
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploiting the radical reactivity of diazaphosphinanes in hydrodehalogenations and cascade cyclizations†
Jingjing
Zhang
a,
Jin-Dong
Yang
 *a and
Jin-Pei
Cheng
*ab
*a and
Jin-Pei
Cheng
*ab
aCenter of Basic Molecular Science, Department of Chemistry, Tsinghua University, Beijing 100084, China. E-mail: jdyang@mail.tsinghua.edu.cn; jinpei_cheng@mail.tsinghua.edu.cn
bState Key Laboratory of Elemento-Organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, China
First published on 23rd April 2020
Abstract
The remarkable reducibility of diazaphosphinanes has been extensively applied in various hydrogenations, based on and yet limited by their well-known hydridic reactivity. Here we exploited their unprecedented radical reactivity to implement hydrodehalogenations and cascade cyclizations originally inaccessible by hydride transfer. These reactions feature a broad substrate scope, high efficiency and simplicity of manipulation. Mechanistic studies suggested a radical chain process in which a phosphinyl radical is generated in a catalytic cycle via hydrogen-atom transfer from diazaphosphinanes. The radical reactivity of diazaphosphinanes disclosed here differs from their well-established hydridic reactivity, and hence, opens a new avenue for diazaphosphinane applications in organic syntheses.
Introduction
As is known, in polar hydride transfer (HT), an adequate thermodynamic compensation is needed to overcome its high kinetic barrier.1 Hence, direct HT usually requires a significant local accumulation of positive charge on hydride acceptors, which are commonly cationic species or some highly polarized compounds. This poses a very challenging, if not insuperable, barrier for reducing less polar substrates via HT. Consequently, hydrodehalogenation of organic halides with a hydride donor is barely achieved via nucleophilic aromatic substitution.2 Compared with polar HT, the radical pathway displays a kinetic superiority because of its considerably lower intrinsic barrier.3 In this regard, hydrogen-atom transfer (HAT) may provide an alternative to enable reduction with hydride donors, which is unlikely to occur through direct HT.N-heterocyclic phosphines have recently attracted particular attention owing to their superior hydricity (Scheme 1a).4 Their hydridic reactivity has been extensively exploited in catalytic or stoichiometric reduction of various unsaturated compounds, such as imines,5 aldehydes and ketones,6 polar olefins7 and pyridines8 (Scheme 1b). A kinetic scale with respect to the HT tendency from N-heterocyclic phosphines was also reported.9 In contrast to the well-studied hydridic reactivity, understanding of their reducibility in hydrogen-atom or electron transfer pathways remains strikingly underdeveloped.
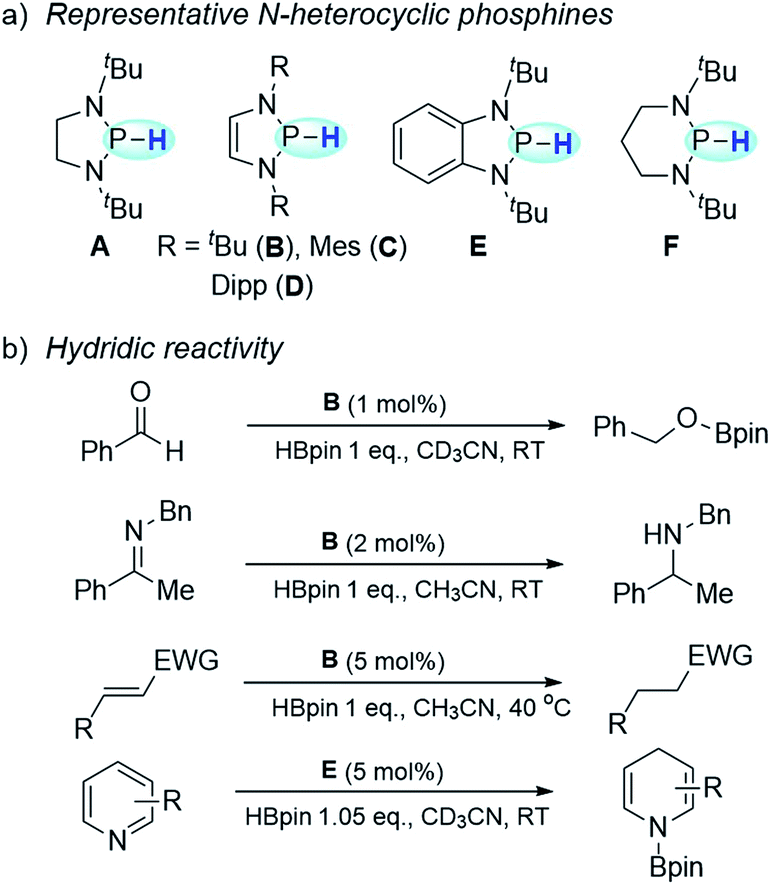 | ||
| Scheme 1 N-heterocyclic phosphines and their applications. (a) Representative N-heterocyclic phosphines. (b) Applications of N-heterocyclic phosphines in hydridic reduction. | ||
Based on the hydrogen-atom donating ability of diazaphosphinanes (DAPs) derived recently,10 we envisioned that trivalent phosphinyl radicals, which could be generated via HAT from DAPs, may allow a chance for radical reduction, and hence, renders a quite different reactivity and selectivity pattern from that by direct HT of DAPs. Aiming to verify this assumption, we have examined here the hydrodehalogenation of organic halides under properly designed conditions in favor of radical initiation. To our delight, the anticipated radical hydrodehalogenation of organohalides was indeed realized with high efficiency and wide substrate coverage, and therefore, could be a superior alternative to substitute some of the traditional approaches (e.g., halogen–metal exchange,11 transition metal catalysis,12 nucleophilic aromatic substitution2 and radical dehalogenation13) where limitation exists for removing halogens. Here, we used 1,3-di-tert-butyl-1,3,2-diazaphosphinane (1a, in Scheme 2a) as a potent hydrogen-atom donor to implement efficient radical hydrodehalogenation as well as cascade cyclization of organic halides, which were found to be inert when reduced by the same diazaphosphinane via direct HT.
 | ||
| Scheme 2 Our work. (a) Radical hydrodehalogenation, and (b) catalytic cycle proposed in this work. | ||
Scheme 2b shows our design, which was proposed based on the following analysis. Comparing the P–H bond dissociation free energy (BDFE) (∼78 kcal mol−1)10 of 1a with that of the isobutyronitrile α–C–H bond (∼85 kcal mol−1),14 we can expect a feasible HAT from 1a to the isobutyronitrile radical, generated from homolysis of azobis(isobutyronitrile) (AIBN), to give the crucial phosphinyl radical 1a-[P]˙ (step a). The in situ formed 1a-[P]˙ with an oxidation potential (Eox) of −2.39 V (vs. ferrocene in acetonitrile, see the ESI† for details) is among the most potent neutral electron donors ever reported.15 It could undergo reversible electron transfer (ET, step b) with bromobenzene16 to give the reactive bromobenzene radical anion and stable phosphenium cation (or alternatively, abstracts a bromine atom (step b′) from bromobenzene). Then, subsequent spontaneous C–Br bond scission (step c) produces the phenyl radical (C–H BDFE of benzene: ∼105 kcal mol−1),14 which readily captures a hydrogen-atom from 1a to regenerate 1a-[P]˙ (step d) and simultaneously initiate the next cycle.
Results and discussion
To evaluate the feasibility of the present design, we chose the bromobenzene 2a as the test substrate (Table 1). As seen, treatment of 2a with 1.2 equiv. of 1a and 10 mol% AIBN in toluene harvested the product benzene 3a in 90% yield (entry 1 in Table 1). Reducing the amount of AIBN to 5 mol% caused an inferior result (78%, entry 2). Further evaluations were performed by varying P–H reductants. Replacement of 1a with structurally similar 1b resulted in a much lower yield (<10%, entry 3). This is primarily because the poor reducing power of the 1b-derived phosphinyl radical (Eox = −1.94 V, Fig. S1†) makes electron transfer to bromobenzene sluggish. The same reason could be applied to rationalize the poor results of 1c and 1d systems (<5%, entry 4 and 5). Nevertheless, employment of the stronger reducing reagent C only gave a moderate yield (77%, entry 6), along with some unidentified byproducts. This is probably due to the phosphinyl radical derived from C being too reactive to allow a sufficient ET (to bromobenzene) to proceed before it is quenched by other components in the system. In the control reactions without either the initiator AIBN or heating, trace amounts of products were obtained (<5%, entries 7 and 8), indicating that nucleophilic aromatic substitution may exist as a background reaction. Eventually, 1.2 equiv. of 1a and 10 mol% AIBN in toluene solvent were used as the standard conditions.|
|
||
|---|---|---|
| Entry | Conditiona | Yieldb |
| a Reactions were conducted using 0.10 mmol of 2a in 0.5 mL toluene. b 1H NMR yields using 1,3,5-trimethoxybenzene as the internal standard. | ||
| 1 | Standard condition | 90% |
| 2 | 5 mol% AIBN | 78% |
| 3 | 1b as reductant | <10% |
| 4 | 1c as reductant | <5% |
| 5 | 1d as reductant | <5% |
| 6 | C as reductant | 77% |
| 7 | No AIBN | <5% |
| 8 | No heat | <5% |

|
||

With the optimized conditions derived, we next evaluated the substrate scope, which covered both aromatic and aliphatic bromides and chlorides (Scheme 3; see the ESI† for details). As shown in Scheme 3, the reduction of 2-methylthio-bromobenzene produced arene 3b in a quantitative yield (98%). 3-Phenyl (2c) and 3,5-di-tert-butyl (2d) analogs provided good results (83% and 73%). Impressively, the sterically hindered 2,4,6-trimethyl-bromobenzene is also a compatible substrate, affording 3e in 93% yield. The present reaction worked well with the labile acetal moiety and furnished 3f in a high yield (88%). Notably, the 2-carbonyl analog can be quantitatively reduced to hydrodehalogenated 3g (99%). Direct hydride transfer to the electrophilic carbonyl group, as previously reported,6 can be completely inhibited. As expected, a moderate yield (60%) was obtained for electron-rich 4-methoxy-bromobenzene (2h) because of its low reduction potential. Furthermore, hydrodebromination of substituted bromonaphthalenes (2i–k) proceeded smoothly and offered moderate to excellent yields (65–96%). Other condensed cyclic bromides (2l and 2m) were also viable substrates which gave corresponding products quantitatively. Besides, electron-rich 5-bromoindole could react with 1a/AIBN and 3n was produced in a moderate yield (70%) after prolonging the reaction time to 12 hours. Other heterocyclic substrates can also be reduced to yield corresponding arenes (3o and 3p in 55% and 90% yields, respectively). Alkenyl bromide can be efficiently hydrodehalogenated as well (3q, 93%). Additionally, all the alkyl bromides tested here were readily debrominated to give 3r–w in excellent yields (>98%). Moreover, alkyl chlorides 2′ were also viable substrates, giving excellent yields under the standard conditions (Scheme 3).
 | ||
| Scheme 3 Substrate scope of hydrodebromination/dechlorination. The reactions were conducted using 0.10 mmol 2 or 2′ in 0.5 mL toluene and isolated yields were given unless otherwise specified. [a] 1H NMR yields using 1,3,5-trimethoxybenzene as the internal standard. [b] Using toluene-d8 as solvent for these low boiling products. [c] Reaction time: 12 hours. | ||
To further apply the 1a/AIBN system to realize a cascade cyclization, a series of substrates bearing ortho-allyl moieties were employed. The results are given in Scheme 4 (see the ESI† for details). As seen, 4a–d were converted to the corresponding 5-exo-trig cyclization products in nearly quantitative yields (88–99%) and high chemo-selectivity (cyclization![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :direct hydrodehalogenation >15:1). Lengthening the side alkyl chain had a trivial effect on the reaction (4b, 95%). The excellent reactivity and chemoselectivity indicated that the present system could avoid the over reduction observed in other similar processes.15b For the substrate bearing the NH group, both cyclization 5e (50%) and direct hydrodehalogenation 5e′ (20%) products were observed with a recovery of about 20% of the starting material. Formation of 5e′ is presumably ascribed to the competitive HAT from the NH moiety or 1a to the aryl radical.
:direct hydrodehalogenation >15:1). Lengthening the side alkyl chain had a trivial effect on the reaction (4b, 95%). The excellent reactivity and chemoselectivity indicated that the present system could avoid the over reduction observed in other similar processes.15b For the substrate bearing the NH group, both cyclization 5e (50%) and direct hydrodehalogenation 5e′ (20%) products were observed with a recovery of about 20% of the starting material. Formation of 5e′ is presumably ascribed to the competitive HAT from the NH moiety or 1a to the aryl radical.
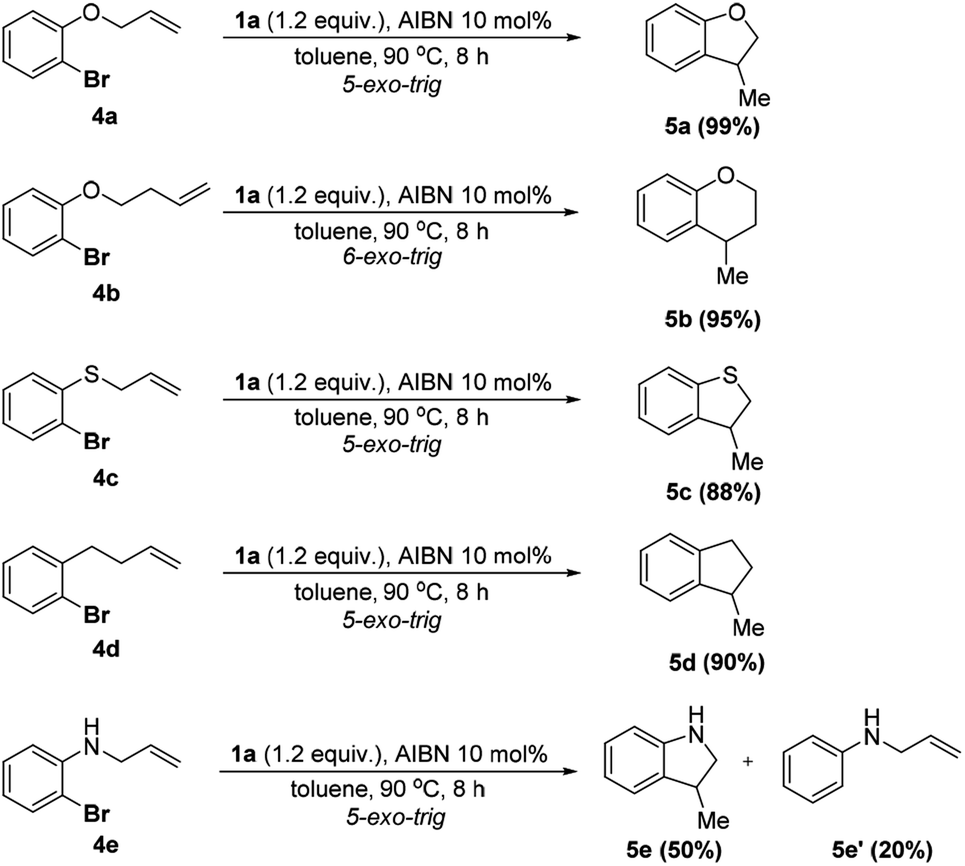 | ||
| Scheme 4 Reductive radical cyclization using 1.0 mmol of 4 in 3.0 mL toluene. Isolated yields were given. | ||
Next, we focused on elucidating the mechanistic details of the present system with particular interest in the process of C–Br bond activation, i.e., to identify whether the phosphinyl radical directly abstracts the bromine atom from bromobenzene like the reactions mediated by tin hydrides,17 silanes,13a silylated cyclohexadienes,13b and N-heterocyclic carbene–borane complexes,13c,18 or the phosphinyl radical transfers an electron to bromobenzene to trigger the cycle as depicted in Scheme 2b. DFT calculations showed that 1a-[P]˙ and 1b-[P]˙ should have a comparable ability (with an energy difference of 1.3 kcal mol−1, see the ESI† for details) in abstracting the bromine atom. This failed to explain the disparate yields of 90% for 1a-[P]˙ and <10% for 1b-[P]˙ (largely from a background reaction, see the text). Besides, according to the redox potentials of 1a-[P]˙ (Eox = −2.39 V vs. Fc in MeCN) and bromobenzene (Ered = −2.8 V),16 the electron transfer from 1a-[P]˙ to bromobenzene is a feasible reversible process, while that for 1b-[P]˙ (Eox = −1.94 V) is thermodynamically prohibited. These findings preferentially support an ET-initiated mechanism rather than a direct bromine abstraction.
On the other hand, we have recently found that 1a reacted smoothly with AIBN to produce the bisphosphine [1a-P]2.10 Considering the facile interconversion between [1a-P]2 and phosphinyl radical 1a-[P]˙,19 bisphosphine was synthesized separately to examine whether it may have functioned as a radical reservoir during hydrodehalogenation. As depicted in Scheme 5 (eqn (1)), no product was detected when [1a-P]2 was mixed with bromobenzene under the standard conditions. This is presumably because the relatively strong P–P bond of [1a-P]2 made it difficult to dissociate to provide the corresponding phosphinyl radical 1a-[P]˙ at the reaction temperature (90 °C, see the ESI† for details). Thus, the possibility of [1a-P]2 as a reaction intermediate was excluded. Because the solvent toluene could be a common hydrogen donor, and if so, it has potential to quench the phenyl radical. To examine this, deuterium labelling experiments were conducted in toluene-d8 (eqn (2) and (3)). The absence of deuterium incorporation indicated that the hydrogen could not come from toluene. Furthermore, replacement of 1a with its deuterated counterparts 1a–d resulted in the desired product with a 90% deuterium abundance (eqn (4)). Hence, 1a is suggested to be the hydrogen source at last.
 | ||
| Scheme 5 Control experiments with 1H NMR yields (using 1,3,5-trimethoxybenzene as the internal standard; see the ESI† for details). | ||
Based on these control experiments, the plausible catalytic mechanism proposed in Scheme 2 can be verified. The in situ generated phosphinyl radical preferentially served as a potent electron donor to activate the bromides. The reaction is most likely to proceed through an ET-initiated radical chain process,20 although a direct bromine abstraction cannot be completely excluded at the present stage.
Conclusions
In conclusion, in this work we unlocked unprecedented radical reactivity of diazaphosphinanes to achieve efficient hydrodehalogenation and cascade cyclization, which is distinguished from their well-established hydridic reactivity. The phosphinyl radical, accessed in a catalytic cycle, is believed to be responsible for activating the carbon–bromine bonds through ET. This new reaction may provide a superior approach to remove halogens from many organohalides in terms of substrate scope, reaction efficiency and chemo-selectivity. Exploitation of other radical reductions using the strategy presented here is ongoing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial grants from the National Natural Science Foundation of China (No. 21973052, 21933008, 21602116, and 91745101), National Science & Technology Fundamental Resource Investigation Program of China (No. 2018FY201200), and Tsinghua University Initiative Scientific Research Program (No. 20181080083).Notes and references
- (a) E.-U. Würthwein, G. Lang, L. H. Schappele and H. Mayr, J. Am. Chem. Soc., 2002, 124, 4084–4092 CrossRef PubMed; (b) D. W. Brinkley and J. P. Roth, J. Am. Chem. Soc., 2005, 127, 15720–15721 CrossRef CAS PubMed; (c) M. Breugst, H. Zipse, J. P. Guthrie and H. Mayr, Angew. Chem., Int. Ed., 2010, 49, 5165–5169 CrossRef CAS PubMed; (d) Y. Li and X.-Q. Zhu, ACS Omega, 2018, 3, 872–885 CrossRef CAS PubMed.
- (a) D. Y. Ong, C. Tejo, K. Xu, H. Hirao and S. Chiba, Angew. Chem., Int. Ed., 2017, 56, 1840–1844 CrossRef CAS PubMed; (b) B. Rösch, T. X. Gentner, H. Elsen, C. A. Fischer, J. Langer, M. Wiesinger and S. Harder, Angew. Chem., Int. Ed., 2019, 58, 5396–5401 CrossRef PubMed.
- (a) S. Fukuzumi, Org. Biomol. Chem., 2003, 1, 609–620 RSC; (b) I. J. Rhile and J. M. Mayer, J. Am. Chem. Soc., 2004, 126, 12718–12719 CrossRef CAS PubMed; (c) J. P. Roth, S. Lovell and J. M. Mayer, J. Am. Chem. Soc., 2000, 122, 5486–5498 CrossRef CAS.
- (a) D. Gudat, A. Haghverdi and M. Nieger, Angew. Chem., Int. Ed., 2000, 39, 3084–3086 CrossRef CAS; (b) S. Burck, D. Gudat, M. Nieger and W.-W. Du Mont, J. Am. Chem. Soc., 2006, 128, 3946–3955 CrossRef CAS PubMed.
- (a) M. R. Adams, C. H. Tien, B. S. N. Huchenski, M. J. Ferguson and A. W. H. Speed, Angew. Chem., Int. Ed., 2017, 56, 6268–6271 CrossRef CAS PubMed; (b) M. R. Adams, T. Chieh-Hung, M. Robert and A. W. H. Speed, Angew. Chem., Int. Ed., 2017, 56, 16660–16663 CrossRef CAS PubMed; (c) C.-H. Tien, M. R. Adams, M. J. Ferguson, E. R. Johnson and A. W. H. Speed, Org. Lett., 2017, 19, 5565–5568 CrossRef CAS PubMed; (d) T. Lundrigan, E. N. Welsh, T. Hynes, C.-H. Tien, M. R. Adams, K. R. Roy, K. N. Robertson and A. W. H. Speed, J. Am. Chem. Soc., 2019, 141, 14083–14088 CrossRef CAS PubMed.
- C. C. Chong, H. Hirao and R. Kinjo, Angew. Chem., Int. Ed., 2015, 54, 190–194 CrossRef CAS PubMed.
- (a) C. C. Chong, B. Rao and R. Kinjo, ACS Catal., 2017, 7, 5814–5819 CrossRef CAS; (b) S. Miaskiewicz, J. H. Reed, P. A. Donets, C. C. Oliveira and N. Cramer, Angew. Chem., Int. Ed., 2018, 57, 4039–4042 CrossRef CAS PubMed.
- (a) B. Rao, C. C. Chong and R. Kinjo, J. Am. Chem. Soc., 2018, 140, 652–656 CrossRef CAS PubMed; (b) T. Hynes, E. N. Welsh, R. McDonald, M. J. Ferguson and A. W. H. Speed, Organometallics, 2018, 37, 841–844 CrossRef CAS.
- J. Zhang, J.-D. Yang and J.-P. Cheng, Angew. Chem., Int. Ed., 2019, 58, 5983–5987 CrossRef CAS PubMed.
- J. Zhang, J.-D. Yang and J.-P. Cheng, Chem. Sci., 2020, 11, 3672–3679 RSC.
- (a) P. Knochel, W. Dohle, N. Gommermann, F. F. Kneisel, F. Kopp, T. Korn, I. Sapountzis and V. A. Vu, Angew. Chem., Int. Ed., 2003, 42, 4302–4320 CrossRef CAS PubMed; (b) F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2002, 102, 4009–4092 CrossRef CAS PubMed.
- (a) B. Sahoo, A.-E. Surkus, M.-M. Pohl, J. Radnik, M. Schneider, S. Bachmann, M. Scalone, K. Junge and M. Beller, Angew. Chem. Int. Ed., 2017, 56, 11242–11247 CrossRef CAS PubMed; (b) V. V. Grushin and H. Alper, Chem. Rev., 1994, 94, 1047–1062 CrossRef CAS.
- (a) C. Chatgilialoglu, Acc. Chem. Res., 1992, 25, 188–194 CrossRef CAS; (b) A. Studer and S. Amrein, Angew. Chem., Int. Ed., 2000, 39, 3080–3082 CrossRef CAS; (c) X. Pan, E. Lacôte, J. Lalevée and D. P. Curran, J. Am. Chem. Soc., 2012, 134, 5669–5674 CrossRef CAS PubMed; (d) A. Dewanji, C. Mück-Lichtenfeld and A. Studer, Angew. Chem., Int. Ed., 2016, 55, 6749–6752 CrossRef CAS PubMed.
- J.-P. Cheng, et al., Internet Bond-energy Databank (iBonD) Home Page, http://ibond.nankai.edu.cn, accessed at Feb. 2020 Search PubMed.
- (a) S. Rohrbach, R. S. Shah, T. Tuttle and J. A. Murphy, Angew. Chem., Int. Ed., 2019, 58, 11454–11458 CrossRef CAS PubMed; (b) S. S. Hanson, E. Doni, K. T. Traboulsee, G. Coulthard, J. A. Murphy and C. A. Dyker, Angew. Chem., Int. Ed., 2015, 54, 11236–11239 CrossRef CAS PubMed.
- (a) C. Costentin, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2004, 126, 16051–16057 CrossRef CAS PubMed; (b) L. Pause, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 1999, 121, 7158–7159 CrossRef CAS.
- P. A. Baguley and J. C. Walton, Angew. Chem., Int. Ed., 1998, 37, 3072–3082 CrossRef CAS PubMed.
- D. P. Curran, A. Solovyev, M. M. Brahmi, L. Fensterbank, M. Malacria and E. Lacôte, Angew. Chem., Int. Ed., 2011, 50, 10294–10317 CrossRef CAS PubMed.
- (a) R. Edge, R. J. Less, E. J. L. McInnes, K. Müther, V. Naseri, J. M. Rawson and D. S. Wright, Chem. Commun., 2009, 1691–1693 RSC; (b) O. Puntigam, D. Förster, N. A. Giffin, S. Burck, J. Bender, F. Ehret, A. D. Hendsbee, M. Nieger, J. D. Masuda and D. Gudat, Eur. J. Inorg. Chem., 2013, 2041–2050 CrossRef CAS; (c) D. Förster, H. Dilger, F. Ehret, M. Nieger and D. Gudat, Eur. J. Inorg. Chem., 2012, 3989–3994 CrossRef; (d) M. Blum, O. Puntigam, S. Plebst, F. Ehret, J. Bender, M. Nieger and D. Gudat, Dalton Trans., 2016, 45, 1987–1997 RSC; (e) D. M. C. Ould, A. C. Rigby, L. C. Wilkins, S. J. Adams, J. A. Platts, S. J. A. Pope, E. Richards and R. L. Melen, Organometallics, 2018, 37, 712–719 CrossRef CAS.
- A. Studer and D. P. Curran, Nat. Chem., 2014, 6, 765–773 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of condition optimization, product characterization and DFT calculations. See DOI: 10.1039/d0sc01352h |
| This journal is © The Royal Society of Chemistry 2020 |