
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExpected and unexpected photoreactions of 9-(10-)substituted anthracene derivatives in cucurbit[n]uril hosts†
Xianchen
Hu
a,
Fengbo
Liu
 a,
Xiongzhi
Zhang
ab,
Zhiyong
Zhao
ab and
Simin
Liu
*ab
a,
Xiongzhi
Zhang
ab,
Zhiyong
Zhao
ab and
Simin
Liu
*ab
aThe State Key Laboratory of Refractories and Metallurgy, School of Chemistry and Chemical Engineering, Wuhan University of Science and Technology, Wuhan 430081, China. E-mail: liusimin@wust.edu.cn
bInstitute of Advanced Materials and Nanotechnology, Wuhan University of Science and Technology, Wuhan 430081, China
First published on 24th April 2020
Abstract
By arranging substrates in a “reaction ready” state through noncovalent interactions, supramolecular nanoreactors/catalysts show high selectivity and/or rate acceleration features. Herein, we report the host–guest complexation of 9-(10-)substituted anthracene derivatives (G1–G3) with cucurbit[n]uril (CB[n], n = 8, 10), and the photoreactions of these derivatives in the presence of CB[n] hosts. Both CB[10] and CB[8] showed no obvious effects on the photoreaction of 9,10-disubstituted derivative G1. For G2 and G3, CB[10] operated as either a nanoreactor or catalyst (10%) for the photodimerization of two compounds with high selectivity and high yield. However, although CB[8] formed a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 complex with G2, as also observed with CB[10], the photosolvolysis product (9-anthracenemethanol) was obtained quantitatively after photoirradiation of the CB[8]·2G2 complex. This unexpected photosolvolysis was rationalized by a plausible catalytic cycle in which anthracene acts as a photoremovable protecting group (PPG) and the carbonium ion intermediate is stabilized by CB[8].
:2 complex with G2, as also observed with CB[10], the photosolvolysis product (9-anthracenemethanol) was obtained quantitatively after photoirradiation of the CB[8]·2G2 complex. This unexpected photosolvolysis was rationalized by a plausible catalytic cycle in which anthracene acts as a photoremovable protecting group (PPG) and the carbonium ion intermediate is stabilized by CB[8].
Introduction
Inspired by natural enzymes, supramolecular nanoreactors/catalysts have been developed to achieve reactions with high selectivity and/or rate acceleration.1,2 In host–guest chemistry, many types of macrocyclic host molecules have been explored as supramolecular nanoreactors/catalysts, such as cyclodextrins,3 calixarenes,4 pillararenes,5 and nanocages, among others.6–9 Photochemical reactions of anthracene and its derivatives, including anthracene dimerization and other unimolecular photoreactions, have been widely investigated over the last 100 years owing to the unique photo-responsive properties of these compounds.10,11 However, the anthracene group acted as photoremovable protecting group (PPG) in only one example.12,13 In supramolecular chemistry, host–guest interactions have been introduced to regulate the photodimerization of anthracene derivatives.14–18 Furthermore, anthracene dimerization has been further used in the design of host–guest-related supramolecular polymers and optical materials/devices.19–25Cucurbit[n]urils (CB[n]s, n = 5–8, 10) are a family of pumpkin-like macrocyclic hosts. CB[n] compounds bear a hydrophobic cavity and two identical portals surrounded by negative carbonyl groups as structural features, and have potential applications in many fields.26–29 Similar to other hosts, CB[n]s have also been employed to promote various organic reactions, especially photodimerization reactions,30–32 but have acted as supramolecular catalysts in only a few examples because the product is usually “trapped” in the CB[n] cavity, inhibiting the catalytic process.33,34
In this work, we investigated host–guest complexation between CB[n] (n = 8, 10) and 9-(10-)substituted anthracene derivatives (G1–G3) (Fig. 1) in aqueous solution, and the CB[n]-mediated (n = 8, 10) photoreaction of these guests. Notably, macrocyclic host-promoted photoreactions of 9-(10-)substituted anthracene derivatives have not been reported previously.35–39 Our results showed that CB[10] and CB[8] had no obvious effects on the photoreaction of G1, despite encapsulating G1. However, for G2 and G3, CB[10] operated as either a nanoreactor or catalyst (10%) for the photodimerization of two compounds with high selectivity and high yield. Although CB[8] formed a 1:2 complex with G2, as also observed with CB[10], photosolvolysis of G2 was unexpectedly observed instead of dimerization after photoirradiation of the CB[8]·2G2 complex (Scheme 1). A plausible reaction mechanism referring to anthracene as a PPG is provided.
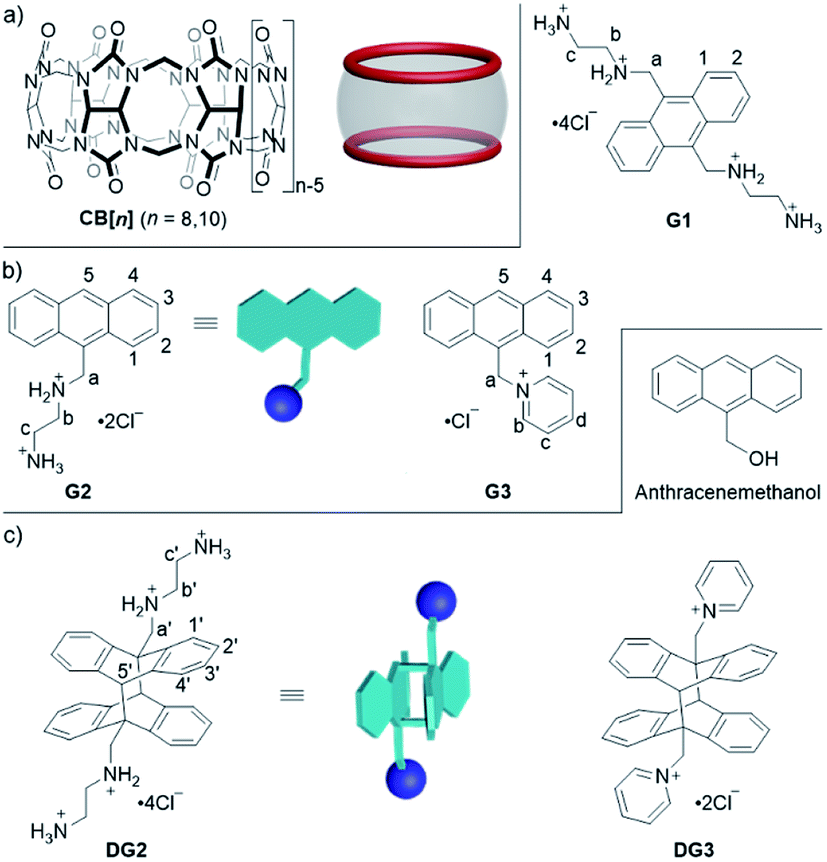 | ||
| Fig. 1 Structures of (a) hosts CB[n] (n = 8, 10), (b) guests G1–G3, and (c) photoreaction products. | ||
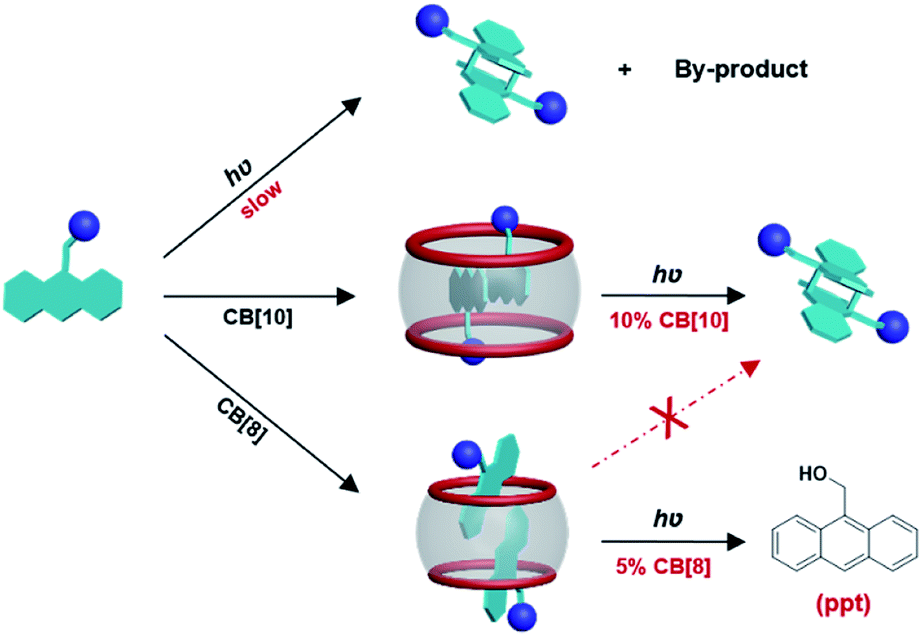 | ||
| Scheme 1 Schematic illustration of CB[n]-mediated photoreactions of 9-substituted anthracene derivative G2. | ||
Results and discussion
Complexation of G1 with CB[n] and photoreactions of complexes
Initially, the host–guest complexation of G1 with CB[n] (n = 8, 10) was investigated by NMR, UV/Vis, and fluorescence spectroscopy, and ESI-MS analysis. 1H NMR titration experiments clearly demonstrated the formation of host–guest complexes CB[10]·G1 and CB[10]·2G1. As shown in Fig. 2b, binding between G1 and CB[10] exhibited slow exchange kinetics on the 1H NMR time scale, because both free and bound proton signals were observed. The slow exchange kinetics also allowed the binding ratio to be calculated as 1:2 (host/guest) from the integrals for peaks of bound guest and host. When the amount of CB[10] added was increased to 0.5 equiv. (Fig. 2c), a new set of bound guest peaks (marked as ‘&’) was observed, along with a tiny amount of free guest (marked as ‘*’). Clearly the new inclusion complex also exhibited slow exchange kinetics on the 1H NMR time scale. When excess CB[10] was added to a solution of G1 (free CB[10] is insoluble in water and complexation with the guest usually allows CB[10] to enter the aqueous phase40), we calculated the binding ratio of CB[10] with G1 to be 1:1, according to 1H NMR peak integration (Fig. 2d). ESI-MS also indicated the existence of two inclusion complexes, CB[10]·G1 and CB[10]·2G1. As shown in Fig. S1a,† ion peaks at m/z 577.5, 769.6, and 781.6 were observed, corresponding to 1:2 complex CB[10]·2G1 ([CB[10] + 2G14+ − 4H+]4+ = 577.5; [CB[10] + 2G14+ − 5H+]3+ = 769.6; [CB[10] + 2G14+ − 4H+ + Cl−]3+ = 781.6). The ion peak at m/z 996.4, corresponding to 1:1 complex CB[10]·G1, was also observed ([CB[10] + G14+ − 2H+]2+ = 996.4) (Fig. S1†).
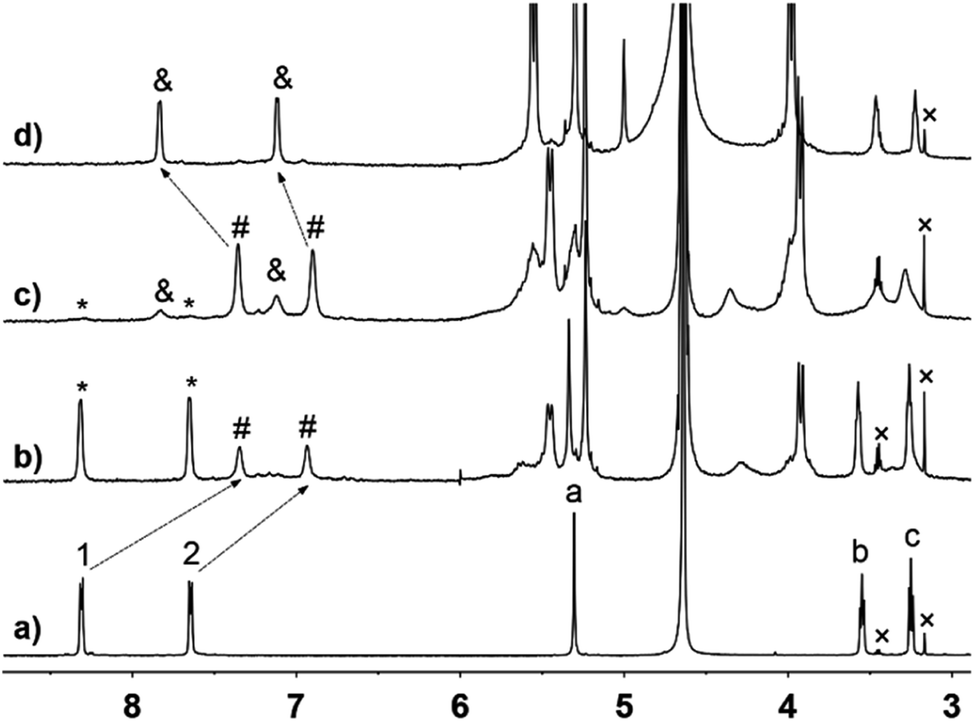 | ||
| Fig. 2
1H NMR spectra (600 MHz, D2O, 298 K): of (a) free G1 (2.5 mM); (b) 4:1 mixture of G1 and CB[10]; (c) 2:1 mixture of G1 and CB[10]; (d) G1 and excess CB[10] (resonances of free G1 are marked with ‘*’; resonances of CB[10]·2G1 are marked with ‘#’, resonances of CB[10]·G1 are marked with ‘&’; proton signals of impurities are marked with ‘×’). | ||
UV/Vis and fluorescence titrations were used to examine the complexation of CB[10] with G1 in aqueous solution. With the addition of CB[10], the long-wavelength absorption of free G1 at λmax = 373 nm showed a bathochromic shift (Fig. S2†). In the absence of CB[10], G1 showed the anthracene emission with a maximum wavelength of 422 nm. The fluorescence intensity of G1 changed when CB[10] was added continuously, owing to host–guest complexation of G1 with the CB[10] host (Fig. 3).41 Furthermore, before the amount of CB[10] was increased to ∼0.5 equiv., the fluorescence intensity of G1 was observed to continuously decrease. However, at 1.0 equiv., the intensity increased about two-fold compared with that of G1 itself, with a slight redshift. Combined with the NMR data, we speculated that CB[10] initially increased π–π stacking of G1 by encapsulating two guest molecules in the cavity, causing fluorescence quenching through formation of the H-dimer of G1.42 The further formation of 1:1 inclusion complex CB[10]·G1 prevented G1 from stacking and exterior aqueous environment, resulting in a stronger G1 emission.41 Meanwhile, host–guest complexation between CB[8] and G1 was examined by 1H NMR (Fig. S3†) and ESI-MS (Fig. S4†). Broadening of all proton signals, including those of CB[8], was observed when the amount of CB[8] was 0.5 equiv. (Fig. S3c†), indicating that binding between CB[8] and G1 underwent intermediate exchange on the 1H NMR time scale.43 ESI-MS showed one ion peak at m/z 826.2, corresponding to 1:1 complex CB[8]·G1 ([CB[8] + G14+ − 2H+]2+ = 826.2).
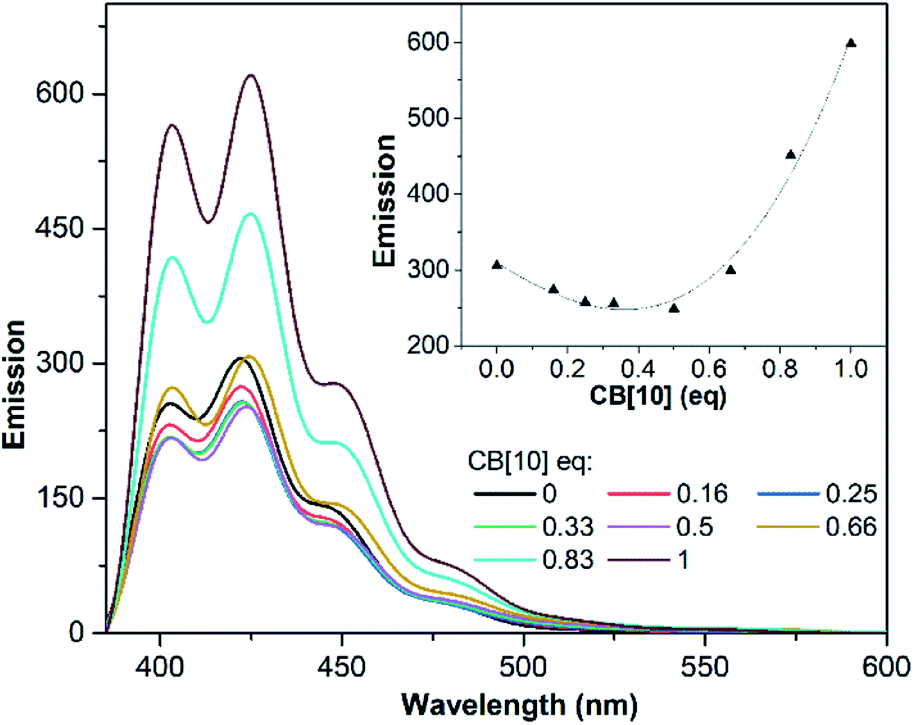 | ||
| Fig. 3 Fluorescence emission spectra recorded for G1 (31.25 μM) upon titration with CB[10]. Inset: emission of G1vs. CB[10] equivalent (λex = 250 nm, λem = 422 nm). | ||
With the binding results in hand, we checked the photoreactions of G1 in the absence and presence of CB[n] (n = 8, 10). Prior to UV irradiation at room temperature, all samples (in D2O or pure water) were degassed with nitrogen for 15 min ([G] = 2.5 mM), and the reaction was monitored by 1H NMR and UV/Vis spectroscopy. As shown in Fig. S5a,† the photoproducts of G1 were complicated and unidentified. In the presence of 0.25 or 0.5 equiv. of CB[10], ratios which benefitted the dimerization reaction, the dimerization product was not observed as expected (Fig. S5b and c†). Molecular modeling (MMFF) suggested two G1 molecules could adopt an “X” shape in the CB[10]·2G1 complex owing to electrostatic repulsion between the 9,10-substituted positive groups (Fig. 4),43 which was not a “reaction ready” state for dimerization. Similar results were obtained for the photoreaction of G1 with various contents of CB[8]. Therefore, guest G2 containing one substituent at the 9-position was designed.
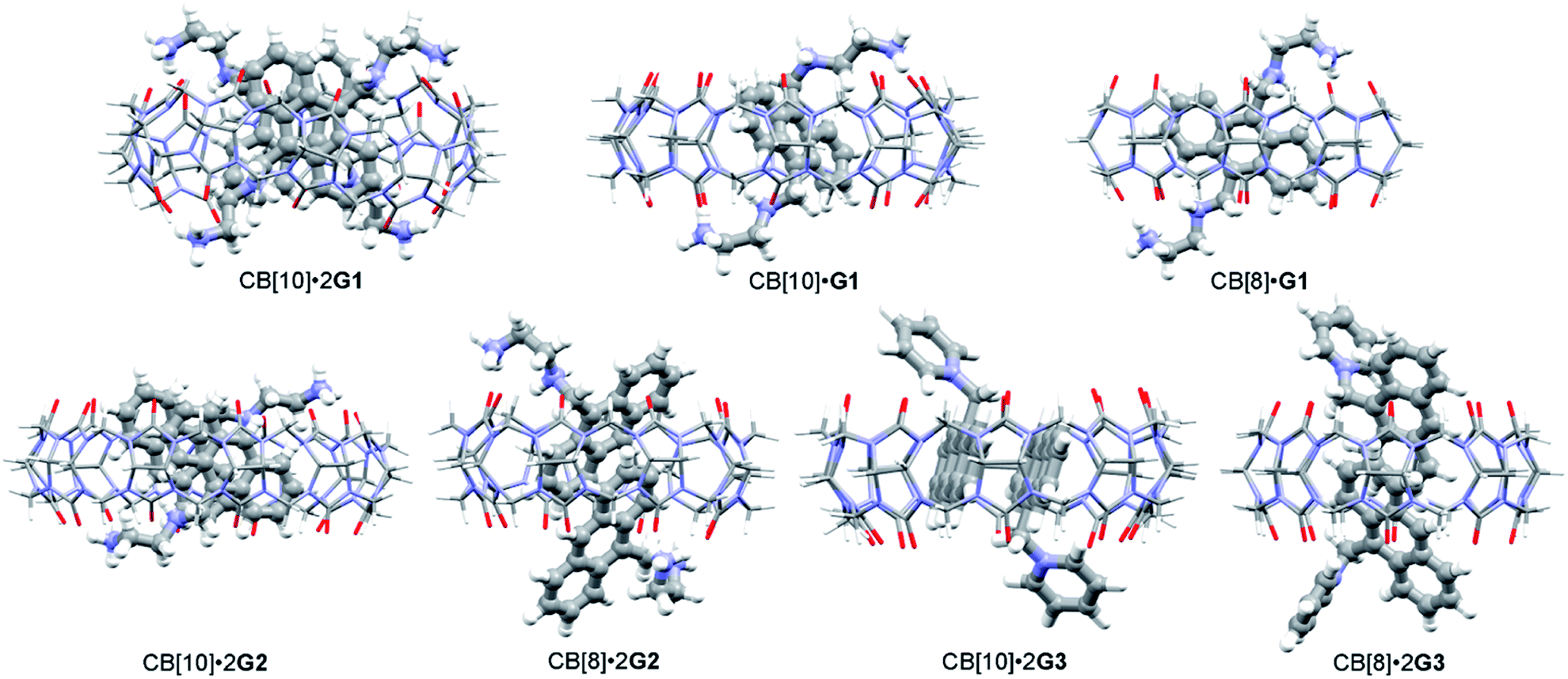 | ||
| Fig. 4 Plausible binding modes of guests with CB[n] (MMFF-minimized). | ||
Complexation of G2 with CB[n] and photoreactions of complexes
Similar to G1, we first investigated the host–guest complexation of G2 with CB[n] hosts, and then conducted the photoreaction of G2 with/without CB[n] (n = 8, 10). 1H NMR titrations showed that the aromatic proton and Ha signals of G2 were shifted upfield (Δδ = 0.97, 0.87, 0.97, 1.28, 1.05, and 1.63 ppm for protons H1–H5 and Ha, respectively) more than the Hb and Hc proton signals (Δδ = 0.47 and 0.28 ppm, respectively) by at least 0.40 ppm, suggesting that the anthracene moiety had advanced deeper and that the substituent was located around the portals of CB[10] (Fig. S6†). Broadening of the guest signals again implied that the binding of CB[10] and G2 underwent intermediate exchange on the 1H NMR time scale.43 Upon adding excess CB[10], the ratio of CB[10] to G2 was calculated to be 1:2 based on the integrals of the host and guest signals (Fig. S6c†). ESI-MS data further confirmed the formation of 1:2 complex CB[10]·2G2. As shown in Fig. S7,† an ion peak was observed at m/z 1081.9, which corresponded to the 1:2 complex ([CB[10] + 2G22+ − 2H+]2+ = 1081.9). UV/Vis and fluorescence titration experiments also corroborated the host–guest interaction of G2 and CB[10] in the aqueous phase. The fluorescence emission intensity of G2 decreased upon adding 0.5 equiv. of CB[10] (Fig. S8†), suggesting the existence of π–π stacking between the encapsulated anthracene units of G2, similar to that of G1. Interestingly, compared with G1, the binding of monosubstituted G2 with CB[8] exhibited slow exchange kinetics on the 1H NMR time scale, allowing the binding ratio to be calculated as 1:2 (host/guest) through integration (Fig. S9†). This 1:2 binding was further verified by ESI-MS (Fig. S10†). The differentiation of proton signals on the left and right phenyl rings in G2, and no splits in the proton signals of CB[8] under slow exchange kinetics, suggested that the anthracene moieties were inserted into the cavity of CB[8] with the two incorporated G2 molecules adopting a head-to-tail (h–t) orientation. Indeed, the MMFF-minimized model of complex CB[8]·2G2 showed partial aromatic rings of two G2 molecules located in the CB[8] cavity. The dihedral angle between the plane of the included anthracene group and the equatorial plane of CB[8] was about 59° (Fig. 4 and S11†). The distance between two included parallel anthracene groups was about 3.42 Å, suggesting the presence of π–π stacking.
Under the same conditions as for G1, photoreactions of G2 were conducted in the absence and presence of CB[n] (n = 8, 10). As shown in Fig. S12,† after UV irradiation for 6 h, h–t dimer product DG2 (purified and identified by irradiating complex CB[10]·2G2) was observed with unidentified side products and unreacted G2. In comparison, in the presence of 0.5 equiv. of CB[10], almost 100% of G2 was converted into a single product within 25 min. As shown in Fig. 5b, under UV irradiation for 10 min, G2 was partially converted into presumed new photoproduct(s). G2 was consumed after 25 min, with only one set of CB[10] host and bound product(s) peaks remaining (Fig. 5c). After the appropriate amount of 3,5-dimethylamantadine hydrochloride (3,5-DMADA) was added to the irradiated solution of CB[10]·2G2 to displace the product(s) from the CB[10] cavity (CB[10]·2(3,5-DMADA) is insoluble in water44), only one exclusive product, characterized as h–t dimer DG2, was obtained in almost quantitative yield (characterization data for DG2 is shown in Fig. S13–S15 in ESI†). By monitoring absorbance changes of G2 at λ = 370 nm, we calculated the yields of DG2 with various irradiation times. As “the intra-complex photodimerization is unimolecular in nature”,18 an apparent rate constant (k1) of 0.1158 ± 0.0079 min−1 for the dimerization reaction of G2 with 50% CB[10] was calculated with first-order kinetics (Fig. S16†). The half-conversion time (t1/2, time for conversion of half the substrate) was used because the rate constant could not be accurately measured in some cases. Clearly, in the presence of 50% CB[10], the dimerization reaction of G2 was accelerated tremendously (Table 1). The high conversion, high selectivity, and rate acceleration of this photoreaction of G2 within CB[10] suggested that two G2 molecules were preorganized by CB[10] to adopt a “reaction ready” h–t orientation, which was in good agreement with the MMFF-minimized model of complex CB[10]·2G2 in Fig. 4.
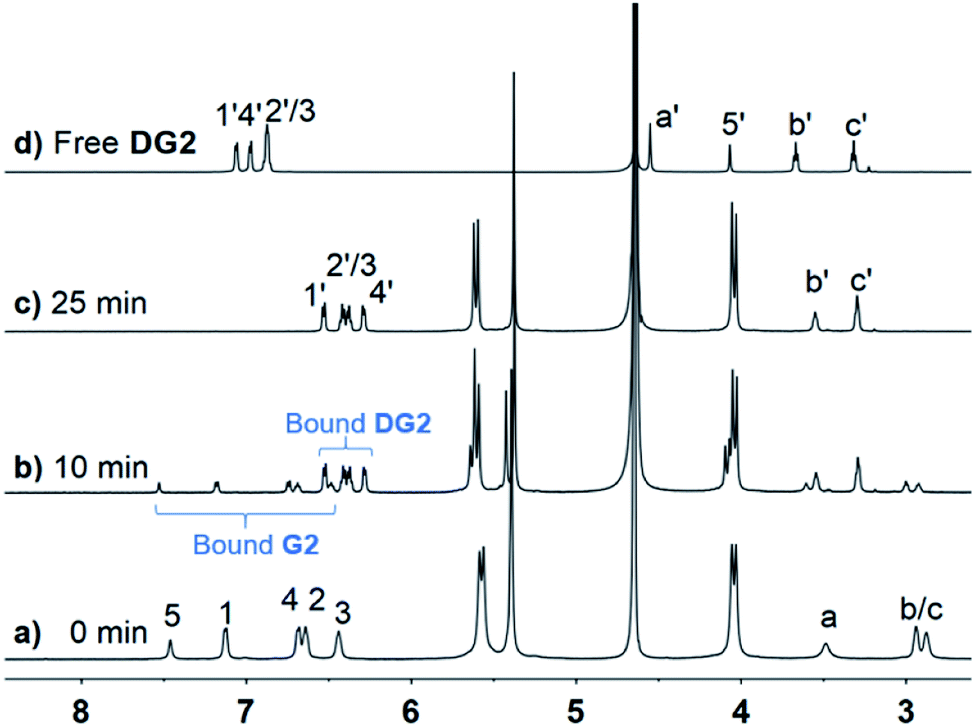 | ||
| Fig. 5 1H NMR spectra (600 MHz, D2O, 298 K) of CB[10]·2G2 ([G2] = 2.5 mM) solution with various UV irradiation times: (a) 0, (b) 10, (c) 25 min, and (d) free dimerization product DG2. | ||
| Host | t 1/2 (G2) | t 1/2 (G3) |
|---|---|---|
| a Estimated from NMR integrals (Fig. S12 and S32). b Calculated by fitting equation (Fig. S16, S18 and S39). c Estimated from diagrams of product yields (Fig. S26). d Not determined. | ||
| None | 1.7 ha | 7.5 ha |
| CB[10] (50%) | 6 minb | 37 minb |
| CB[10] (10%) | 44 minb | 110 minb |
| CB[8] (50%) | 8 minc | —d |
| CB[8] (5%) | 35 minc | —d |
Inspired by a previous sample,33 we investigated whether CB[10] could act as a supramolecular catalyst to convert G2 into DG2 exclusively. In the presence of 0.1 equiv. of CB[10] (10%), almost 100% of G2 was converted into photoproduct DG2 with trace amounts of side products within 120 min (Fig. S17†). The t1/2 of 44 min was calculated from the first-order kinetics equation (Fig. S18†). In comparison, the t1/2 of G2 without CB[10] was estimated to be 1.7 hours (Table 1). All data suggested that CB[10] operated as the supramolecular catalyst in this case, although the photoreaction took longer to achieve than that with 0.5 equiv. of CB[10] (Scheme 1). As mentioned above, effective replacement of the product (DG2) with the starting material (G2) was necessary for host (CB[10]) to operate as a catalyst. To verify this, 1H NMR competition experiments between DG2 and G2 with CB[10] were performed.
As predicted, the mixture of G2, DG2, and CB[10] with a 2:1:1 ratio resulted in most CB[10] being occupied by G2 (Fig. S19c†). An approximate equilibrium constant (Keq) was calculated as 8.8 × 105 M−1 (Fig. S20 and S21†), suggesting that CB[10] operated as a supramolecular catalyst owing to the spontaneous replacement of product DG2 with starting material G2. The results showed that, as either the supramolecular nanoreactor or catalyst, CB[10] not only shortened the photoreaction time, but also tremendously improved the reaction selectivity.
When the host molecule was switched to CB[8], an unexpected photoreaction of G2 occurred, although CB[8] bound G2 with the same binding ratio as CB[10]. In the presence of 0.5 equiv. of CB[8], precipitate formation was observed when the G2 solution was irradiated with UV light. With extended irradiation time, additional yellow precipitate was generated. Therefore, the solutions after centrifugation were carefully monitored by 1H NMR. By analyzing the stacking NMR spectra (Fig. 6) and the precipitate, the bound peaks of G2 were found to fully disappear in 60 min, while a singlet peak at 3.1 ppm appeared and remained, and the yellow precipitate contained no CB[8] and was soluble in DMSO and CDCl3. First, we suspected that the precipitate was anthraquinone, because anthracene derivatives can easily be oxidized,45,46 despite the photoreaction being conducted under N2 atmosphere. However, the NMR signals for the precipitate did not match those of anthraquinone. Further hypotheses included the photosolvolysis of G2. Eventually, the yellow precipitate was verified to be 9-anthracenemethanol by EI-MS and 1H NMR (Fig. S22a and S23†). When irradiated in air, G2 first generated 9-anthracenemethanol as a precipitate (suspended), which was then converted to anthraquinone (Fig. S22b†), further confirming the occurrence of photosolvolysis prior to photooxidation. The singlet peak in the aqueous phase represented the ethylene proton signal of ethylenediamine (EDA). And the binding of protonated EDA in CB[8] helped solubilize CB[8] in water at high concentrations (maximum solubility of CB[8] in water is about 0.2 mM). Indeed, NMR titration experiments proved that small EDA molecules could be encapsulated by CB[8] (Fig. S24†). Furthermore, CB[8] was tested to determine whether it could act as a supramolecular catalyst in the photosolvolysis of G2. In the presence of 0.05 equiv. of CB[8] (5%), G2 was almost all converted to 9-anthracenemethanol within 4 h (Fig. S25†). In comparison, no precipitate was observed without CB[8]. The t1/2 of G2 in the presence of CB[8] was estimated because the data could not be ideally fitted with first-order kinetics (Table 1 and Fig. S26†). CB[8] doubtless operated as a supramolecular catalyst for the photosolvolysis of G2. Therefore, why photosolvolysis occurred when G2 itself did not show the same reactivity required further investigation.
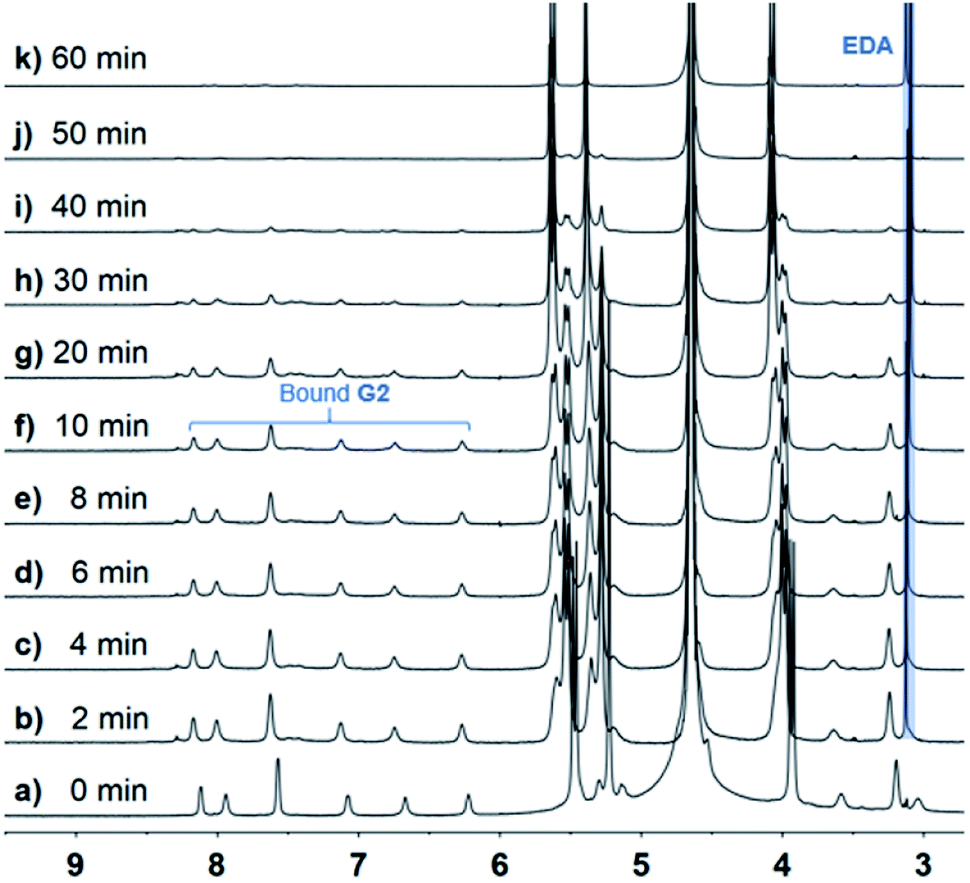 | ||
| Fig. 6
1H NMR spectra (600 MHz, D2O, 298 K) of 2:1 mixture of G2 (2.5 mM) and CB[8] with various UV irradiation times. | ||
After a broad search of the photosolvolysis literature, we noted that a “photoremovable protecting group (PPG)” could be involved in this reaction.12,47 To date, only one example of an anthracene unit acting as a PPG has been reported, with the plausible mechanism involving heterolysis and/or homolysis of the CH2–X bond in PPG–CH2–X (X, leaving group) to produce a carbonium ion intermediate. CB[n] hosts are known to be capable of stabilizing active carbocations.34,48,49 Therefore, a plausible mechanism for the photosolvolysis of G2 with CB[8] as the supramolecular catalyst in aqueous solution was proposed. As shown in Scheme 2, the initial step is light-induced heterolytic cleavage of the C–N bond of encapsulated G2, and/or homolytic cleavage of the C–N bond followed by rapid electron transfer (ET), to give the carbonium ion intermediate and EDA. The CB[8]-stabilized carbonium ion is then attacked by solvent molecule H2O to give product 9-anthracenemethanol. Owing to its weak binding with CB[8] and insolubility, 9-anthracenemethanol is displaced by G2 or EDA and precipitated out of solution, allowing the catalytic process to continue. Compared with the observation of radical side products (caused by homolysis) in most reported examples,12 the generation of a single product with quantitative yield in this case suggested that, with the assistance of CB[8], either C–N bond heterolysis was favored over homolysis or the electron transfer step after homolysis was accelerated.
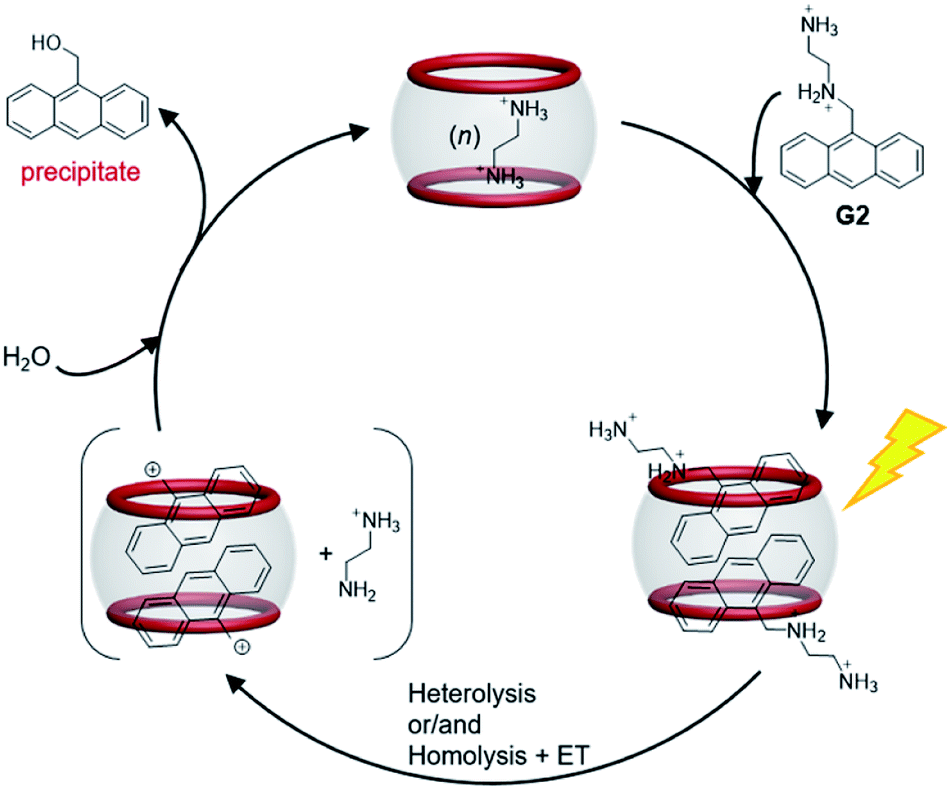 | ||
| Scheme 2 Plausible reaction mechanism for the photosolvolysis reaction of G2 in the presence of CB[8] host (5%). | ||
The high conversion and high selectivity of this photosolvolysis of G2 suggested that the orientation of the two G2 molecules in CB[8] was not only unfavorable for dimerization, but also favorable for stabilization of the carbonium ion intermediate by CB[8]. In addition to acting as the supramolecular catalyst, the most interesting role of CB[8] in this case was efficiently switching the reaction pathway of G2 from familiar photodimerization to unusual photosolvolysis.
Complexation of G3 with CB[n] and photoreactions of complexes
Guest molecule G3 was synthesized to explore the effect of different substituent groups on the photochemical reaction. The host–guest complexation of G3 with CB[n] (n = 8, 10) was investigated by 1H NMR, UV/Vis, and fluorescence spectroscopy, and ESI-MS analysis (Fig. S27–S31†). The ESI-MS data indicated that the complexation ratio of CB[n] with G3 was 1:2 (ion peaks at m/z 1100.9 and 934.8 corresponding to 1:2 complexes CB[10]·2G3 and CB[8]·2G3, respectively) (Fig. S29 and S31†). Similar to G2, the photoreaction of G3 slowly produced h–t dimerization product DG3 and unidentified side products (Fig. S32†). Furthermore, the dimerization of G3 with CB[10] operating as a supramolecular nanoreactor and catalyst, exclusively gave DG3 (Fig. S33–S39†). No bound peaks of DG3 were observed in the NMR competition experiments, indicating that the binding of G3 with CB[10] was much stronger than that of DG3 (Fig. S38†). The dimerization reaction of G3 was also greatly accelerated with assistance from CB[10] (Table 1 and Fig. S39†). However, in the presence of CB[8], no precipitate and an extremely complicated 1H NMR spectrum of photoproducts were observed, suggesting the negative effect of CB[8] on the photoreaction of G3 (Fig. S40†). It is unclear whether the lack of photosolvolysis of G3 was due to the deactivating effect of the pyridinium substituent group or the unfavorable orientation of G3 in CB[8].
Conclusions
In summary, we have reported the effects of CB[n] (n = 8, 10) hosts on the photoreaction of 9-(10-)anthracene derivatives. Both CB[8] and CB[10] operated as supramolecular nanoreactors and catalysts in the photoreaction of 9-substituted anthracene derivative G2. However, CB[10] promoted the photodimerization of G2, which occurred slowly with lower selectivity without a host. In contrast, CB[8] exclusively induced the photosolvolysis of G2, which did not occur without a host. Furthermore, the photoreactions of two anthracene derivatives, G1 and G3, were investigated in the presence of CB[n] to compare the effect of different CB[n] on the reactivity of anthracene derivatives with various substituents. The results showed that small differences in the host structures could cause large different effects on the host-involved reaction selectivity.As the first example of CB[10] operating as a supramolecular nanoreactor/catalyst, we anticipate that more reactions, involving large-sized or more than two substrate molecules, can be promoted by the large cavity of CB[10]. The selectivity for photosolvolysis of aromatic substrates with a PPG is also expected to be improved by the ability of CB[n] to stabilize cations. These finding will further benefit CB[n] chemistry and supramolecular catalysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21871216, 21472143).Notes and references
- P. W. N. M. van Leeuwen, in Supramolecular catalysis, Wiley-VCH, 2008 Search PubMed.
- J. Meeuwissen and J. N. H. Reek, Nat. Chem., 2010, 2, 615–621 CrossRef CAS PubMed.
- U. H. Brinker and J.-L. Mieusset, in Molecular Encapsulation Organic Reactions in Constrained Systems, Wiley, 2010, pp. 43–115 Search PubMed.
- P. Neri, J. L. Sessler and M.-X. Wang, in Calixarenes and beyond, Springer, 2016, pp. 691–742 Search PubMed.
- E. H. Wanderlind, D. G. Liz, A. P. Gerola, R. F. Affeldt, V. Nascimento, L. C. Bretanha, R. Montecinos, L. Garcia-Rio, H. D. Fiedler and F. Nome, ACS Catal., 2018, 8, 3343–3347 CrossRef CAS.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS PubMed.
- S. Zarra, D. M. Wood, D. A. Roberts and J. R. Nitschke, Chem. Soc. Rev., 2015, 44, 419–432 RSC.
- A. Galan and P. Ballester, Chem. Soc. Rev., 2016, 45, 1720–1737 RSC.
- S. Sadjadi, in Organic Nanoreactors: From Molecular to Supramolecular Organic Compounds, Academic Press, 2016 Search PubMed.
- H. D. Becker, Chem. Rev., 1993, 93, 145–172 CrossRef CAS.
- H. Bouas-Laurent, A. Castellan, J.-P. Desvergne and R. Lapouyade, Chem. Soc. Rev., 2000, 29, 43–55 RSC.
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef PubMed.
- A. K. Singh and P. K. Khade, Tetrahedron Lett., 2005, 46, 5563–5566 CrossRef CAS.
- H. Wang, F. Liu, R. C. Helgeson and K. N. Houk, Angew. Chem., Int. Ed., 2013, 52, 655–659 CrossRef CAS PubMed.
- A. Tron, H.-P. Jacquot de Rouville, A. Ducrot, J. H. R. Tucker, M. Baroncini, A. Credi and N. D. McClenaghan, Chem. Commun., 2015, 51, 2810–2813 RSC.
- J.-C. Gui, Z.-Q. Yan, Y. Peng, J.-G. Yi, D.-Y. Zhou, D. Su, Z.-H. Zhong, G.-W. Gao, W.-H. Wu and C. Yang, Chin. Chem. Lett., 2016, 27, 1017–1021 CrossRef CAS.
- X. Wei, W. Wu, R. Matsushita, Z. Yan, D. Zhou, J. J. Chruma, M. Nishijima, G. Fukuhara, T. Mori, Y. Inoue and C. Yang, J. Am. Chem. Soc., 2018, 140, 3959–3974 CrossRef CAS PubMed.
- J. Ji, W. Wu, W. Liang, G. Cheng, R. Matsushita, Z. Yan, X. Wei, M. Rao, D.-Q. Yuan, G. Fukuhara, T. Mori, Y. Inoue and C. Yang, J. Am. Chem. Soc., 2019, 141, 9225–9238 CrossRef PubMed.
- J.-F. Xu, Y.-Z. Chen, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, Org. Lett., 2013, 15, 6148–6151 CrossRef CAS PubMed.
- P. Wei, X. Yan and F. Huang, Chem. Commun., 2014, 50, 14105–14108 RSC.
- X. Zhang, Y. Gao, Y. Lin, J. Hu and Y. Ju, Polym. Chem., 2015, 6, 4162–4166 RSC.
- W. Guan, G. Wang, J. Ding, B. Li and L. Wu, Chem. Commun., 2019, 55, 10788–10791 RSC.
- Q. Zhou, B. Zhang, D. Han, R. Chen, F. Qiu, J. Wu and H. Jiang, Chem. Commun., 2015, 51, 3124–3126 RSC.
- W. Zhou, Y. Chen, Q. Yu, P. Li, X. Chen and Y. Liu, Chem. Sci., 2019, 10, 3346–3352 RSC.
- M. Tu, H. Reinsch, S. Rodríguez-Hermida, R. Verbeke, T. Stassin, W. Egger, M. Dickmann, B. Dieu, J. Hofkens, I. F. J. Vankelecom, N. Stock and R. Ameloot, Angew. Chem., Int. Ed., 2019, 58, 2423–2427 CrossRef CAS PubMed.
- J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44, 4844–4870 CrossRef CAS PubMed.
- E. Masson, X. Ling, R. Joseph, L. Kyeremeh-Mensah and X. Lu, RSC Adv., 2012, 2, 1213–1247 RSC.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS PubMed.
- X. Yang, F. Liu, Z. Zhao, F. Liang, H. Zhang and S. Liu, Chin. Chem. Lett., 2018, 29, 1560–1566 CrossRef CAS.
- K. I. Assaf and W. M. Nau, Chem. Soc. Rev., 2015, 44, 394–418 RSC.
- S. Sadjadi, in Organic Nanoreactors: From Molecular to Supramolecular Organic Compounds, Elsevier, 2016, pp. 43–84 Search PubMed.
- K. Kim, in Cucurbiturils and Related Macrocycles, The Royal Society of Chemistry, 2019, pp. 86–120 Search PubMed.
- Y. Kang, X. Tang, H. Yu, Z. Cai, Z. Huang, D. Wang, J.-F. Xu and X. Zhang, Chem. Sci., 2017, 8, 8357–8361 RSC.
- L. Scorsin, J. A. Roehrs, R. R. Campedelli, G. F. Caramori, A. O. Ortolan, R. L. T. Parreira, H. D. Fiedler, A. Acuña, L. García-Río and F. Nome, ACS Catal., 2018, 8, 12067–12079 CrossRef CAS.
- C. Yang, T. Mori, Y. Origane, Y. H. Ko, N. Selvapalam, K. Kim and Y. Inoue, J. Am. Chem. Soc., 2008, 130, 8574–8575 CrossRef CAS PubMed.
- F. Biedermann, I. Ross and O. A. Scherman, Polym. Chem., 2014, 5, 5375–5382 RSC.
- C. P. Carvalho, Z. Domínguez, J. P. Da Silva and U. Pischel, Chem. Commun., 2015, 51, 2698–2701 RSC.
- C.-H. Tung and J.-Q. Guan, J. Org. Chem., 1998, 63, 5857–5862 CrossRef CAS PubMed.
- D.-Y. Wu, B. Chen, X.-G. Fu, L.-Z. Wu, L.-P. Zhang and C.-H. Tung, Org. Lett., 2003, 5, 1075–1077 CrossRef CAS PubMed.
- W. Gong, X. Yang, P. Y. Zavalij, L. Isaacs, Z. Zhao and S. Liu, Chem.–Eur. J., 2016, 22, 17612–17618 CrossRef CAS PubMed.
- R. N. Dsouza, U. Pischel and W. M. Nau, Chem. Rev., 2011, 111, 7941–7980 CrossRef CAS PubMed.
- N. Barooah, J. Mohanty and A. C. Bhasikuttan, Chem. Commun., 2015, 51, 13225–13228 RSC.
- X. Zhao, F. Liu, Z. Zhao, H. Karoui, D. Bardelang, O. Ouari and S. Liu, Org. Biomol. Chem., 2018, 16, 3809–3815 RSC.
- X. Yang, Z. Zhao, X. Zhang and S. Liu, Sci. China: Chem., 2018, 61, 787–791 CrossRef CAS.
- K. I. Assaf, M. A. Alnajjar and W. M. Nau, Chem. Commun., 2018, 54, 1734–1737 RSC.
- P. Anzenbacher, T. Niwa, L. M. Tolbert, S. R. Sirimanne and F. P. Guengerich, Biochemistry, 1996, 35, 2512–2520 CrossRef CAS PubMed.
- P. Wang, Asian J. Org. Chem., 2013, 2, 452–464 CrossRef CAS.
- R. Wang and D. H. Macartney, Tetrahedron Lett., 2008, 49, 311–314 CrossRef CAS.
- N. Basilio, L. García-Río, J. A. Moreira and M. Pessêgo, J. Org. Chem., 2010, 75, 848–855 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, binding 1H NMR spectra, UV/Vis spectra of guests with CB[n], and relevant COSY, ESI-MS, 13C NMR spectra, kinetics fitting. See DOI: 10.1039/d0sc00409j |
| This journal is © The Royal Society of Chemistry 2020 |