
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facilitating the reduction of V–O bonds on VOx/ZrO2 catalysts for non-oxidative propane dehydrogenation†
Yufei
Xie
 ,
Ran
Luo
,
Guodong
Sun
,
Sai
Chen
,
Zhi-Jian
Zhao
,
Rentao
Mu
and
Jinlong
Gong
*
,
Ran
Luo
,
Guodong
Sun
,
Sai
Chen
,
Zhi-Jian
Zhao
,
Rentao
Mu
and
Jinlong
Gong
*
Key Laboratory for Green Chemical Technology of Ministry of Education, School of Chemical Engineering and Technology, Tianjin University, Collaborative Innovation Center of Chemical Science and Engineering, Tianjin 300072, China. E-mail: jlgong@tju.edu.cn
First published on 16th March 2020
Abstract
Supported vanadium oxide is a promising catalyst in propane dehydrogenation due to its competitive performance and low cost. Nevertheless, it remains a grand challenge to understand the structure–performance correlation due to the structural complexity of VOx-based catalysts in a reduced state. This paper describes the structure and catalytic properties of the VOx/ZrO2 catalyst. When using ZrO2 as the support, the catalyst shows six times higher turnover frequency (TOF) than using commercial γ-Al2O3. Combining H2-temperature programmed reduction, in situ Raman spectroscopy, X-ray photoelectron spectroscopy and theoretical studies, we find that the interaction between VOx and ZrO2 can facilitate the reduction of V–O bonds, including V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, V–O–V and V–O–Zr. The promoting effect could be attributed to the formation of low coordinated V species in VOx/ZrO2 which is more active in C–H activation. Our work provides a new insight into understanding the structure–performance correlation in VOx-based catalysts for non-oxidative propane dehydrogenation.
O, V–O–V and V–O–Zr. The promoting effect could be attributed to the formation of low coordinated V species in VOx/ZrO2 which is more active in C–H activation. Our work provides a new insight into understanding the structure–performance correlation in VOx-based catalysts for non-oxidative propane dehydrogenation.
Introduction
Propylene is one of the most important chemical building blocks and its high-value derivatives are of great demand in the chemical industry. Due to the large-scale exploitation of shale gas, direct propane dehydrogenation becomes a particularly important method to produce propylene using propane as feedstock. Commercialized propane dehydrogenation plants generally utilize Pt-based and CrOx-based catalysts, however these two kinds of catalysts are either expensive or toxic.1 Alternatively, supported vanadium oxide is a promising catalyst compared with Pt and CrOx for its competitive performance, low cost and low toxicity.2,3It is well established that VOx-based catalysts can be utilized in propane oxidative dehydrogenation (ODH).4–8 However, there is a trade-off effect between conversion and selectivity associated with over oxidation to COx, which prevents the ODH process from achieving high propylene yield. In contrast, better reactivity and selectivity, especially excellent regeneration stability can be achieved when supported VOx catalysts are used in the non-oxidative propane dehydrogenation (PDH) process.9–13 Nevertheless, the structure–performance correlation of VOx-based catalysts is still unclear due to the structural complexity of the supported VOx catalytic system. Previous studies have confirmed that several factors could influence the performance of VOx-based catalysts such as polymerization forms of VOx, chemical states of V and the identity of the support.14–18
The support effect is an essential parameter because VOx can be bonded to a support through the V–O–support interaction. The variety of supports could lead to significant changes in the catalytic properties of VOx. It is generally accepted that the support could affect reactivity by stabilizing the active sites and/or altering the electron state of active sites.19–22 The support effect has been extensively studied over VOx-based catalysts in the ODH process. Researchers have concluded that the lattice oxygen in VO and V–O–support (V–O–S) bonds is consumed in C–H activation and H2O is formed. Thus, support identities could affect the reaction by tuning the oxygen vacancy formation energy, which is confirmed by experiments and DFT calculations.23–25
However, the support effect works in an entirely different way in the PDH process because not only active sites but also reaction mechanisms are distinct from those of the ODH process. V–O bonds directly catalyze C–H activation forming H2 rather than H2O. Previous studies found that the reactivity of PDH is dependent on the bond strength of V–O–S.22 It is proposed that V–O–S bonds are active sites. In addition, the support identity also influences the behavior of carbon deposition on VOx based catalysts, which inversely affects activity and on-stream stability.12,18 Nevertheless, the effect of a support is still not clear because the structure of VOx and the VOx–support interface in a reduced state is still ambiguous.
Herein, we explore the support effect on the catalytic performance of VOx-based catalysts for PDH and provide a new insight into understanding how support identity matters. We discovered that VOx/ZrO2, a well-known catalyst for ODH,26–28 has a much more superior PDH performance than commonly used VOx/Al2O3. The turnover frequency (TOF) is six times higher by loading VOx on ZrO2 than on Al2O3. The remarkable improvement of reactivity has not been reported in previous studies. Rate measurement of catalysts with gradient V loadings was employed to identify the active site of the VOx/ZrO2 catalyst. In situ Raman spectroscopic measurements, X-ray photoelectron spectroscopy (XPS) and density functional theory (DFT) calculations were used to determine the structure evolution of VOx under a reducing atmosphere, and they show that many more V–O bonds on ZrO2 (VO, V–O–V and V–O–Zr) are consumed during reduction. On this basis, we proposed that the facile reducing nature of V–O bonds promotes the formation of lower coordinated V species which accounts for C–H activation enhancement.
Results and discussion
Catalyst structure
A series of characterization techniques were used to determine the bulk and surface structure of the catalysts (VOx loaded on ZrO2 and Al2O3 are denoted as xVZr and xVAl where x represents the mass percentage of V, metal base). The bare ZrO2 support, 1VZr and 1VAl were characterized by X-ray diffraction (XRD). As shown in Fig. 1a, ZrO2 is mainly composed of monoclinic phase (m-ZrO2, JCPDS 72-1669) with small amount of a tetragonal phase (t-ZrO2, JCPDS 79-1771), which is consistent with the previous report using the same preparation method.29 The same XRD pattern of 1VZr and pure ZrO2 indicates that VOx does not change the crystalline structure of ZrO2. In addition, for VOx loaded catalysts, only diffraction lines of the support could be detected (Fig. 1a and S1a†), which means amorphous VOx is well dispersed on these catalysts.30,31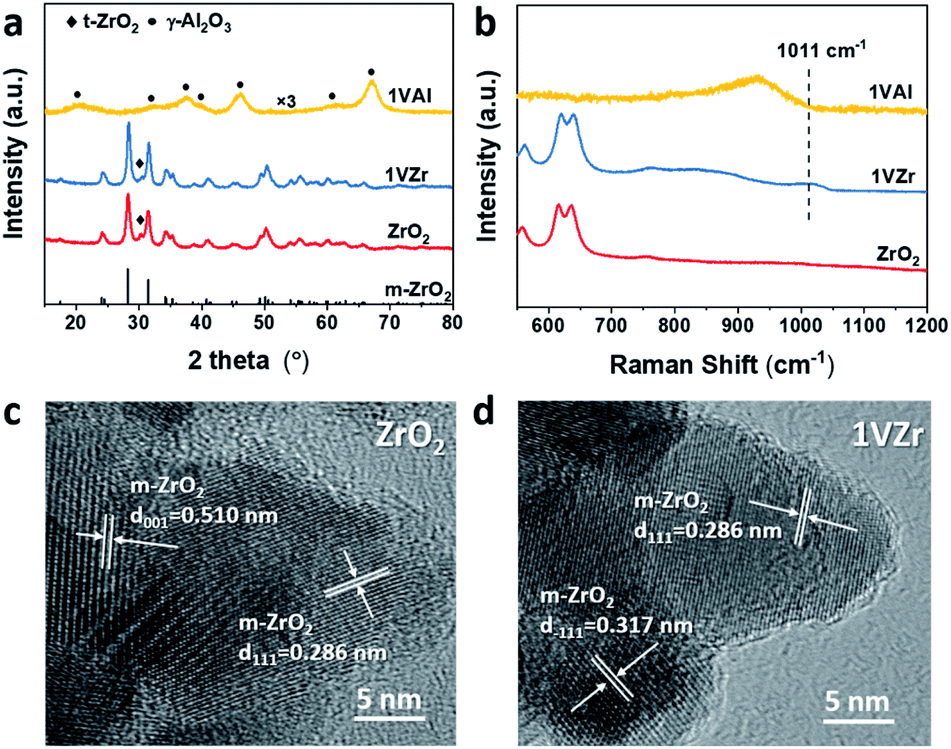 | ||
| Fig. 1 (a) XRD patterns of the ZrO2 support and VOx supported on ZrO2 and Al2O3. (b) Raman spectra (532 nm excitation) of the ZrO2 support and VOx supported on ZrO2 and Al2O3. TEM images of (c) ZrO2 and (d) VOx/ZrO2. | ||
Vis-Raman spectroscopic measurements, which are sensitive to the presence of V2O5, were performed over the catalysts to gain insight into the kind of vanadium species. Raman spectra of the ZrO2 support, 1VZr and 1VAl are displayed in Fig. 1b. A band at 1011 cm−1 is attributed to vanadyl stretching of surface-dispersed VOx. No sharp band at 995 cm−1, assigned to the stretching vibration of VO in crystal V2O5, is detected. With the V loading increasing, a band characteristic of V2O5 at 995 cm−1 appears after the loading reached 3 wt% (Fig. S1b†). This demonstrates the existence of crystal V2O5 in 3VZr and 4VZr. From the enlarged region on the left from 75 to 160 cm−1, a small band which corresponds to V2O5 at around 150 cm−1 can be seen in 2.5VZr. This implies that VOx begins to crystalize at 2.5VZr. Besides, the absence of a peak at around 770 cm−1 suggests no ZrV2O7 is formed in our samples.32 The Raman spectra demonstrate that VOx species are well dispersed as oligomeric species when the loading is less than 2 wt% and V2O5 crystals are formed after the loading reaches 2.5 wt%.
Surface density of V was calculated through the BET surface area and V loading. The detailed results are listed in Table S1.† 2.5VZr has a V density of 6.9 nm−2 which is a bit lower than the theoretical monolayer coverage. It has been proved that the presence of V2O5 is inevitable below theoretical monolayer coverage by a simple incipient wetness impregnation method.10 This agrees with the observation of the V2O5 peak in 2.5VZr through Raman spectroscopic measurements.
The morphologies of ZrO2 and 1VZr were also characterized by TEM (Fig. 1c and d). ZrO2 is in the form of a nanoparticle with a relevant uniform size of around 30 nm and its morphology does not change after loaded with VOx. Lattice fringes of V2O5 could not be found because amorphous VOx is well dispersed on the surface of ZrO2. The TEM results are in good agreement with the observations of XRD and Raman results.
Catalytic performance
Catalytic performance of VOx supported on ZrO2 and γ-Al2O3 for the PDH reaction was studied. The initial propane conversion and propylene selectivity based on all products are illustrated in Fig. 2a and b. The initial conversion of 1VZr is approximately five times higher than that of 1VAl and pure ZrO2, which shows a significant support effect. The total selectivity towards propylene is around 80% and gas-phase selectivity towards it is >90% (Fig. S2a†) for all catalysts, which are at a similar level with respect to those of the VOx-based catalysts in previous studies.10,14,18,33 In addition, we performed 120 min of the on stream reaction and studied the reaction–regeneration cycles over 1VZr (Fig. S2b and S3†). The propane conversion and propylene selectivity exhibit little change during six cycles, implying outstanding regeneration stability.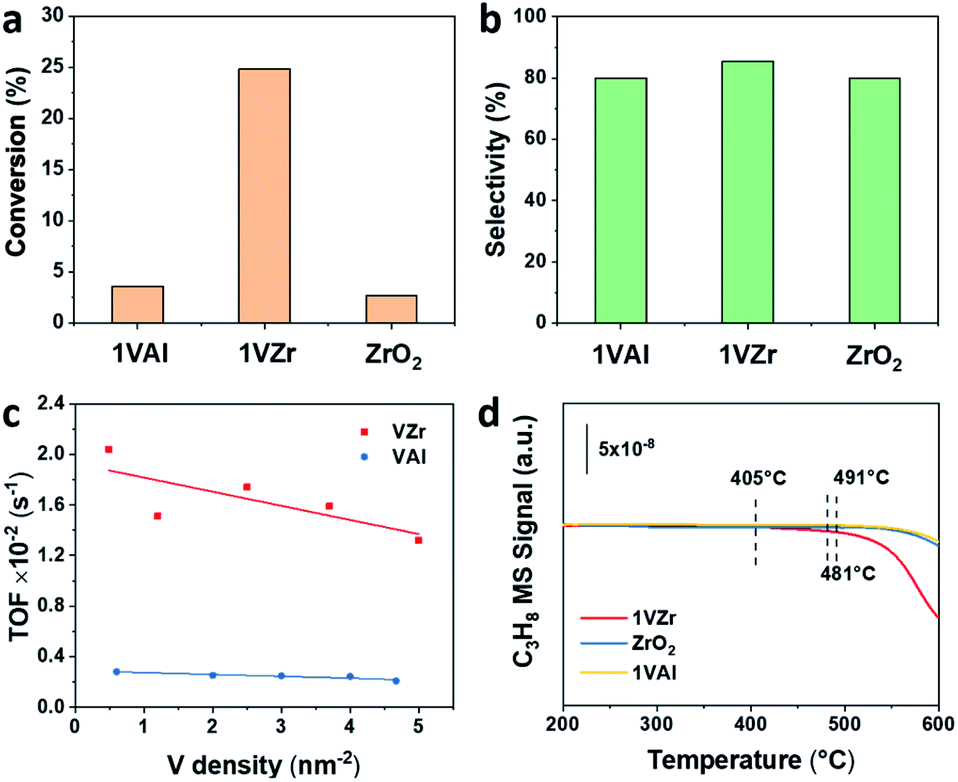 | ||
Fig. 2 (a) Initial propane conversion and (b) propylene selectivity (based on all products) of 1VAl, 1VZr and ZrO2. Reaction conditions: mcat = 0.4 g; C3H8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N2:H2 = 7:36:7; T = 550 °C; inlet flow = 50 mL min−1. (c) Comparison of TOF values between VZr and VAl with different V densities. (d) C3H8-TPSR of 1VZr, ZrO2 and 1VAl. :N2:H2 = 7:36:7; T = 550 °C; inlet flow = 50 mL min−1. (c) Comparison of TOF values between VZr and VAl with different V densities. (d) C3H8-TPSR of 1VZr, ZrO2 and 1VAl. | ||
Because the surface areas of ZrO2 and Al2O3 are different, when the V loadings of VZr and VAl are identical, they have different V densities. Considering the activity is also influenced by V density, the TOFs of VZr and VAl were also compared based on V density (Fig. 2c and Table S2†). The calculated TOF of VZr is almost 6-fold higher than that of VAl and only slightly decreases with the V surface density, which implies that the activity is strongly support-dependent. This phenomenon is similar to that of the GaOx catalytic system, where the activity is not a consequence of Ga nuclearities, but of the Ga–O–support interaction.34
To further evaluate the intrinsic activity in C–H activation, propane temperature-programmed surface reaction (C3H8-TPSR) experiments were carried out over 1VZr, ZrO2 and 1VAl. The temperature where the C3H8 signal begins to drop is determined as the C–H activation temperature. As shown in Fig. 2d, the initial C–H activation temperature of 1VZr is around 405 °C, which is almost 80 and 90 °C lower than that of ZrO2 (481 °C) and 1VAl (491 °C). Moreover, the C–H activation temperature of 3VAl (similar V density to 1VZr, Fig. S4†) was also tested, which is 100 °C higher than that of 1VZr. Along with the TOF difference between VZr and VAl, we can conclude that the intrinsic C–H activation ability of 1VZr is distinguishable.
Active phase identification
It has been proved that pure ZrO2 can also catalyze propane dehydrogenation through coordinatively unsaturated Zr (Zrcus).35–37 In addition, Jeon et al. prepared V–Zr mix oxide and found that V incorporation into the ZrO2 bulk phase would promote generation of more Zrcus.38 Although in our VOx/ZrO2 system, VOx is supported on the surface of a support, not in the bulk phase, there still remains different possibilities for this superior performance of the VOx/ZrO2 catalyst: ZrO2 enhances the activity of VOx, VOx enhances the activity of ZrO2 (Zrcus), or a combination of the two. Therefore, it is essential to verify if the active phase is VOx or Zrcus in order to understand the origin of this dramatic performance improvement. To this end, various experiments were performed to figure out whether the catalytic performance is due to VOx or Zrcus or both of them.The propane consumption rate versus V loading is shown in Fig. 3. The reaction rate rises linearly as the V loading increases until the V loading reaches 2 wt%, indicating VOx should be the active component. After this linearly increasing period, the C3H8 consumption rate does not change with increasing V loading because full VOx overlayers are formed at 2 wt% and crystalized V2O5 appears afterward as confirmed by the Raman spectra (Fig. S1b†). In addition, previous articles reported a shift of XPS Zr 3d peaks and reduction peaks of ZrO2 seen in the H2 temperature programmed reduction (H2-TPR) profile if Zrcus is the active site.37,39,40 However, these phenomena cannot be observed in the VOx/ZrO2 system. No ZrO2 reduction peak can be observed up to 600 °C (Fig. 4a), while our reactions were conducted at 550 °C. In addition, the XPS test also confirms that the binding energy of Zr 3d in ZrO2 and 1VZr has no difference (Fig. S5†). These results provide evidence that ZrO2 is not the active phase in VZr catalysts. Along with catalytic activity being linearly related to the amount of V, which directly proves VOx is responsible for the activity, we suggest VOx rather than Zrcus to be the main active phase. However, it is a pity that these experiments cannot identify which kind of V–O bond (VO, V–O–V, V–O–Zr or all of them) is responsible for C–H activation. A potential method to solve this problem is Surface Organometallic Chemistry (SOMC),41–43 which provides access to producing well-defined isolated VOx only possessing VO and V–O–support. This direction still needs further investigation.
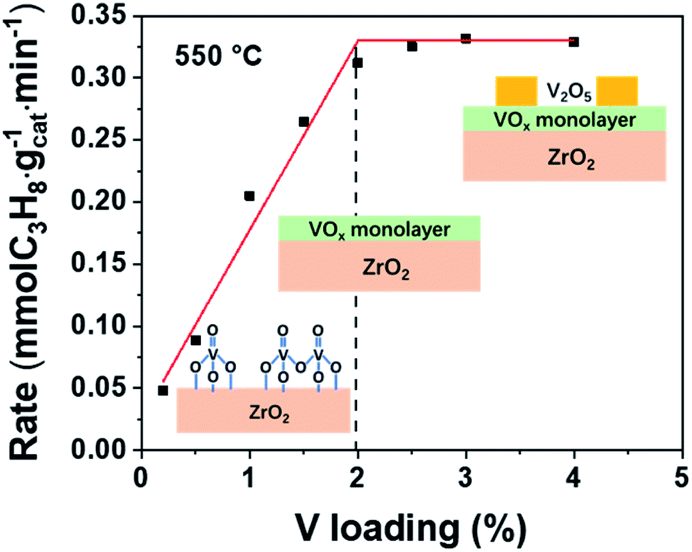 | ||
| Fig. 3 C3H8 reaction rate versus different V loadings on VZr catalysts at 550 °C. | ||
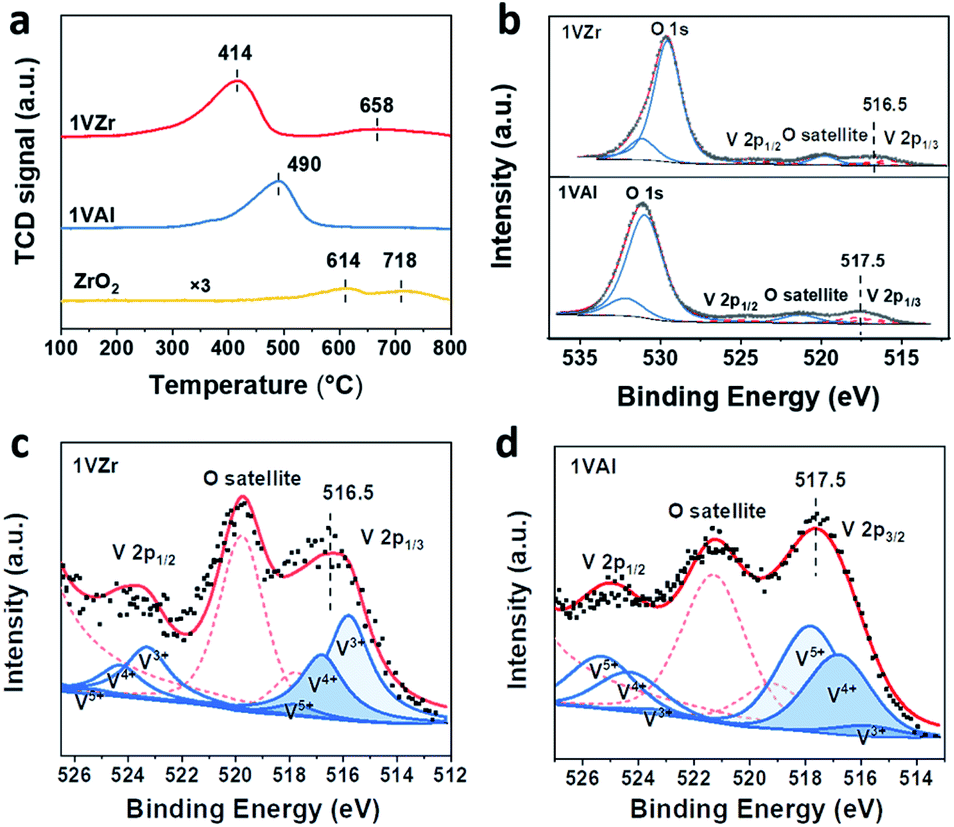 | ||
| Fig. 4 (a) H2-TPR profiles of 1VZr, 1VAl and pure ZrO2. (b) XPS O 1s and V 2p peaks for 1VZr and 1VAl. Deconvolution results of V 2p3/2 for (c) 1VZr and (d) 1VAl after reduction for 30 minutes. | ||
Structure–performance correlation
H2-TPR experiments were performed to test the reducibility and VOx–support interaction of the catalysts (Fig. 4a). The main peak at around 400–500 °C is attributed to the reduction of VOx and peaks above 600 °C are ascribed to a partial reduction of ZrO2.36,44–46 The reduction temperature of VOx for 1VZr is lower than that for 1VAl. Considering that the reduction temperature is influenced by the dispersion of VOx,15,47 we performed H2-TPR on a series of VAl and VZr catalysts (Fig. S6†). The reduction temperatures of VZr catalysts are at about 400 °C while those of VAl catalysts are at about 500 °C. Thus, the low reduction temperature of the VZr catalyst implies that the interaction between VOx and ZrO2 is weaker than that between VOx and Al2O3. This phenomenon is further discussed in the in situ Raman spectroscopy section below. Alternatively, the peak area of 1VZr is larger than that of 1VAl. We calculated the H:V ratio through the peak area and estimated the average oxidation state (AOS) of V. As listed in Table S3,† the AOS of V in 1VZr and 1VAl corresponds to 3.5 and 4.0, which indicates that VOx can be more readily and deeply reduced on ZrO2.
To further identify the valance state of V, XPS investigation was carried out over 1VZr and 1VAl after H2 reduction for 30 min. V 2p core levels of 1VZr and 1VAl are displayed in Fig. 4b. We compare the binding energy of V in different catalysts qualitatively. The binding energy of V 2p2/3 centers at 516.5 eV in 1VZr and 517.5 eV in 1VAl. This indicates that the valance state of V in 1VZr is lower than that in 1VAl. According to the deconvolution results of V 2p shown in Fig. 4c, d and Table S4,† the oxidation state of V is a mixture of V5+, V4+ and V3+, which is consistent with previous results.33,44,48 The fraction of V3+ is 48.6% in 1VZr, which is higher than 6.6% in 1VAl.
It has been elucidated in the former articles that V3+ is more active in the PDH reaction.14,33 However, seldom detailed analyzes have been conducted to identify the structure of VOx in a reduced state and the structure–performance correlation is not clear. Therefore, in situ UV-Raman spectroscopy, which is more sensitive to the surface VOx structure,49 was performed to monitor the evolution of VO, V–O–V, and the V–O–support.
The evolution of V–O–V, V–O–Zr and VO in 1VZr during H2 reduction is shown in Fig. 5a and b. A broad band at 750–950 cm−1 is ascribed to V–O–V and V–O–Zr bonds.32,50 The band at around 1020 cm−1 is assigned to VO in dispersed VOx.27,32,51 The number of V–O–V and V–O–Zr bonds decreases substantially which implies that plenty of these bonds are consumed during reduction. In addition, nearly 60% of VO in 1VZr disappeared after reduction for 30 min. The comparative experiment of 1VAl is displayed in Fig. 5c and d. The bands attributed to V–O–V and V–O–Al on the Al2O3 support are located at 400–600 cm−1 and 910 cm−1 respectively.49 A sharp band at 1018 cm−1 is ascribed to VO.49 In contrast with V–O–Zr and V–O–V in the 1VZr sample, the V–O–Al band only decreases to 60% of that for fresh 1VZr and no change of the V–O–V band is detected which implies V–O–V bonds on the Al2O3 support cannot be reduced under H2 treatment. Meanwhile, 40% of VO in 1VAl is reduced, which is less than that in 1VZr. The result of in situ Raman spectroscopy confirms that not only VO and V–O–Zr, but also V–O–V can be deeply reduced in 1VZr which results in the deeper reduction degree of 1VZr observed in H2-TPR and XPS studies.
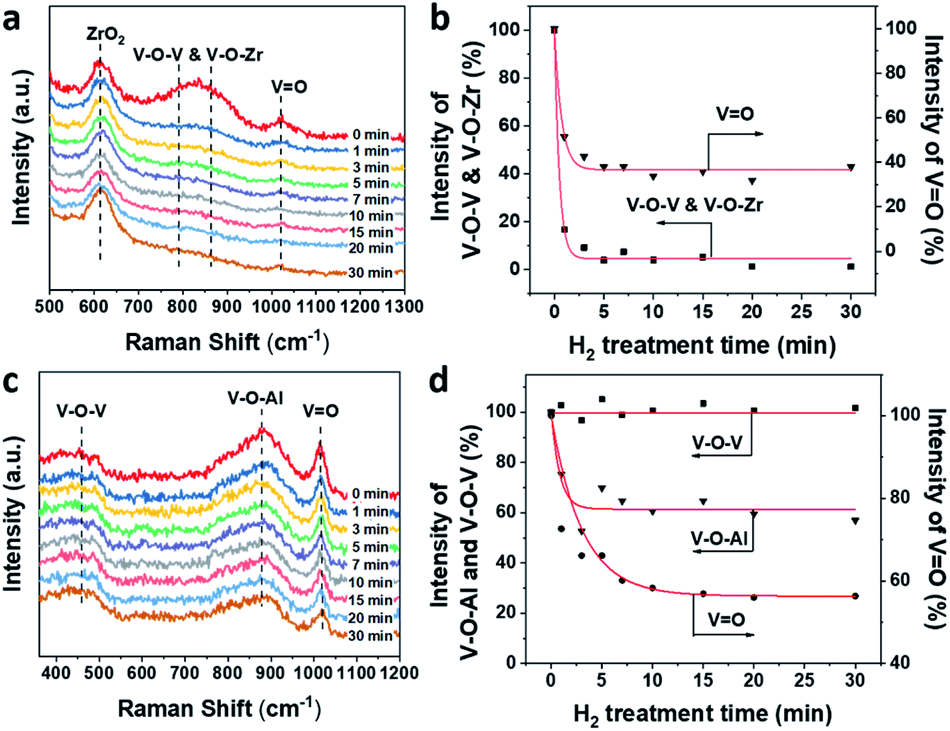 | ||
| Fig. 5 (a) In situ UV-Raman (325 nm excitation) spectra of VZr during H2 reduction at 550 °C. (b) Intensities of V–O–V, V–O–Zr, and VO versus H2 treatment time. (c) In situ UV-Raman (325 nm excitation) spectra of 1VAl during H2 reduction at 550 °C. (d) Intensities of V–O–V, V–O–Al and VO versus H2 treatment time. | ||
The facile reduction nature of V–O bonds in the VZr catalyst can be interpreted by the lower electronegativity of Zr compared with Al, which causes the difference of the interaction between VOx and the support. It has been shown that a lower support cation electronegativity can result in a higher electron density of the V–O–S bond, which lead to these bonds being readily reduced.52,53 Along with the lower reduction temperature of VZr shown in the H2-TPR profile, it can be deduced that the interaction between VOx and ZrO2 weakens the V–O bonds thus resulting in the facile reduction nature of the VZr catalyst.
Note that when O atoms are removed during the reduction period, the chemical environment of V changes, which leads to the formation of coordinatively unsaturated V. It is established that metal cations (Ga3+, Zn2+, Zr4+, etc.) with a low coordination number are more active for the reaction.54 As more V–O bonds are reduced in the VZr catalyst, we could deduce that lower coordinated V sites are more active for the C–H activation. Our recent DFT calculations also proved that coordinatively unsaturated VOx was more active in C–H activation55 while this work gives experimental evidence.
DFT calculations
DFT calculations were conducted to gain further insight into the structure of the active site and its influence on catalytic performance. Noting that the TOF difference between VZr and VAl exists with all the V densities, we constructed dimeric vanadium oxide species on the m-ZrO2(![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 11) and γ-Al2O3(100) surface as representatives36,55 (Fig. S7†).
11) and γ-Al2O3(100) surface as representatives36,55 (Fig. S7†).
We calculated oxygen vacancy formation energy of different oxygen atoms, with the energy of H2O(g) and H2(g) as the reference, in these vanadium oxide dimers to confirm the different reducibilities they showed in our experiment results. As listed in Table 1, oxygen atoms are removed from VO, V–O–V and the V–O–support. Four of five V–O bonds in VZr (VO, V–O–V and two V–O–Zr) have relatively low oxygen formation energies while only two of five in VZr do (VO, V–O–Al). The calculated oxygen vacancy formation energies are in good agreement with the Raman spectroscopy result that VO, V–O–V and V–O–Zr bonds in VZr can be reduced under a H2 atmosphere while only VO and part of V–O–Al are reduced in VAl.
11) and γ-Al2O3(100)
| Oxygen vacancy formation energies (eV) | |||||
|---|---|---|---|---|---|
| VO |
V–O–V | V–O–support | |||
| V2O5/m-ZrO2(11) |
0.13 | −0.13 | 0.03 | 0.14 | 0.34 |
| V2O5/γ-Al2O3(100) | −0.07 | 0.44 | 0.36 | 0.35 | −0.25 |
Since VZr and VAl have different degrees of reduction, we constructed V2O2/m-ZrO2(11) and V2O3/γ-Al2O3(100) models to represent the catalyst structure in a reduced state (Fig. 6a and b). In terms of the in situ Raman results and calculated oxygen vacancy formation energies, three (one in each VO, V–O–Zr and V–O–V) and two (one VO and one V–O–Al) V–O bonds with the lowest oxygen vacancy formation energies were removed from the initial dimeric V2O5 structure respectively. The propane dehydrogenation barriers over partially reduced VZr and VAl were computed and transition state (TS) geometries are shown in Fig. 6c. The first and second step C–H activation barriers in VZr is 0.66 eV and 0.85 eV, respectively, while in VAl they are 0.99 eV and 1.26 eV, respectively. DFT calculations also confirm that low coordinated V species are more active in C–H activation, which is consistent with the highest TOF and the lowest C–H activation temperature for VZr observed in the catalytic performance and C3H8-TPSR tests.
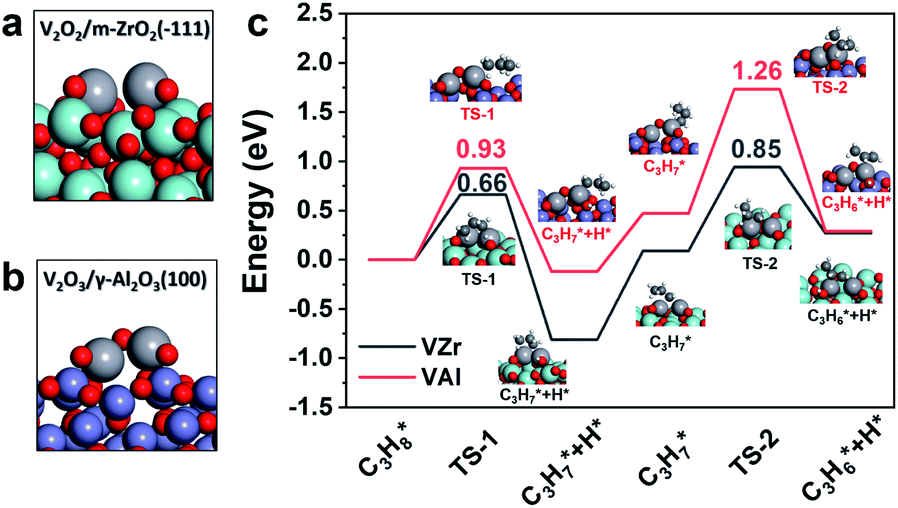 | ||
| Fig. 6 Reduced catalyst models of (a) V2O2/m-ZrO2(11) and (b) V2O3/γ-Al2O3(100). (c) Calculated potential energy diagrams of the first and second propane dehydrogenation step. | ||
Based on the experimental and theoretical results, we propose a structure–performance correlation of VZr and VAl catalysts, as shown in Scheme 1. VOx can be readily reduced on ZrO2 because the interaction between VOx and ZrO2 facilitates the reduction of VO, V–O–V and V–O–Zr bonds. However, for the VAl catalyst, only VO and some V–O–Al bonds can be reduced. Thus, more low coordinated V species form in the VZr catalyst during reduction, and these low coordinated V species exhibit a better performance in PDH.
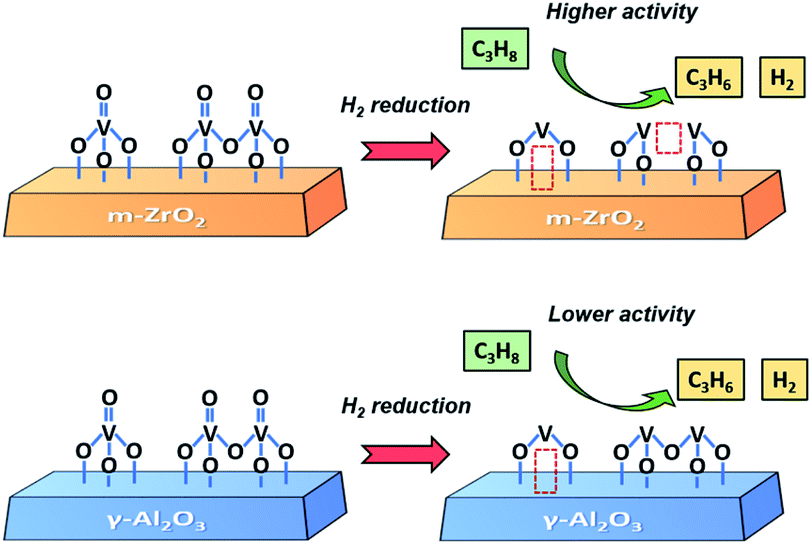 | ||
| Scheme 1 Propane dehydrogenation on VZr and VAl catalysts with different V structures and VOx reduction degrees. | ||
Conclusions
In summary, we prepared VOx loaded on ZrO2 through a simple incipient wetness impregnation method, which exhibits a dramatically improved performance compared with VOx loaded on Al2O3 for propane dehydrogenation. The TOFC3H8 of 1VZr is determined to be 0.0161 s−1, which is almost six times higher than that of 1VAl.We further prove that the remarkable reactivity of the 1VZr catalyst was attributed to the promotion of C–H activation over VOx species rather than it over ZrO2. Besides, combining in situ Raman and XPS spectroscopy results, we propose the enhanced C–H activation on VZr results from the facile reduction of VO, V–O–V and V–O–Zr bonds, thus producing deeply reduced and lower coordinated V species. DFT calculations also confirm that the C–H rupture energy barrier is lower for partially reduced VZr with low coordinated V species. Considering the dramatic performance achieved through the interaction between VOx and ZrO2, our work provides a new insight into high-performance VOx-based catalysts for propane dehydrogenation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the National Natural Science Foundation of China (21525626, and 51761145012), and the Program of Introducing Talents of Discipline to Universities (B06006) for financial support.Notes and references
- J. J. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS PubMed.
- S. Sokolov, M. Stoyanova, U. Rodemerck, D. Linke and E. V. Kondratenko, J. Catal., 2012, 293, 67–75 CrossRef CAS.
- B. M. Weckhuysen and D. E. Keller, Catal. Today, 2003, 78, 25–46 CrossRef CAS.
- S. Barman, N. Maity, K. Bhatte, S. Ould-Chikh, O. Dachwald, C. Haeßner, Y. Saih, E. Abou-Hamad, I. Llorens, J.-L. Hazemann, K. Köhler, V. D'Elia and J.-M. Basset, ACS Catal., 2016, 6, 5908–5921 CrossRef CAS.
- B. Beck, M. Harth, N. G. Hamilton, C. Carrero, J. J. Uhlrich, A. Trunschke, S. Shaikhutdinov, H. Schubert, H.-J. Freund, R. Schlögl, J. Sauer and R. Schomäcker, J. Catal., 2012, 296, 120–131 CrossRef CAS.
- C. A. Carrero, R. Schloegl, I. E. Wachs and R. Schomaecker, ACS Catal., 2014, 4, 3357–3380 CrossRef CAS.
- K. Chen, A. T. Bell and E. Iglesia, J. Catal., 2002, 209, 35–42 CrossRef CAS.
- F. Cavani, N. Ballarini and A. Cericola, Catal. Today, 2007, 127, 113–131 CrossRef CAS.
- P. Hu, W.-Z. Lang, X. Yan, L.-F. Chu and Y.-J. Guo, J. Catal., 2018, 358, 108–117 CrossRef CAS.
- T. Wu, G. Liu, L. Zeng, G. Sun, S. Chen, R. Mu, S. Agbotse Gbonfoun, Z.-J. Zhao and J. Gong, AIChE J., 2017, 63, 4911–4919 CrossRef CAS.
- P. Bai, Z. Ma, T. Li, Y. Tian, Z. Zhang, Z. Zhong, W. Xing, P. Wu, X. Liu and Z. Yan, ACS Appl. Mater. Interfaces, 2016, 8, 25979–25990 CrossRef CAS PubMed.
- S. Sokolov, V. Y. Bychkov, M. Stoyanova, U. Rodemerck, U. Bentrup, D. Linke, Y. P. Tyulenin, V. N. Korchak and E. V. Kondratenko, ChemCatChem, 2015, 7, 1691–1700 CrossRef CAS.
- Y.-P. Tian, P. Bai, S.-M. Liu, X.-M. Liu and Z.-F. Yan, Fuel Process. Technol., 2016, 151, 31–39 CrossRef CAS.
- G. Liu, Z.-J. Zhao, T. Wu, L. Zeng and J. Gong, ACS Catal., 2016, 6, 5207–5214 CrossRef CAS.
- U. Rodemerck, S. Sokolov, M. Stoyanova, U. Bentrup, D. Linke and E. V. Kondratenko, J. Catal., 2016, 338, 174–183 CrossRef CAS.
- U. Rodemerck, M. Stoyanova, E. V. Kondratenko and D. Linke, J. Catal., 2017, 352, 256–263 CrossRef CAS.
- M. E. Harlin, V. M. Niemi and A. O. I. Krause, J. Catal., 2000, 195, 67–78 CrossRef CAS.
- S. Sokolov, M. Stoyanova, U. Rodemerck, D. Linke and E. V. Kondratenko, Catal. Sci. Technol., 2014, 4, 1323–1332 RSC.
- M. Ahmadi, H. Mistry and B. Roldan Cuenya, J. Phys. Chem. Lett., 2016, 7, 3519–3533 CrossRef CAS PubMed.
- S. Hanukovich, A. Dang and P. Christopher, ACS Catal., 2019, 9, 3537–3550 CrossRef CAS.
- S. L. Nauert, L. Savereide and J. M. Notestein, ACS Catal., 2018, 8, 7598–7607 CrossRef CAS.
- D. E. Keller, S. M. K. Airaksinen, A. O. Krause, B. M. Weckhuysen and D. C. Koningsberger, J. Am. Chem. Soc., 2007, 129, 3189–3197 CrossRef CAS PubMed.
- M. V. Ganduglia-Pirovano, C. Popa, J. Sauer, H. Abbott, A. Uhl, M. Baron, D. Stacchiola, O. Bondarchuk, S. Shaikhutdinov and H. J. Freund, J. Am. Chem. Soc., 2010, 132, 2345–2349 CrossRef CAS PubMed.
- Y. Li, Z. Wei, F. Gao, L. Kovarik, R. A. L. Baylon, C. H. F. Peden and Y. Wang, ACS Catal., 2015, 5, 3006–3012 CrossRef CAS.
- T. Kropp, J. Paier and J. Sauer, J. Am. Chem. Soc., 2014, 136, 14616–14625 CrossRef CAS PubMed.
- A. Khodakov, J. Yang, S. Su, E. Iglesia and A. T. Bell, J. Catal., 1998, 177, 343–351 CrossRef CAS.
- C. L. Pieck, M. A. Bañares and J. L. G. Fierro, J. Catal., 2004, 224, 1–7 CrossRef CAS.
- A. Adamski, Z. Sojka, K. Dyrek, M. Che, G. Wendt and S. Albrecht, Langmuir, 1999, 15, 5733–5741 CrossRef CAS.
- T. P. Otroshchenko, V. A. Kondratenko, U. Rodemerck, D. Linke and E. V. Kondratenko, Catal. Sci. Technol., 2017, 7, 4499–4510 RSC.
- I. E. Wachs, Dalton Trans., 2013, 42, 11762–11769 RSC.
- I. E. Wachs and B. M. Weckhuysen, Appl. Catal., A, 1997, 157, 67–90 CrossRef CAS.
- A. Christodoulakis, J. Catal., 2004, 222, 293–306 CrossRef CAS.
- Z. J. Zhao, T. Wu, C. Xiong, G. Sun, R. Mu, L. Zeng and J. Gong, Angew. Chem., Int. Ed., 2018, 57, 6791–6795 CrossRef CAS PubMed.
- K. C. Szeto, Z. R. Jones, N. Merle, C. Rios, A. Gallo, F. Le Quemener, L. Delevoye, R. M. Gauvin, S. L. Scott and M. Taoufik, ACS Catal., 2018, 8, 7566–7577 CrossRef CAS.
- T. Otroshchenko, S. Sokolov, M. Stoyanova, V. A. Kondratenko, U. Rodemerck, D. Linke and E. V. Kondratenko, Angew. Chem., Int. Ed., 2015, 54, 15880–15883 CrossRef CAS PubMed.
- Y. Zhang, Y. Zhao, T. Otroshchenko, H. Lund, M. M. Pohl, U. Rodemerck, D. Linke, H. Jiao, G. Jiang and E. V. Kondratenko, Nat. Commun., 2018, 9, 3794 CrossRef PubMed.
- Y. Zhang, Y. Zhao, T. Otroshchenko, S. Han, H. Lund, U. Rodemerck, D. Linke, H. Jiao, G. Jiang and E. V. Kondratenko, J. Catal., 2019, 371, 313–324 CrossRef CAS.
- N. Jeon, H. Choe, B. Jeong and Y. Yun, Catal. Today, 2019 DOI:10.1016/j.cattod.2019.12.012.
- T. Otroshchenko, O. Bulavchenko, H. V. Thanh, J. Rabeah, U. Bentrup, A. Matvienko, U. Rodemerck, B. Paul, R. Kraehnert, D. Linke and E. V. Kondratenko, Appl. Catal., A, 2019, 585, 117189 CrossRef CAS.
- Z. Xie, Y. Ren, J. Li, Z. Zhao, X. Fan, B. Liu, W. Song, L. Kong, X. Xiao, J. Liu and G. Jiang, J. Catal., 2019, 372, 206–216 CrossRef CAS.
- C. Copéret, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Núñez-Zarur and P. A. Zhizhko, Chem. Rev., 2016, 116, 323–421 CrossRef PubMed.
- M. K. Samantaray, V. D'Elia, E. Pump, L. Falivene, M. Harb, S. Ould Chikh, L. Cavallo and J.-M. Basset, Chem. Rev., 2020, 120, 734–813 CrossRef CAS PubMed.
- M. K. Samantaray, E. Pump, A. Bendjeriou-Sedjerari, V. D'Elia, J. D. A. Pelletier, M. Guidotti, R. Psaro and J.-M. Basset, Chem. Soc. Rev., 2018, 47, 8403–8437 RSC.
- M. E. Harlin, V. M. Niemi, A. O. I. Krause and B. M. Weckhuysen, J. Catal., 2001, 203, 242–252 CrossRef CAS.
- J. M. Kanervo, M. E. Harlin, A. O. I. Krause and M. A. Bañares, Catal. Today, 2003, 78, 171–180 CrossRef CAS.
- T. P. Otroshchenko, U. Rodemerck, D. Linke and E. V. Kondratenko, J. Catal., 2017, 356, 197–205 CrossRef CAS.
- X. Gao, M. A. Bañares and I. E. Wachs, J. Catal., 1999, 188, 325–331 CrossRef CAS.
- Y.-P. Tian, Y.-A. Liu, X.-M. Liu and Z.-F. Yan, Catal. Sci. Technol., 2018, 8, 5473–5481 RSC.
- Z. Wu, H. S. Kim, P. C. Stair, S. Rugmini and S. D. Jackson, J. Phys. Chem. B, 2005, 109, 2793–2800 CrossRef CAS PubMed.
- J. L. Male, H. G. Niessen, A. T. Bell and T. Don Tilley, J. Catal., 2000, 194, 431–444 CrossRef CAS.
- S. C. Su and A. T. Bell, J. Phys. Chem. B, 1998, 102, 7000–7007 CrossRef CAS.
- I. E. Wachs, Catal. Today, 2005, 100, 79–94 CrossRef CAS.
- C. Zhao and I. E. Wachs, J. Catal., 2008, 257, 181–189 CrossRef CAS.
- U. Das, G. Zhang, B. Hu, A. S. Hock, P. C. Redfern, J. T. Miller and L. A. Curtiss, ACS Catal., 2015, 5, 7177–7185 CrossRef CAS.
- C. Xiong, S. Chen, P. Yang, S. Zha, Z.-J. Zhao and J. Gong, ACS Catal., 2019, 9, 5816–5827 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc00690d |
| This journal is © The Royal Society of Chemistry 2020 |