
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceH-bond donor-directed switching of diastereoselectivity in the Michael addition of α-azido ketones to nitroolefins†
Pei-Gang
Ding
a,
Feng
Zhou
 a,
Xin
Wang
*b,
Qiu-Hua
Zhao
a,
Jin-Sheng
Yu
ac and
Jian
Zhou
*ad
a,
Xin
Wang
*b,
Qiu-Hua
Zhao
a,
Jin-Sheng
Yu
ac and
Jian
Zhou
*ad
aShanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, Shanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, 3663N Zhongshan Road, Shanghai 200062, China
bCollege of Chemistry, Sichuan University, Chengdu, Sichuan 610064, China
cKey Laboratory of Tropical Medicinal Resource Chemistry of Ministry of Education, Hainan Normal University, Haikou 571158, China
dState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China. E-mail: wangxin@scu.edu.cn; jzhou@chem.ecnu.edu.cn
First published on 11th March 2020
Abstract
The development of catalyst-controlled stereodivergent asymmetric catalysis is important for providing facile access to all stereoisomers of chiral products with multiple stereocenters from the same starting materials. Despite progress, new design strategies for diastereodivergent asymmetric catalysis are still highly desirable. Here we report the potency of H-bond donors as the governing factor to tune diastereoselectivity in a highly diastereoselective switchable enantioselective Michael addition of α-azido ketones to nitroolefins. While a newly developed bifunctional tertiary amine, phosphoramide, preferentially afforded syn-adducts, an analogous squaramide catalyst selectively gave anti-adducts. The resulting multifunctional tertiary azides can be converted to spiro-pyrrolidines with four continuous stereocenters in a one-pot operation. Mechanistic studies cast light on the control of diastereoselectivity by H-bond donors. While the squaramide-catalyzed reaction proceeded with a transition state with both squaramide N–H bonds binding to an enolate intermediate, an unprecedented model was proposed for the phosphoramide-mediated reaction wherein an amide N–H bond and an alkylammonium ion formed in situ interact with nitroolefins, with the enolate stabilized by nonclassical C–H⋯O hydrogen-bonding interactions.
Introduction
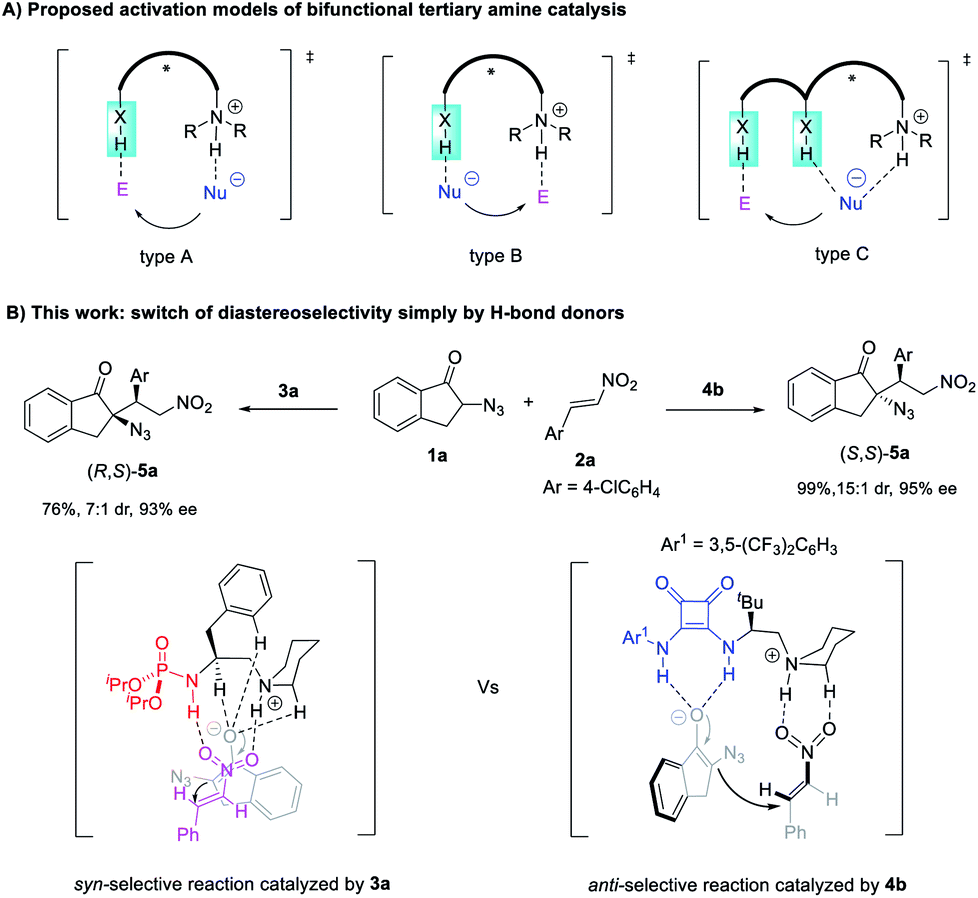
Chiral molecules with multiple stereocenters in principle exist as enantiomeric pairs of diastereomers.1 It is important to access all the possible stereoisomers because the absolute and relative configurations often influence their properties such as bioactivity.2 However, the synthesis of a single diastereomer and enantiomer is inherently favored in most catalytic enantioselective reactions forming multiple stereocenters.3 Developing catalyst-controlled stereodivergent processes to access all the possible stereoisomers from the same starting materials is challenging.4 Despite the progress made by varying the reaction conditions5 or chiral catalysts6,7 or by dual catalysis,8 new strategies to realize diastereodivergent asymmetric catalysis (DAC) are still highly sought after.By contrast, while H-bond donor-containing bifunctional catalysts have found wide application,9 controlling the diastereoselectivity simply by varying the H-bond donor of such a catalyst is largely unexplored.10 For bifunctional tertiary amine catalysis, theoretical studies have proposed three types of working models that differ in how H-bond donors of the catalysts interact with the nucleophile and electrophile (Scheme 1A).11–15 The ion pair-hydrogen bonding model (type A) was initially proposed by Wynberg11a and supported by theoretical studies by Cucinotta and Gervasio.11b The Brønsted acid-hydrogen bonding model (type B) was revealed by Houk et al. via quantum mechanical calculations.12 The type A model differs from type B in that the H-bond donor of the catalyst is used to activate the electrophile and to stabilize the nucleophilic intermediate, respectively, with the simultaneously formed alkylammonium ion acting as a Brønsted acid to interact with the rest of the nucleophiles or electrophiles, respectively.
 | ||
| Scheme 1 DAC enabled by H-bond donors. | ||
When dual H-bond donors such as (thio)urea are involved, the reaction may proceed via a transition state of the type A model with both N–H bonds interacting with the electrophile, as suggested by Takemoto via experimental studies13a and supported by theoretical studies,13b–d or via model B with both N–H bonds binding to nucleophiles, as revealed by the theoretical calculation by Pápai14a,b and Houk.14c A more complicated model (type C) was also proposed by Wang et al., with the synergistic action of alkylammonium ions and one N–H bond of (thio)urea on the nucleophile.15
Based on these mechanistic insights, we speculate that it is possible to control the diastereoselectivity simply by altering the H-bond donors because they differ greatly from each other in terms of the structure and number of X–H bonds. The interplay of different H-bond donors with alkylammonium ions may create distinct chiral pockets capable of controlling the relative spatial position of both reaction partners to accomplish a diastereodivergent bond-forming process. Here we report the successful reversal of diastereoselectivity in an unprecedented Michael addition of α-azido indanones to nitroolefins catalyzed by bifunctional tertiary amines, simply by varying the H-bond donor of the catalyst from phosphoramide to squaramide (Scheme 1B).
Results and discussion
Catalyst preparation
During our efforts to construct a fully substituted carbon stereocenter by bifunctional tertiary amine catalysis,16 we initiated a plan to develop phosphoramide-based bifunctional catalysts. This is because phosphoramide is seldom present in chiral ligands or organocatalysts, even though it is a unique single H-bond donor featuring two amide substituents as the shielding group. We have developed two phosphoramide-amine catalysts 617a–c and 717d (Scheme 2), which can achieve better enantioselectivity than their analogues with other H-bond donors such as amides, sulfonamides, (thio)urea, and squaramide in two distinct Michael addition reactions, respectively. Inspired by these results, we hope to take advantage of the structural differences between phosphoramide and another H-bond donor to develop diastereodivergent reactions.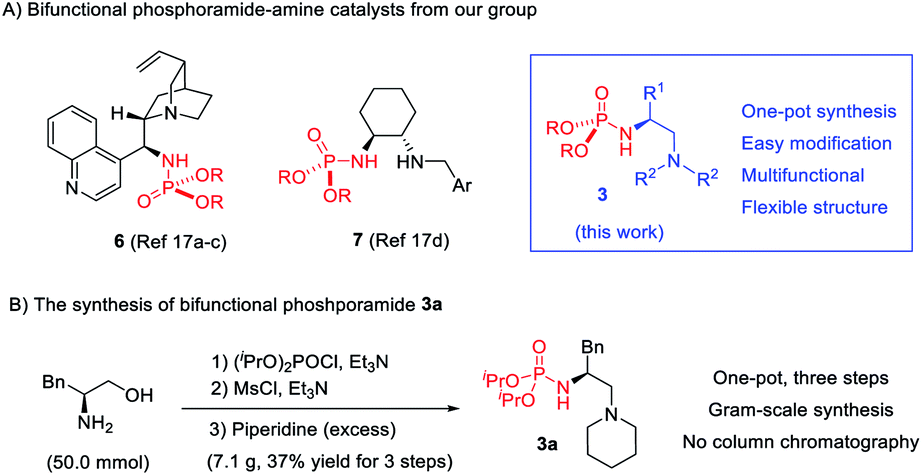 | ||
| Scheme 2 Bifunctional phosphoramide-amine catalysts. | ||
A new phosphoramide-tertiary amine catalyst 3 with a flexible acyclic structure is then developed, the three substituents (R, R1, and R2) of which can be modified to improve the stereoselectivity. Given the sufficient structural differences, phosphoramide and the alkylammonium ion formed in situ may organize the reaction partners in an orientation substantially different from that effected by another H-bond donor, leading to reversal of diastereoselectivity. Notably, since phosphoramide can serve as an N-protecting group, the synthesis of 3 is extremely easy, a simple one-pot three-step operation without column chromatography purification, as exemplified by the access of 3a in 37% overall yield from (S)-phenylalaninol (for details, see the ESI†). The corresponding bifunctional squaramide 4, urea 8, and sulfonamide 9 are known catalysts.18
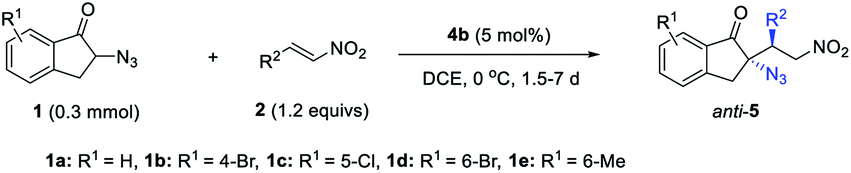
Recently, the deprotonative activation of α-azido carbonyl compounds emerged as a fruitful approach for catalytic asymmetric synthesis of α-chiral azides.19 However, despite achievements, including an elegant diastereodivergent asymmetric aldol reaction of α-azido 7-azaindoline acetamides and ortho-substituted benzaldehydes developed by Shibasaki et al.,20 this method has not been applied in the diastereodivergent synthesis of chiral tertiary azides. This lack of application is possibly because, upon treating with a base, α-azido carbonyl compounds may undergo decomposition.21 In the construction of tetrasubstituted carbon stereocenters, the reactivity is generally lower than that of tertiary ones,22 so this side reaction may compete with the desired asymmetric reaction. In view of this challenge, along with some successful DAC based on indanone derivatives and nitroolefins,23 we consider exploiting an unprecedented Michael reaction of α-azido ketones 1 and nitroolefin 2 to test the idea of varying the H-bond donor of bifunctional tertiary amines to alter the diastereoselectivity (Table 1). We speculate that the simultaneous activation of both α-azido indanones and nitroolefins by bifunctional tertiary amines should facilitate the desired Michael addition, while minimizing unwanted side reactions. It should be noted that protocols for the catalytic enantioselective synthesis of chiral tertiary azides are still limited.24 This new reaction would provide facile access to multifunctional chiral tertiary azides 5 of high synthetic value.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Cat. | X (mol%) | Solvent | Time (d) | Yielda (%) |
syn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :antib :antib |
eec (%) |
|
a Isolated yield.
b Determined by 1H NMR analysis of the crude mixture.
c Determined by HPLC analysis.
d Room temperature.
e 0.3 mmol, with 90 mg powdered 5 Å MS, [1a] = 0.3 M.
f [1a] = 0.1 M. [1a] = 0.2 M without noted. DCM: CH2Cl2. DCE:CH2ClCH2Cl.
|
|||||||
| 1d | DABCO | 20 | DCM | 2 | 45 | 1:1.2 |
— |
| 2 | 3a | 20 | Toluene | 4 | 80 | 5.4:1 |
88 |
| 3e | 3a | 20 | Toluene | 7 | 76 | 7.0:1 |
93 |
| 4 | 4a | 20 | Toluene | 4 | 80 | 1:11 |
56 |
| 5f | 4a | 5 | DCE | 2 | 95 | 1:24 |
70 |
| 6f | 4b | 5 | DCE | 2 | 90 | 1:15 |
95 |
| 7 | 8 | 20 | Toluene | 4 | 52 | 3.9:1 |
72 |
| 8 | 9 | 20 | Toluene | 4 | 51 | 3.5:1 |
73 |
| 9 | 6a | 20 | Toluene | 4 | 33 | 1.6:1 |
56 |
| 10 | 10 | 20 | Toluene | 4 | 79 | 1:5.2 |
63 |
| 11 | 11 | 20 | Toluene | 1 | 89 | 1:3.0 |
61 |
| 12 | 12 | 20 | Toluene | 1 | 63 | 1.2:1 |
84 |

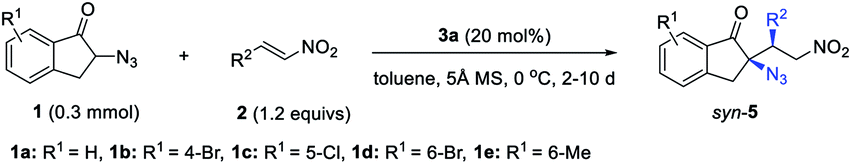
Gratifyingly, the switching of diastereoselectivity of the Michael addition of 1a to 2a is indeed observed by varying the H-bond donor of the bifunctional amine catalysts. The model addition reaction of 1a to 2a was performed at 0 °C, to minimize the decomposition of α-azido indanone 1a in the presence of a basic catalyst21 because the racemic version mediated by 1,4-diaza[2.2.2]bicyclooctane (DABCO) gave anti-5a in 1.2:1 dr and around 45% yield, with a substantial amount of side products (entry 1, Table 1). The poor dr also suggested trivial substrate bias4i,7g in the diastereoselectivity of this reaction. To our delight, phosphoramide catalyst 3a gave syn-5a in 88% ee and 5.4:1 dr (entry 2). The addition of MS 5 Å improved the result to 93% ee and 7.0:1 dr (entry 3). When replacing the phosphoramide with a squaramide, the resulting catalyst 4a afforded anti-5a in 11:1 dr (entry 4) and up to 24:1 dr with 70% ee by varying the solvent from toluene to DCE (entry 5). This result unambiguously shows the possibility of tuning diastereoselectivity simply by altering the H-bond donor in bifunctional tertiary amine-catalyzed reactions. Further studies revealed that squaramide catalyst 4 was more active, and the catalyst loading could be decreased to 5 mol% (entry 5). The use of 5 mol% tert-butyl-substituted squaramide 4b allowed the reaction to work well in DCE, affording anti-5a in up to 15:1 dr and 95% ee (entry 6). Bifunctional catalysts 8 and 9 bearing urea or sulfonamide as the H-bond donor were also examined and gave syn-5a as the major product, albeit with inferior results (entries 7 and 8 vs. 2). To examine the role of the catalyst skeleton in switching the diastereoselectivity, the cinchona alkaloid-derived phosphoramide 6a and squaramide 10 were also used. Again, a reversal of diastereoselectivity was observed (entry 9 vs. 10), although the dr values were less effective than those obtained by using catalysts 3a and 4b (entries 2 and 6). This further supports that phosphoramide and squaramide are mainly responsible for this diastereodivergent reaction but that the catalyst structure also has an impact. Furthermore, catalysts 11 and 12 used by Deng to develop diastereodivergent Michael addition of α-cyanoindanone to 2-chloroacrylonitrile10 proved to be less effective in the control of diastereoselectivity (entries 11 vs. 12). These results justified the necessity of developing new bifunctional tertiary amines for stereodivergent asymmetric catalysis.
In the following, the scope of bifunctional phosphoramide 3a mediated syn-selective Michael addition reactions was evaluated. All reactions were carried out in toluene at 0 °C, in the presence of 20 mol% 3a and powdered MS 5 Å, as shown in Table 2. A broad range of nitroolefins 2a–g with different β-aryl substituents all gave the desired syn-5a–g in 73–85% yield and with 7:1 to 10:1 dr values, with 93–96% ee (entries 1–7, Table 2). β-Alkenyl nitroolefins 2h–l furnished adducts syn-5h–l in >12:1 dr and 92–94% ee (entries 8–12). Differently substituted α-azido indanones 1b–e also worked well to give syn-5m–p with high dr and ee values (entries 13–16). The relative and absolute configurations of syn-5k were assigned by single-crystal X-ray diffraction analysis (see the ESI†). Notably, β-CF3 or CF2H nitroolefins 2m–n25 are also viable substrates, affording fluorinated azides syn-5q–s with high to excellent ee values, albeit in moderate dr (entries 17–19). Their reactivity appeared to be lower than that of β-aryl olefins, but the addition of 20 mol% K2HPO4·3H2O facilitated the reaction.26 In addition, α-azido chromanone 1f was a viable substrate giving syn-5t in 9:1 dr and 93% ee. α-Azido 2,3-dihydroquinolinone 1g could also work, but the desired product syn-5u was obtained in only 3:1 dr and 65% ee. However, α-azido benzosuberone 1h and cyclic ketones 1i–k failed to react with nitroolefin 2i.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | 1 | 2: R2 | Time (d) | syn-5 | Yielda (%) | drb | eec (%) |
| a Isolated yield. b Determined by 1H NMR analysis of the crude reaction mixture. c Determined by HPLC analysis. d −20 °C. e 5 Å MS was replaced by 20 mol% K2HPO4·3H2O. | |||||||
| 1 | 1a | 2a: 4-ClC6H4 | 7 | syn-5a | 76 | 7:1 |
93 |
| 2 | 1a | 2b: 3-ClC6H4 | 8 | syn-5b | 79 | 7:1 |
93 |
| 3 | 1a | 2c: 2-FC6H4 | 7 | syn-5c | 73 | 9:1 |
95 |
| 4 | 1a | 2d: Ph | 9 | syn-5d | 85 | 9:1 |
96 |
| 5 | 1a | 2e: 4-MeC6H4 | 10 | syn-5e | 79 | 9:1 |
96 |
| 6 | 1a | 2f: 2-naphthyl | 7 | syn-5f | 81 | 8:1 |
93 |
| 7 | 1a | 2g: 2-thienyl | 7 | syn-5g | 85 | 10:1 |
96 |
| 8 | 1a |
2h: (E)-PhCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH CH |
8 | syn-5h | 76 | 15:1 |
94 |
| 9 | 1a |
2i: (E)-4-BrC6H4CHCH |
5 | syn-5i | 70 | 13:1 |
93 |
| 10 | 1a |
2j: (E)-3-FC6H4CHCH |
6 | syn-5j | 68 | 12:1 |
92 |
| 11 | 1a |
2k: (E)-2-FC6H4CHCH |
8 | syn-5k | 81 | 12:1 |
94 |
| 12 | 1a |
2l: (E)-2-MeC6H4CHCH |
9 | syn-5l | 80 | 13:1 |
94 |
| 13d | 1b |
2h: (E)-PhCHCH |
4 | syn-5m | 88 | 12:1 |
90 |
| 14 | 1c |
2h: (E)-PhCHCH |
2 | syn-5n | 79 | 12:1 |
88 |
| 15d | 1d |
2h: (E)-PhCHCH |
4 | syn-5o | 72 | 13:1 |
93 |
| 16 | 1e |
2h: (E)-PhCHCH |
8 | syn-5p | 69 | 14:1 |
95 |
| 17e | 1c | 2m: CF3 | 3 | syn-5q | 75 | 3:1 |
90 |
| 18e | 1d | 2m: CF3 | 2 | syn-5r | 76 | 4:1 |
92 |
| 19e | 1d | 2n: CF2H | 6 | syn-5s | 52 | 2:1 |
87 |
|
|||||||
The scope of the anti-selective Michael addition was examined by running the reaction in DCE at 0 °C, under the catalysis of 5 mol% squaramide 4b. As shown in Table 3, β-aryl nitroolefins 2a–g worked well with indanone 1a to give the desired anti-5a–g in 90–99% yield, 8:1 to 25:1 dr and 91–97% ee (entries 1–7). Unexpectedly, β-alkenyl nitroolefins were less active under these conditions. For example, it took 10 mol% catalyst 4b to mediate the reaction of 2h and 1a, affording anti-5h in a diminished 6:1 dr with 90% ee (entry 8). Different α-azido indanones 1b–e reacted smoothly with nitroolefin 2a to deliver anti-5v–y in 86–99% yield, 10:1 to 30:1 dr, and 93–96% ee (entries 9–12). The relative and absolute configurations of product anti-5g were assigned by X-ray diffraction analysis (see the ESI†). The reversal of diastereoselectivity was also achieved when β-CF3 or CF2H nitroethylene 2m–n was used, affording the desired trifluoromethylated anti-5q–r or difluoromethylated anti-5s in 88–93% yield, 7:1 to 19:1 dr and 94% ee (entries 13–15). These results constitute a rare example of diastereodivergent asymmetric trifluoro- and difluoromethylation reactions.6f,27 Considering the importance of molecules bearing a CF3 or CF2H group at the chiral center for medicinal research,28,29 this protocol may be potentially useful for drug discovery.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | 1 | 2: R2 | T (d) | anti-5 | Yielda (%) | drb | eec (%) |
| a Isolated yield. b Determined by 1H NMR analysis of the crude mixture. c Determined by HPLC analysis. d 10 mol% 4b. e 20 mol% 4b and 20 mol% K2HPO4·3H2O. f 10 mol% 4b and 10 mol% K2HPO4·3H2O. g 20 mol% 4b. | |||||||
| 1 | 1a | 2a: 4-ClC6H4 | 2.5 | anti-5a | 99 | 15:1 |
95 |
| 2 | 1a | 2b: 3-ClC6H4 | 2.5 | anti-5b | 99 | 14:1 |
95 |
| 3 | 1a | 2c: 2-FC6H4 | 3 | anti-5c | 95 | 8:1 |
91 |
| 4 | 1a | 2d: Ph | 5 | anti-5d | 90 | 20:1 |
96 |
| 5 | 1a | 2e: 4-MeC6H4 | 5 | anti-5e | 91 | 25:1 |
95 |
| 6 | 1a | 2f: 2-naphthyl | 5 | anti-5f | 99 | 22:1 |
95 |
| 7 | 1a | 2g: 2-thienyl | 5 | anti-5g | 97 | 20:1 |
97 |
| 8d | 1a |
2h: (E)-PhCHCH |
5 | anti-5h | 78 | 6:1 |
90 |
| 9 | 1b | 2a: 4-ClC6H4 | 1.5 | anti-5v | 89 | 10:1 |
93 |
| 10 | 1c | 2a: 4-ClC6H4 | 3 | anti-5w | 86 | 17:1 |
95 |
| 11 | 1d | 2a: 4-ClC6H4 | 2 | anti-5x | 91 | 30:1 |
96 |
| 12 | 1e | 2a: 4-ClC6H4 | 4 | anti-5y | 99 | 14:1 |
94 |
| 13e | 1c | 2m: CF3 | 3.5 | anti-5q | 88 | 11:1 |
94 |
| 14f | 1d | 2m: CF3 | 3.5 | anti-5r | 91 | 19:1 |
94 |
| 15e | 1d | 2n: CF2H | 3 | anti-5s | 93 | 7:1 |
94 |
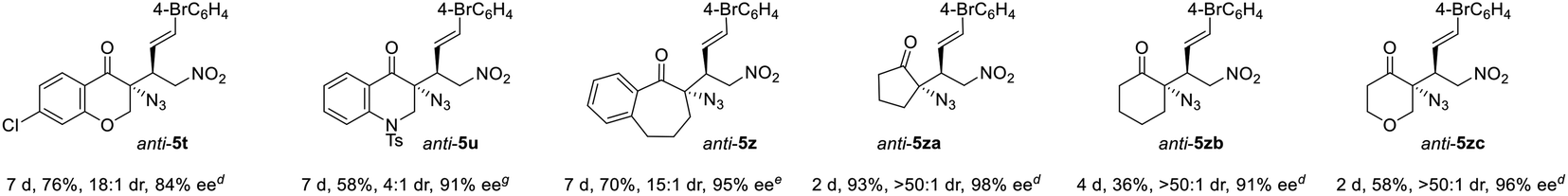
|
|||||||
To our delight, owing to the higher activity of the squaramide catalyst, four other types of α-azido ketones readily underwent the desired anti-selective addition reaction. α-Azido chromanone 1f gave product anti-5t in 18:1 dr and 84% ee, and α-azido 2,3-dihydroquinolinone 1g furnished anti-5u in 4:1 dr and 91% ee. Notably, α-azido benzosuberone 1h and cyclic ketones 1i–k readily afforded the corresponding tertiary azides anti-5z and anti-5za–zc in 15:1 to >50:1 dr and 91–98% ee.
This stereodivergent protocol could be extended to acyclic α-azido ketones (Scheme 3), as exemplified by the reversal of diastereoselectivity of the reaction of α-azido acetophenone 13 with nitroolefin 2i. The syn-14 was obtained in 3:1 dr and 81% ee by using phosphoramide 3a as the catalyst, whilst anti-14 was furnished in 5:1 dr and 92% ee under the catalysis of squaramide 4b. Nevertheless, due to the low activity of α-azido acetophenone, the reaction conversion is low in both cases. The relative and absolute configurations of product anti-14 were assigned by X-ray diffraction analysis (see the ESI†).
 | ||
| Scheme 3 DAC reaction of acyclic α-azido ketones. | ||
Product elaboration
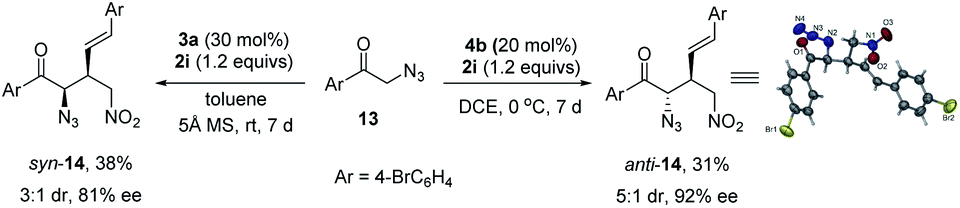
These multifunctional tertiary azides 5 are versatile synthons for aza-spirocycles. For example, by using both enantiomers of catalysts 3a and 4b, diastereodivergent asymmetric synthesis of four possible stereoisomers of 5a was achieved with high yield, dr value, and >90% ee (Scheme 4). They can be converted to the four stereoisomers of spirocycle 15 with acceptable overall yield by a one-pot three-step operation. Structurally diverse spirocycles are privileged scaffolds in natural products, drugs, and bioactive compounds. The thus obtained spiro-pyrrolidines 15 with four continuous stereocenters are of interest for medicinal research, and their synthesis via asymmetric [3+2] cycloaddition of nitroolefins and azomethine ylides is unknown.30 In addition, the skeleton 15 may be used for developing new chiral ligands or organocatalysts.31 | ||
| Scheme 4 Synthesis of all stereoisomers of 5a and their conversion to spiro-pyrrolidines 15. | ||
Mechanistic studies
The reversal of diastereoselectivity simply by varying the H-bond donor of bifunctional tertiary amines is intriguing. Accordingly, mechanistic investigations were conducted. First, to confirm the importance of bifunctional tertiary amine catalysis, the corresponding N-methylated catalyst 3b and phosphoramide 3f were subjected to the reaction of 1a and 2a (Scheme 5). Without free N–H bonds, tertiary amine 3b exhibited very poor catalytic properties, and monofunctional phosphoramide 3f was almost inert. This confirmed that both the phosphoramide N–H bond and tertiary amine were crucial for the syn-selective Michael addition. In the case of squaramide catalyst 4b, its two N–H bonds seemed to have different roles in controlling the stereoselectivity, and both were indispensable for the observed reactivity. While the squaramide catalyst 4b afforded anti-5a in a high 15:1 dr, the shielding of the C3 N–H bond of 4b by a methyl group led to the reversal of diastereoselectivity because the corresponding 4c gave syn-5a in 5.4:1 dr, identical to that obtained using phosphoramide 3a. This finding further supported our idea of controlling the diastereoselectivity by varying the structure of H-bond donors. By contrast, with the methylated C4 N–H bond, catalyst 4d furnished anti-5a in a poor 1.4:1 dr and 41% ee, much lower than that obtained by squaramide 4b, suggesting that the C4 N–H bond is important for achieving high anti selectivity and enantioselectivity.
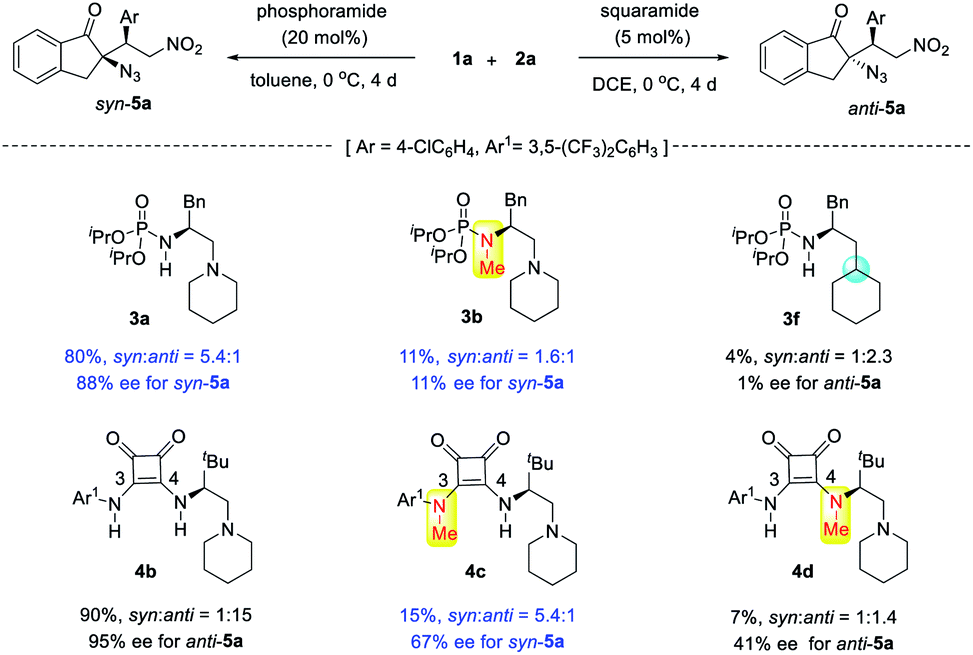 | ||
| Scheme 5 Control experiments. | ||
Next, whether nonlinear effects (NLE) were operative in the reaction course was also studied to gain further insight into the behavior of bifunctional tertiary amines.
When the reaction of 1a and 2a was conducted with 20 mol% catalyst 3a prepared with reduced enantiomeric excess, a linear relationship between the catalyst ee value and that of the product syn-5a was observed (Fig. 1), suggesting a monomeric catalytic species.32 If the linear relationship was not followed, the association of chiral phosphoramide 3a might be expected to produce a diastereomeric active species. Unfortunately, the study of the NLE of squaramide 4b-mediated anti-selective reaction failed due to the poor solubility of 4b in DCE.
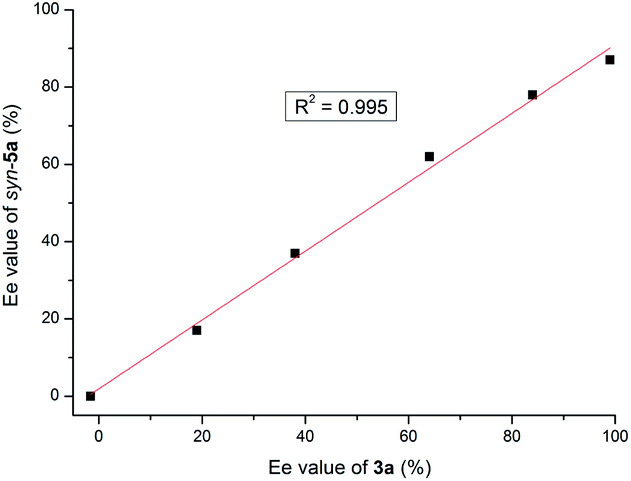 | ||
| Fig. 1 Result of the nonlinear experiment of 3a with syn-5a. | ||
To probe the possible mechanism for this DAC, NMR analysis was also carried out (for details, see Section 7 of the ESI†). However, although phosphoramide catalyst 3a could form H-bonding interactions with α-azido indanone 1a and nitroolefin 2e, no useful information about the catalyst-substrate recognition model of the transition state was obtained. In addition, the NMR analysis of squaramide 4b-mediated reaction failed, due to its extremely poor solubility in CD2Cl2 or toluene-d8. Therefore, theoretical studies were carried out to cast light on the mechanism of the above syn- and anti-selective Michael reactions, and computed transition state (TS) structures stabilized by different H-bonding interactions are shown below (for details, see the ESI†).
The optimized model for the phosphoramide 3a-catalyzed reaction of 1a and 2d, TSI2A, is characterized by the double H-bonding interactions between nitroolefin 2d with the phosphoramide N–H bond and the in situ-generated alkylammonium ion, as well as three kinds of nonclassical C–H⋯O hydrogen-bonding interactions33 between the oxygen anion of enolate derived from 1a with the N+–C–H of the quininium ion,34 the C–H bond of the phenyl ring,35 and the α-H of the amide nitrogen.36 The relatively short H⋯O distances observed for the three kinds of C–H⋯O contacts were 2.29, 2.31 and 2.25 Å, respectively, obviously shorter than the sum of van der Waals radii of an oxygen atom (1.5 Å) and a carbon-bonded hydrogen (∼1.2 Å).34a These important H-bonding interactions stabilized the TS and directed the Re face nucleophilic addition of enolate to the Si face of nitroolefin to give the syn-adduct (Fig. 2).
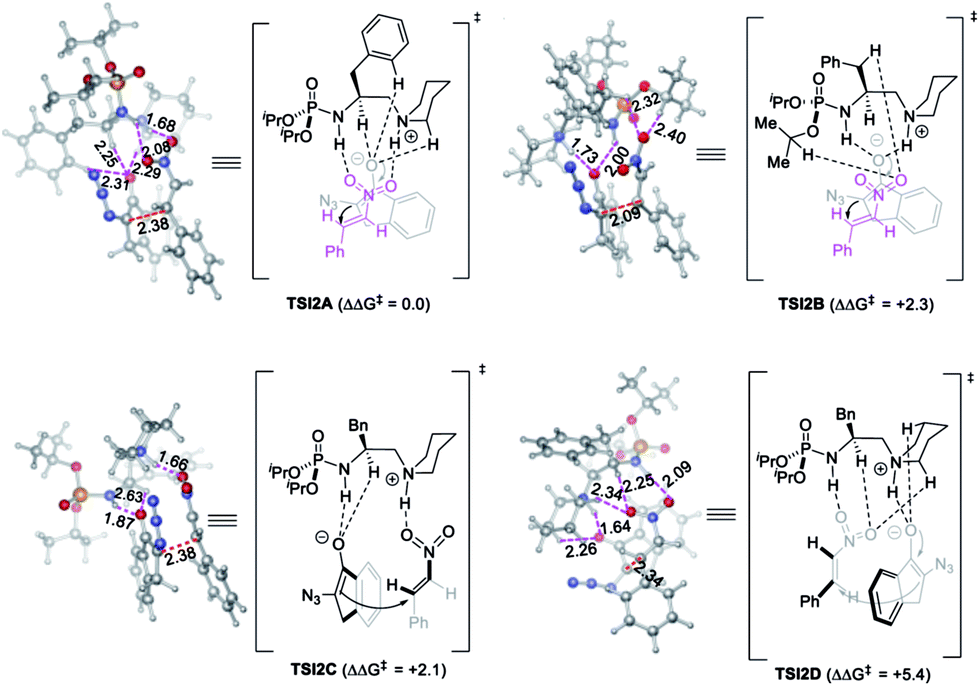 | ||
| Fig. 2 The optimized structures for TS modes of phosphoramide 3a-catalyzed α-azido indanone 1a and nitroolefin 2d. The relative Gibbs free energies are in kcal mol−1, which were calculated at the IEFPCM-M06-2X-D3/6-311++G(d,p)//IEFPCM-M06-2X/6-31G(d,p) level. The bond distances are given in Å. | ||
Notably, this model is unprecedented in the literature because the electrophile interacts with both the H-bond donor and alkylammonium ion, whilst the enolate is stabilized by the nonclassical C–H⋯O hydrogen-bonding interactions. This is distinct from the three models shown in Scheme 1A. The alternative model of double H-bonding interactions with the enolate intermediate (TSI2B), type B Brønsted acid-hydrogen bonding model (TSI2C), and type A ion pair-hydrogen bonding model (TSI2D) are higher in free energy by 2.3, 2.1 and 5.4 kcal mol−1, respectively. This is possibly because the strong steric repulsion of the two isopropoxy groups of the phosphoramide prevents the formation of H-bonding interaction of the enolate with either the phosphoramide or the alkylammonium ion. This model to some extent agreed with the observation that the NMR analysis of the mixture of 3a, 1a, and 2e showed a significantly bigger change in the chemical shift of the phosphoramide N–H bond than that by mixing of 3a with either 1a or 2e (g vs. e and f, Fig. S2†). This finding further suggests that it is interesting to employ phosphoramide, the only single H-bond donor bearing two amide shielding groups, to develop new chiral ligands or organocatalysts.
On the other hand, the chiral squaramide 4b-catalyzed reaction proceeded via a typical Brønsted acid-hydrogen bonding model (TSII2A, Fig. 3): both N–H bonds of the squaramide interacted with α-azido enolate via H-bonding interactions, with a simultaneously generated ammonium moiety bound to nitroolefin, leading to a favorable transition state that enabled the Si face nucleophilic attack of α-azido enolate to the Si face of nitroolefin to afford the anti-product. This working model could also reasonably explain why N-methylated squaramide 4c gave syn-adduct 5 as the major diastereomer (Scheme 5), because the lack of a C3 N–H bond made it impossible for squaramide 4c to form the calculated Brønsted acid-hydrogen bonding model in the transition state, but offered the possibility of forming a transition state resembling that developed by phosphoramide catalyst 3a. The ion pair-hydrogen bonding model (TSII2B) is also obtained, but is higher in free energy by 6.8 kcal mol−1. A transition state with an azido group as an H-bond acceptor37 was also observed (TSII2C) but with a much higher free energy (14.2 kcal mol−1).
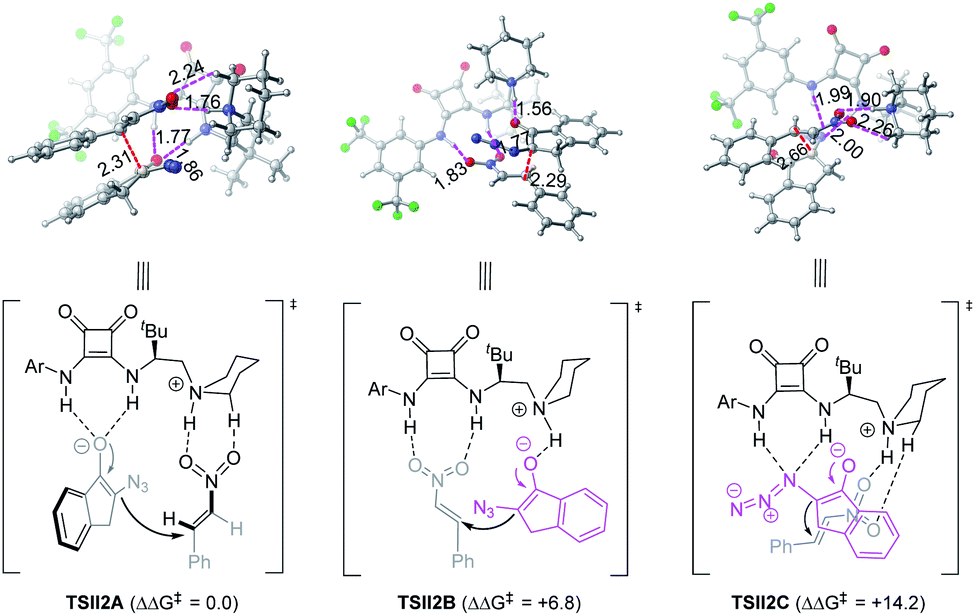 | ||
| Fig. 3 The optimized structures for TS modes of squaramide 4b-catalyzed α-azido indanone 1a and nitroolefin 2d. The relative Gibbs free energies are in kcal mol−1, which were calculated at the IEFPCM-M06-2X-D3/6-311++G(d,p)//IEFPCM-M06-2X/6-31G(d,p) level. The bond distances are given in Å. | ||
Conclusions
In summary, we have demonstrated that H-bond donors can play a key role in controlling diastereoselectivity in a catalytic enantioselective reaction. Accordingly, an unprecedented highly enantioselective diastereodivergent asymmetric Michael addition of α-azido ketones to nitroolefins was developed by using our newly developed bifunctional phosphoramide catalyst 3a or the analogous squaramide catalyst 4b. Both cyclic and acyclic α-azido ketones, along with a broad range of nitroolefins, including β-CF3 or CF2H nitroolefins, can work under the established conditions. The resulting multifunctional tertiary azides 5 could be used to synthesize aza-spirocycles bearing four continuous stereocenters without a loss of enantioselectivity. Mechanistic studies revealed that the reversal of diastereoselectivity originates from the alternative catalyst-substrate recognition model due to the variation of the H-bond donor of the catalysts from phosphoramide to squaramide. The development of new DAC reactions by using other types of H-bond donors containing bifunctional catalysts is ongoing in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the NSFC (21725203 and 21971067), the Ministry of Education (PCSIRT) and the Fundamental Research Funds for the Central Universities.Notes and references
- G. E. K. Branch and T. L. Hill, J. Org. Chem., 1940, 5, 86 CrossRef CAS.
- T. L. Lemke, D. A. Williams, V. F. Roche and S. W. Zito, Foye's Principles of Medicinal Chemistry, Lippincott Williams & Wilkins: Wolters Kluwer, Baltimore, 7th edn, 2013 Search PubMed.
- R. S. Atkinson, Stereoselective Synthesis, Wiley, Chichester, 1995 Search PubMed.
- For selected reviews: (a) L. C. Miller and R. Sarpong, Chem. Soc. Rev., 2011, 40, 4550 RSC; (b) C. S. Schindler and E. N. Jacobsen, Science, 2013, 340, 1052 CrossRef CAS PubMed; (c) M. T. Oliveira, M. Luparia, D. Audisio and N. Maulide, Angew. Chem., Int. Ed., 2013, 52, 13149 CrossRef CAS PubMed; (d) G. Zhan, W. Du and Y.-C. Chen, Chem. Soc. Rev., 2017, 46, 1675 RSC; (e) L. Lin and X. Feng, Chem.–Eur. J., 2017, 23, 6464 CrossRef CAS PubMed; (f) M. Bihania and J. C.-G. Zhao, Adv. Synth. Catal., 2017, 359, 534 CrossRef; (g) S. Krautwald and E. M. Carreira, J. Am. Chem. Soc., 2017, 139, 5627 CrossRef CAS PubMed; (h) I. P. Beletskaya, C. Nájera and M. Yus, Chem. Rev., 2018, 118, 5080 CrossRef CAS PubMed; (i) S. Masamune, W. Choy, J. S. Petersen and L. R. Sita, Angew. Chem., Int. Ed., 1985, 24, 1 CrossRef.
- For selected examples by varying temperature: (a) Z.-Z. Huang, Y.-B. Kang, J. Zhou, M.-C. Ye and Y. Tang, Org. Lett., 2004, 6, 1677 CrossRef CAS PubMed; by varying additives: (b) X. Tian, C. Cassani, Y. Liu, A. Moran, A. Urakawa, P. Galzerano, E. Arceo and P. Melchiorre, J. Am. Chem. Soc., 2011, 133, 17934 CrossRef CAS PubMed; by varying solvents: (c) X. Wu, Z. Chen, Y.-B. Bai and V. M. Dong, J. Am. Chem. Soc., 2016, 138, 12013 CrossRef CAS PubMed; (d) Q. Cheng, F. Zhang, Y. Cai, Y.-L. Guo and S.-L. You, Angew. Chem., Int. Ed., 2018, 57, 2134 CrossRef CAS PubMed.
- Selected examples by varying the chiral ligand of the metal complex: (a) X.-X. Yan, Q. Peng, Q. Li, K. Zhang, J. Yao, X.-L. Hou and Y.-D. Wu, J. Am. Chem. Soc., 2008, 130, 14362 CrossRef CAS PubMed; (b) M. Luparia, M. T. Oliveira, D. Audisio, F. Frébault, R. Goddard and N. Maulide, Angew. Chem., Int. Ed., 2011, 50, 12631 CrossRef CAS PubMed; (c) X. Hao, L. Lin, F. Tan, C. Yin, X. Liu and X. Feng, ACS Catal., 2015, 5, 6052 CrossRef CAS; (d) H.-L. Teng, Y. Luo, M. Nishiura and Z. Hou, J. Am. Chem. Soc., 2017, 139, 16506 CrossRef CAS PubMed; (e) T. Itoh, Y. Kanzaki, Y. Shimizu and M. Kanai, Angew. Chem., Int. Ed., 2018, 57, 8265 CrossRef CAS PubMed; (f) R. Pluta, N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2019, 58, 2459 CrossRef CAS PubMed; selected examples by varying the metal of chiral metal catalysis: (g) D. A. Evans, D. W. C. MacMillan and K. R. Campos, J. Am. Chem. Soc., 1997, 119, 10859 CrossRef CAS; (h) A. Nojiri, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 3779 CrossRef CAS PubMed; (i) J. Lv, L. Zhang, S. Luo and J.-P. Cheng, Angew. Chem., Int. Ed., 2013, 52, 9786 CrossRef CAS PubMed.
- Selected examples by organocatalysis: (a) S. Mitsumori, H. Zhang, P. H.-Y. Cheong, K. N. Houk, F. Tanaka and C. F. Barbas III, J. Am. Chem. Soc., 2006, 128, 1040 CrossRef CAS PubMed; (b) X. Feng, Z. Zhou, R. Zhou, Q.-Q. Zhou, L. Dong and Y.-C. Chen, J. Am. Chem. Soc., 2012, 134, 19942 CrossRef CAS PubMed; (c) X. Li, M. Lu, Y. Dong, W. Wu, Q. Qian, J. Ye and D. J. Dixon, Nat. Commun., 2014, 5, 4479 CrossRef CAS PubMed; (d) D. Uraguchi, K. Yoshioka and T. Ooi, Nat. Commun., 2017, 8, 14793 CrossRef PubMed; (e) S. B. J. Kan, H. Maruyama, M. Akakura, T. Kano and K. Maruoka, Angew. Chem., Int. Ed., 2017, 56, 9487 CrossRef CAS PubMed; (f) L. Zhang, H. Yuan, W. Lin, Y. Cheng, P. Li and W. Li, Org. Lett., 2018, 20, 4970 CrossRef CAS PubMed. For organocatalytic stereodivergent synthesis of atropisomers: (g) D. Lotter, A. Castrogiovanni, M. Neuburger and C. Sparr, ACS Cent. Sci., 2018, 4, 656 CrossRef CAS PubMed.
- Selected examples for cooperative catalysis: (a) J. Jiang, H.-D. Xu, J.-B. Xi, B.-Y. Ren, F.-P. Lv, X. Guo, L.-Q. Jiang, Z.-Y. Zhang and W.-H. Hu, J. Am. Chem. Soc., 2011, 133, 8428 CrossRef CAS PubMed; (b) S. Krautwald, D. Sarlah, M. A. Schafroth and E. M. Carreira, Science, 2013, 340, 1065 CrossRef CAS PubMed; (c) S. Krautwald, M. A. Schafroth, D. Sarlah and E. M. Carreira, J. Am. Chem. Soc., 2014, 136, 3020 CrossRef CAS PubMed; (d) N. K. Rana, H. Huang and J. C.-G. Zhao, Angew. Chem., Int. Ed., 2014, 53, 7619 CrossRef CAS PubMed; (e) X. Huo, R. He, X. Zhang and W. Zhang, J. Am. Chem. Soc., 2016, 138, 11093 CrossRef CAS PubMed; (f) X. Jiang, J. J. Beiger and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 87 CrossRef CAS PubMed; (g) F. A. Cruz and V. M. Dong, J. Am. Chem. Soc., 2017, 139, 1029 CrossRef CAS PubMed; (h) L. Wei, Q. Zhu, S.-M. Xu, X. Chang and C.-J. Wang, J. Am. Chem. Soc., 2018, 140, 1508 CrossRef CAS PubMed; (i) X. Huo, J. Zhang, J. Fu, R. He and W. Zhang, J. Am. Chem. Soc., 2018, 140, 2080 CrossRef CAS PubMed; (j) M.-M. Zhang, Y.-N. Wang, B.-C. Wang, X.-W. Chen, L.-Q. Lu and W.-J. Xiao, Nat. Commun., 2019, 10, 2716 CrossRef PubMed; (k) Z.-T. He, X. Jiang and J. F. Hartwig, J. Am. Chem. Soc., 2019, 141, 13066 CrossRef CAS PubMed; for tandem catalysis: (l) Y. Huang, A. M. Walji, C. H. Larsen and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 15051 CrossRef CAS PubMed; (m) S.-L. Shi, Z. L. Wong and S. L. Buchwald, Nature, 2016, 532, 353 CrossRef CAS PubMed; (n) B. M. Trost, C.-I. Hung, T. Saget and E. Gnanamani, Nat. Catal., 2018, 1, 523 CrossRef CAS.
- (a) J. Zhou, Multicatalyst system in asymmetric catalysis, Wiley, New York, 2014, ch. 3 Search PubMed; (b) Y. Takemoto, Org. Biomol. Chem., 2005, 3, 4299 RSC; (c) S. J. Connon, Chem. Commun., 2008, 44, 2499 RSC; (d) T. Kano and K. Maruoka, Chem. Commun., 2008, 44, 5465 RSC; (e) X. Liu, L. Lin and X. Feng, Chem. Commun., 2009, 45, 6145 RSC; (f) A. Grossmann and D. Enders, Angew. Chem., Int. Ed., 2012, 51, 314 CrossRef CAS PubMed; (g) S. Shirakawa and K. Maruoka, Tetrahedron Lett., 2014, 55, 3833 CrossRef CAS; (h) H. Ni, W.-L. Chan and Y. Lu, Chem. Rev., 2018, 118, 9344 CrossRef CAS PubMed.
- To our knowledge, only Deng et al. reported a closely related example of switching the diastereoselectivity of a tandem Michael-protonation reaction by using cinchona alkaloid derivatives 11 and 12 (with a C6' OH group or a C9 thiourea moiety), wherein both the H-bond donors and the catalyst backbone play a role. (a) Y. Wang, X. Liu and L. Deng, J. Am. Chem. Soc., 2006, 128, 3928 CrossRef CAS PubMed; (b) B. Wang, F. Wu, Y. Wang, X. Liu and L. Deng, J. Am. Chem. Soc., 2007, 129, 768 CrossRef CAS PubMed.
- (a) H. Hiemstra and H. Wynberg, J. Am. Chem. Soc., 1981, 103, 417 CrossRef CAS; (b) C. S. Cucinotta, M. Kosa, P. Melchiorre, A. Cavalli and F. L. Gervasio, Chem.–Eur. J., 2009, 15, 7913 CrossRef CAS PubMed.
- M. N. Grayson and K. N. Houk, J. Am. Chem. Soc., 2016, 138, 1170 CrossRef CAS PubMed.
- (a) T. Okino, Y. Hoashi, T. Furukawa, X. Xu and Y. Takemoto, J. Am. Chem. Soc., 2005, 127, 119 CrossRef CAS PubMed; (b) S. J. Zuend and E. N. Jacobsen, J. Am. Chem. Soc., 2007, 129, 15872 CrossRef CAS PubMed; (c) P. Hammar, T. Marcelli, H. Hiemstra and F. Himo, Adv. Synth. Catal., 2007, 349, 2537 CrossRef CAS; (d) B. Zhu, W. Zhang, R. Lee, Z. Han, W. Yang, D. Tan, K.-W. Huang and Z. Jiang, Angew. Chem., Int. Ed., 2013, 52, 6666 CrossRef CAS PubMed.
- (a) A. Hamza, G. Schubert, T. Soós and I. Pápai, J. Am. Chem. Soc., 2006, 128, 13151 CrossRef CAS PubMed; (b) B. Kótai, G. Kardos, A. Hamza, V. Farkas, I. Pápai and T. Soós, Chem.–Eur. J., 2014, 20, 5631 CrossRef PubMed; (c) M. N. Grayson and K. N. Houk, J. Am. Chem. Soc., 2016, 138, 9041 CrossRef CAS PubMed.
- J.-L. Zhu, Y. Zhang, C. Liu, A.-M. Zheng and W. Wang, J. Org. Chem., 2012, 77, 9813 CrossRef CAS PubMed.
- (a) Z.-Y. Cao, F. Zhou and J. Zhou, Acc. Chem. Res., 2018, 51, 1443 CrossRef CAS PubMed; (b) Y.-L. Liu, B.-L. Wang, J.-J. Cao, L. Chen, Y.-X. Zhang, C. Wang and J. Zhou, J. Am. Chem. Soc., 2010, 132, 15176 CrossRef CAS PubMed; (c) Y.-L. Liu, T.-D. Shi, F. Zhou, X.-L. Zhao, X. Wang and J. Zhou, Org. Lett., 2011, 13, 3826 CrossRef CAS PubMed; (d) Y.-L. Liu and J. Zhou, Chem. Commun., 2013, 49, 4421 RSC; (e) X.-P. Yin, X.-P. Zeng, Y.-L. Liu, F.-M. Liao, J.-S. Yu, F. Zhou and J. Zhou, Angew. Chem., Int. Ed., 2014, 53, 13740 CrossRef CAS PubMed.
- (a) M. Ding, F. Zhou, Y.-L. Liu, C.-H. Wang, X.-L. Zhao and J. Zhou, Chem. Sci., 2011, 2, 2035 RSC; (b) W.-M. Gao, J.-S. Yu, Y.-L. Zhao, Y.-L. Liu, F. Zhou, H.-H. Wu and J. Zhou, Chem. Commun., 2014, 50, 15179 RSC; (c) Z.-Y. Cao, Y.-L. Zhao and J. Zhou, Chem. Commun., 2016, 52, 2537 RSC; (d) J.-S. Yu, F.-M. Liao, W.-M. Gao, K. Liao, R.-L. Zuo and J. Zhou, Angew. Chem., Int. Ed., 2015, 54, 7381 CrossRef CAS PubMed.
- (a) P. Li, Z. Chai, S.-L. Zhao, Y.-Q. Yang, H.-F. Wang, C.-W. Zheng, Y.-P. Cai, G. Zhao and S.-Z. Zhu, Chem. Commun., 2009, 45, 7369 RSC; (b) K. Hu, A. Lu, Y. Wang, Z. Zhou and C. Tang, Tetrahedron: Asymmetry, 2013, 24, 953 CrossRef CAS; (c) L. Jiao, X. Zhao, H. Liu, X. Ye, Y. Li and Z. Jiang, Org. Chem. Front., 2016, 3, 470 RSC.
- For Mannich reactions: (a) N. S. Chowdari, M. Ahmad, K. Albertshofer, F. Tanaka and C. F. Barbas III, Org. Lett., 2006, 8, 2839 CrossRef CAS PubMed; (b) Z. Sun, K. Weidner, N. Kumagai and M. Shibasaki, Chem.–Eur. J., 2015, 21, 17574 CrossRef CAS PubMed; (c) X. Ye, Y. Pan and X. Yang, Chem. Commun., 2020, 56, 98 RSC; for aldol reactions: (d) Á. Martínez-Castañeda, K. Kędziora, I. Lavandera, H. Rodríguez-Solla, C. Concellón and V. del Amo, Chem. Commun., 2014, 50, 2598 RSC; (e) S. Okumuş, C. Tanyeli and A. S. Demir, Tetrahedron Lett., 2014, 55, 4302 CrossRef; (f) H. Noda, F. Amemiya, K. Weidner, N. Kumagai and M. Shibasaki, Chem. Sci., 2017, 8, 3260 RSC; for Michael reactions: (g) J. McNulty and C. Zepeda-Velázquez, Angew. Chem., Int. Ed., 2014, 53, 8450 CrossRef CAS PubMed; (h) J. McNulty, L. D'Aiuto, Y. Zhi, L. McClain, C. Zepeda-Velázquez, S. Ler, H. A. Jenkins, M. B. Yee, P. Piazza, R. H. Yolken, P. R. Kinchington and V. L. Nimgaonkar, ACS Med. Chem. Lett., 2016, 7, 46 CrossRef CAS PubMed.
- K. Weidner, Z. Sun, N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2015, 54, 6236 CrossRef CAS PubMed.
- α-Azido ketones with α protons are highly base-sensitive, see:
(a) J. H. Boyer and F. C. Canter, Chem. Rev., 1954, 54, 1 CrossRef CAS. Hoffman et al. found that treating azide I with TEA in acetone led to the formation of 3-aminochromone II and a dimeric adduct III, see:
(b) T. Patonay and R. V. Hoffman, J. Org. Chem., 1995, 60, 2368 CrossRef CAS
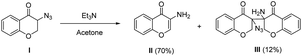 The decomposition of α-azido indanone 1a at room temperature in the presence of bifunctional tertiary amine catalysts 3a or 4b was also detected by TLC analysis, showing that the desired Michael addition competed with the side decomposition of α-azido ketones. This gave a reasonable explanation for why the reaction of less active α-azido tetralone with nitroolefin 2a ended in a mess under standard conditions, without the formation of the desired Michael adduct.
The decomposition of α-azido indanone 1a at room temperature in the presence of bifunctional tertiary amine catalysts 3a or 4b was also detected by TLC analysis, showing that the desired Michael addition competed with the side decomposition of α-azido ketones. This gave a reasonable explanation for why the reaction of less active α-azido tetralone with nitroolefin 2a ended in a mess under standard conditions, without the formation of the desired Michael adduct. - (a) P. G. Cozzi, R. Hilgraf and N. Zimmermann, Eur. J. Org. Chem., 2007, 5969 CrossRef CAS; (b) M. Shibasaki and M. Kanai, Chem. Rev., 2008, 108, 2853 CrossRef CAS PubMed.
- (a) C. Verrier and P. Melchiorre, Chem. Sci., 2015, 6, 4242 RSC; (b) J. I. Martínez, L. Villar, U. Uria, L. Carrillo, E. Reyes and J. L. Vicario, Adv. Synth. Catal., 2014, 356, 3627 CrossRef; (c) J. I. Martínez, U. Uria, M. Muñiz, E. Reyes, L. Carrillo and J. L. Vicario, Beilstein J. Org. Chem., 2015, 11, 2577 CrossRef PubMed; (d) T. Sekikawa, T. Kitaguchi, H. Kitaura, T. Minami and Y. Hatanaka, Org. Lett., 2016, 18, 646 CrossRef CAS PubMed; (e) X.-N. Ping, P.-S. Wei, X.-Q. Zhu and J.-W. Xie, J. Org. Chem., 2017, 82, 2205 CrossRef CAS PubMed.
- For a review: (a) P.-G. Ding, X.-S. Hu, F. Zhou and J. Zhou, Org. Chem. Front., 2018, 5, 1542 RSC; (b) A. S. Carlson and J. J. Topczewski, Org. Biomol. Chem., 2019, 17, 4406 RSC; for selected examples: (c) P. Gu, Y. Su, X.-P. Wu, J. Sun, W. Liu, P. Xue and R. Li, Org. Lett., 2012, 14, 2246 CrossRef CAS PubMed; (d) Q.-H. Deng, T. Bleith, H. Wadepohl and L. H. Gade, J. Am. Chem. Soc., 2013, 135, 5356 CrossRef CAS PubMed; (e) M. V. Vita, P. Caramenti and J. Waser, Org. Lett., 2015, 17, 5832 CrossRef CAS PubMed; (f) A. A. Ott, C. S. Goshey and J. J. Topczewski, J. Am. Chem. Soc., 2017, 139, 7737 CrossRef CAS PubMed; (g) P. Zhou, L. Lin, L. Chen, X. Zhong, X. Liu and X. Feng, J. Am. Chem. Soc., 2017, 139, 13414 CrossRef CAS PubMed; (h) F. J. Seidl, C. Min, J. A. Lopez and N. Z. Burns, J. Am. Chem. Soc., 2018, 140, 15646 CrossRef CAS PubMed; (i) X. Zhang, J. Ren, S. M. Tan, D. Tan, R. Lee and C.-H. Tan, Science, 2019, 363, 400 CrossRef CAS PubMed; (j) N. Thirupathi, F. Wei, C.-H. Tung and Z. Xu, Nat. Commun., 2019, 10, 3158 CrossRef PubMed; (k) R. S. Gomes and E. J. Corey, J. Am. Chem. Soc., 2019, 141, 20058 CrossRef PubMed.
- J.-H. Lin and J.-C. Xiao, Eur. J. Org. Chem., 2011, 4536 CrossRef CAS.
- The use of basic additives to improve the yield was inspired by the following studies, but the mechanism was unclear until now. (a) Y. Zhang, C. Yu, Y. Ji and W. Wang, Chem.–Asian J., 2010, 5, 1303 CAS; (b) J. Deng, S. Zhang, P. Ding, H. Jiang, W. Wang and J. Li, Adv. Synth. Catal., 2010, 352, 833 CrossRef CAS.
- F. Cheng, S. J. Kalita, Z.-N. Zhao, X. Yang, Y. Zhao, U. Schneider, N. Shibata and Y.-Y. Huang, Angew. Chem., Int. Ed., 2019, 58, 16637 CrossRef CAS PubMed.
- For enantioselective trifluoro- and difluoro-methylation, see: (a) Y.-L. Liu, J.-S. Yu and J. Zhou, Asian J. Org. Chem., 2013, 2, 194 CrossRef CAS; (b) J.-H. Lin and J.-C. Xiao, Tetrahedron Lett., 2014, 55, 6147 CrossRef CAS; (c) X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826 CrossRef CAS PubMed; (d) Y. Gong, J.-S. Yu, Y.-J. Hao, Y. Zhou and J. Zhou, Asian J. Org. Chem., 2019, 8, 610 CrossRef CAS.
- For our efforts in enantioselective fluoroalkylation: (a) Y.-L. Liu and J. Zhou, Chem. Commun., 2012, 48, 1919 RSC; (b) Y.-L. Liu, X. Wang, Y.-L. Zhao, F. Zhu, X.-P. Zeng, L. Chen, C.-H. Wang, X.-L. Zhao and J. Zhou, Angew. Chem., Int. Ed., 2013, 52, 13735 CrossRef CAS PubMed; (c) J.-S. Yu and J. Zhou, Org. Chem. Front., 2016, 3, 298 RSC; (d) Z.-Y. Cao, W. Wang, K. Liao, X. Wang, J. Zhou and J. Ma, Org. Chem. Front., 2018, 5, 2960 RSC.
- For reviews: (a) K. V. Gothelf and K. A. Jørgensen, Chem. Rev., 1998, 98, 863 CrossRef CAS PubMed; (b) T. Hashimoto and K. Maruoka, Chem. Rev., 2015, 115, 5366 CrossRef CAS PubMed; (c) S. Tang, X. Zhang, J. Sun, D. Niu and J. J. Chruma, Chem. Rev., 2018, 118, 10393 CrossRef CAS PubMed.
- (a) J.-M. Tian, A.-F. Wang, J.-S. Yang, X.-J. Zhao, Y.-Q. Tu, S.-Y. Zhang and Z.-M. Chen, Angew. Chem., Int. Ed., 2019, 58, 11023 CrossRef CAS PubMed; (b) Q. Zhang, F.-M. Zhang, C.-S. Zhang, S.-Z. Liu, J.-M. Tian, S.-H. Wang, X.-M. Zhang and Y.-Q. Tu, Nat. Commun., 2019, 10, 2507 CrossRef PubMed; (c) Y.-H. Yuan, X. Han, F.-P. Zhu, J.-M. Tian, F.-M. Zhang, X.-M. Zhang, Y.-Q. Tu, S.-H. Wang and X. Guo, Nat. Commun., 2019, 10, 3394 CrossRef PubMed. Also see: (d) I. Arrastia, A. Arrieta and F. P. Cossío, Eur. J. Org. Chem., 2018, 5889 CrossRef CAS PubMed.
- C. Girard and H. B. Kagan, Angew. Chem., Int. Ed., 1998, 37, 2922 CrossRef PubMed.
- For selected reviews: (a) P. H.-Y. Cheong, C. Y. Legault, J. M. Um, N. Çelebi-Ölçüm and K. N. Houk, Chem. Rev., 2011, 111, 5042 CrossRef CAS PubMed; (b) R. C. Johnston and P. H.-Y. Cheong, Org. Biomol. Chem., 2013, 11, 5057 RSC; (c) Y. Lam, M. N. Grayson, M. C. Holland, A. Simon and K. N. Houk, Acc. Chem. Res., 2016, 49, 750 CrossRef CAS PubMed; (d) Q. Peng and R. S. Paton, Acc. Chem. Res., 2016, 49, 1042 CrossRef CAS PubMed; (e) G. Tanriver, B. Dedeoglu, S. Catak and V. Aviyente, Acc. Chem. Res., 2016, 49, 1250 CrossRef CAS PubMed; (f) D. M. Walden, O. M. Ogba, R. C. Johnston and P. H.-Y. Cheong, Acc. Chem. Res., 2016, 49, 1279 CrossRef CAS PubMed.
- For examples of the nonclassical C–H⋯O hydrogen-bond with the cationic tetraalkylammonium unit of the catalyst, see: (a) C. E. Cannizzaro and K. N. Houk, J. Am. Chem. Soc., 2002, 124, 7163 CrossRef CAS PubMed; (b) E. Gomez-Bengoa, A. Linden, R. López, I. Múgica-Mendiola, M. Oiarbide and C. Palomo, J. Am. Chem. Soc., 2008, 130, 7955 CrossRef CAS PubMed; (c) W. Zhang, D. Tan, R. Lee, G. Tong, W. Chen, B. Qi, K.-W. Huang, C.-H. Tan and Z. Jiang, Angew. Chem., Int. Ed., 2012, 51, 10069 CrossRef CAS PubMed; (d) C. Yang, W. Zhang, Y.-H. Li, X.-S. Xue, X. Li and J.-P. Chen, J. Org. Chem., 2017, 82, 9321 CrossRef CAS PubMed; (e) S.-L. Li, Q. Wu, C. Yang, X. Li and J.-P. Cheng, Org. Lett., 2019, 21, 5495 CrossRef CAS PubMed. For experimental support: (f) M. T. Reetz, S. Hütte and R. Goddard, J. Am. Chem. Soc., 1993, 115, 9339 CrossRef CAS; (g) E. J. Corey, F. Xu and M. C. Noe, J. Am. Chem. Soc., 1997, 119, 12414 CrossRef CAS. Also see: (h) K. Brak and E. N. Jacobsen, Angew. Chem., Int. Ed., 2013, 52, 534 CrossRef CAS PubMed.
- For examples of the C–H⋯O hydrogen-bond with the aromatic C–H bond of the catalyst, see: (a) W. Xu, X. Shen, Q. Ma, L. Gong and E. Meggers, ACS Catal., 2016, 6, 7641 CrossRef CAS; (b) T. J. Seguin and S. E. Wheeler, Angew. Chem., Int. Ed., 2016, 55, 15889 CrossRef CAS PubMed; (c) Y. Li, C. Q. He, F.-X. Gao, Z. Li, X.-S. Xue, X. Li, K. N. Houk and J.-P. Cheng, Org. Lett., 2017, 19, 1926 CrossRef CAS PubMed . Also see ref. 13d..
- For examples of the C–H⋯O hydrogen-bond with the α C–H bond of amide nitrogen of the catalyst, see: (a) S. Bahmanyar and K. N. Houk, J. Am. Chem. Soc., 2001, 123, 12911 CrossRef CAS PubMed; (b) M. Drees, L. Kleiber, M. Weimer and T. Strassner, Eur. J. Org. Chem., 2002, 2405 CrossRef CAS; (c) M. Pareek and R. B. Sunoj, Org. Lett., 2016, 18, 5932 CrossRef CAS PubMed.
- For a review: (a) R. Adhikary, J. Zimmermann and F. E. Romesberg, Chem. Rev., 2017, 117, 1927 CrossRef CAS PubMed; for selected examples: (b) K.-I. Oh, J.-H. Lee, C. Joo, H. Han and M. Cho, J. Phys. Chem. B, 2008, 112, 10352 CrossRef CAS PubMed; (c) J.-H. Choi, K.-I. Oh and M. Cho, J. Chem. Phys., 2008, 129, 174512 CrossRef PubMed; (d) J.-H. Choi, D. Raleigh and M. Cho, J. Phys. Chem. Lett., 2011, 2, 2158 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure and characterization data. CCDC 1874277–1874279, 1874281 and 1874283. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc00475h |
| This journal is © The Royal Society of Chemistry 2020 |