DOI:
10.1039/D0SC00404A
(Edge Article)
Chem. Sci., 2020,
11, 3836-3844
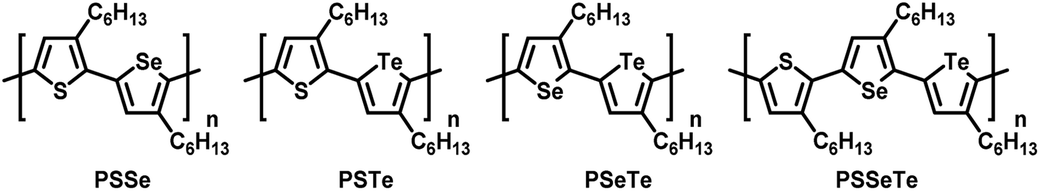
Synthesis of side-chain regioregular and main-chain alternating poly(bichalcogenophene)s and an ABC-type periodic poly(terchalcogenophene)†
Received
22nd January 2020
, Accepted 9th March 2020
First published on 10th March 2020
Abstract
Three unsymmetrical diiodobichalcogenophenes SSeI2, STeI2, and SeTeI2 and a diiodoterchalcogenophene SSeTeI2 were prepared. Grignard metathesis of SSeI2, STeI2, SeTeI2, and SSeTeI2 occurred regioselectively at the lighter chalcogenophene site because of its relatively lower electron density and less steric bulk. Nickel-catalyzed Kumada catalyst-transfer polycondensation of these Mg species provided a new class of side-chain regioregular and main-chain AB-type alternating poly(bichalcogenophene)s—PSSe, PSTe, and PSeTe—through a chain-growth mechanism. The ring-walking of the Ni catalyst from the lighter to the heavier chalcogenophene facilitated subsequent oxidative addition, thereby suppressing the possibility of chain-transfer or chain-termination. More significantly, the Ni catalyst could walk over the distance of three rings (ca. 1 nm)—from a thiophene unit via a selenophene unit to a tellurophene unit—to form PSSeTe, the first ABC-type regioregular and periodic poly(terchalcogenophene) comprising three different types of 3-hexylchalcogenophenes.
Introduction
Conjugated polymers featuring continuous sp2/sp-hybridized backbones for extensive delocalization of electrons have been explored widely for several decades.1–5 Regioregular poly(3-hexylthiophene) (P3HT) exhibiting high charge mobility has been widely applied in organic light-emitting diodes,6,7 transistors,8–14 and polymer solar cells15–18 due to its high crystallinity and well-ordered packing structure in the thin film.19,20 As a result, extensive research has been focused on Kumada catalyst-transfer polycondensation (KCTP) for the synthesis of highly regioregular P3HT having a low polydispersity index (PDI).21–36 However, an intrinsic drawback of P3HT and other thiophene-based polymers is the limited absorption window from approximately 300 to 550 nm, thereby hindering their various optoelectronic applications. Structurally similar to thiophene, two other five-membered heterocycles in the chalcogenophene family—selenophene and tellurophene, featuring group-16 Se and Te elements, respectively—have attracted considerable interest because (1) Se and Te atoms are larger and have d-orbitals of higher polarizability (relative to S) to induce strong Se⋯Se and Te⋯Te attractions, potentially strengthening their interpolymer interactions,37–41 and (2) as the chalcogen becomes heavier, the π-electrons in selenophene and tellurophene tend to adopt a more quinoidal character with higher coplanarity, giving rise to narrower band gaps and bathochromic shifts in their absorption.42–50 The homopolymers 3-alkylselenophene (P3AS)51,52 and 3-alkyltellurophene (P3ATe)53,54 have also been prepared using the KCTP. We were interested in studying the effects of incorporating two different chalcogenophenes into a single polymer to generate a new class of poly(bichalcogenophene)s. By integrating thiophene, selenophene, and tellurophene units in a single polymer in a controlled sequence, the properties of the polymer could presumably be tailored specifically. In particular, we envisaged that positioning selenophene and tellurophene units, having five-membered ring structures similar to thiophene, into the polymers would allow fine-tuning of the optical and electronic properties without substantially affecting the conformation of the main chain, thereby maintaining high crystallinity. To date, the synthesis of random55–59 and block poly(bichalcogenophene)s60–63 [e.g., poly(thiophene-block-selenophene)] have been realized simply by controlling the sequence of addition of the monomer. Nevertheless, the synthesis of main-chain alternating and side-chain regioregular AB-type poly(bichalcogenophene)s remains a challenge, and these materials have not been well explored.64–66 In particular, tellurophene-incorporated alternating poly(bichalcogenophene)s have never been described previously. In this present study, we developed a new class of AB-type alternating poly(bichalcogenophene)s constructed from 3-hexylthiophene, 3-hexylselenophene, and 3-hexyltellurophene units (Fig. 1). After careful design of the unsymmetrical bichalcogenophene monomers, we synthesized poly(3-hexylthiophene-alt-3-hexylselenophene) (PSSe), poly(3-hexylthiophene-alt-3-hexyltellurophene) (PSTe), and poly(3-hexylselenophene-alt-3-hexyltellurophene) (PSeTe) through KCTP operated in a chain-growth manner. Furthermore, we report the first example of a regioregular ABC-type periodic poly(terchalcogenophene), poly(3-hexylthiophene-per-3-hexylselenophene-per-3-hexyltellurophene) (PSSeTe).
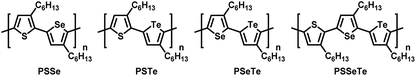 |
| | Fig. 1 Chemical structures of the alternating poly(bichalcogenophene)s PSSe, PSTe, and PSeTe and the periodic poly(terchalcogenophene) PSSeTe. | |
Results and discussion
Molecular design
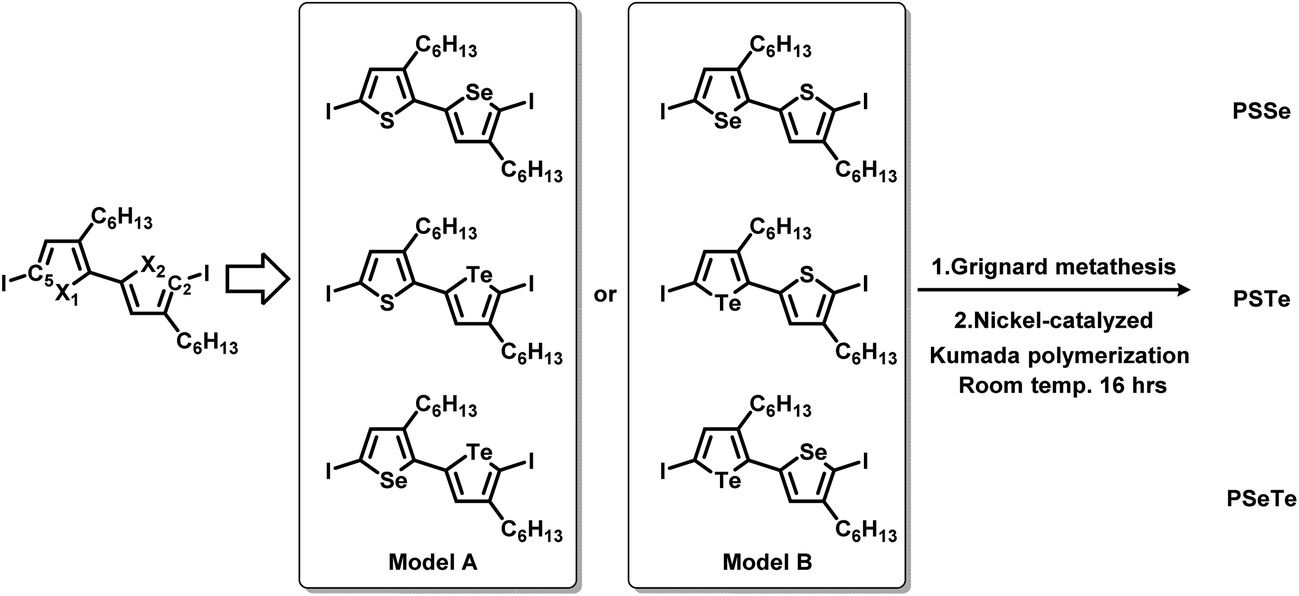
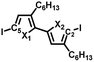
To synthesize the three alternating polymers PSSe, PSTe, and PSeTe through KCTP, we designed three corresponding unsymmetrical diiodobichalcogenophene monomers having a head-to-tail arrangement of their hexyl side chains. There are two possible isomeric arrangements for the unsymmetrical monomers (Fig. 2): one with the heavier chalcogen placed at X2 (i.e., model A: X1 = S, X2 = Se; X1 = S, X2 = Te; X1 = Se, X2 = Te) and the other with the heavier chalcogen placed at X1 (i.e., model B: X1 = Se, X2 = S; X1 = Te, X2 = S; X1 = Te, X2 = Se). Achieving high selectivity between the two reactive carbon atoms (C2 and C5) during Grignard metathesis would be an important criterion toward obtaining polymers of high head-to-tail regioregularity after the nickel-catalyzed KCTP.27,67 We used Hirshfeld charge analysis to calculate the charge distributions at C2 and C5 of the diiodobichalcogenophene monomers in models A and B (Table 1). Because of the proximity of the electron-donating aliphatic substituent and the heavier chalcogen, C2 bore a more negative charge than C5 in model A; in other words, a pronounced charge difference existed between C5 and C2 in model A and, furthermore, C5 is less sterically hindered than C2. Considering both the electronic and steric effects, we envisioned that C5 in model A would be more susceptible to Mg/I exchange (Grignard metathesis) than C2, potentially leading to highly regioregular polymers. In contrast, the C2 and C5 atoms in model B had very similar charge densities, suggesting that Grignard metathesis might occur with no selectivity. Consequently, we chose the three monomers in model A for polymerization.
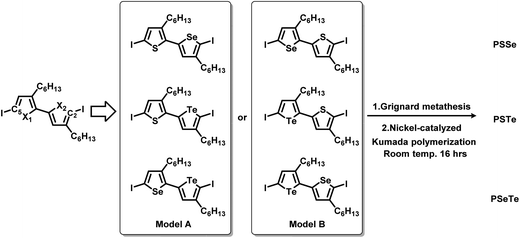 |
| | Fig. 2 Design of six unsymmetrical monomers, in models A and B, for the synthesis of PSSe, PSTe, and PSeTe. | |
Table 1 Hirshfeld charge analysis of the charge distributions at atoms C2 and C5 of six unsymmetrical diiodonated bichalcogenophenes, calculated at the cam-B3LYP/6-311G(d,P) and LanL2DZ(d,p) levels of theory
|
|
C2 |
C5 |
| X1 = S; X2 = Se |
−0.036 |
−0.025 |
| X1 = S; X2 = Te |
−0.042 |
−0.026 |
| X1 = Se; X2 = Te |
−0.043 |
−0.032 |
| X1 = Se; X2 = S |
−0.030 |
−0.030 |
| X1 = Te; X2 = S |
−0.031 |
−0.036 |
| X1 = Te; X2 = Se |
−0.038 |
−0.036 |
Synthetic procedures
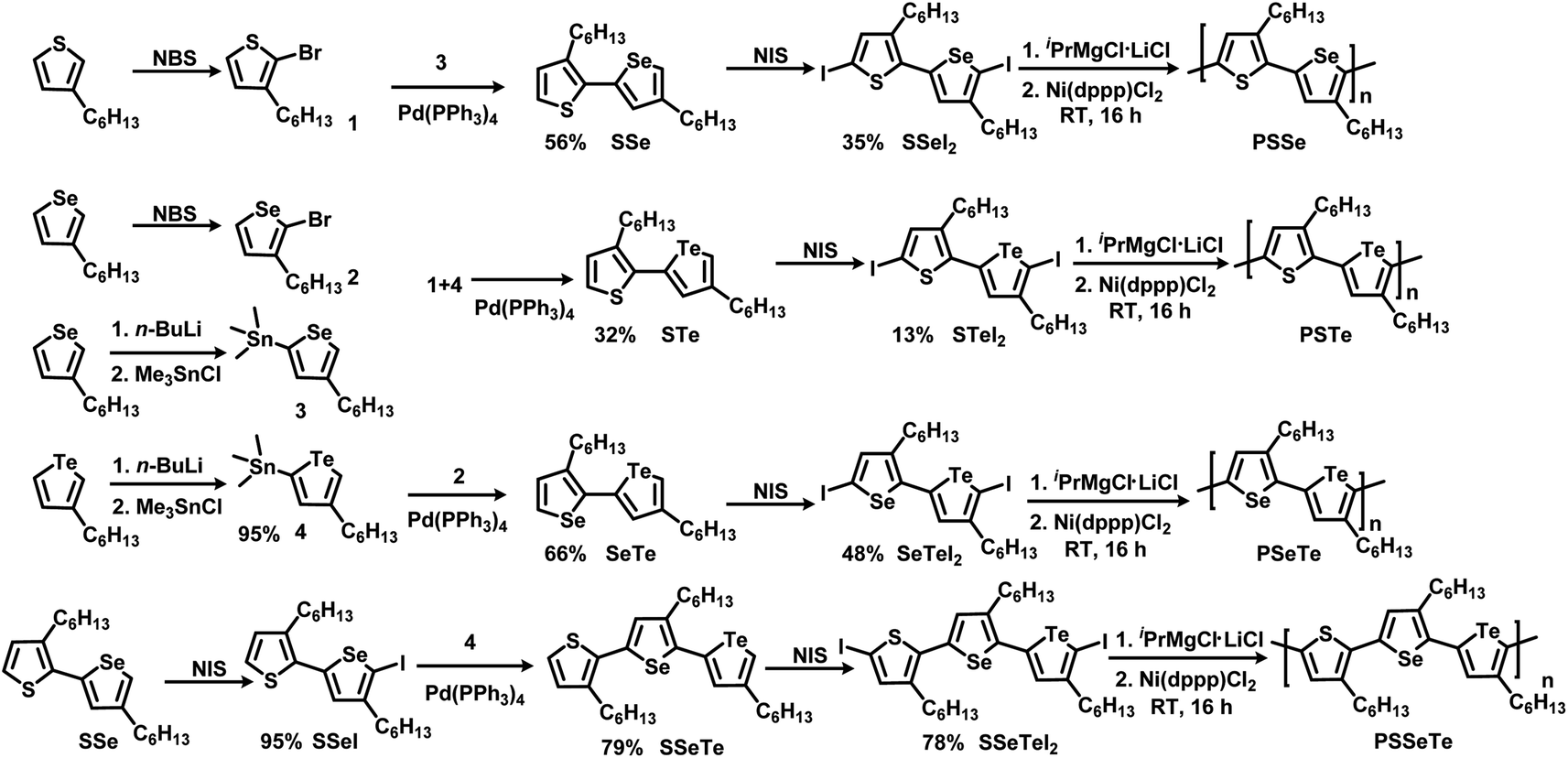
Scheme 1 illustrates the synthesis of the three monomers SSeI2, STeI2, and SeTeI2. Bromination of 3-hexylthiophene and 3-hexylselenophene in the presence of N-bromosuccinimide (NBS) afforded compounds 1 and 2, respectively. Stannylation of 3-hexylselenophene and 3-hexyltellurophene using n-BuLi and Me3SnCl furnished compounds 3 and 4, respectively. The three unsymmetrical bichalcogenophenes SSe, STe, and SeTe were obtained through Stille coupling of 1 and 3, 1 and 4, and 2 and 4, respectively. The three SSeI2, STeI2, and SeTeI2 monomers in model A were prepared through iodination of SSe, STe, and SeTe, respectively, with N-iodosuccinimide (NIS). Scheme 1 also presents the synthesis of the terchalcogenophene monomer SSeTeI2. Iodination of SSe with one equivalent of NIS afforded SSeI selectively; it was Stille coupled with compound 4 to yield SSeTe. Iodination of SSeTe with NIS provided SSeTeI2 in a yield of 78%.
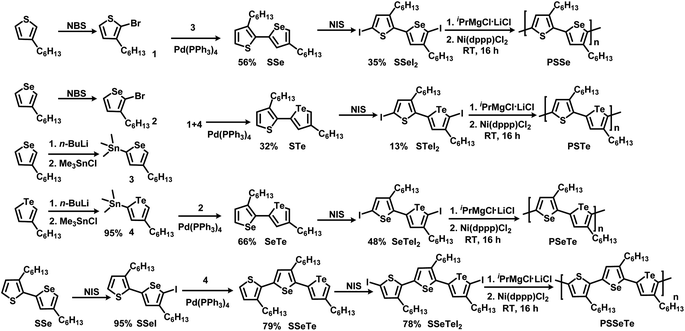 |
| | Scheme 1 Synthesis of the monomers SSeI2, STeI2, SeTeI2, and SSeTeI2 and their polymerization. | |
Selectivity of Grignard metathesis
To confirm the regioselectivity of Grignard metathesis, we conducted model studies prior to performing the polymerizations. After treatment of SSeI2, STeI2, and SeTeI2 with one equivalent of isopropylmagnesium chloride lithium chloride complex (iPrMgCl·LiCl) to induce Grignard metathesis, the reactions were quenched with aqueous NH4Cl to replace the reaction site with a proton. We used 1H NMR spectroscopy to examine the crude products (Fig. S1a†). If the Grignard metathesis had occurred at the C2 position, three singlets should have appeared in the aromatic region of the 1H NMR spectrum; if it had occurred at the C5 position, one singlet and two doublets should have been expected. We observed one singlet and two doublets in the spectra of each of the model monomers, revealing that the Grignard metathesis had taken place predominantly at the C5 atom. This phenomenon also occurred for the SSeTeI2 monomer, the 1H NMR spectrum of which featured two singlets and two doublets (Fig. S1b†), implying that its Grignard metathesis also occurred at the C5 position, rather than at the C2 position. For reference, we synthesized another isomeric SSeI2 monomer, of the model B type, and performed the same experiment. Fig. S2† reveals two major products, resulting from protonation at both the C2 and C5 positions, after quenching with NH4Cl(aq), confirming that the unsymmetrical bichalcogenophenes in model B did not exhibit selectivity toward Grignard metathesis. Thus, we were correct in choosing model A to achieve high selectivity in Grignard metathesis.
Polymer synthesis and structure identification
We treated the monomers SSeI2, STeI2, SeTeI2, and SSeTeI2 with 1.0 equivalent of iPrMgCl·LiCl and then performed Ni(dppp)Cl2-catalyzed KCTP at room temperature to yield the desired polymers PSSe, PSTe, PSeTe, and PSSeTe, respectively (Scheme 1). All the polymers were precipitated using 6 M HCl/MeOH solution and then they were washed with MeOH. Because of the limited solubility of the polymers, we used high-temperature gel permeation chromatography (GPC), with trichlorobenzene as the eluent, to determine their molecular weights. The GPC data confirmed the excellent control over the molecular weights with the relatively low polydispersity (Table 2). The molecular weight of PSTe increased linearly (6800, 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400, and 30800 g mol−1) upon increasing the monomer/catalyst molar ratio (25, 50 and 100, respectively), confirming that the reaction had the characteristics of catalyst-transfer polycondensation.27,35 The polydispersity (PDI) of PSSe is a little bit larger than the previous report.66 It should be noted that we used diiodobichalcogenophene monomers for polymerization. Compared to bromine or chlorine, the larger atomic radius of iodine may increase the activation energy for transmetalation during the polymerization, resulting in the higher PDI.68
400, and 30800 g mol−1) upon increasing the monomer/catalyst molar ratio (25, 50 and 100, respectively), confirming that the reaction had the characteristics of catalyst-transfer polycondensation.27,35 The polydispersity (PDI) of PSSe is a little bit larger than the previous report.66 It should be noted that we used diiodobichalcogenophene monomers for polymerization. Compared to bromine or chlorine, the larger atomic radius of iodine may increase the activation energy for transmetalation during the polymerization, resulting in the higher PDI.68
Table 2 Molecular weights, PDIs, and regioregularities of the polymers
| Polymer |
M/cat. |
M
n
|
PDI |
RR |
|
Measured through high-temperature GPC at 160 °C, using polystyrene standards and 1,2,4-trichlorobenzene as the eluent. All the concentrations of polymeric solutions are 1 mg mL−1.
|
|
PSSe
|
50 |
16500 |
1.38 |
92% |
|
PSeS
|
20 |
4700 |
2.59 |
85% |
|
PSTe
|
25 |
6800 |
1.34 |
94% |
| 50 |
14400 |
1.33 |
| 100 |
30800 |
1.49 |
|
PSeTe
|
50 |
23900 |
1.47 |
91% |
|
PSSeTe
|
50 |
20600 |
1.57 |
94% |
We used 1H and 13C NMR spectroscopy to investigate the chemical structures and regioregularities of the new poly(bichalcogenophene)s and poly(terchalcogenophene). The 1H NMR spectra of the polymers PSSe, PSTe, and PSeTe all featured two well-defined aromatic singlets representing the two kinds of protons on the two different 3-hexylchalcogenophene rings (Fig. S3†). Similarly, the spectrum of PSSeTe featured three singlets representing the three different 3-hexylchalcogenophene rings. The protons on the first carbon atoms of the hexyl groups appeared at 2.6–2.8 ppm; those on the terminal carbon atoms of the hexyl groups appeared at 0.91–0.93 ppm. Through integration of the signals of the first sets of protons, we estimated the head-to-tail regioregularity to be greater than 90%.67 The 13C NMR spectra (Fig. S4†) featured eight well-defined aromatic peaks for PSSe, PSTe, and PSeTe and 12 for PSSeTe, suggesting that these new polychalcogenophenes featured alternating and periodic main chains characterized by regioregular head-to-tail hexyl groups. Dependence of the number average molecular weight (Mn) and PDI on monomer conversion for PSTe is shown in Table S1 and Fig. S5† where the Mn increased linearly with the conversion, indicating the controlled polymerization process. MALDI-TOF mass spectrometry was used to characterize the end-groups of PSSeTe (Fig. S6†). The major peaks of PSSeTe matched those expected for the (SSeTe)n structure (where n is the number of repeat units) terminated with an iodine and a hydrogen (I/H) atom. Thus, the MALDI-TOF-MS was also in good agreement with a mechanism involving catalyst-transfer polycondensation.
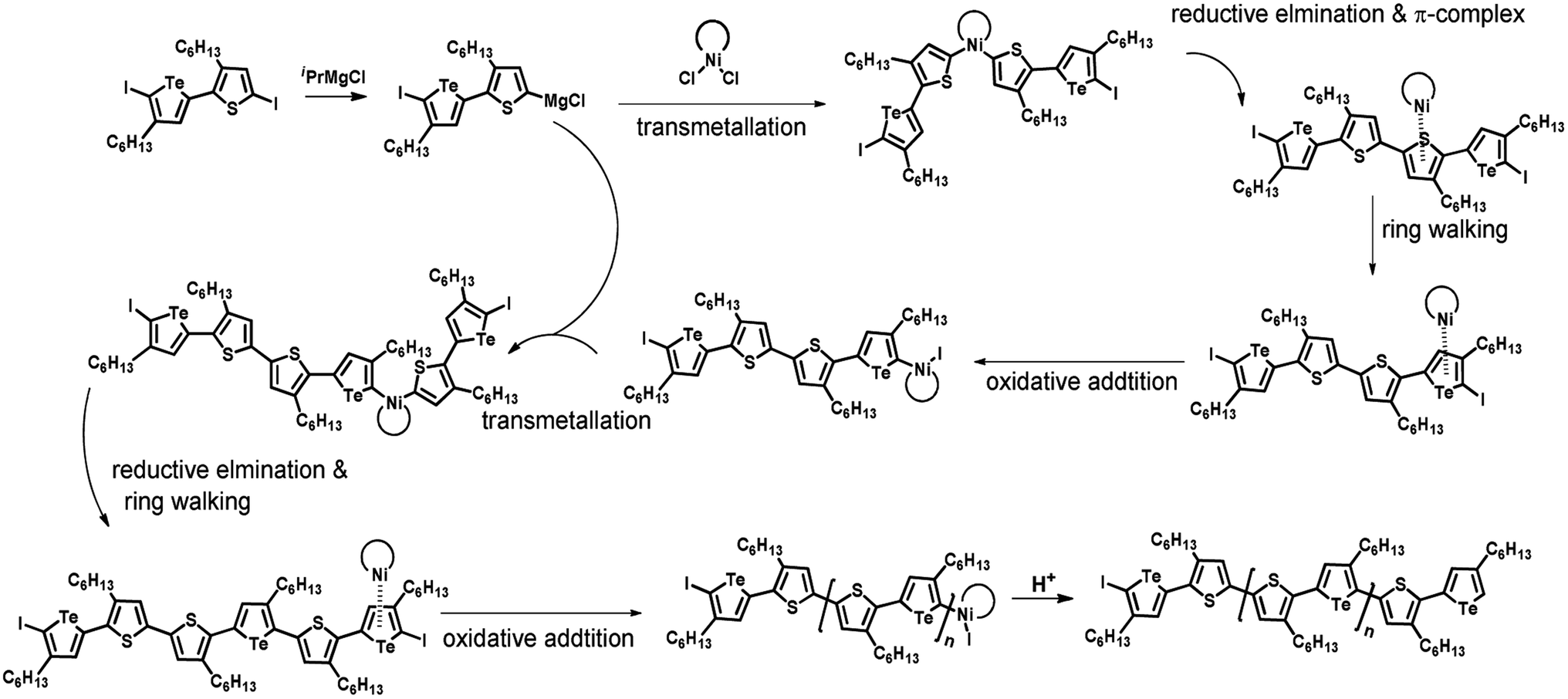
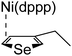
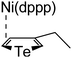
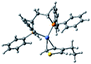
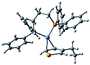
Plausible mechanism
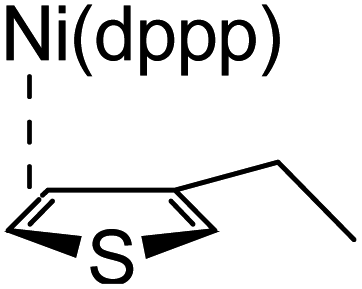
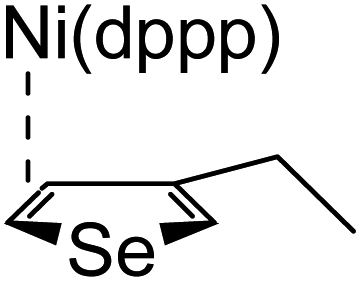
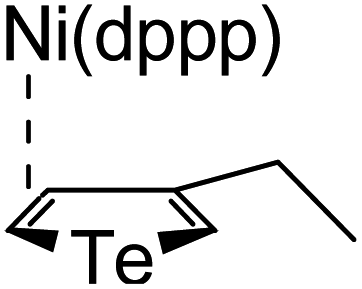
According to the mechanism of catalyst-transfer polycondensation proposed by Yokozawa et al.27 and research through theoretical calculations, Scheme 2 presents our suggested mechanism for the polymerization, taking STeI2 as an example. Initially, STeI2 undergoes regioselective Grignard metathesis to form a nucleophilic ITeS-MgCl intermediate, which doubly attacks the Ni(dppp)Cl2 catalyst for ligand transfer. Following reductive elimination, the Ni species is coordinated to and stabilized by the π-electron bichalcogenophenes.69 We suspect that the stronger chelation ability of the heavier Te atom (relative to a S atom) drives the Ni atom to shift from thiophene to tellurophene through a selective “ring-walking” process. The chain grows through a repetitive sequence of transmetalation, reductive elimination, Ni π-complexation, ring-walking, and oxidative addition. Eventually, the polymerization is terminated by a proton to give the final polymer chain. Based on this mechanism, the polymer would contain repeating biaryl or triaryl units with I and H atoms at the chain ends [I(biaryl)nH or I(triaryl)nH, respectively]. Notably, the successful synthesis of PSSeTe suggests that the Ni catalyst could walk smoothly over three rings: from the thiophene unit, via the selenophene unit, to the tellurophene unit. To support this hypothesis, we performed theoretical calculations to estimate the complexation energies of the Ni catalyst with the independent chalcogenophenes. Table 3 reveals that the complexation energies of a Ni(dppp) species with 2-ethylthiophene (complex 1), 2-ethylselenophene (complex 2), and 2-ethyltellurophene (complex 3) were approximately −15.1, −17.3, and −19.3 kcal mol−1, respectively. With increased complexation energy, the migration of the Ni complex would be thermodynamically favored from the thiophene unit via the selenophene unit to the tellurophene unit, in turn facilitating the subsequent oxidative addition at the iodotellurophene chain end. Thus, our rational design of the four unsymmetrical monomers in model A not only allowed regioselective Grignard metathesis but also favored selective ring-walking, leading to highly regioregular polychalcogenophenes through the KCTP mechanism. In contrast, the polymer PSeS prepared from the isomeric model-B monomer SSeI2 featured (Table 2) much higher polydispersity (2.59) and lower regioregularity (85%), suggesting that the Ni catalyst was more likely to dissociate from the polymer, leading to chain-transfer and chain-termination reactions.
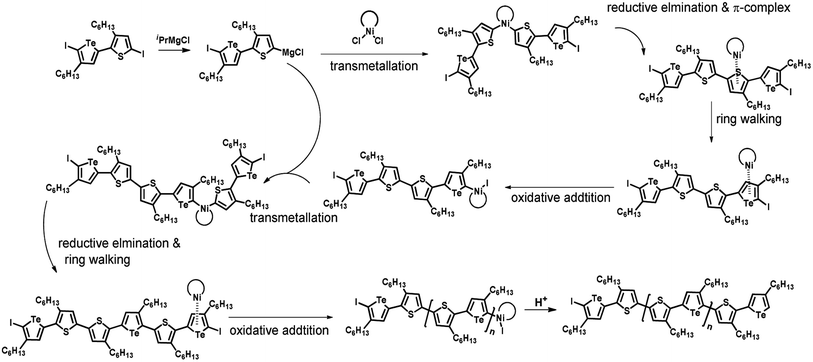 |
| | Scheme 2 Proposed mechanism for catalyst-transfer polycondensation of PSTevia ring-walking. | |
Table 3 Association energies (ΔG) for the interactions of Ni(dppp) with 2-ethylthiophene, 2-ethylselenophene, and 2-ethyltellurophene
|
|
Complex 1 |
Complex 2 |
Complex 3 |
|
|
|
|
|
|
|
|
| ΔG |
−15.1 kcal mol−1 |
−17.3 kcal mol−1 |
−19.3 kcal mol−1 |
Optical properties
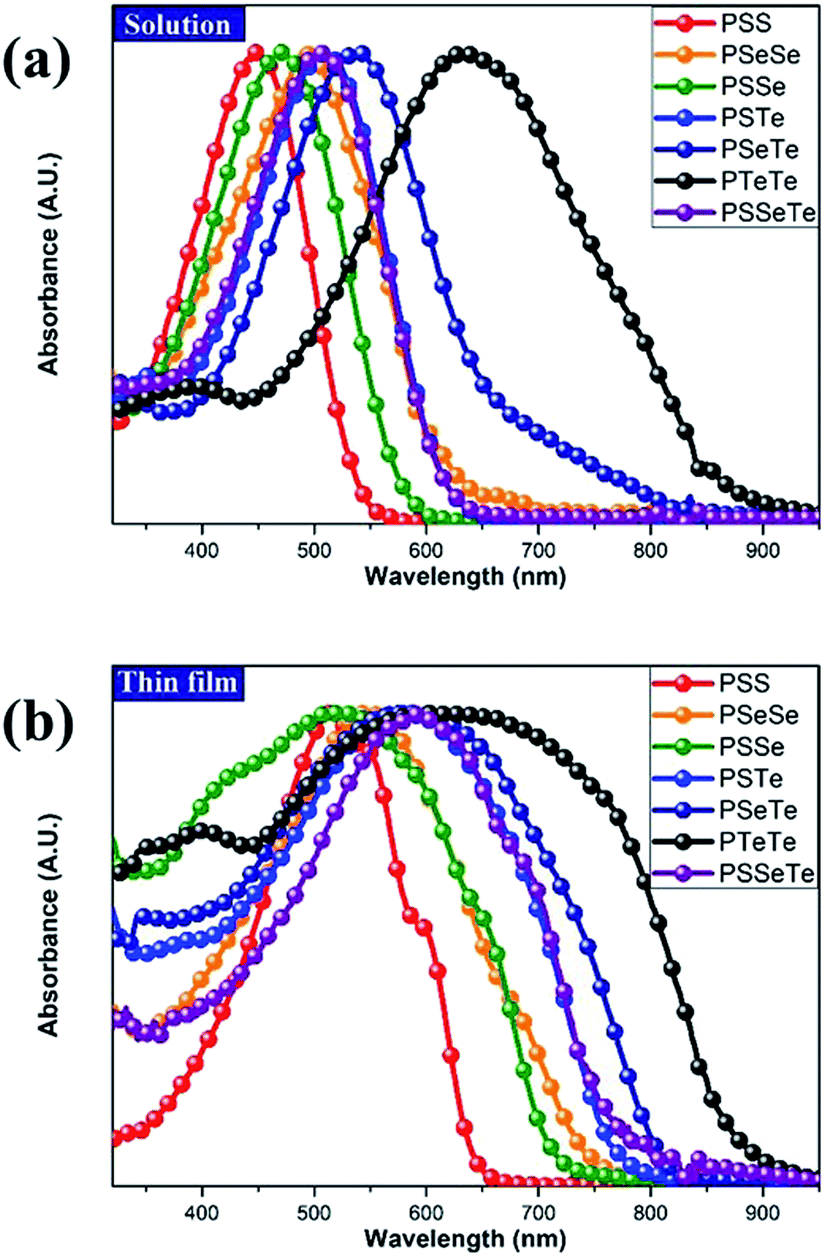
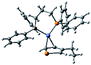
To investigate the effect of the chalcogen on the optical absorption behavior of the polychalcogenophenes, we recorded the UV-Vis spectra of our four new polymers PSSe, PSTe, PSeTe, and PSSeTe as well as those of the three homopolymers PSS (P3HT), PSeSe (P3HS), and PTeTe (P3HTe). Fig. 3 presents the UV-Vis spectra of all of these polymers in solution (o-dichlorobenzene) and in the solid state; Table 4 summarizes the absorption parameters and the band gaps. In solution, the values of λmax of the polymers were highly dependent on the chemical composition: PTeTe (631 nm) > PSeTe (541 nm) > PSTe (511 nm) > PSeSe (494 nm) > PSSe (471 nm) > PSS (448 nm). Upon incorporating heavier chalcogenophenes (selenophene and tellurophene) into the polymers, the absorption red-shifted with concomitant broadening of the full width at half maximum (FWHM) of the band. The ability of the chalcogenophene to red-shift the absorption of the polymer followed the order tellurophene > selenophene > thiophene. Thus, the absorption of the polychalcogenophene in the range from 300 to 800 nm could be tuned subtly by carefully selecting the combination of chalcogenophenes in a single polymer. Furthermore, these polymers exhibited significant red-shifting and broadening of their absorptions upon proceeding from solution to the solid state, indicating that strong intermolecular interactions existed between the chalcogenophene moieties. Notably, the tellurophene-containing polymer PSeTe, without featuring any electron-deficient units, possessed a relatively small band gap and had an onset wavelength of 800 nm.
 |
| | Fig. 3 Absorption spectra of PTeTe, PSeTe, PSSeTe, PSTe, PSeSe, PSSe, and PSS (a) in oDCB solution and (b) as thin films. | |
Table 4 UV-Vis absorption spectral properties of the polymers
|
|
λ
max (nm) |
λ
onset (nm) |
E
optg (eV) |
|
oDCBa (FWHM) |
Film |
|
o-Dichlorobenzene.
|
|
PSS
|
448 (119) |
514 |
643 |
1.93 |
|
PSSe
|
471 (140) |
523 |
711 |
1.74 |
|
PSeSe
|
494 (167) |
547 |
746 |
1.66 |
|
PSTe
|
511 (143) |
576 |
770 |
1.61 |
|
PSSeTe
|
497 (147) |
593 |
777 |
1.60 |
|
PSeTe
|
541 (167) |
589 |
811 |
1.53 |
|
PTeTe
|
631 (246) |
632 |
877 |
1.41 |
Electrochemical properties
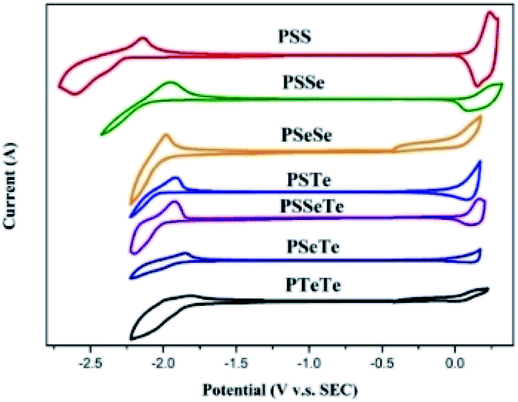
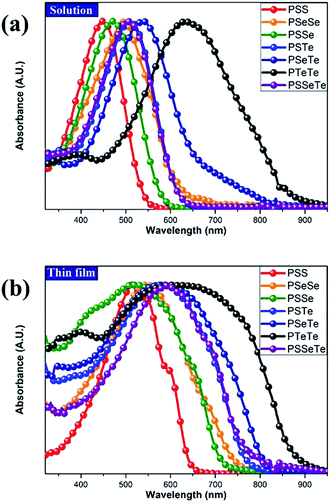
We used cyclic voltammetry to evaluate the electrochemical properties of the polymers and determine their HOMO and LUMO energy levels (Fig. 4, Table 5). When a heavier chalcogenophene was incorporated, the HOMO was slightly elevated (higher-lying) while the LUMO had descended (lower-lying) more significantly. Thus, the HOMO and LUMO energy levels, along with the band gaps, could be controlled by combining different chalcogenophenes into a single polymer.
 |
| | Fig. 4 CV plots of the polymer films, relative to ferrocene, revealing their oxidation and reduction onsets. | |
Table 5 CV data of the polymers
|
|
LUMO (eV) |
HOMO (eV) |
E
eleg (eV) |
|
PSS
|
−2.46 |
−4.89 |
2.43 |
|
PSSe
|
−2.70 |
−4.88 |
2.18 |
|
PSTe
|
−2.76 |
−4.82 |
2.06 |
|
PSeSe
|
−2.78 |
−4.81 |
2.03 |
|
PSSeTe
|
−2.80 |
−4.86 |
2.06 |
|
PSeTe
|
−2.92 |
−4.81 |
1.89 |
|
PTeTe
|
−2.96 |
−4.77 |
1.81 |
Thin film morphologies
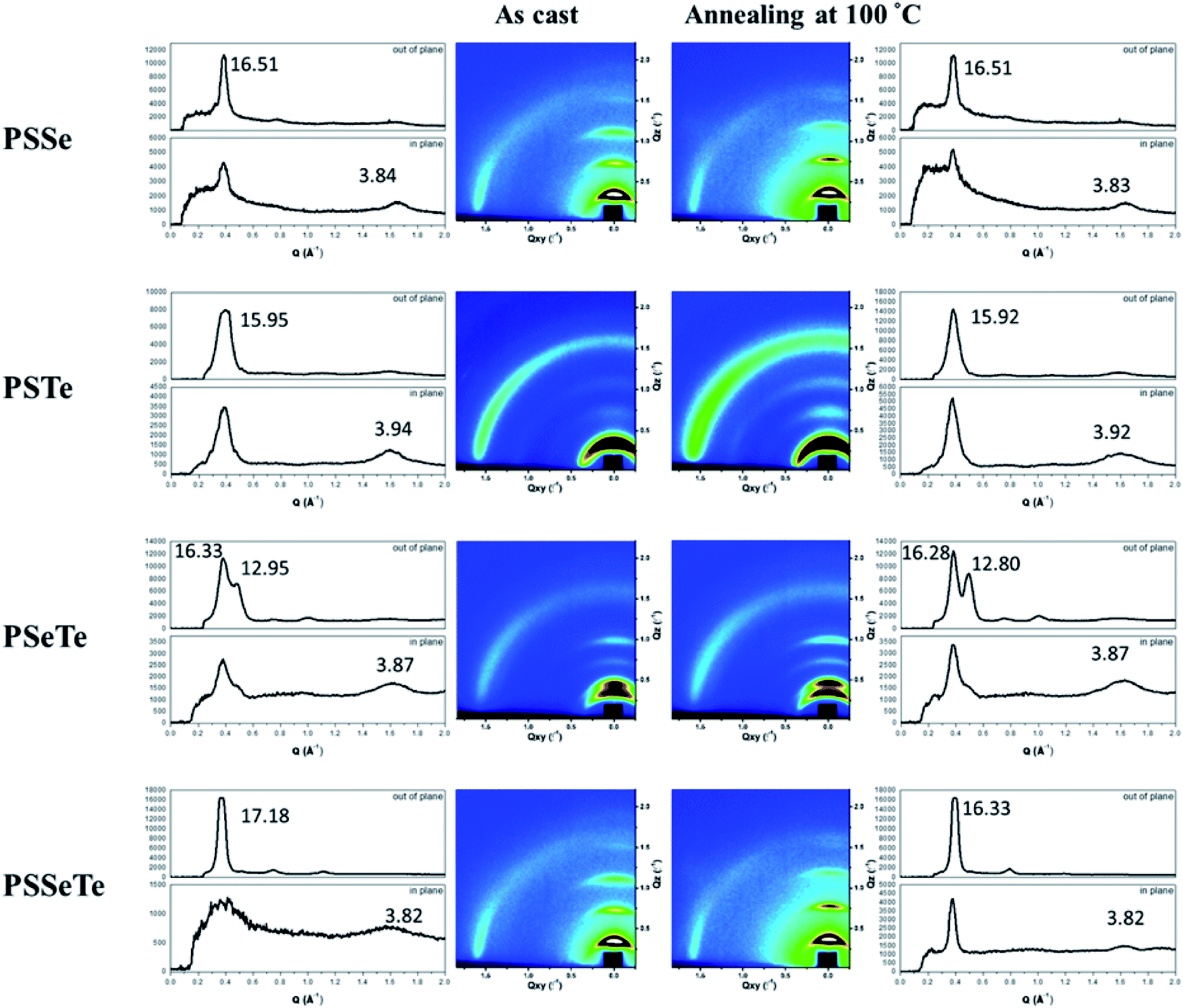
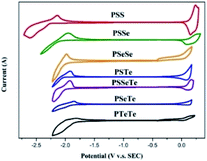
We used grazing-incidence wide-angle X-ray scattering (GIWAXS) to investigate the morphologies of the polychalcogenophenes. Because of the relatively poor solubility of the polychalcogenophenes, we selected o-dichlorobenzene as the processing solvent. Fig. 5 presents the two-dimensional (2D) images and corresponding one-dimensional (1D) patterns. All of the polymer films exhibited out-of-plane (h00) signals corresponding to side-chain interdigitation. We also observed obvious (010) in-plane peaks corresponding to periodic π-stacking between the two facing conjugated backbones. These in-plane (010) peaks revealed that the polymers adopted predominately edge-on orientations with the backbone plane approximately perpendicular to the substrate. The out-of-plane lamellar spacing (dl) and the in-plane π-stacking spacing (dπ) of the polymers were estimated using the Bragg equation (Table 6). The π-stacking spacing of the poly(bichalcogenophene)s increased upon increasing the tellurophene content (dπ: 3.84 Å for PSSe, 3.87 Å for PSeTe, and 3.94 Å for PSTe), presumably because of the larger size of the Te atom. In contrast, the lamellar spacing decreased upon increasing the tellurophene content (dl: 15.95 Å for PSTe, 16.33 Å for PSeTe, and 16.51 Å for PSSe). It is likely that the increased π-stacking distance provided more volume for the hexyl groups to interdigitate, thereby contracting the lamellar spacing.70 Nevertheless, it is interesting that the poly(terchalcogenophene) PSSeTe, featuring the lowest tellurophene content (one third), possessed the shortest π-stacking spacing (3.82 Å) and, thus, the largest lamellar spacing (17.18 Å). After thermal annealing at 100 °C for 30 min, the diffraction patterns were essentially unchanged, but with enhanced signal intensities and slightly lower values of dπ and dl. Interestingly, PSeTe exhibited two lamellar interlayer spacings (16.33 and 12.95 Å), indicating the formation of two crystal phases with different types of side-chain interdigitation.52,71
 |
| | Fig. 5 2D GIWAXS images of the polymers and their corresponding 1D in-plane and out-of-plane patterns, recorded before and after thermal annealing at 100 °C. | |
Table 6 Lamellar spacings and π-stacking spacings of the polymers
|
|
As-cast |
Thermal annealing at 100 °C |
| Lamellar spacing (Å) |
π-stacking spacing (Å) |
Lamellar spacing (Å) |
π-stacking spacing (Å) |
|
PSSe
|
16.51 |
3.84 |
16.51 |
3.83 |
|
PSTe
|
15.95 |
3.94 |
15.92 |
3.92 |
|
PSeTe
|
16.33, 12.95 |
3.87 |
16.28, 12.80 |
3.87 |
|
PSSeTe
|
17.18 |
3.82 |
16.55 |
3.82 |
OFET devices
To evaluate the p-type mobilities of PSSe, PSTe, PSeTe, and PSSeTe, we fabricated OFET devices incorporating ODTS-treated SiO2/Si dielectric substrates in a bottom-gate/top-contact configuration. The spin-cast polymer thin films were thermally annealed at 100 °C for 10 min. When the applied gate-to-source voltage (VGS) was ramped from 0 to −75 V in steps of −15 V, the OFETs exhibited typical p-type behavior (Fig. S7†). Table 7 summarizes the saturation organic field-effect mobilities (μsat), threshold voltages (Vth), and current on/off ratios (Ion/Ioff) calculated from the transfer characteristics (Fig. S8†) of these devices. The edge-on π-stacking orientations of the polymers were favorable for horizontal carrier mobility, because the direction of π-stacking was the same as the direction of current flow. The mobility of the poly(bichalcogenophene)s decreased when incorporating the two heavier chalcogenophenes: 7.2 × 10−3 cm2 V−1 s−1 for PSSe > 2.7 × 10−3 cm2 V−1 s−1 for PSTe > 5.2 × 10−4 cm2 V−1 s−1 for PSeTe. Because of the larger radius of the Te atom, PSTe and PSeTe possessed larger π-stacking spacings, which were presumably related to their lower mobilities. Notably, however, the tellurophene-containing polymers had relatively poorer solubilities, which might also have influenced their film quality and, thus, mobility. Interestingly, the poly(terchalcogenophene) PSSeTe exhibited the highest mobility (1.2 × 10−2 cm2 V−1 s−1), consistent with it having the shortest π-stacking distance. This mobility is one of the highest ever reported for a tellurophene-chalcogenophene-based polymer.70
Table 7 Charge carrier mobilities, threshold voltages, and on/off ratios for OFETs incorporating spin-coated PSSe, PSTe, PSeTe, and PSSeTe films
| Polymer |
I
on/Ioff |
V
th (V) |
Mobility (cm2 V−1 s−1) |
|
PSSe
|
8.5 × 103 |
−7.5 |
7.2 × 10−3 |
|
PSTe
|
8.1 × 102 |
−3.6 |
2.7 × 10−3 |
|
PSeTe
|
4.9 × 103 |
−19.6 |
5.2 × 10−4 |
|
PSSeTe
|
8.0 × 103 |
−23.1 |
1.2 × 10−2 |
Conclusions
We have developed a new class of side-chain regioregular and main-chain alternating polychalcogenophenes having precisely controlled sequences. Our success in obtaining these highly regular polychalcogenophenes relied on the synthesis of corresponding unsymmetrical monomers: the diiodobichalcogenophenes SSeI2, STeI2, and SeTeI2 and the diiodoterchalcogenophene SSeTeI2 (in model A). Because of greater electron-deficiency and less steric hindrance, Grignard metathesis of the monomers SSeI2, STeI2, and SeTeI2 occurred regioselectively at the lighter chalcogenophene unit (i.e., the thiophene units for SSeI2, STeI2, and SSeTeI2; the selenophene unit for SeTeI2). All of the regioselectively prepared Mg species underwent Ni(dppp)Cl2-catalyzed KCTP. The key step during the KCTP was the Ni complex undergoing thermodynamically favorable ring-walking from the lighter to the heavier chalcogenophene, with corresponding higher complexation energy with the Ni catalyst. The ring-walking facilitated subsequent oxidative addition, suppressing the possibility of chain-transfer or chain-termination. Our first synthesis of an ABC-type periodic poly(terchalcogenophene) PSSeTe also reveals that the Ni catalyst could walk efficiently over a distance of three rings (ca. 1 nm) from a thiophene unit via a selenophene unit to a tellurophene unit, all with a gradual increase in complexation energy, thereby preserving the mechanism of catalyst-transfer polycondensation. The thin-film morphologies and the optical, electrochemical, conformational, and OFET properties of the polymers could be tailored systematically by combining the different chalcogenophenes at various ratios—a promising feature for a wide range of potential applications. This paper provides a design concept for the use of single unsymmetrical monomers to create new AB- and ABC-type alternating and periodic conjugated polymers with high regioregularity.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is supported by the Ministry of Science and Technology, Taiwan (grant no. MOST107-3017-F009-003) and Ministry of Education, Taiwan (SPROUT Project-Center for Emergent Functional Matter Science of National Chiao Tung University). We thank the National Center of High-Performance Computing (NCHC) in Taiwan for computer time and facilities. We also thank the National Synchrotron Radiation Research Center (NSRRC), and Dr U-Ser Jeng and Dr Chun-Jen Su at BL23A1 station for the help with the GIXS experiments.
Notes and references
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS PubMed.
- L. Dou, Y. Liu, Z. Hong, G. Li and Y. Yang, Chem. Rev., 2015, 115, 12633–12665 CrossRef CAS PubMed.
- A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS.
- J. Roncali, Chem. Rev., 1997, 97, 173–206 CrossRef CAS PubMed.
- H. A. M. van Mullekom, J. A. J. M. Vekemans, E. E. Havinga and E. W. Meijer, Mater. Sci. Eng., R, 2001, 32, 1–40 CrossRef.
- D. Braun, G. Gustafsson, D. McBranch and A. J. Heeger, J. Appl. Phys., 1992, 72, 564–568 CrossRef CAS.
- H. Nishino, G. Yu, A. J. Heeger, T. A. Chen and R. D. Rieke, Synth. Met., 1995, 68, 243–247 CrossRef CAS.
- Z. Bao, A. Dodabalapur and A. J. Lovinger, Appl. Phys. Lett., 1996, 69, 4108–4110 CrossRef CAS.
- D. H. Kim, Y. D. Park, Y. Jang, H. Yang, Y. H. Kim, J. I. Han, D. G. Moon, S. Park, T. Chang, C. Chang, M. Joo, C. Y. Ryu and K. Cho, Adv. Funct. Mater., 2005, 15, 77–82 CrossRef CAS.
- R. J. Kline, M. D. McGehee, E. N. Kadnikova, J. Liu and J. M. J. Fréchet, Adv. Mater., 2003, 15, 1519–1522 CrossRef CAS.
- H. Sirringhaus, P. J. Brown, R. H. Friend, M. M. Nielsen, K. Bechgaard, B. M. W. Langeveld-Voss, A. J. H. Spiering, R. A. J. Janssen, E. W. Meijer, P. Herwig and D. M. de Leeuw, Nature, 1999, 401, 685–688 CrossRef CAS.
- H. Sirringhaus, N. Tessler and R. H. Friend, Science, 1998, 280, 1741–1744 CrossRef CAS PubMed.
- H. Yang, T. J. Shin, L. Yang, K. Cho, C. Y. Ryu and Z. Bao, Adv. Funct. Mater., 2005, 15, 671–676 CrossRef CAS.
- R. Zhang, B. Li, M. C. Iovu, M. Jeffries-El, G. Sauvé, J. Cooper, S. Jia, S. Tristram-Nagle, D. M. Smilgies, D. N. Lambeth, R. D. McCullough and T. Kowalewski, J. Am. Chem. Soc., 2006, 128, 3480–3481 CrossRef CAS PubMed.
- S. Holliday, R. S. Ashraf, A. Wadsworth, D. Baran, S. A. Yousaf, C. B. Nielsen, C.-H. Tan, S. D. Dimitrov, Z. Shang, N. Gasparini, M. Alamoudi, F. Laquai, C. J. Brabec, A. Salleo, J. R. Durrant and I. McCulloch, Nat. Commun., 2016, 7, 11585 CrossRef CAS PubMed.
- Z. Xu, L.-M. Chen, G. Yang, C.-H. Huang, J. Hou, Y. Wu, G. Li, C.-S. Hsu and Y. Yang, Adv. Funct. Mater., 2009, 19, 1227–1234 CrossRef CAS.
- C. J. Brabec, N. S. Sariciftci and J. C. Hummelen, Adv. Funct. Mater., 2001, 11, 15–26 CrossRef CAS.
- S. Lilliu, T. Agostinelli, E. Pires, M. Hampton, J. Nelson and J. E. Macdonald, Macromolecules, 2011, 44, 2725–2734 CrossRef CAS.
- D. H. Kim, J. T. Han, Y. D. Park, Y. Jang, J. H. Cho, M. Hwang and K. Cho, Adv. Mater., 2006, 18, 719–723 CrossRef CAS.
- M. Brinkmann and J.-C. Wittmann, Adv. Mater., 2006, 18, 860–863 CrossRef CAS.
- R. D. McCullough, Adv. Mater., 1998, 10, 93–116 CrossRef CAS.
- R. S. Loewe, P. C. Ewbank, J. Liu, L. Zhai and R. D. McCullough, Macromolecules, 2001, 34, 4324–4333 CrossRef CAS.
- E. E. Sheina, J. Liu, M. C. Iovu, D. W. Laird and R. D. McCullough, Macromolecules, 2004, 37, 3526–3528 CrossRef CAS.
- R. Miyakoshi, A. Yokoyama and T. Yokozawa, Macromol. Rapid Commun., 2004, 25, 1663–1666 CrossRef CAS.
- A. Yokoyama, R. Miyakoshi and T. Yokozawa, Macromolecules, 2004, 37, 1169–1171 CrossRef CAS.
- Z. J. Bryan and A. J. McNeil, Macromolecules, 2013, 46, 8395–8405 CrossRef CAS.
- R. Miyakoshi, A. Yokoyama and T. Yokozawa, J. Am. Chem. Soc., 2005, 127, 17542–17547 CrossRef CAS PubMed.
- T. Beryozkina, V. Senkovskyy, E. Kaul and A. Kiriy, Macromolecules, 2008, 41, 7817–7823 CrossRef CAS.
- M. C. Iovu, E. E. Sheina, R. R. Gil and R. D. McCullough, Macromolecules, 2005, 38, 8649–8656 CrossRef CAS.
- R. Tkachov, V. Senkovskyy, H. Komber, J.-U. Sommer and A. Kiriy, J. Am. Chem. Soc., 2010, 132, 7803–7810 CrossRef CAS PubMed.
- A. K. Leone, P. K. Goldberg and A. J. McNeil, J. Am. Chem. Soc., 2018, 140, 7846–7850 CrossRef CAS PubMed.
- S. Tamba, K. Shono, A. Sugie and A. Mori, J. Am. Chem. Soc., 2011, 133, 9700–9703 CrossRef CAS PubMed.
- R. Grisorio and G. P. Suranna, Polym. Chem., 2015, 6, 7781–7795 RSC.
- T. Yokozawa and Y. Ohta, Chem. Rev., 2016, 116, 1950–1968 CrossRef CAS PubMed.
- T. Yokozawa and A. Yokoyama, Chem. Rev., 2009, 109, 5595–5619 CrossRef CAS PubMed.
- A. Kiriy, V. Senkovskyy and M. Sommer, Macromol. Rapid Commun., 2011, 32, 1503–1517 CrossRef CAS PubMed.
- M. Planells, B. C. Schroeder and I. McCulloch, Macromolecules, 2014, 47, 5889–5894 CrossRef CAS.
- A. Linden, Y. Zhou and H. Heimgartner, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2014, 70, 482–487 CrossRef CAS PubMed.
- K. Takimiya, Y. Konda, H. Ebata, N. Niihara and T. Otsubo, J. Org. Chem., 2005, 70, 10569–10571 CrossRef CAS PubMed.
- C.-E. Tsai, R.-H. Yu, F.-J. Lin, Y.-Y. Lai, J.-Y. Hsu, S.-W. Cheng, C.-S. Hsu and Y.-J. Cheng, Chem. Mater., 2016, 28, 5121–5130 CrossRef CAS.
- M. Jeffries-El, B. M. Kobilka and B. J. Hale, Macromolecules, 2014, 47, 7253–7271 CrossRef CAS.
- X. Wu, L. Lv, L. Hu, Q. Shi, A. Peng and H. Huang, ChemPhysChem, 2019, 20, 2600–2607 CrossRef CAS PubMed.
- R. S. Ashraf, I. Meager, M. Nikolka, M. Kirkus, M. Planells, B. C. Schroeder, S. Holliday, M. Hurhangee, C. B. Nielsen, H. Sirringhaus and I. McCulloch, J. Am. Chem. Soc., 2015, 137, 1314–1321 CrossRef CAS PubMed.
- F.-Y. Cao, C.-C. Tseng, F.-Y. Lin, Y. Chen, H. Yan and Y.-J. Cheng, Chem. Mater., 2017, 29, 10045–10052 CrossRef CAS.
- J. Casado, M. Moreno Oliva, M. C. Ruiz Delgado, R. Ponce Ortiz, J. Joaquín Quirante, J. T. López Navarrete, K. Takimiya and T. Otsubo, J. Phys. Chem. A, 2006, 110, 7422–7430 CrossRef CAS PubMed.
- Y.-Y. Lai, H.-H. Chang, Y.-Y. Lai, W.-W. Liang, C.-E. Tsai and Y.-J. Cheng, Macromolecules, 2015, 48, 6994–7006 CrossRef CAS.
- F.-Y. Cao, F.-Y. Lin, C.-C. Tseng, K.-E. Hung, J.-Y. Hsu, Y.-C. Su and Y.-J. Cheng, ACS Appl. Mater. Interfaces, 2019, 11, 11674–11683 CrossRef CAS PubMed.
- J. G. Manion, S. Ye, A. H. Proppe, A. W. Laramée, G. R. McKeown, E. L. Kynaston, S. O. Kelley, E. H. Sargent and D. S. Seferos, ACS Appl. Energy Mater., 2018, 1, 5033–5042 CrossRef CAS.
- M. Al-Hashimi, Y. Han, J. Smith, H. S. Bazzi, S. Y. A. Alqaradawi, S. E. Watkins, T. D. Anthopoulos and M. Heeney, Chem. Sci., 2016, 7, 1093–1099 RSC.
- H. Ebata, E. Miyazaki, T. Yamamoto and K. Takimiya, Org. Lett., 2007, 9, 4499–4502 CrossRef CAS PubMed.
- M. Heeney, W. Zhang, D. J. Crouch, M. L. Chabinyc, S. Gordeyev, R. Hamilton, S. J. Higgins, I. McCulloch, P. J. Skabara, D. Sparrowe and S. Tierney, Chem. Commun., 2007, 5061–5063 RSC.
- Y. Wang, H. Cui, M. Zhu, F. Qiu, J. Peng and Z. Lin, Macromolecules, 2017, 50, 9674–9682 CrossRef CAS.
- A. A. Jahnke, B. Djukic, T. M. McCormick, E. Buchaca Domingo, C. Hellmann, Y. Lee and D. S. Seferos, J. Am. Chem. Soc., 2013, 135, 951–954 CrossRef CAS PubMed.
- S. Ye, M. Steube, E. I. Carrera and D. S. Seferos, Macromolecules, 2016, 49, 1704–1711 CrossRef CAS.
- J. H. Bannock, M. Al-Hashimi, S. H. Krishnadasan, J. J. M. Halls, M. Heeney and J. C. de Mello, Mater. Horiz., 2014, 1, 214–218 RSC.
- H. Yan, J. Hollinger, C. R. Bridges, G. R. McKeown, T. Al-Faouri and D. S. Seferos, Chem. Mater., 2014, 26, 4605–4611 CrossRef CAS.
- E. F. Palermo and A. J. McNeil, Macromolecules, 2012, 45, 5948–5955 CrossRef CAS.
- J. Hollinger, A. A. Jahnke, N. Coombs and D. S. Seferos, J. Am. Chem. Soc., 2010, 132, 8546–8547 CrossRef CAS PubMed.
- D. Gao, J. Hollinger, A. A. Jahnke and D. S. Seferos, J. Mater. Chem. A, 2014, 2, 6058–6063 RSC.
- J. Hollinger, P. M. DiCarmine, D. Karl and D. S. Seferos, Macromolecules, 2012, 45, 3772–3778 CrossRef CAS.
- D. Gao, J. Hollinger and D. S. Seferos, ACS Nano, 2012, 6, 7114–7121 CrossRef CAS PubMed.
- E. L. Kynaston, Y. Fang, J. G. Manion, N. K. Obhi, J. Y. Howe, D. F. Perepichka and D. S. Seferos, Angew. Chem., Int. Ed., 2017, 56, 6152–6156 CrossRef CAS PubMed.
- J. A. Amonoo, A. Li, G. E. Purdum, M. E. Sykes, B. Huang, E. F. Palermo, A. J. McNeil, M. Shtein, Y.-L. Loo and P. F. Green, J. Mater. Chem. A, 2015, 3, 20174–20184 RSC.
- Y.-Y. Lai, T.-C. Tung, W.-W. Liang and Y.-J. Cheng, Macromolecules, 2015, 48, 2978–2988 CrossRef CAS.
- Y. Qiu, A. Fortney, C.-H. Tsai, M. A. Baker, R. R. Gil, T. Kowalewski and K. J. T. Noonan, ACS Macro Lett., 2016, 5, 332–336 CrossRef CAS.
- C.-H. Tsai, A. Fortney, Y. Qiu, R. R. Gil, D. Yaron, T. Kowalewski and K. J. T. Noonan, J. Am. Chem. Soc., 2016, 138, 6798–6804 CrossRef CAS PubMed.
- T.-A. Chen, X. Wu and R. D. Rieke, J. Am. Chem. Soc., 1995, 117, 233–244 CrossRef CAS.
- S. Ye, S. M. Foster, A. A. Pollit, S. Cheng and D. S. Seferos, Chem. Sci., 2019, 10, 2075–2080 RSC.
- S. K. Sontag, J. A. Bilbrey, N. E. Huddleston, G. R. Sheppard, W. D. Allen and J. Locklin, J. Org. Chem., 2014, 79, 1836–1841 CrossRef CAS PubMed.
- S. Ye, L. Janasz, W. Zajaczkowski, J. G. Manion, A. Mondal, T. Marszalek, D. Andrienko, K. Müllen, W. Pisula and D. S. Seferos, Macromol. Rapid Commun., 2019, 40, 1800596 CrossRef PubMed.
- L. Li, J. Hollinger, A. A. Jahnke, S. Petrov and D. S. Seferos, Chem. Sci., 2011, 2, 2306–2310 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc00404a |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  c,
Yen-Chen
Su
a,
Hau-Ren
Yang
c,
Kuang-Yi
Cheng
a,
Sheng-Cih
Huang
a and
Yen-Ju
Cheng
c,
Yen-Chen
Su
a,
Hau-Ren
Yang
c,
Kuang-Yi
Cheng
a,
Sheng-Cih
Huang
a and
Yen-Ju
Cheng