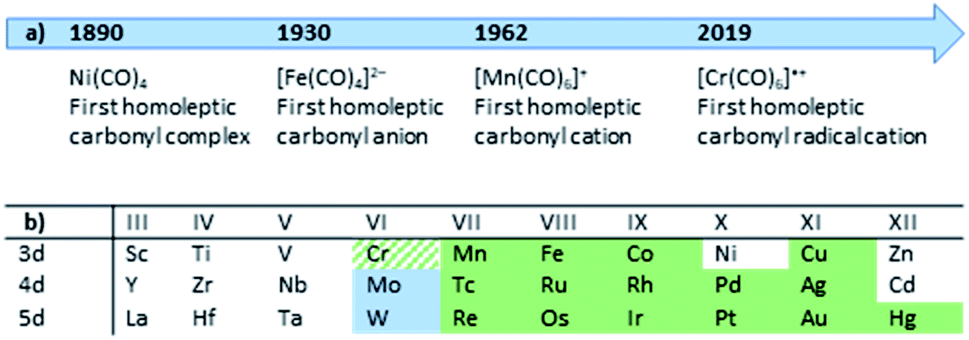 | ||
| Fig. 1 (a) Timeline of the milestones in the synthesis of homoleptic carbonyl complexes; (b) Overview to literature-known and fully characterized homoleptic carbonyl cations (green; our work: green-hatched)3–12 and this work (pale blue). | ||
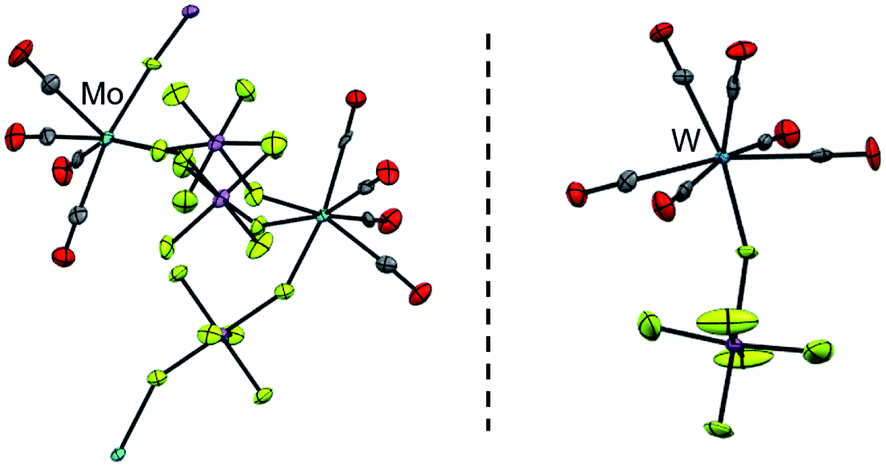 | ||
| Fig. 2 The cationic coordination polymer [{Mo(CO)4}2(cis-μ-F2SbF4)3]x+ (left, d(Mo–C) = 206.6(10) pm, d(C–O) = 111.3(11) pm and d(Mo–F) = 216.8(5) pm; av. = average values) and the [W(CO)6(FSbF5)]+ cation (right, d(W–C) = 209.3(8) pm, d(C–O) = 112.5(9) pm and d(W–F) = 210.9(5) pm; av.) known from the literature;17 the counterions were omitted for clarity. | ||
Our approach is an oxidative one starting from the commercially available neutral carbonyl complexes Mo(CO)6 and W(CO)6 and the Ag+/0.5 Halogen2 or [NO]+ oxidants as salts of the WCAs [Al(ORF)4]− and [F-{Al(ORF)3}2]− (RF = C(CF3)3). Both anions have proven their value in the stabilization of numerous reactive cations.18 The larger and even less coordinating [F-{Al(ORF)3}2]− anion has a higher stability towards strong electrophiles19 and was essential for the stabilization of [W(CO)6]+˙. Furthermore, we make use of standard Schlenk-techniques as well as easy-to-handle solvents and reagents, which should increase the accessibility to a broad scientific community.
Results and discussion
Previous findings on [Cr(CO)6]+˙
Recently, we published our discovery of the surprising formation of [Cr(CO)6][WCA] when Cr(CO)6 was oxidized with NO[WCA].2 Although thermodynamically favored by about 180 kJ mol−1, the heteroleptic substitution product [Cr(CO)5(NO)][WCA] is only quantitatively gained after one to two weeks of stirring in a closed vessel. The NO/CO exchange therefore is slow enough for the kinetic product [Cr(CO)6]+˙ to be selectively synthesized at −78 °C under removal of the evolving NO(g). By assuming an associative reaction mechanism, an energetically highly disfavored transition state with a coordination number of >6 for Cr may account for this. The [Cr(CO)6]+˙ radical cation features the same structural fluctuation as the isoelectronic vanadium hexacarbonyl V(CO)6: the D3d ground state with a low-lying (∼1 kJ mol−1 barrier) D2h transition state leads to a fluctuating structure even at low temperatures. This is reflected in a broad Eg CO vibration in Raman spectroscopy as well as a (pseudo-)isotropic EPR signal at 100 K. Only at 4 K, the D3d ground state manifests itself by a rhombic EPR spectrum. Now, the question arose, whether the synthetic methodology could be expanded to the heavier group 6 metal carbonyls of Mo and W, or if other oxidants than [NO]+ had to be used.[NO]+ as oxidant: ternary carbonyl/nitrosyl cations
Since the gas-phase ionization energies (IE) of the heavier homologues are similar to that of Cr(CO)6 (8.2 eV Cr(CO)6; 8.25 eV Mo(CO)6; 8.0 eV W(CO)6),20 the use of [NO]+ as oxidant (IE: 9.26 eV)21 was our starting point. However, already on physical contact of Mo(CO)6/W(CO)6 with the solid NO[Al(ORF)4], the distinctive reactivity of the heavier group 6 homologues became apparent. The orange substitution products [Mo(CO)5(NO)][Al(ORF)4] (1) or [W(CO)5(NO)][Al(ORF)4] (2) were immediately generated, even without solvent or also in inert solvents (such as perfluorohexane, C6F14) at −78 °C in a dynamic vacuum. This observation is in accordance with our above-mentioned assumption of the kinetic hindrance of Cr(CO)6 towards a preferred associative CO/NO substitution mechanism via a seven-coordinate transition state.¶ Because of the ready tendency of Mo and W to adopt a coordination number of 7, it was not surprising that the reaction to the mere oxidation products [Mo/W(CO)6]+˙ was not observed. Instead, the CO/NO exchange is inevitable and only the heteroleptic carbonyl/nitrosyl cation salts 1 and 2 were obtained according to eqn (1). | (1) |
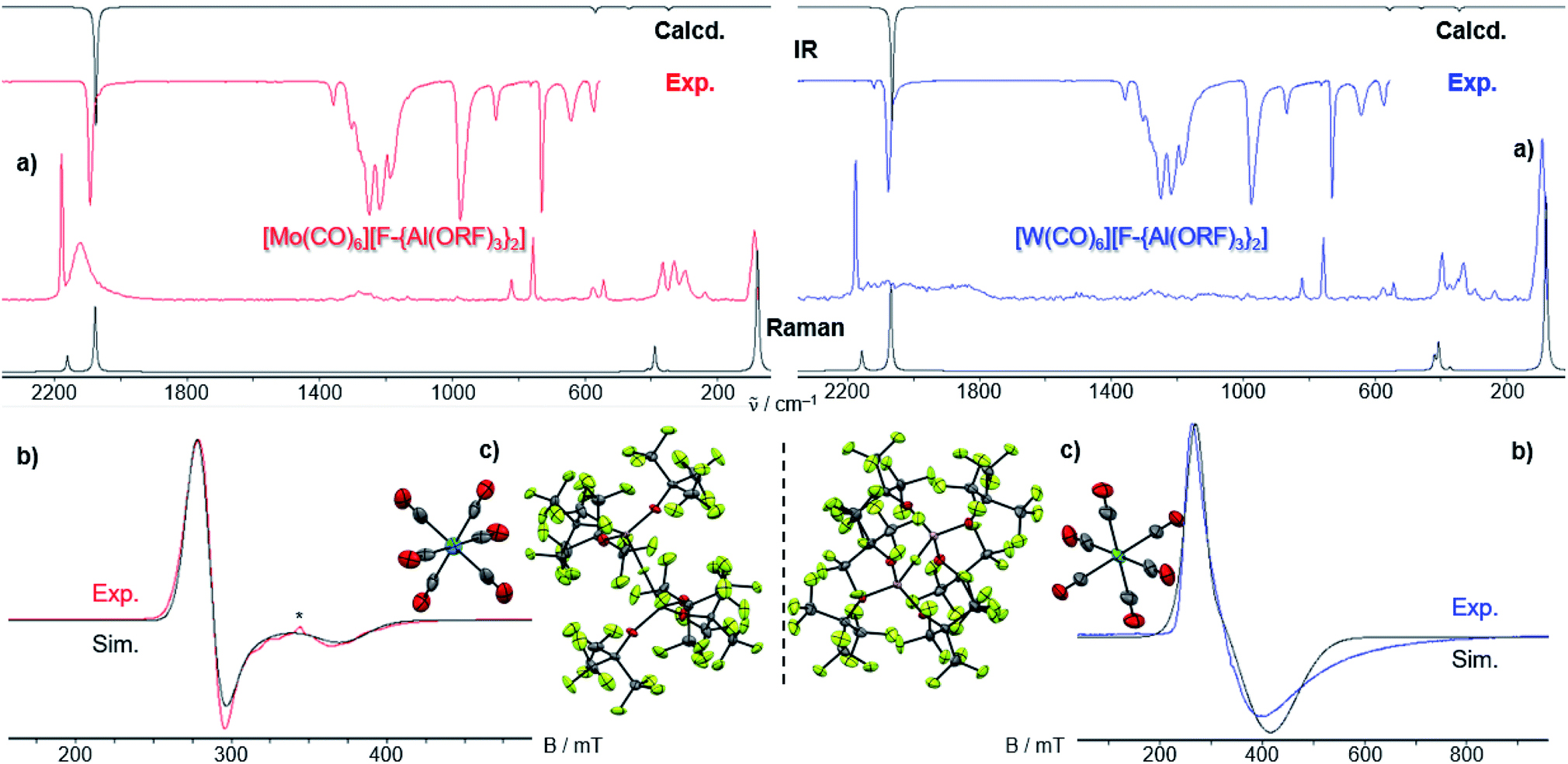
By vapor diffusion of n-pentane into an ortho-difluorobenzene (oDFB) solution of 1 and 2, the complexes can be crystallized in yields of around 90%.‡ They are isostructural to their lighter homologue [Cr(CO)5(NO)][Al(ORF)4] and the cations feature the same undistorted local C4v symmetry in the solid state. This reflects in good agreement of the experimental vibrational spectra with the simulation from the respective calculated gas-phase cations (Fig. 3).
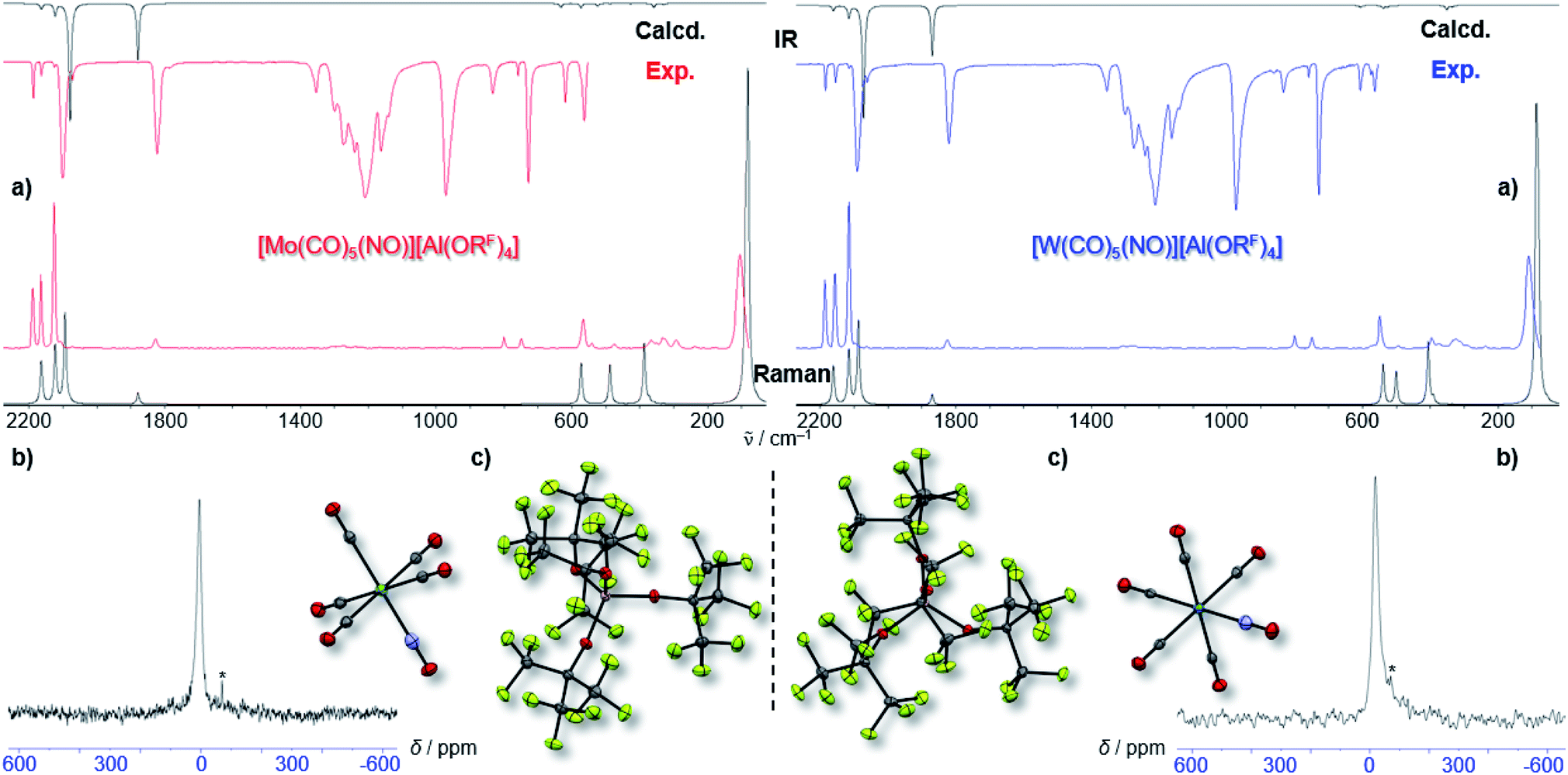 | ||
| Fig. 3 (a) Experimental (Exp., red or blue respectively) and calculated (calcd., black, C4v symmetry @BP86def2/TZVPP-D3BJ, no scaling factor was applied) vibrational spectra of 1 and 2; (b) 14N-NMR-spectra (oDFB, rt, 28.92/21.69 MHz); * signal from N2 atmosphere; (c) crystal structures of 1 (P4/n, R1 = 2.3%, wR2 = 5.7%) and 2 (P4/n, R1 = 2.4%, wR2 = 7.4%) – note that the NO ligand is crystallographically indistinguishable from CO and is only colorized for visual purposes. | ||
Fully characterized ternary transition metal carbonyl/nitrosyl cations are surprisingly scarce in literature.22 The only example, apart from [Cr(CO)5(NO)]+ recently reported by us,2 is the [Co(CO)2(NO)2]+ cation reported in 2006.23 For structurally characterized ternary Mo and W carbonyl/nitrosyls, no entry is found in the CCDC to date (neither anionic, neutral, nor cationic).||
The synergistic oxidative system Ag+/0.5 I2 and its complications
Obviously, a different oxidant than [NO]+ was necessary here. It should be noted as a side that the reaction of [Cr(CO)6][Al(ORF)4] with the neutral W(CO)6 in TFB (=1,2,3,4-tetrafluorobenzene) did not yield any apparent reaction (e.g. color change of the homogenous solution) over the course of several weeks, despite the about 0.2 eV lower IE of the tungsten compound. Only [Cr(CO)6][Al(ORF)4] was visible after work up by IR spectroscopy. Note that related studies with the isoelectronic V(CO)6 showed its capability to oxidize carbonyl anions such as [Mn(CO)5]− and [Co(CO)4]− to the respective neutral carbonyls and [V(CO)6]−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 24 or Nb(mes)2 to [Nb(mes)2(CO)][V(CO)6] (mes: C9H12).25
24 or Nb(mes)2 to [Nb(mes)2(CO)][V(CO)6] (mes: C9H12).25
Since also Ag[WCA] showed no reaction with Cr(CO)6, the additional use of halogens seemed promising. The resulting synergistic Ag+/0.5 Hal2 (Hal = Cl2, Br2, I2) system is known to be very strongly oxidizing26,27 and the respective silver halides should simply precipitate after the oxidation took place as in eqn (2).
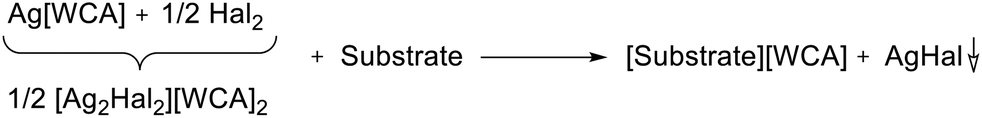 | (2) |
In terms of reactivity, it did not matter, whether the (Ag+)2/Hal2 complex was formed in situ or was isolated prior to use – indicating that the same active species is present in solution. The exact 2:1 stoichiometry of 2Ag+:Hal2 is crucial to prevent any excess halogen from reacting with the desired products. In terms of practicability, this meant that the heavy diiodine was the dihalogen of choice, since (a) [Ag2I2][Al(ORF)4]2, shown in Fig. 4, is the only one of the Cl2/Br2/I2 triad to be stable enough to be isolated and stored as starting material and (b) the in situ generation of 2 Ag+/Hal2 with an exact 2:1 stoichiometry is more tedious (to say the least) with small amounts of gaseous Cl2 and volatile Br2.** Furthermore, it should be noted that already†† the 2 Ag+/I2 mixture is strongly oxidizing enough to react with solvents up to an IE of 11.4 eV such as oDFB or CH2Cl2, as indicated by the immediate precipitation of yellow AgI.27
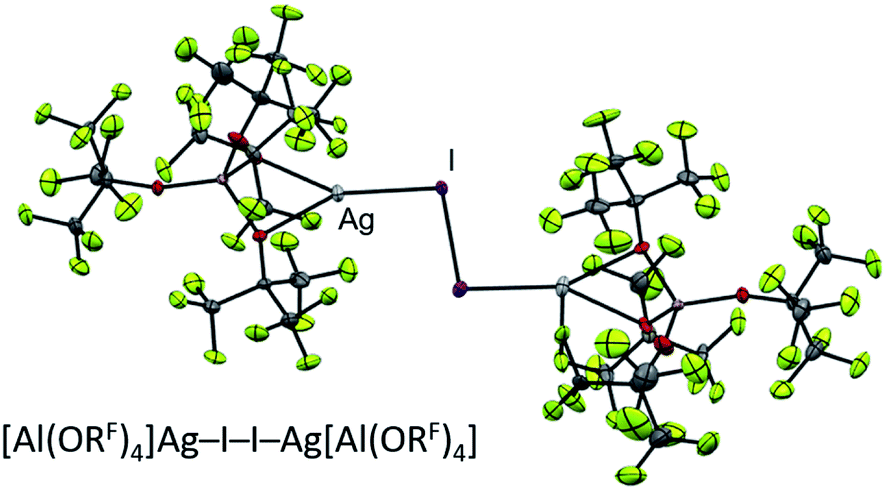 | ||
| Fig. 4 Molecular structure of the known27 [Ag2I2][Al(ORF)4]2 oxidant. | ||
Impact of anion and solvents
The limitations of the [Al(ORF)4]− anion became clear when we switched from Cr(CO)6 to the Mo(CO)6 system. Ag[Al(ORF)4]/0.5 I2 reacted with Cr(CO)6 according to eqn (3) cleanly and quantitatively to [Cr(CO)6][Al(ORF)4] and AgI precipitate in any solvent that we used (SO2, oDFB, C6F14). Thus, this new route is the most convenient and preferred one for its synthesis.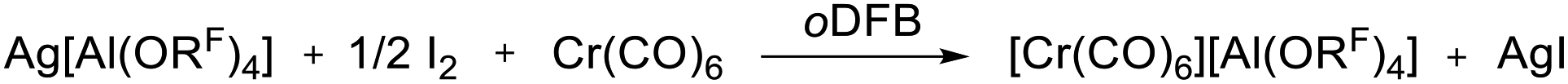 | (3) |
However, the accessibility of the coordination number 7 for Mo became evident: when applying the same reaction conditions to Mo(CO)6, always an inseparable orange mixture of the as [Al(ORF)4]− salts co-crystallizing cations [Mo(CO)6]+˙ and [Mo2(CO)8I3]+ was obtained. Leftover Ag[Al(ORF)4] contaminated the product and proved difficult to be separated by crystallization (Scheme 1). It soon became apparent that the solvent played a crucial role. In ‘well-dissolved’ systems (such as CH2Cl2, oDFB, TFB or SO2), the formation of different Mo-iodide species could never be satisfyingly suppressed. In low polarity inert solvents (perfluorohexane C6F14 or perfluorobenzene C6F6), the reaction mixture was a suspension,‡‡ which induced long reaction times (e.g. several days in C6F14), but at the same time also little or no iodide-containing side products (vide infra). In C6F6, an inseparable mixture of [Mo(CO)6][Al(ORF)4], [Mo(CO)6]/[Mo2(CO)8I3][Al(ORF)4]2 and [{Mo(CO)4I}2][F-{Al(ORF)3}2] was gained upon crystallization. The presence of [F-{Al(ORF)3}2]− is proof for the decomposition of the [Al(ORF)4]− anion,§§ and demonstrated the limitation of the [Al(ORF)4]− WCA in these systems. However, when the reaction of Ag[Al(ORF)4]/0.5 I2 with Mo(CO)6 was carried out in perfluorohexane, the desired [Mo(CO)6][Al(ORF)4] salt could be obtained as the main product with only minor impurities. The iodo-bridged and in other solvents co-crystallizing [Mo2(CO)8I3]+ species was not observed (Scheme 1). Conveniently, if the more stable Ag[F-{Al(ORF)3}2] salt was used from the beginning, [Mo(CO)6][F-{Al(ORF)3}2] was formed as the main product independently from the solvent, even though inseparable side products are still problematic and often appeared as additional precipitate upon crystallization of the filtered reaction mixture.
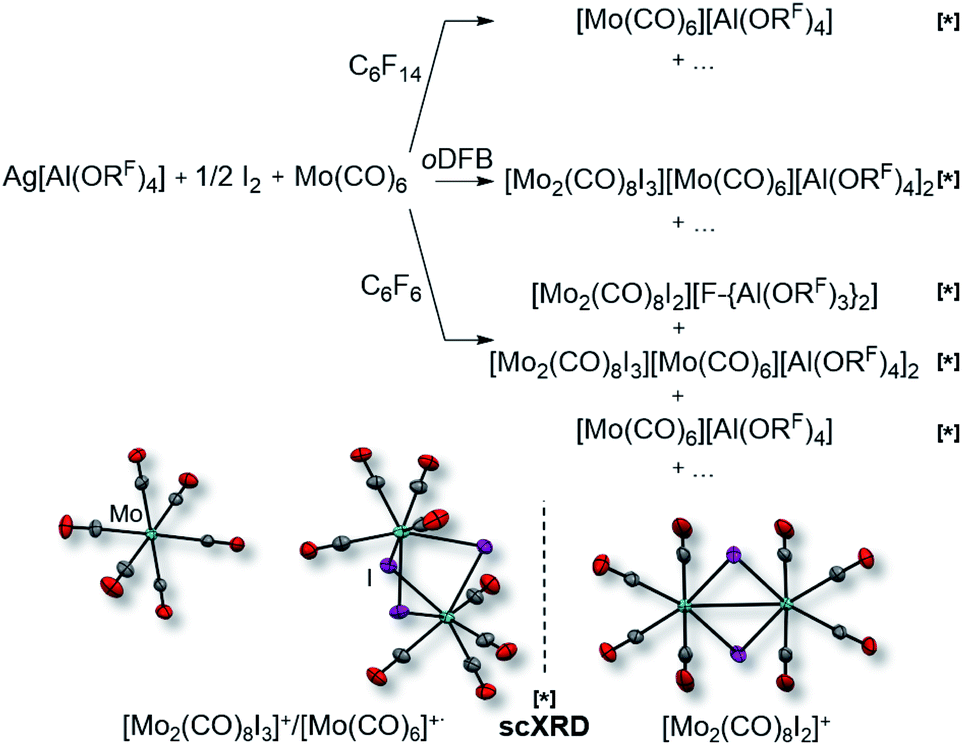 | ||
| Scheme 1 Impact of the solvent on the formation of [Mo(CO)6]+˙; bottom: molecular structures of [Mo(CO)6]+˙ (d(Mo–C) = 211.2(3) pm, d(C–O) = 111.8(4) pm)/[Mo2(CO)8I3]+ (d(Mo–Mo) = 355.0(1) pm), d(Mo–C) = 203.7(3) pm, d(C–O) = 112.5(4) pm, d(Mo–I) = 287.1(3) pm; av.) co-crystals and [Mo2(CO)8I2]+ (d(Mo–Mo) = 312.4(1) pm, d(Mo–C) = 208.0(9) pm, d(C–O) = 112.5(10) pm, d(Mo–I) = 271.5(1) pm; av.), the anions were omitted for clarity; all complexes marked with [*] were identified by single-crystal XRD (scXRD); thermal ellipsoids were drawn at 50% probability level; note that the “+…” stands for additional (unidentified) side-products; AgI was formed in all reactions, leftover Ag[Al(ORF)4] was also commonly observed, albeit not for the best reaction in C6F14. | ||
It became more and more apparent that the synthesis would be even more fickle for the heavier homologue W(CO)6. The first initial experiments soon showed that [F-{Al(ORF)3}2]− had to be the anion of choice. Depending on the solvent (oDFB, C6F6), either only seven-coordinate [W(CO)6I][Al(ORF)4] or decomposition products of the [Al(ORF)4]− ion such as [W(CO)6(ORF)][F-{Al(ORF)3}2], [W(CO)4(ORF)2][F-{Al(ORF)3}2] and a grey precipitate were obtained (Scheme 2). All of those include W in oxidation state +II or +III. However, the last reaction also showed traces of the desired [W(CO)6][F-{Al(ORF)3}2], which confirmed our assumptions that the highly reactive [W(CO)6]+˙ would only be compatible with the more robust [F-{Al(ORF)3}2]− anion.
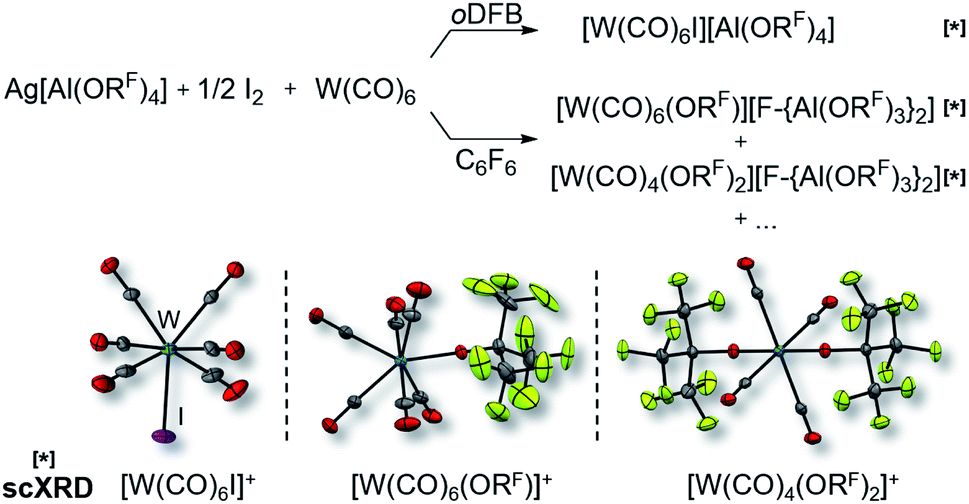 | ||
| Scheme 2 Impact of the solvent on the formation of the undesired (side-)products [W(CO)6I]+ (d(W–C) = 209.3(7) pm, d(C–O) = 112.5(8) pm, d(W–I) = 276.7(1) pm), [W(CO)6(ORF)]+ (d(W–C) = 210.3(3) pm, d(C–O) = 113.5(3) pm, d(W–O) = 201.4(2) pm) and [W(CO)4(ORF)2]+ (d(W–C) = 214.5(3) pm, d(C–O) = 111.4(3) pm, d(W–O) = 189.6(2) pm; av.) as well as their molecular structures (anions are omitted for clarity); all complexes marked with [*] were identified by single-crystal XRD (scXRD); thermal ellipsoids were drawn at 50% probability level; note that the “+…” stands for additional (unidentified) side-products; AgI was formed in all reactions, which turned to a grey precipitate after some time. | ||
In order to evaluate the role of the halogen in the Ag+/0.5 Hal2 oxidant and its ability to coordinate to the metal center, we reacted Ag[F-{Al(ORF)3}2]/0.5 Cl2 with W(CO)6 in perfluorohexane. Solely the chlorido-bridged tungsten(II) salt [W2(CO)8Cl3][F-{Al(ORF)3}2] could be isolated and crystallized (Scheme 3), underlining the importance of I2 as an oxidant – especially since with gaseous Cl2 and liquid Br2 the spatial separation of dihalogen and the volatile carbonyl substrate is difficult to realize in the reaction vessel. Thus, it is not clear, if the neutral carbonyl already reacts with the dihalogen without presence of the silver salt.
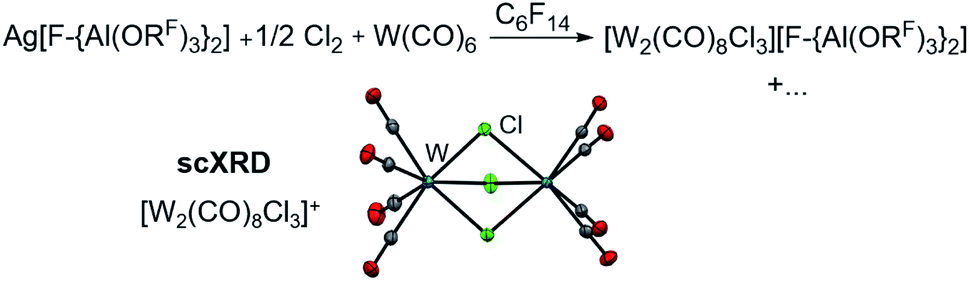 | ||
| Scheme 3 Use of Cl2 as oxidant and the molecular structure of the [W2(CO)8Cl3]+ cation (d(W–W) = 350.7(1) pm, d(W–C) = 203.1(3) pm, d(C–O) = 113.0(3) pm, d(W–Cl) = 251.1(3) pm, av.; anions are omitted for clarity) identified by single-crystal XRD. Note that the “+…” stands for additional (unidentified) side-products. | ||
The conclusions from these insights were that (a) [F-{Al(ORF)3}2]− was our anion of choice, (b) iodine was the most viable halogen and (c) the reaction was best carried out in an inert solvent. We believe that the solvation of the molecular complex [WCA]Ag–I–I–Ag[WCA] (Fig. 4), (“[Ag2I2][WCA]2”) plays a crucial role in the reaction behavior: ideally, to ensure a mere oxidation, an intact Ag2I2-moiety is necessary. This is only the case for an inert solvent (such as C6F14 or 1,2,3,4-tetrafluorobenzene,¶¶ see eqn (4a)), since coordinating solvents (such as oDFB) solvate Ag+ and therefore promote an asymmetric dissociation of the [Ag2I2][WCA]2 complex. The resulting molecular [WCA]Ag–I–I species, which was crystallographically characterized27 with the WCA [Al(ORF)4]−, can then act as an I+ source upon formation of AgI, which eventually leads to the mixed carbonyl/halide side-products as in eqn (4b). We believe this behavior to be similar for both WCAs [Al(ORF)4]− and [F-{Al(ORF)3}2].
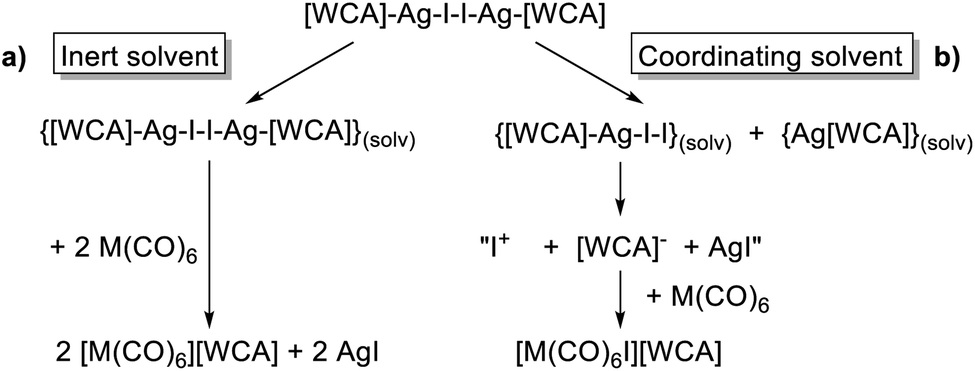 | (4) |
When carrying out the reaction in C6F14, in order to separate the desired products from the precipitated AgI, the crude reaction mixture needs to be dissolved, extracted and then crystallized for purification. The dissolution in oDFB or TFB then often led to precipitation of grey solids, presumably due to incomplete reactions even after numerous days, which also complicated reproducibility.
Best reaction conditions towards [M(CO)6]+˙
A number of diverse reaction conditions led us to the conclusion that the best oxidizer is [Ag2I2][F-{Al(ORF)3}2]2 in the solvent TFB, since especially [W(CO)6]+˙ is not stable in oDFB. Furthermore, all manipulations in solution were carried out at lower temperatures (∼0 °C) due to the temperature-sensitive nature of [W(CO)6]+˙ and the side-products. This led to the curious point, where crystallization at room temperature was actually beneficial, due to the decomposition and precipitation of undesired by-products, which otherwise would have contaminated the crystalline [W(CO)6][F-{Al(ORF)3}2]. Overall, the best procedure was to stir the crude reaction mixture over night thawing from 0 °C to room temperature (RT), after which the color of the suspension changed from green to grey and after filtration, a clear yellow TFB solution was obtained in the W(CO)6 case given in eqn (5). Unfortunately, this precipitation of undesired side products also led to a partial decomposition even of the robust [F-{Al(ORF)3}2]− anion and the reproducible formation of [W(CO)6(ORF)][F-{Al(ORF)3}2] was observed as a side product. Again, this was difficult to separate from [W(CO)6][F-{Al(ORF)3}2] by crystallization – especially, since both species are of similar yellow color and equally soluble in TFB. Careful manual sorting of the two crystalline species is necessary, leading to a quite low total yield (35%) for M = W. However, the procedure for M = Mo given in eqn (5) yields a rather clean product at 78% yield.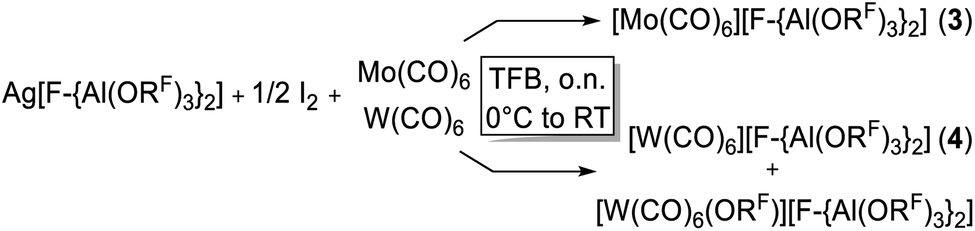 | (5) |
Both [F-{Al(ORF)3}2]− salts 3 and 4 crystallize isostructural to the [Cr(CO)6]+˙ analogue in the cubic space group Pa![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) (Fig. 5). The M atoms of the [M(CO)6]+˙ cation reside on a −3 position and feature only one symmetry-independent CO ligand. They exhibit crystallographic D3d symmetry – as in the undistorted gas phase and thus underlining the claim of the [F-{Al(ORF)3}2]− anion being “least-coordinating” and providing exceptional pseudo gas phase conditions.18
(Fig. 5). The M atoms of the [M(CO)6]+˙ cation reside on a −3 position and feature only one symmetry-independent CO ligand. They exhibit crystallographic D3d symmetry – as in the undistorted gas phase and thus underlining the claim of the [F-{Al(ORF)3}2]− anion being “least-coordinating” and providing exceptional pseudo gas phase conditions.18
 | ||
| Fig. 5 (a) Experimental (Exp., red or blue respectively) and calculated (calcd., black, D3d symmetry @BP86def2/TZVPP-D3BJ, no scaling factor was applied) vibrational spectra of 3 and 4, the additional small band in the IR of [W(CO)6][F-{Al(ORF)3}2] at 2117 cm−1 is either the Raman-active Eg vibration or a contamination which coincidentally vibrates at that same wavenumber; (b) experimental (4 K, red/blue, Exp.) and simulated (Sim., black) EPR-spectra of 3 and 4, the * marks an unknown impurity; (c) crystal structures of 3 (Pa, R1 = 5.4, wR2 = 15.5%) and 4 (Pa, R1 = 7.6%, wR2 = 14.7%), thermal ellipsoids were drawn at 50% probability level. | ||
In summary, a completely satisfying route to the heavier group VI carbonyl cations is still to be found. By principle, our system allows access to and first insights of the properties of these compounds. However, the limitations of the synergistic 2 Ag[WCA]/Hal2 oxidant are also clear: if the coordination number of seven is somewhat easily attainable, coordination of a halide to the Lewis acidic metal center is always problematic.
It also has to be noted here that all the identified and structurally characterized side-products were previously unknown from the literature and should – in principle – be accessible selectively. However, an extensive study of the novel mixed group 6 carbonyl/halide cations was not part of our project and would by far exceed the scope of this report.
Comparison of the [M(CO)6]+˙ and [M(CO)5(NO)]+ triad (M = Cr, Mo, W)
For the first time, it is possible to study a complete isostructural triad of homoleptic radical carbonyl and heteroleptic carbonyl/nitrosyl cations for their properties.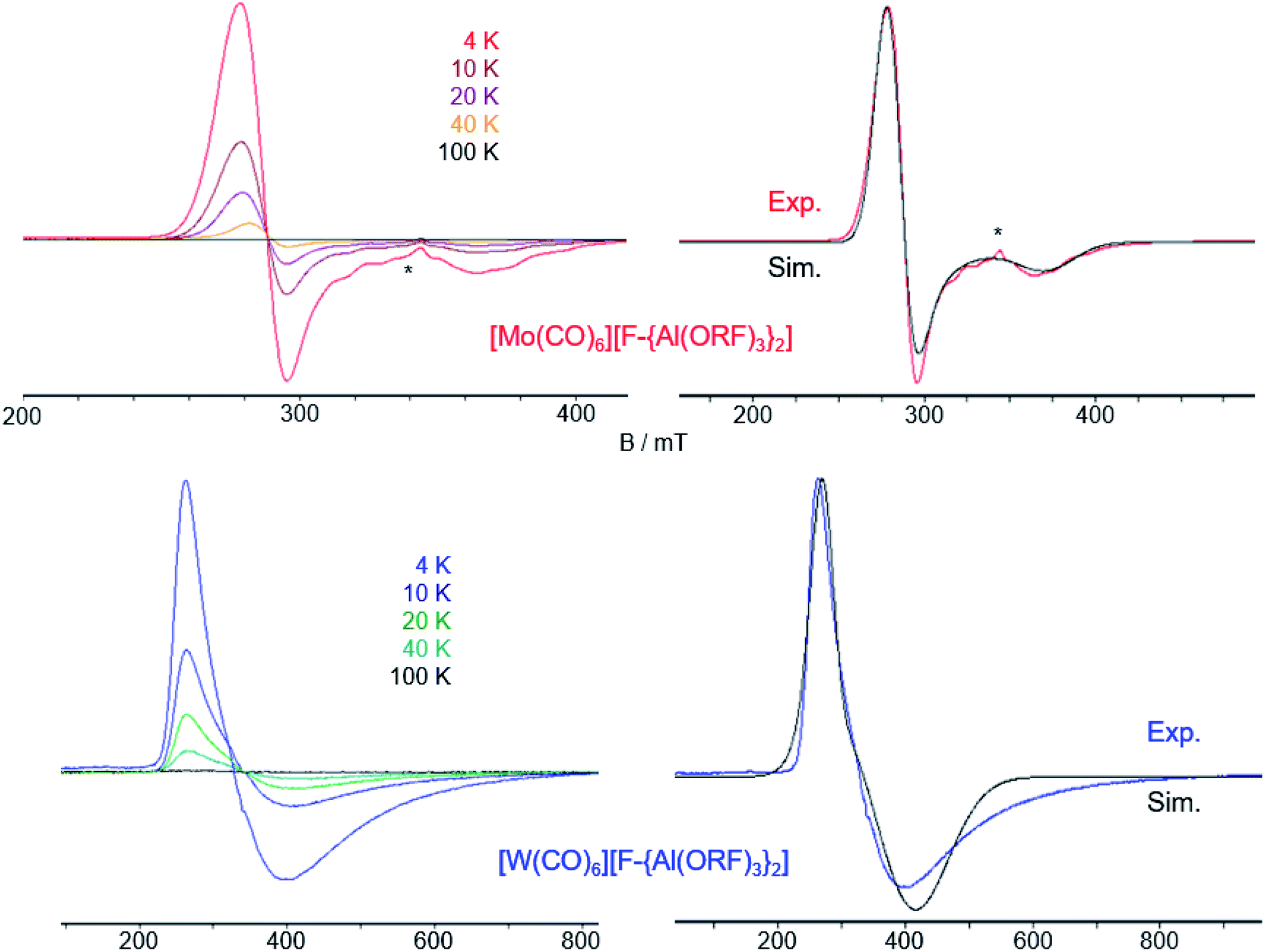 | ||
| Fig. 6 Temperature dependence of the CW X-band EPR spectra of 3 (top left) and 4 (bottom left) in frozen TFB. Experimental and simulated EPR spectra of 3 (top right) and 4 (bottom right) at 4 K. Simulation parameters for 3: g⊥ = 2.374, g∥ = 1.800; simulation parameters for 4: g⊥ = 1.722, g∥ = 2.427. The asterisk (*) denotes an impurity (see ESI Fig. 22‡). | ||
As for the [Cr(CO)6]+˙ [WCA]− spectra, at higher temperatures (100 K) we detected only weak (pseudo)isotropic signals indicating a fluctuating structure of the radical cations. At lower temperatures we could clearly detect an anisotropic signature of the EPR signals of both radical cations in agreement with an axial Jahn–Teller distortion of the d5 complexes as it was already seen in the crystal structure of 3 and 4 and described for the isoelectronic [Cr(CO)6]+˙ and V(CO)6˙.30 In contrast to [Cr(CO)6]+˙ and [Mo(CO)6]+˙, where g⊥ > g∥, for [W(CO)6]+˙ it is opposed, i.e. g⊥ < g∥. Reports on the EPR spectra of isoelectronic Nb(CO)6˙ and Ta(CO)6˙ generated in CO matrix at 2 K31 reveal that these complexes also adopt a linear distorted octahedral structure, however, their specific molecular symmetry could not be determined yet. DFT studies32 suggest the neutral Ta(CO)6˙ radical should not exist as a monomer but form CO-bridged dimeric structures. Interestingly, the hypothetic Ta(CO)6˙ monomer is predicted to take a C2h symmetric structure in contrast to the D3d structure found for [Mo(CO)6]+˙ and [W(CO)6]+˙, which is not even a local minimum structure in the case of Ta(CO)6˙.
We performed SA–CAS–SCF calculations33 on the isolated radical cations both in D3d and D4h symmetry and compared the obtained g values with the experimentally determined ones (Table 1). Although there are deviations between calculated and experimental values, the results show that the axial EPR signature of the radical cation [Mo(CO)6]+˙ is rooted in a D3d symmetric ground state analogously to [Cr(CO)6]+˙. For [Mo(CO)6]+˙, our calculations predict a smaller g-anisotropy of the hypothetic D4h symmetric state compared to the D3d symmetric one, whilst the opposite is true for [Cr(CO)6]+˙.
| [Cr(CO)6]+˙ | [Mo(CO)6]+˙ | [W(CO)6]+˙ | |||||
|---|---|---|---|---|---|---|---|
| D 3d | D 4h | D 3d | D 4h | D 3d | D 4h | ||
| a For details of the g-value calculations please refer to theESI. Values for [Cr(CO)6]+˙ taken from ref. 2. b Electronic energies (in cm−1, in brackets: kJ mol−1) were calculated at the DLPNO-CCSD(T)/def2-TZVPP level of theory using structures optimized with TPSSh-D3BJ/def2-TZVPP. c g iso = (2g⊥ + g∥)/3. d g iso = (g⊥ + 2g∥)/3. e The better accordance of the isotropic g-value of D4h with experiment must not be misinterpreted: deviations in the calculation of the perpendicular component g⊥ enter twofold. | |||||||
| ΔEb | 0.00 | 136 (1.6) | 0.00 | 204 (2.4) | 0.00 | 710 (8.5) | |
| g ⊥ | Exp. | 2.185 | 2.374 | 1.722 | |||
| Calc. | 2.173 | 2.434 | 2.408 | 2.216 | 2.701 | 2.164 | |
| g ∥ | Exp. | 1.947 | 1.800 | 2.427 | |||
| Calc. | 1.971 | 1.761 | 1.848 | 1.956 | 0.977 | 1.212 | |
| g iso | Exp. | 2.106c | 2.058c | 2.192d | |||
| Calc.e | 2.106c | 2.210c | 2.221c | 2.129c | 2.126 | 1.847 | |
For [W(CO)6]+˙, the calculations predict the g⊥ > g∥ like for the other cations, however, we were not able to get an acceptable fit of the EPR spectrum of 4 using this g factor ratio. Instead, only a ratio g⊥ < g∥ led to reasonable accordance of experimental and simulated EPR spectra. We were not able to uncover the underlying reasons for this difficult to calculate heavy element and avoid speculative proposals.
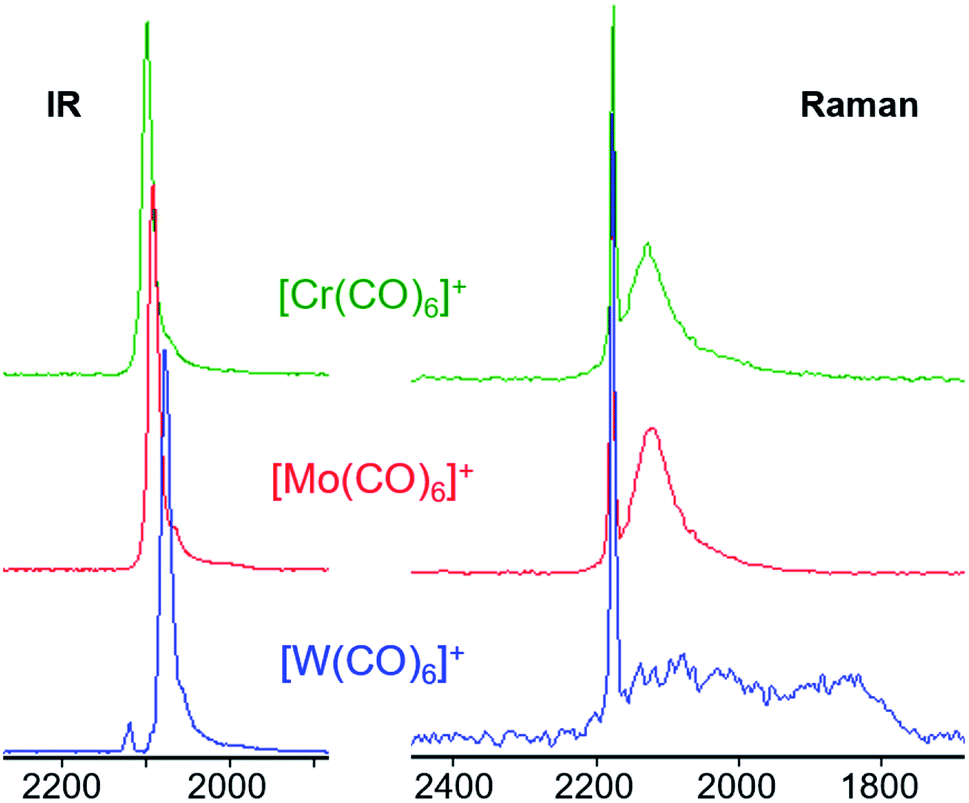 | ||
| Fig. 7 Comparison of the IR (left) and Raman (right) spectra of the [M(CO)6][F-{Al(ORF)3}2] triad. All three Raman spectra feature the same sharp A1g CO vibration at 2173 cm−1 and a broad Eg vibration indicating a fluctuating structure, even more so for [W(CO)6]+˙ where two very broad bands spanning over more than 300 cm−1 are visible between 1800 and almost 2200 cm−1. | ||
| [Cr(CO)6]+˙ | [Mo(CO)6]+˙ | [W(CO)6]+˙ | Cr(CO)6 | Mo(CO)6 | W(CO)6 | |
|---|---|---|---|---|---|---|
| a IR: overlap of A2u and Eu CO vibration. b Fluoride ion affinities FIAs referenced to [Si(CH3)3]+ = 958 kJ mol−1.39 | ||||||
| IR ν(CO)a/cm−1 A2u/Eu | 2096 | 2089 | 2075 | |||
| DFT ν(CO)a/cm−1 (102 N m−1) A2u/Eu | 2081(33.78) | 2072(33.78) | 2062(33.46) | |||
| Raman ν(CO)/cm−1 A1g | 2173 | 2173 | 2173 | 2110 | 2115 | 2115 |
| DFT ν(CO)/cm−1 (102 N m−1) A1g | 2158(36.39) | 2158(36.33) | 2155(36.18) | 2104(34.50) | 2105(34.51) | 2104(34.41) |
| DFT ν(M–C)/cm−1 (102 N m−1) A1g | 364(1.07) | 385(1.20) | 403(1.31) | 406(1.34) | 418(1.43) | 435(1.56) |
| XRD d(M–C)/pm | 198.2(2) | 210.5(7) | 210.5(9) | 191.5(1)36 | 205.9(4)37 | 204.9(5)38 |
| XRD d(C–O)/pm | 112.2(2) | 112.0(8) | 110.8(11) | 114.2(1)36 | 113.4(6)37 | 113.6(9)38 |
| DFT d(M–C)/pm | 197.5 | 209.9 | 210.4 | 190.2 | 205.5 | 206.7 |
| DFT d(C–O)/pm | 113.9 | 113.9 | 114.1 | 115.2 | 115.2 | 115.3 |
| FIAb/kJ mol−1 | 711 | 715 | 741 | |||
The broad Eg Raman-active vibration at around 2125 cm−1 indicates the same structural properties for the heavier homologue Mo as already shown for the [Cr(CO)6]+˙ or V(CO)6˙ cases. At room temperature, a very low-lying D2h symmetric transition state (M = Cr, Mo) or intermediate (M = W) probably allows for equilibration of the two different D3d symmetric ground states (see Scheme 4 and S.I. Section 4‡ for an in-depth discussion) – even on the fast time scale of vibrational spectroscopy.
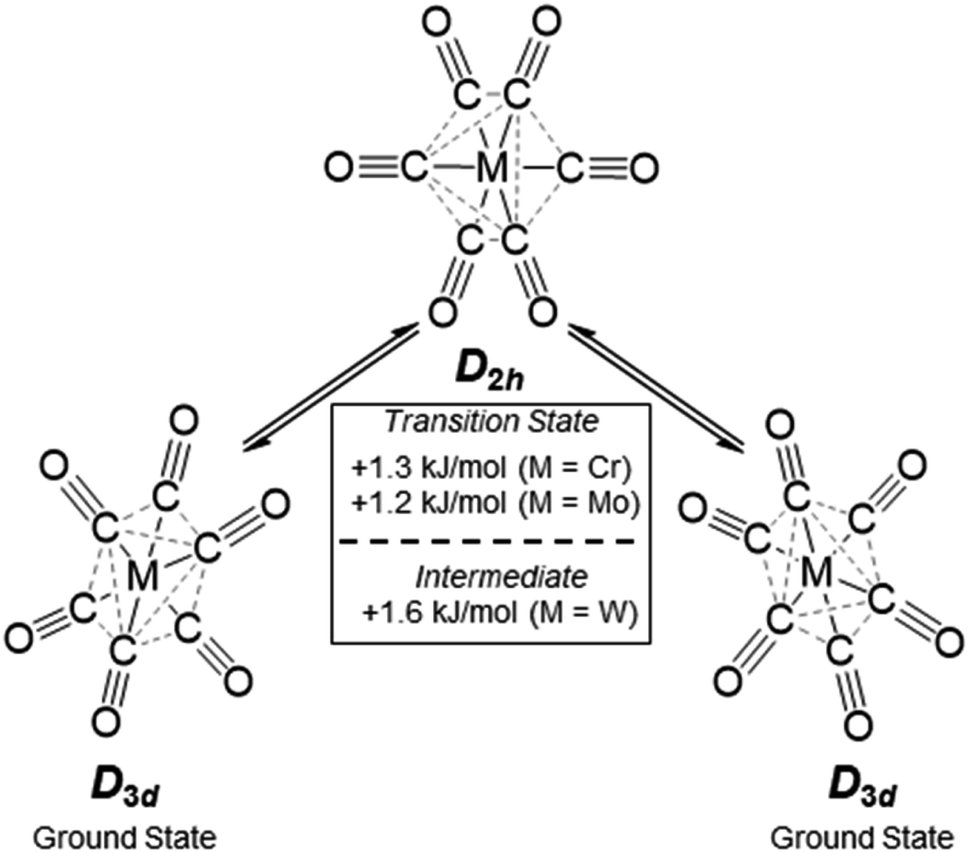 | ||
| Scheme 4 Structural fluctuation of the [Cr/Mo/W(CO)6]+˙ triad, energy differences from single point calculations at DPLNO-CCSD(T)/def2-TZVPP level of theory on TPSSH-D3BJ/def2-TZVPP structures. | ||
This does not affect the all-symmetric A1g CO stretch. A very broad band, spanning over 100 cm−1, is the result. [W(CO)6]+˙ shows two very broad bands which cover over 300 cm−1 in its Raman spectrum, reaching as far down as 1800 cm−1, which is in the range of bridging carbonyl ligands. However, all spectra were measured from crystalline complexes [M(CO)6][F-{Al(ORF)3}2], so an intermolecular exchange is impossible – whereas in solution a dimerization-equilibrium of [W(CO)6]+˙ and [W2(CO)12]2+ with bridging μ-CO entities could be imaginable, but appears unlikely based on orienting DFT calculations.
The calculated force constants imply an increase in the M–C bond strength as well as a decrease in the C–O bond strengths from Cr to W (Table 2). The dicationic group 7 triad [M′(CO)6]2+ (M′ = Fe, Ru, Os) features the same trend in their CO force constants.||||35 For the all-symmetric A1g stretching vibration, the force constants are in contrast to the observed Raman values for the group 6 triad, be it the neutral or cationic carbonyls. This might indicate a trend in the ability for π-back donation that does not reflect in the CO vibration. Yet, more data points are required to allow for a proper interpretation of these observations.
For the heteroleptic nitrosyl complexes, the two different coupled M–N/M–C A1 vibrations do not follow a clear trend (Table 3). The CO/NO vibrations, however, decrease just as the shifts in 13C NMR (202/187; 193/180; 186/180 ppm) and 14N NMR (17; 3; −15 ppm) do (Fig. 8, Table 3 and S.I‡). The same trend in 13C NMR shifts is seen for the M(CO)6 ‘parent’ compounds (212; 204; 192 ppm).40 According to a previous DFT study on homoleptic hexacarbonyls,41 within a triad, the chemical shift is decreasing as σ-donation becomes more important for the 5d homologues. This might also be the case for the [M(CO)5(NO)]+ cations, although we tend to interpret this with caution: more recent studies show no direct relation between NMR chemical shifts and carbon charges for selected transition metal alkylidene complexes43 – further analysis is needed here.
| [Cr(CO)5(NO)]+ | [Mo(CO)5(NO)]+ | [W(CO)5(NO)]+ | |
|---|---|---|---|
| a Only the most intense CO vibration (E) is shown, force constants (in brackets) were calculated with Gaussian. b In oDFB solution, 298 K, CH3NO2 as reference. c Average of all M–C/N and C/N–O bonds due to the indistinguishable NO position in the experimental data. | |||
| IR ν(CO)a/cm−1 E | 2108 | 2099 | 2090 |
| DFT ν(CO)a/cm−1 (102 N m−1) E | 2085(34.16) | 2078(33.98) | 2070(33.71) |
| IR ν(NO)/cm−1 A1 | 1841 | 1820 | 1818 |
| DFT ν(NO)/cm−1 (102 N m−1) A1 | 1899(31.22) | 1877(30.42) | 1868(30.09) |
| DFT ν(M–N)/cm−1 (102 N m−1) A1 | 667(5.55) | 570(3.48) | 537(2.73) |
| 506(2.03) | 485(1.87) | 498(1.96) | |
| NMR (14N) δb/ppm | 17 | 3 | −15 |
| DFT (14N) δb/ppm | 35 | 43 | −30 |
| XRD d(M–C)c/pm | 195.6(4) | 208.4(3) | 207.0(6) |
| XRD d(C–O)c/pm | 112.6(5) | 112.5(3) | 113.0(8) |
| DFT d(M–C)c/pm | 193.0 | 208.3 | 207.5 |
| DFT d(C–O)c/pm | 113.9 | 114.1 | 114.2 |
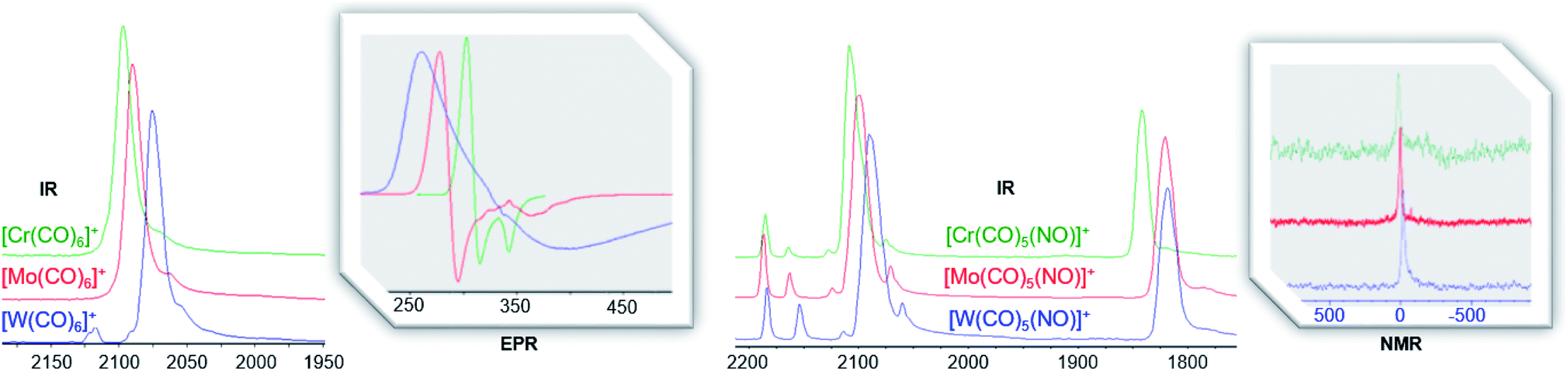 | ||
| Fig. 8 Comparison of IR (CO and NO region respectively) and 14N NMR (298 K, oDFB, ref. CH3NO2)/EPR (4 K, oDFB/TFB solution) spectra of the group 6 triad as homoleptic carbonyl cation [F-{Al(ORF)3}2]− salts (left) and heteroleptic carbonyl/nitrosyl cation [Al(ORF)4]− salts (right). | ||
| Reaction | M |
|
|
|||
|---|---|---|---|---|---|---|
| BP86 | B3-LYP | ε = 8.93 | ε = 13.8 | |||
| a The F− is weakly bound to the metal (Cr–F distance: 260 pm) – in the actual minimum structure (Δ ≈−30 kJ mol−1), the F− is bound to the CO as an acyl fluoride. b COSMO solution thermodynamics in CH2Cl2 (ε = 8.93) and oDFB (ε = 13.8) with the BP86 functional. c FIA referenced to [Si(CH3)3]+ (gas-phase FIA = 958 kJ mol−1, solution FIA = 404 (CH2Cl2)/370 (oDFB) kJ mol−1). | ||||||
| 6 | M(CO)6 + [NO]+ ⇌ [M(CO)6]+ + NO | Cr | −71 | −143 | ||
| Mo | −82 | −121 | ||||
| W | −84 | −119 | ||||
| 7 | M(CO)6 + [NO]+ ⇌ [M(CO)5(NO)]+ + CO | Cr | −240 | −245 | ||
| Mo | −252 | −255 | ||||
| W | −254 | −255 | ||||
| 8 | M(CO)6 + [Agl2]+ ⇌ [M(CO)6l]+ + Agl | Cr | 58 | 66 | 64 | 63 |
| Mo | 8 | 14 | 14 | 13 | ||
| W | −8 | −6 | −1 | −1 | ||
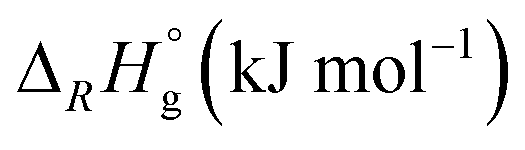
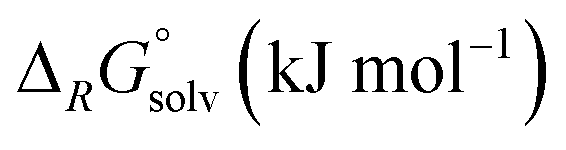
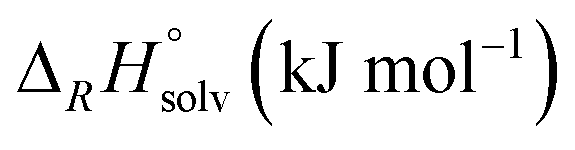
In reaction (8), the formal transfer of an iodine cation (I+) as a possible explanation for the observed carbonyl/iodide cations was examined: the reaction of the heavier congeners is about 50–60 kJ mol−1 more exothermic. Gas-phase and solution thermodynamics (calculated with the COSMO42 model using respective dielectric constants of the solvents CH2Cl2 (ε = 8.93) and oDFB (ε = 13.8)) are here in accordance. For Cr(CO)6, the equilibrium is far to the left so that the effective concentration for AgI would the less than its solubility constant. In contrast, for Mo(CO)6 and W(CO)6 the equilibrium constants ranges around 1, so if any [AgI2]+ is present, molecular AgI is initially formed, which then precipitates and further promotes the reaction. This means experimentally that the availability of [AgI2]+ needs to be sufficiently suppressed in order to prevent the availability of the formal “I+” species. Keeping the [Ag2I2][WCA]2 complex intact underlines the importance of non-coordinating solvents.
Interestingly, the difference in the actual iodide affinity for of the homoleptic hexacarbonyl cations is nonexistent (BP86) or only about 30 kJ mol−1 (B3-LYP). The gas-phase reaction of [M(CO)6]+˙ with AgI to Ag+ and M(CO)6 is equally disfavored (ca. 480 kJ mol−1) for all three metals (these and additional reaction enthalpies are deposited in the S.I‡). This both indicates that the reaction to the iodide containing side-products probably happens before and (not after) [M(CO)6]+˙ is formed.
The fluoride ion affinities (FIAs) of the [M(CO)6]+˙ species were calculated in reaction (9). If referenced to [Si(CH3)3]+ (gas-phase FIA = 958 kJ mol−1),39 the values increase from 711 for [Cr(CO)6]+˙, over 715 for [Mo(CO)6]+˙ to 741 kJ mol−1 for [W(CO)6]+˙. To put this into a general perspective, these species are 120 to 150 kJ mol−1 more Lewis acidic than gaseous SbF5 and are in the range of phosphenium cations.44 In solution, the FIA values with respect to [Si(CH3)3]+ are generally lower, yet also in solution the expected trend of an increasing Lewis acidity from Cr to W is visible.
It shall be noted here that the calculated seven-coordinate Cr(CO)6F complex features an imaginary vibration, if the fluoride ion is bound to Cr. In the actual minimum structure, the fluorine is bound to a CO ligand as an acyl-fluoride. This underlines the incapability of Cr to adopt coordination numbers >6 and shows that the CO ligand is the actual Lewis-acidic center, not the metal – similarly seen in the Hieber base reaction.45 For consistency reasons, the Cr–F bound structure was used in the calculations to compare only the ‘metal-centered’ Lewis acidity.
In regard to the Lewis acidity of our complexes, we carried out a natural bond orbital (NBO) and atoms in molecules (AIM) charge analysis (Table 5). Although the absolute values of both methods differ greatly, the charge on the metal centers increases from Cr to W in both cases – in agreement with experiment and theory. Furthermore, for [Cr(CO)6]+˙ the charge on the carbon atom of the CO group is the highest of the triad, with only a small charge difference between Cr and CO in AIM. For [W(CO)6]+˙, the high reactivity and Lewis acidity also reveals in NBO and AIM charges. It is not only the highest amongst the [M(CO)6]+˙ triad, but also the AIM charge is very high for the W atom – a truly sharp and reactive metal center. We note that the absolute values of the partial charges differ enormously between AIM and NBO. This reiterates our notion that partial charges are no physical observables and their magnitude has to be used with caution.46
| Charge (NBO) | Charge (AIM) | ||
|---|---|---|---|
| [Cr(CO)6]+˙ | Cr | −0.95 | +1.29 |
| C | +0.67 | +1.05 | |
| O | −0.35 | −1.10 | |
| [Mo(CO)6]+˙ | Mo | −0.57 | +1.49 |
| C | +0.60 | +1.01 | |
| O | −0.34 | −1.09 | |
| [W(CO)6]+˙ | W | −0.34 | +1.66 |
| C | +0.56 | +0.98 | |
| O | −0.34 | −1.09 |
In a final analysis, we compared the experimental CO stretch vibrations of all homoleptic hexacarbonyl complexes known to date (Fig. 9), ranging from 1748 cm−1 for [Ti(CO)6]2− to 2254 cm−1 for [Ir(CO)6]3+. The electron deficient 17 VE species V(CO)6˙ and the [M(CO)6]+˙ triad all showcase (slightly) higher CO vibrational frequencies than the general trend for the 18 VE (truly octahedral) complexes.
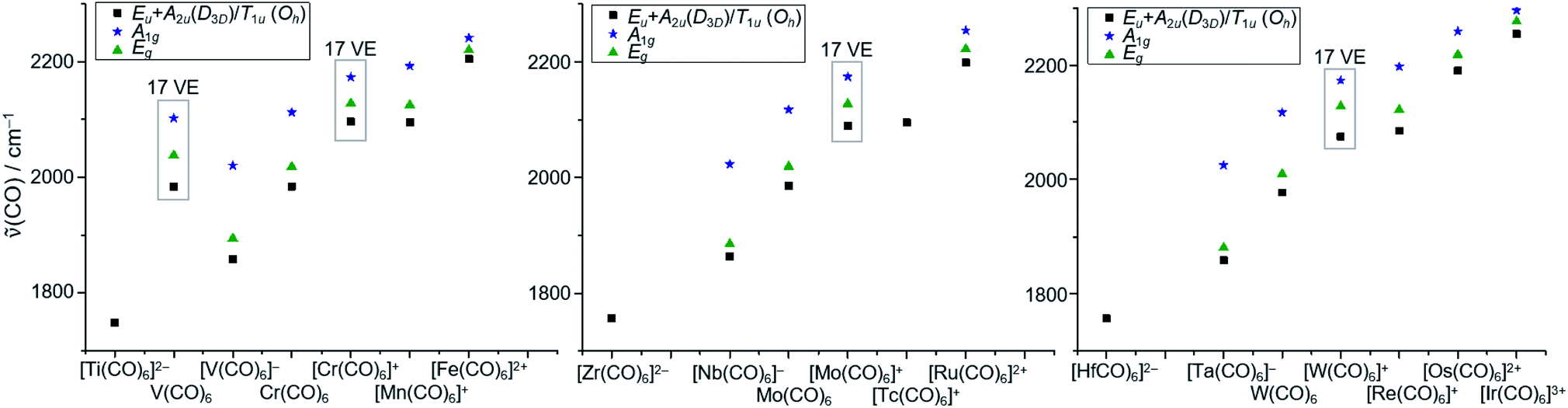 | ||
| Fig. 9 Comparison of the CO stretch vibrations of all homoleptic hexacarbonyl complexes known to date. Literature values taken from13,47 and references therein; Eu/A2u/T1u vibrations are IR active (black squares), A1g and Eg vibrations are Raman active (blue stars and green triangles) note: the methods of measurement of these data differ (e.g. solution/solid state IR); a detailed table can therefore be found in the S.I.‡ | ||
Conclusion
By oxidation of the neutral group VI hexacarbonyl precursors M(CO)6 (M = Cr, Mo, W) with NO[Al(ORF)4], the CO/NO exchange gives the novel heteroleptic carbonyl nitrosyl cations [M(CO)5(NO)]+ as [Al(ORF)4]− salts. They can be accessed selectively and (near) quantitatively and were fully characterized by single-crystal and powder XRD, as well as NMR and vibrational spectroscopy. The synergistic oxidant system Ag[F-{Al(ORF)3}2]/0.5 I2 leads to the formation of the homoleptic radical cations [M(CO)6]+˙ as [F-{Al(ORF)3}2]− salts. However, for Mo/W and due to the accessibility of coordination number 7, the formation of side-products such as heteroleptic carbonyl/iodides proved difficult to suppress. Especially for the more reactive [W(CO)6]+˙, partial abstraction of an alkoxide moiety from the anion leads to [W(CO)6(ORF)][F-{Al(ORF)3}2], which can only be separated by careful manual sorting of both crystalline species. Although improvable, the synthesis in TFB described here is the best compromise between feasibility and yield to date.The study of an isostructural and isoelectronic triad of complexes allows for new insights on the bonding situation for homologous carbonyl complexes, which are in agreement with the [Fe/Ru/Os(CO)6]2+ sequence. The paramagnetic nature of [M(CO)6]+˙ leads to D3d symmetric ground state (instead of Oh for the diamagnetic ‘true’ octahedra), which readily fluctuate to give broad Raman vibrations and a pseudo-octahedral crystal structure. EPR studies support these findings and proof the identity of d5 metal-centered radical cations described in this work.
Overall, our results show the capabilities as well as the limitations of the Ag[WCA]/0.5 I2 oxidant system for the formation of reactive cations. Innocence and non-innocence is an inherent problem (or feature!) of all strong oxidants that are currently available and the search for truly innocent oxidants is still ongoing. As is, on the other hand, the search for the perfect WCA in order to tame reactive cations and access them in the solid state: [Al(ORF)4]− is easily available on a large scale with numerous oxidants, yet lacks stability towards the strongest electrophiles. [F-{Al(ORF)3}2]− is very robust against electrophiles but unstable towards Lewis-basic sites or solvents. This limits their use to the respective problem at hand. Especially and in regard to the development and discoveries in recent decades, further improvements to the current [Ox][WCA] systems are necessary… Then, one might fantasize of access to unprecedented and deemed-impossible cations such as [Nb/Ta(CO)7][WCA] or even [Ti/Zr/Hf(CO)8][WCA]2.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors would like to thank Dr Michael Daub for powder-XRD, Dr Harald Scherer and Fadime Bitgül for NMR. J. B. gratefully acknowledges financial support by the LGFG Graduate Funding. W. F. gratefully acknowledges the Carl-Zeiss-Stiftung for financial and the Studienstiftung des Deutschen Volkes e.V. for general support. IK thanks the Deutsche Forschungsgemeinschaft DFG for support of this project. PJM and JC are grateful for financial support from the Foundation for Polish Science (Homing Programme, agreement No. POIR.04.04.00-00-1D24/16).References
- L. Mond, C. Langer and F. Quincke, J. Chem. Soc., Trans., 1890, 57, 749 RSC.
- J. Bohnenberger, W. Feuerstein, D. Himmel, M. Daub, F. Breher and I. Krossing, Nat. Commun., 2019, 10, 624 CrossRef CAS PubMed.
- H. Willner and F. Aubke, Organometallics, 2003, 22, 3612 CrossRef CAS.
- H. Willner and F. Aubke, Chem.–Eur. J., 2003, 9, 1668 CrossRef CAS PubMed.
- Q. Xu, Coord. Chem. Rev., 2002, 231, 83 CrossRef CAS.
- H. Willner and F. Aubke, Angew. Chem., Int. Ed., 1997, 36, 2402 CrossRef CAS.
- J. Schaefer, A. Kraft, S. Reininger, G. Santiso-Quinones, D. Himmel, N. Trapp, U. Gellrich, B. Breit and I. Krossing, Chem.–Eur. J., 2013, 19, 12468 CrossRef CAS PubMed.
- V. V. Gurzhiy, A. E. Miroslavov, G. V. Sidorenko, A. A. Lumpov, S. V. Krivovichev and D. N. Suglobov, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, 1145 CrossRef PubMed.
- P. K. Hurlburt, O. P. Anderson and S. H. Strauss, J. Am. Chem. Soc., 1991, 113, 6277 CrossRef CAS.
- J. Geier, H. Willner, C. W. Lehmann and F. Aubke, Inorg. Chem., 2007, 46, 7210 CrossRef CAS PubMed.
- Y. Souma, J. Iyoda and H. Sano, Inorg. Chem., 1976, 15, 968 CrossRef CAS.
- S. M. Ivanova, S. V. Ivanov, S. M. Miller, O. P. Anderson, K. A. Solntsev and S. H. Strauss, Inorg. Chem., 1999, 38, 3756 CrossRef CAS.
- J. E. Ellis, Organometallics, 2003, 22, 3322 CrossRef CAS.
- W. Hieber and E. Becker, Ber. Dtsch. Chem. Ges., 1930, 63, 1405 CrossRef.
- G. Bistoni, S. Rampino, N. Scafuri, G. Ciancaleoni, D. Zuccaccia, L. Belpassi and F. Tarantelli, Chem. Sci., 2016, 7, 1174 RSC.
- R. Bröchler, D. Freidank, M. Bodenbinder, I. H. T. Sham, H. Willner, S. J. Rettig, J. Trotter and F. Aubke, Inorg. Chem., 1999, 38, 3684 CrossRef PubMed.
- R. Bröchler, I. H. T. Sham, M. Bodenbinder, V. Schmitz, S. J. Rettig, J. Trotter, H. Willner and F. Aubke, Inorg. Chem., 2000, 39, 2172 CrossRef PubMed.
- I. M. Riddlestone, A. Kraft, J. Schaefer and I. Krossing, Angew. Chem., Int. Ed., 2017, 57, 13982 CrossRef PubMed.
- A. Martens, P. Weis, M. C. Krummer, M. Kreuzer, A. Meierhöfer, S. C. Meier, J. Bohnenberger, H. Scherer, I. Riddlestone and I. Krossing, Chem. Sci., 2018, 9, 7058 RSC.
- G. Cooper, J. C. Green, M. P. Payne, B. R. Dobson and I. H. Hillier, J. Am. Chem. Soc., 1987, 109, 3836 CrossRef CAS.
- G. Reiser, W. Habenicht, K. Müller-Dethlefs and E. W. Schlag, Chem. Phys. Lett., 1988, 152, 119 CrossRef CAS.
- T. W. Hayton, P. Legzdins and W. B. Sharp, Chem. Rev., 2002, 102, 935 CrossRef CAS PubMed.
- E. Bernhardt, M. Finze, H. Willner, C. W. Lehmann and F. Aubke, Chem.–Eur. J., 2006, 12, 8276 CrossRef CAS PubMed.
- F. Calderazzo and G. Pampaloni, J. Chem. Soc., Chem. Commun., 1984, 1249 RSC.
- F. Calderazzo, G. Pampaloni, L. Rocchi, J. Strähle and K. Wurst, J. Organomet. Chem., 1991, 413, 91 CrossRef CAS.
- (a) V. Zhuravlev and P. J. Malinowski, Angew. Chem., Int. Ed., 2018, 130, 11871 CrossRef; (b) P. J. Malinowski, D. Himmel and I. Krossing, Angew. Chem., Int. Ed., 2016, 55, 9259 CrossRef CAS PubMed.
- P. J. Malinowski, D. Himmel and I. Krossing, Angew. Chem., Int. Ed., 2016, 55, 9262 CrossRef CAS PubMed.
- M. Rohde, L. O. Müller, D. Himmel, H. Scherer and I. Krossing, Chem.–Eur. J., 2014, 20, 1218 CrossRef CAS PubMed.
- A. Bihlmeier, M. Gonsior, I. Raabe, N. Trapp and I. Krossing, Chem.–Eur. J., 2004, 10, 5041 CrossRef CAS PubMed.
- E. Bernhardt, H. Willner, A. Kornath, J. Breidung, M. Bühl, V. Jonas and W. Thiel, J. Phys. Chem. A, 2003, 107, 859 CrossRef CAS.
- S. H. Parrish and W. Weltner, J. Phys. Chem. A, 1999, 103, 1025 CrossRef CAS.
- T. F. Miller, D. L. Strout and M. B. Hall, Organometallics, 1998, 17, 4164 CrossRef CAS.
- T. Helgaker, P. Jørgensen and J. Olsen, Molecular electronic-structure theory, Wiley, Chichester, 2004 Search PubMed.
- F. A. Cotton and C. S. Kraihanzel, J. Am. Chem. Soc., 1962, 84, 4432 CrossRef CAS.
- B. Bley, H. Willner and F. Aubke, Inorg. Chem., 1997, 36, 158 CrossRef CAS.
- B. Rees and A. Mitschler, J. Am. Chem. Soc., 1976, 98, 7918 CrossRef CAS.
- T. C. W. Mak, Z. Kristallogr. - Cryst. Mater., 1984, 166, 277 CAS.
- F.-W. Grevels, J. Jacke, W. E. Klotzbücher, F. Mark, V. Skibbe, K. Schaffner, K. Angermund, C. Krüger, C. W. Lehmann and S. Özkar, Organometallics, 1999, 18, 3278 CrossRef CAS.
- H. Böhrer, N. Trapp, D. Himmel, M. Schleep and I. Krossing, Dalton Trans., 2015, 44, 7489 RSC.
- B. E. Mann, J. Chem. Soc., Dalton Trans., 1973, 2012 RSC.
- A. W. Ehlers, Y. Ruiz-Morales, E. J. Baerends and T. Ziegler, Inorg. Chem., 1997, 36, 5031 CrossRef CAS.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799 RSC.
- S. Halbert, C. Copéret, C. Raynaud and O. Eisenstein, J. Am. Chem. Soc., 2016, 138, 2261 CrossRef CAS PubMed.
- J. M. Slattery and S. Hussein, Dalton Trans., 2012, 41, 1808 RSC.
- W. Hieber and F. Leutert, Naturwissenschaften, 1931, 19, 360 CrossRef CAS.
- (a) P. Weis, I. M. Riddlestone, H. Scherer and I. Krossing, Chem.–Eur. J., 2019, 25, 12159 CrossRef CAS PubMed; (b) I. M. Riddlestone, P. Weis, A. Martens, M. Schorpp, H. Scherer and I. Krossing, Chem.–Eur. J., 2019, 25, 10546 CrossRef CAS PubMed.
- (a) R. K. Szilagyi and G. Frenking, Organometallics, 1997, 16, 4807 CrossRef CAS; (b) V. Jonas and W. Thiel, Organometallics, 1998, 17, 353 CrossRef CAS.
Footnotes |
| † Dedicated to Prof. Dr Manfred Scheer on occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available: A complete analysis of all bands in IR and Raman spectroscopy (Section 4) as well as powder-XRD diffractograms (Section 6), NMR spectra (Section 3) and additional details on the crystal structures (Section 7 + 8) and DFT calculations (Section 9) are provided in the ESI. CCDC 1952378–1952389. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc06445a |
| § Those type of complexes are therefore also called ‘σ-carbonyls’ or ‘σ-only-bonded’. |
| ¶ Exemplarily, both seven-coordinate complexes W(CO)6(NO) and [W(CO)6(NO)]+ are not stable in our gas phase DFT (BP86def2-TZVPP/D3BJ) calculations and loose a NO/CO ligand respectively without an energy barrier. |
| || CCDC search (ConQuest 2.0.4), as of 12/2019. |
| ** E.g. the scale of a typical reaction (∼250–300 mg silver salt) requires an exact stoichiometric amount in the range of 6 mg of Cl2. |
†† The combined system potentials 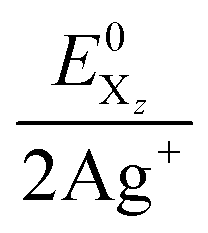 of the 2 Ag+/Hal2 systems are +1.49 V (I2), +1.82 V (Br2) and +1.93 V (Cl2) in aqueous solution.27 of the 2 Ag+/Hal2 systems are +1.49 V (I2), +1.82 V (Br2) and +1.93 V (Cl2) in aqueous solution.27 |
| ‡‡ Only Ag[WCA]/I2 is somewhat soluble in C6F14 and moderately soluble in C6F6. The neutral carbonyls as well as all products of the reaction are insoluble in these media and the reaction is effectively carried out as a suspension. |
| §§ Typically, [F-{Al(ORF)3}2]− is obtained, when [Al(ORF)4]− is decomposed in the presence of extreme electrophiles, which either abstract a fluoride (e.g. small silylium ions like [Me3Si]+)28 or a perfluorinated alkoxide group (e.g. [PCl2]+).29 |
| ¶¶ Ag[F-{Al(ORF)3}2] can be crystallized “naked” (without solvent molecules) from TFB solutions. By contrast, a salt [Ag(oDFB)3]+[F-{Al(ORF)3}2]− including three coordinated oDFB molecules crystallizes from oDFB solution. |
| |||| It shall be noted here that the calculations in literature are done with the Cotton–Kraihanzel method,34 which is based on the experimental values of the CO stretch. |
| *** The different kinetics also reflect in the surprisingly high stability of [Cr(CO)6]+˙: unlike the heavier homologues, it only very slowly decomposes on air or in NO atmosphere. |
| This journal is © The Royal Society of Chemistry 2020 |