
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multifunctional streptavidin–biotin conjugates with precise stoichiometries†
Dongdong
Xu
a and
Seraphine V.
Wegner
 *ab
*ab
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany
bUniversity of Münster, Institute for Physiological Chemistry and Pathobiochemistry, Waldeyerstr. 15, 48149 Münster, Germany. E-mail: wegnerse@exchange.wwu.de
First published on 20th April 2020
Abstract
Streptavidin is ubiquitously used to link different biotinylated molecules thanks to its tetravalent binding to biotin. An unwanted side-effect is the resulting statistical mixtures of products. Here, a general approach to form multifunctional streptavidin conjugates with precise stoichiometries and number of open binding pockets is reported. This method relies on an iminobiotin-polyhistidine tag, which allows separating streptavidin conjugates with different numbers of tags, and later reopening binding pockets at lowered pH to introduce a second functionality. Pure fluorescently labelled mono-, di- and trivalent streptavidin–biotin conjugates prepared in this way were used for imaging biotinylated cell surface molecules with controlled clustering. Furthermore, these conjugates were functionalized with a second biotinylated molecule, folic acid–biotin, to investigate the importance of multivalent binding in targeted delivery of cancer cells. These streptavidin–biotin conjugates with precise stoichiometries combined with a variety of biotinylated molecules render this method a diverse and powerful tool for molecular biology and biotechnology.
Introduction
The binding of streptavidin to biotin is well known for the strong noncovalent interaction with femtomolar affinity (Kd = 10−14).1 The high affinity, slow exchange rate, and good specificity of the biotin–streptavidin interaction has resulted in a wide range of biotechnological applications including extracellular and in vitro labelling,2,3 therapeutics, biosensing and biofunctionalization.4,5 The large range of biotinylated small molecules, peptides, proteins, antibodies and nucleic acids as well as materials adds to the diversity of the biotin–streptavidin chemistry. Each streptavidin tetramer has four independent biotin binding sites. This tetravalence poses a conundrum. On the one hand, it allows streptavidin to be used as a linker between a wide variety of biotinylated molecules with targeting, sensing, diagnostic and therapeutic functionalities and assembling them into one molecule in a modular fashion. On the other hand, tetravalence can be a disadvantage as it leads to statistical mixtures of conjugates when multiple biotinylated molecules are conjugated as well as unwanted cross-linking and aggregation.6,7 A partial solution to the problems arising from the multivalence are genetically engineered monovalent versions of streptavidin with only one active biotin binding site per tetramer, as well as a monomeric biotin binder, rhizavidin, which can almost achieve multimeric streptavidin-like binding stability for biotin conjugates.6–10 Alternatively, (strep)-avidins composed of different subunits have been used to integrate different functionalities within one conjugate.11,12 These streptavidins have been used to form structurally defined one-to-one streptavidin–biotin conjugates and image biotinylated cell surface receptors without the formation of artificial receptor clustering. However, the monovalent streptavidins are unfit as linkers to assemble streptavidin conjugates with multiple functional groups, or with multiple copies of one functional group. Given this, a method of assembling multifunctional streptavidin–biotin conjugates with precise stoichiometries still remains a big challenge and if successful, it would significantly expand the potential of streptavidin–biotin based technologies.In this study, we report a new strategy to assemble multifunctional streptavidin–biotin conjugates with precise stoichiometries and different valencies. Using native streptavidin we were able to produce streptavidin (S) conjugates with one (SA1), two (SA2) or three (SA3) copies of a biotinylated molecule (A), where the residual biotin binding pockets remain open to introduce a second biotin conjugated functionality. Using this method, we were able to produce precise fluorescent streptavidin conjugates for cell surface labelling and to investigate how the number of targeting ligands per conjugate affects cellular uptake. These initial illustrations show how this method can be used to address a wide range of questions in molecular and cell biology.
Results and discussion
The assembly of multifunctional streptavidin conjugates with precise stoichiometries relies on two well-established chemistries; the separation of proteins with different numbers of His-tags using Ni2+-NTA (nitrilotriacetic acid) columns, and secondly, the pH-dependent binding of iminobiotin to streptavidin (basic pH, Kd ∼ 10−11 M; acidic pH, Kd ∼ 10−3 M).13 In this method, varying numbers of biotin or iminobiotin conjugated His-tags (Bio-His-Tag or Ibio-His-Tag, respectively) were first introduced on to streptavidin (Fig. 1). This allowed for the easy separation of species with differing numbers of His-tags and open biotin binding pockets on a Ni2+-NTA column using an imidazole gradient. Subsequently, a biotin conjugated molecule of choice (A) was coupled to the open binding pockets yielding pure streptavidin conjugated with precise stoichiometries (S(His-Tag)3A1, S(His-Tag)2A2, S(His-Tag)1A3). In the case of the iminobiotin conjugated His-tag complexes, the His-tag was released by decreasing the pH to 3.5 and a second biotin conjugated molecule (B) was added with a precise stoichiometry (SA1B3, SA2B2, SA3B1).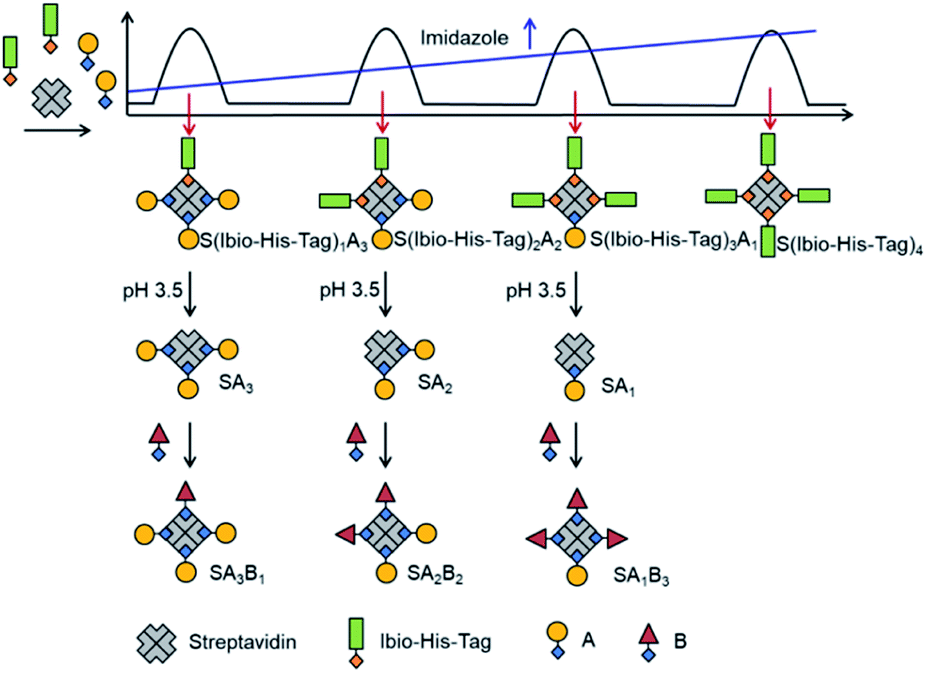 | ||
| Fig. 1 Strategy for obtaining multifunctional streptavidin–biotin conjugates with defined stoichiometries (SA3B1, SA2B2, SA1B3) using the Ibio-His-Tag (iminobiotin-His-tag). The statistical mixture of the four streptavidin conjugates with different numbers of Ibio-His-Tags and the biotin-conjugated molecule, A, can be separated on a Ni2+-NTA column based on their number of tags with an imidazole gradient. Once isolated, the Ibio-His-Tag can be removed from each conjugate at pH 3.5 to open the biotin binding pockets and introduce a second biotin conjugated molecule, B. | ||
In a first step, to produce stoichiometrically defined streptavidin conjugates with one functionality, streptavidin, (S = 30 μM) was incubated with a biotin conjugated His6-tag peptide (biotin-(His)6, Bio-His-Tag = 90 μM), which yielded a statistical mixture of S, S(Bio-His-Tag)1, S(Bio-His-Tag)2, S(Bio-His-Tag)3 and S(Bio-His-Tag)4. Subsequently, the mixture was passed over a Ni2+-NTA agarose column; unbound molecules without His-tags such as S were washed off and different species were eluted using an imidazole gradient, where species bearing more tags eluted at higher imidazole concentrations. In the chromatogram there were four clearly separated peaks that eluted at different imidazole concentrations (relative integrated areas: 19.4% 1st peak, 14.3% 2nd peak, 20.6% 3rd peak, 45.7% 4th peak) (Fig. 2a). Presumably the ratio of these peaks can be changed using different S to Bio-His-tag ratios. To demonstrate how each of these peaks corresponds to a single species with a defined stoichiometry, the open biotin binding pockets of S(Bio-His-Tag)1-4 were titrated with biotin-5-fluorescein where the fluorescence is quenched upon streptavidin binding (Fig. 2b–e).14,15 The streptavidin conjugate in the first peak required 3 equivalents of the dye to saturate all biotin binding sites and therefore was assigned as the S(Bio-His-Tag)1. Likewise, the molecules in the second, third and fourth peaks required 2, 1 and 0 equivalents of dye to saturate all biotin binding sites, respectively, and they corresponded to S(Bio-His-Tag)2, S(Bio-His-Tag)3 and S(Bio-His-Tag)4, respectively. This assignment is also consistent with streptavidin conjugates eluting at higher imidazole concentrations having more Bio-His-Tags.
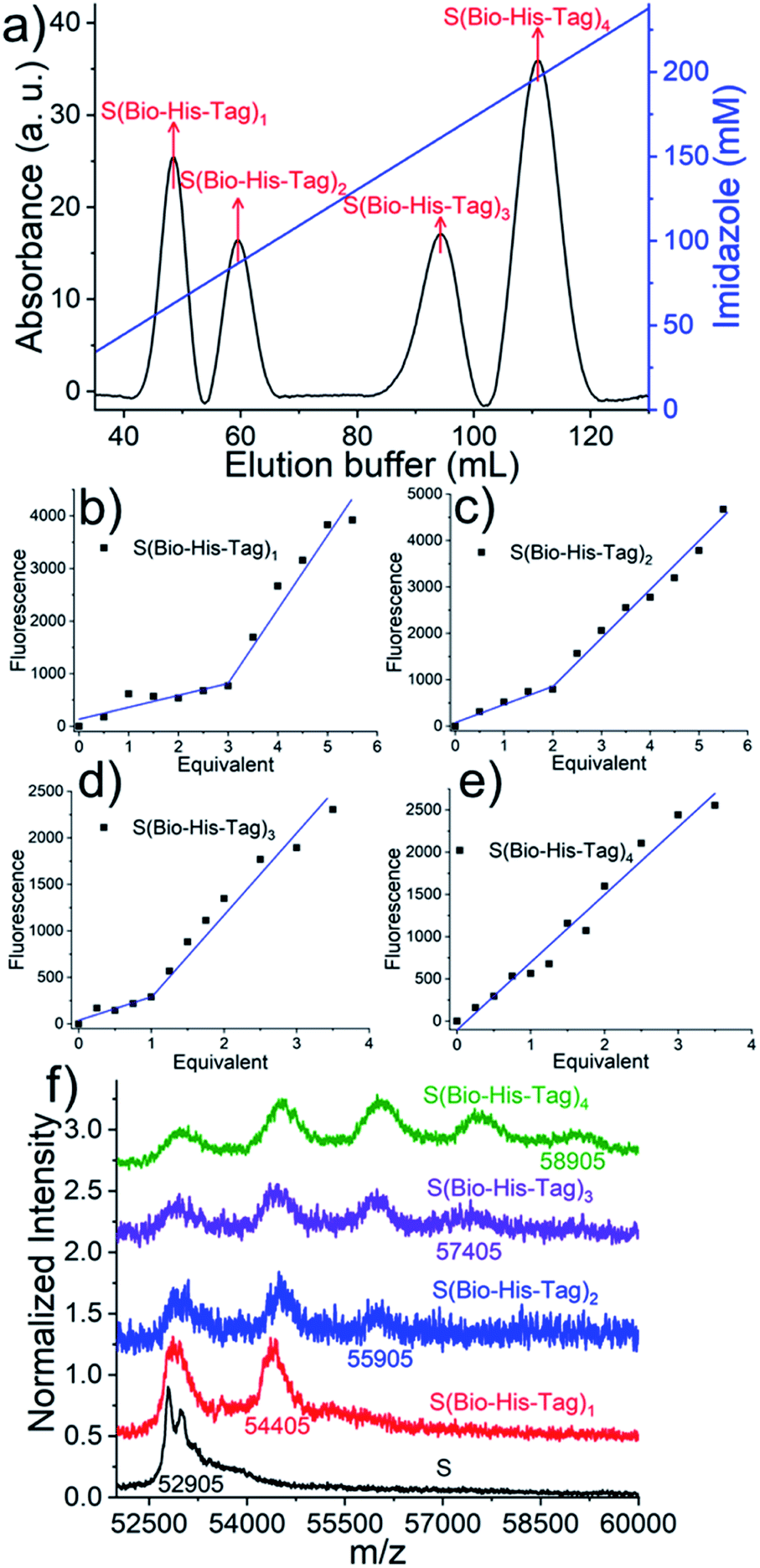 | ||
Fig. 2 (a) Samples are separated by different concentrations of imidazole solution, typically, 30 μM streptavidin was mixed with 90 μM Bio-His-Tag for 15 min, the mixture was loaded on a Ni2+-NTA column and washed with a concentration gradient of imidazole, S(Bio-His-Tag)1 (the first peak) occurs at 62.4 mM imidazole and S(Bio-His-Tag)2 (the second peak) was separated at 86.3 mM imidazole, peak 3 (S(Bio-His-Tag)3) was detected at 161.2 mM imidazole and S(Bio-His-Tag)4 (peak 4) occurs at 196.3 mM imidazole. The open biotin binding pockets of S(Bio-His-Tag)1 (b), S(Bio-His-Tag)2 (c), and S(Bio-His-Tag)3 (d) were titrated with biotin-5-fluorescein where the fluorescence is quenched upon streptavidin binding. Result indicates that S(Bio-His-Tag)1, S(Bio-His-Tag)2, and S(Bio-His-Tag)3 required three, two and one equivalents of the dye to saturate all biotin binding sites, respectively. (e) No quenching happens in the S(Bio-His-Tag)4 titration experiment, which indicates that all biotin binding pockets have been occupied by the Bio-His-Tag. (f) MALDI-TOF mass spectra of pure streptavidin (S: m/z 52![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 905), S(Bio-His-Tag)1 (m/z 54405), S(Bio-His-Tag)2 (m/z 55905), S(Bio-His-Tag)3 (m/z 57405) and S(Bio-His-Tag)4 (m/z 58905). 905), S(Bio-His-Tag)1 (m/z 54405), S(Bio-His-Tag)2 (m/z 55905), S(Bio-His-Tag)3 (m/z 57405) and S(Bio-His-Tag)4 (m/z 58905). | ||
The identity of the different species was further confirmed with MALDI-TOF mass spectroscopy. While S and Bio-His-tag have molecular weights of 52905 and 1500 Da (Fig. S1†), respectively, the different conjugates of S and Bio-His-Tag have higher molecular weights (Fig. 2f). The isolated species with one to four Bio-His-tags had maximal peaks 54405 Da (S(Bio-His-Tag)1), 55905 Da (S(Bio-His-Tag)2), 57405 Da (S(Bio-His-Tag)3) and 58905 Da (S(Bio-His-Tag)4), respectively, which are well in agreement with the theoretically expected values. Moreover, for species with different numbers of Bio-His-Tags lower molecular weight peaks were observed due to the dissociation of the Bio-His-Tag from S in the mass spectrometer. More specifically, there were two peaks for S(Bio-His-Tag)1 (S and S(Bio-His-Tag)1) and three peaks for S(Bio-His-Tag)2 (S, S(Bio-His-Tag)1 and S(Bio-His-Tag)2), while no peak of S(Bio-His-Tag)2 was observed for S(Bio-His-Tag)1. These results confirm and fully resolve the molecular structure of the prepared streptavidin conjugates with defined stoichiometries.
The isolated S(Bio-His-Tag)1, S(Bio-His-Tag)2 and S(Bio-His-Tag)3 conjugates are equivalent to trivalent, divalent and monovalent streptavidin and can be used to form precise conjugates with a large variety of biotinylated molecules including small molecules, peptides, proteins, DNA and antibodies. As an example, biotinylated mOrange (orange fluorescent proteins, O, 31.3 kDa) was reacted with the streptavidin conjugates of varying valences. Each of the conjugates eluted at a different volume on a size exclusion column, and the molecular weights determined, based on a protein standard, were in agreement with the theoretically expected values (S(Bio-His-Tag)1O3 (theo. 148.4 kDa, exp. 141.0 kDa), S(Bio-His-Tag)2O2 (theo. 118.6 kDa, exp. 108.6 kDa) and S(Bio-His-Tag)3O1 (theo. 88.8 kDa, exp. 81.3 kDa)) (Fig. S2†). The agreement between theoretical and experimental values supports the assigned structures and shows the great potential of this approach to form precise protein conjugates.
For the success of this approach, the slow exchange rate of the Bio-His-Tag with streptavidin is required so that species isolated in different peaks do not interconvert. To test the kinetic stability, purified S(Bio-His-Tag)2 was stored at 4 °C, room temperature (RT), and at 37 °C. After 1 and 5 days, the samples were loaded onto the Ni2+-NTA agarose column and eluted using an imidazole gradient (Fig. 3). The S(Bio-His-Tag)2 stored at 4 °C and RT for up to five days eluted as a single peak like the fresh sample (0 day), demonstrating the long-term stability of isolated species. Even at 37 °C after one day, there was no significant change for S(Bio-His-Tag)2, while after five days at 37 °C, 30% of S(Bio-His-Tag)2 converted into S(Bio-His-Tag)1 and S(Bio-His-tag)3. Therefore, a single species prepared using this method is kinetically stable enough to be used in future studies.
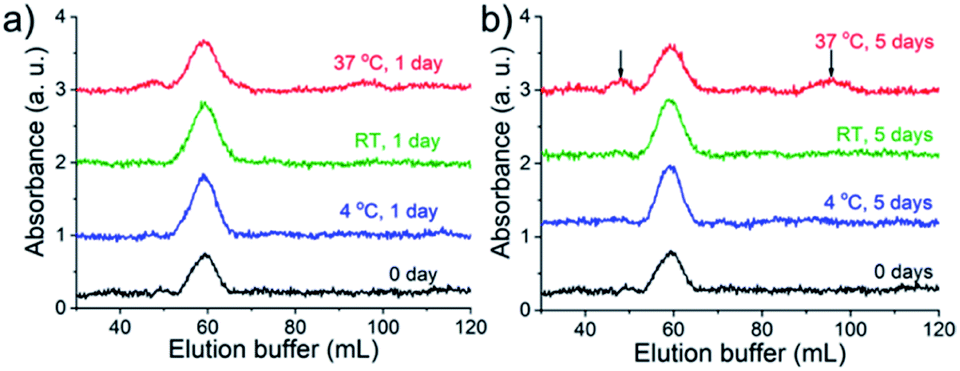 | ||
| Fig. 3 Long time stability test at the absorbance wavelength of 280 nm. (a) S(Bio-His-Tag)2 was stored at 4 °C, room temperature (RT) and 37 °C separately for one day, and then was loaded on the Ni2+-NTA column and washed with elution buffer. There is no change for samples stored at 4 °C compared with the sample stored at room temperature, and there is a very little change at 37 °C compared with the fresh sample (0 day). (b) There is no change for samples stored at 4 °C and room temperature after five days, while 70% of S(Bio-His-Tag)2 remained stable when stored at 37 °C for five days, and 8.4% and 21.6% of S(Bio-His-Tag)2 changed to S(Bio-His-Tag)1 and S(Bio-His-Tag)3, respectively (black arrow). | ||
In the second step, in order to produce precise streptavidin conjugates with two different functional groups, we used an iminobiotin conjugated His-tag (Ibio-His-Tag) instead of the Bio-His-Tag. The Ibio-His-Tag is removable from the streptavidin complex at a lower pH (pH 3.5) and can be used to reopen biotin binding pockets in isolated streptavidin conjugates with precise stoichiometry and a first functionality. The reopened biotin binding pockets can then be used to introduce a second biotin conjugated molecule. In initial experiments, we noticed that the streptavidin-Ibio-His-Tag complexes had a lower affinity to the Ni2+-NTA column. A potential reason for this could be the different orientations of biotin and iminobiotin in the streptavidin binding pocket. While serine 27 of streptavidin acts as a H-bond donor for biotin, it is a H-bond acceptor for iminobiotin.16 Consequently, the exposure of the connected His-tags could also be affected. To increase the affinity to the column and later separation of different species, the His6-tag was extended to a His12-tag, which increases the number of ligands that can bind to the column and the Ni2+-NTA column was replaced with a Cu2+-NTA column, as Cu2+ has a higher binding affinity towards His-tags than Ni2+.17 To produce the Ibio-His-tagged streptavidins, streptavidin (S = 30 μM) was incubated with the Ibio-His-Tag (iminobiotin-(His)12 = 90 μM) for 15 minutes. Additionally, to simplify the procedure, the first biotin conjugated molecule, atto-565-biotin (A = 50 μM), was subsequently added to the reaction mixture and incubated for 15 minutes before the separation of different species (S(Ibio-His-Tag)1A3, S(Ibio-His-Tag)2A2, S(Ibio-His-Tag)3A1, S(Ibio-His-Tag)4) over the Cu2+-NTA column using an imidazole gradient. In the chromatogram, four peaks were visible in the absorbance channel at 280 nm, but only the first three absorbed at 563 nm, where atto-565 absorbs (Fig. S4a,† relative integrated areas at 280 nm: 5.1% 1st peak, 42.6% 2nd peak, 26.7% 3rd peak and 25.6% 4th peak). To confirm the identity of the species in each peak, first the Ibio-His-Tag was removed by lowering the pH to 3.5 and by dialysis, and subsequently the streptavidin species were titrated with biotin-5-fluorescein as described above (Fig. S3b–e†). The streptavidin species in the first peak reacted with 1 equivalent of biotin-5-fluorescein and was therefore identified as SA3, which carried 1 Ibio-His-Tag before the acidification. Similarly, the species in the following peaks reacted with 2, 3 and 4 equivalents of biotin-5-fluorescein, respectively, and were identified as SA2 (two open biotin sites), SA1 (three open biotin sites) and S (four open biotin sites). Moreover, this assignment was also supported by the relative absorbance of the conjugates in the UV-vis at 280 nm where streptavidin and atto-565 absorb and 532 nm where only atto-565 absorbs (Table S1†). Using the Ibio-His-Tag, we were able to prepare fluorescently labelled mono- (SA3), di- (SA2) and tri- (SA1) valent streptavidins without genetic manipulation. Moreover, the atto-565-biotin used in this protocol can easily be replaced by another biotin conjugated molecule of choice. The straightforward preparation of these functionalized streptavidin derivatives with defined valences for the introduction of a second biotin coupled molecule offers us a new domain to expand and develop the many applications of biotin–streptavidin chemistry.
One area where fluorescently labelled monovalent streptavidins are especially useful is in the imaging of biotinylated cell surface molecules. Unlike monovalent streptavidins, multivalent streptavidins lead to artificial clustering of the biotinylated surface molecules, resulting in altered biological responses and cell uptake. Given this, the atto-565-biotin labelled monovalent streptavidin SA3 described above is ideal for the visualization of biotinylated cell surface molecules. For this purpose, the cell membranes of MDA-MB-231 cells were first randomly biotinylated using sulfo-NHS-LC-biotin10 and subsequently the cell membranes were labeled with SA3, SA2 or SA1, which was visible under the confocal microscope (Fig. 4, Movies S1–S3†). However, the fluorescence staining appeared different for different atto-565-biotin–streptavidin conjugates over time, as membrane proteins quickly internalize when they are cross-linked.7,10 The cells labeled with monovalent SA3 were stained at the membrane's periphery and even after 30 minutes hardly any labeled protein was internalized. On the other hand, cells incubated with divalent SA2 and trivalent SA1 showed significant protein internalization after just 10 minutes, where this effect was more pronounced for SA1. The conclusions were also supported quantitatively based on the intracellular fluorescence intensity (Fig. S4†). The fluorescently labelled monovalent streptavidin, SA3, can actually be used at concentrations as low as 0.05 μM for clearly cell surface labelling with high sensitivity (Fig. S5 and S6†). Thus, it is a good alternative to current monovalent streptavidins, which require an additional labelling step with the fluorophore and are only labelled statistically.
 | ||
| Fig. 4 Confocal microscopy images of biotinylated cell-surface proteins using sulfo-NHS-LC-biotin which were fluorescently labelled with trivalent (SA1) (a–c), divalent (SA2) (d–f) and monovalent (SA3) (g–i) streptavidin–atto-565-biotin conjugates. Artificial protein clustering induced by trivalent (SA1) and divalent (SA2) conjugated results in protein internalization over time but not the monovalent (SA3) conjugate. Cell nuclei were stained with TO-PRO-3. Scale bars are 10 μm. | ||
The open biotin binding pockets in the fluorescently labelled streptavidins can also be used to introduce a second functionality with precise molecular stoichiometry. For instance, we added folic acid, an active and selective targeting molecule for aggressive cancer cells that overexpress the folic acid receptor on their surfaces.19–21 The folic acid and atto-565 labelled streptavidins allowed us to determine whether the number of folic acid groups per molecule impacts cellular uptake by using the signal from the fluorescent label. For this purpose, pure SA3, SA2 and SA1 were each incubated first with excess folic acid–PEG–biotin (F) for 20 minutes and then excess F was removed by dialysis. The final conjugates, SA3F1, SA2F2 and SA1F3 were tested on two different cell lines: folate receptor-positive MDA-MB-231 and folate receptor-negative MCF-7. After 4 hours of incubation the fluorescence signal from the atto-565 was much brighter in the MDA-MB-231 cells than in the MCF-7 cells (Fig. 5a). The uptake was clearly due to the folic acid as cells incubated with SA1, SA2 and SA3 were not significantly fluorescent (Fig. S7 and S8†). For quantification, the fluorescence intensities of SA1F3, SA2F2 and SA3F1 in both MDA-MB-231 and MCF-7 cells were measured and normalized to the relative brightness of the streptavidin-atto-565 species (Fig. 5b and Table S2†). This analysis shows that all three streptavidin-folic acid conjugates were taken up equally well by the folic acid receptor positive cell line MDA-MB-231 independent of the number of folic acids in its structure.
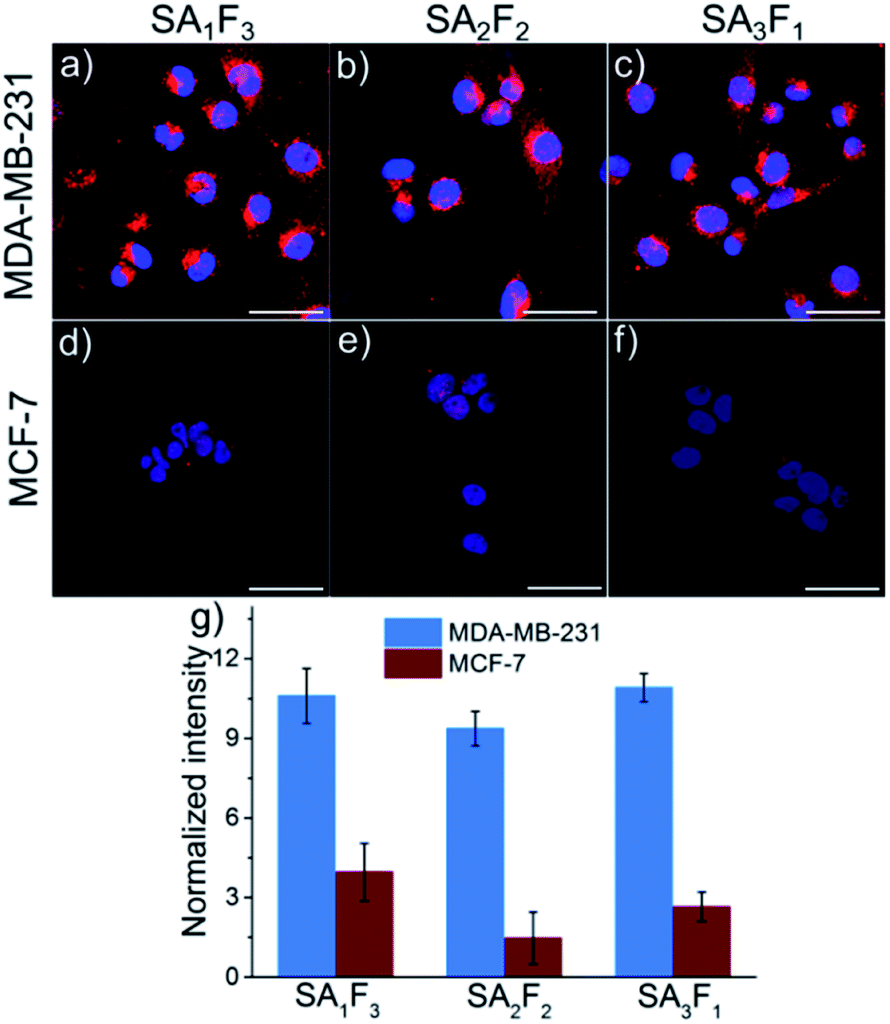 | ||
| Fig. 5 Confocal microscopy images of folate-receptor positive MDA-MB-231 cells (a–c) and folate-receptor negative MCF-7 cells (d–f) incubated with atto-565 (A) (shown red) labelled folic acid (F) streptavidin conjugates with different stoichiometries. Cells were incubated with 1 μM SA1F3 (a and d), SA2F2 (b and e) or SA3F1 (c and f) in RPMI-1640 (no folic acid) medium for 4 h at 37 °C. Cell nuclei were stained with DAPI (shown blue). Scale bars are 50 μm. (g) The fluorescence intensities of different streptavidin conjugates in MDA-MB-231 and MCF-7 cells, normalized to the relative brightness of the conjugates. The fluorescence intensity for all streptavidin conjugates is higher in MDA-MB-231 cells than in MCF-7 cells but increasing numbers of folic acid in the conjugates do not result in higher uptake. | ||
Conclusions
In summary, we demonstrated that the Bio-His-Tag and Ibio-His-Tag can both be used to prepare multifunctional streptavidin–biotin conjugates with precise stoichiometry and structure. This method, as demonstrated by two examples, can be widely applied and is highly adaptable. It is an ideal approach to answer questions in molecular biology and for biotechnological applications. We used fluorescently labelled monovalent (SA3), divalent (SA2) and trivalent (SA1) streptavidin for imaging biotinylated cell surface molecules and investigated the importance of multivalent cell receptor interactions with folic acid in the cellular uptake and targeting (SA3F1, SA2F2 and SA1F3). The wide variety of commercially available biotinylated molecules ranging from small molecules and peptides to proteins, nucleic acids and antibodies, as well as the Bio-His-Tag and Ibio-His-Tag, all make this method extremely versatile and accessible. The potential diversity in precise streptavidin–biotin conjugates opens the door to building new bio- and nanostructures and will play a significant role in expanding the well-established status of streptavidin–biotin chemistry in biotechnology.22,23Experimental
Materials
Streptavidin (MW 53361 g mol−1) was purchased from Cedarlane Laboratories. The Bio-His-Tag (biotin-(His)6, MW 1500 g mol−1, sequence: biotin-GSGSGSHHHHHH) was synthesized by Peptide Specialty Laboratories GmbH and the Ibio-His-Tag (MW 2322 g mol−1, iminobiotin-(His)12, sequence: iminobiotin-GSGSGSHHHHHHHHHHHH) was synthesized by Pepscan. Folic acid–PEG–biotin (MW 2000 g mol−1) was purchased from Nanocs. Sulfo-NHS-LC-biotin (MW 558 g mol−1) was purchased from AdooQ Bbioscience. BL21 (DE3) E. coli was purchased from New England Biolabs. pET Biotin His6 mOrange LIC cloning vector (H6-mOrange) was a gift from Scott Gradia (Addgene plasmid # 29723) and BirA in pET28a (w400-2) was a gift from Eric Campeau (Addgene plasmid # 26624). DMEM and RPMI-1640 without folic acid medium were purchased from Thermo Fisher Scientific. The Ni2+-NTA column (HisTrap™ HP, column volume 5 mL) was purchased from GE Healthcare Life Sciences. All other chemicals were purchased from Sigma-Aldrich. Buffers and aqueous solutions were prepared with Milli-Q grade water.Streptavidin sequence: MAEAGITGTWYNQLGSTFIVTAGADGALTGTYESAVGNAESRYVLTGRYDSAPATDGSGTALGWTVAWKNNTRNAHSATTWSGQYVGGAEARINTQWLLTSGTTEANAWKSTLVGHDTFTKVKPSAAS
Separation of streptavidin Bio-His-Tag conjugates
30 μM streptavidin was mixed with 90 μM Bio-His-Tag (biotin-(His)6) for 15 min at room temperature. A Ni2+-NTA column (HisTrap™ HP, column volume 5 mL) was pre-equilibrated with 50 mL buffer A (50 mM Tris–HCl, 300 mM NaCl, pH 7.4) using a FPLC system (GE healthcare, AKTA explorer). Then, 0.5 mL streptavidin Bio-His-Tag reaction mixture was loaded on the Ni2+-NTA column and the column was washed with 25 mL buffer A. Finally, the different streptavidin Bio-His-Tag conjugates were eluted with a linear imidazole gradient from 0 to 260 mM imidazole in buffer A over 130 mL. The elution of different species was monitored by the absorbance at 280 nm, the curves were corrected for the absorbance of imidazole and each peak was collected in a separate fraction. A flow rate of 0.5 mL min−1 was used throughout the experiment. The collected samples were dialyzed (10 kDa molecular weight cut-off) against 2 L of buffer A at 4 °C for at least 6 h to remove imidazole. Subsequently, the samples were concentrated using a centrifugal filtration device (10 kDa molecular weight cut-off). The protein concentration was determined by UV-Vis spectroscopy.To analyze the kinetic stability of S(Bio-His-Tag)2, different samples were incubated at 4 °C, room temperature and at 37 °C for one and five days and analyzed on a Ni2+-NTA column using the same imidazole gradient as described above.
Separation of streptavidin Ibio-His-Tag conjugates
The Cu2+-NTA column was prepared by removing Ni2+ ions from a Ni2+-NTA column (HisTrap™ HP, column volume 5 mL) with ethylenediaminetetraacetic acid (EDTA) and reloading it with Cu2+ ions. Firstly, 30 μM streptavidin was mixed with 90 μM Ibio-His-Tag for 15 min at room temperature to ensure the binding of iminobiotin to streptavidin. Then, 50 μM atto-565-biotin was added to the reaction mixture and incubated for another 15 min. 0.5 mL of the reaction mixture was loaded on a Cu2+-NTA column, which was pre-equilibrated with 50 mL buffer A and then washed with 25 mL buffer A. At last, the different streptavidin conjugates were eluted with a linear imidazole gradient from 0 to 20 mM imidazole in buffer A over 100 mL. The elution of different species was monitored by the absorbance at 280 nm (streptavidin and atto-565-biotin) and 563 nm (atto-565-biotin), the curves were corrected for the absorbance of imidazole and different peaks were collected separately. A flow rate of 0.5 mL min−1 was used throughout the experiment. Each peak was first dialyzed against (10 kDa molecular weight cut-off) 2 L buffer pH = 3.5, 50 mM Tris–HCl, 300 mM NaCl solution for 1 h at 4 °C to remove the Ibio-His-Tag and then dialyzed twice against 2 L buffer A for at least 6 h to remove the imidazole. The samples were concentrated using a centrifugal filtration device (10 kDa molecular weight cut-off) for further studies. The protein concentration was determined by UV-Vis spectroscopy.Determination of open biotin binding pockets
The number of open biotin binding pockets was determined using biotin-5-fluorescein, whose fluorescence is quenched upon binding to streptavidin. Typically, 200 μL of 10 nM of a streptavidin conjugate in buffer A was added in a transparent 96-well plate (Greiner bio-one, F-bottom), different concentrations (0 to 50 nM) of biotin-5-fluorescein were added to each well and the samples were incubated for 10 min at room temperature. The fluorescence intensity of each well was measured (excitation wavelength 490 nm, emission wavelength 524 nm) using a plate reader (TECAN, infinite M1000).Mass spectrometry
MALDI-TOF was used to test the molecular weight of the samples and performed on a Bruker Daltonics Reflex III spectrometer. A saturated solution of sinapinic acid dissolved in 1:1 water:acetonitrile with 0.1% trifluoroacetic acid was used as the matrix solution. Typically, a sample solution (10 μM) was mixed (1:1) with the matrix solution and spotted on the steel plate. Then an additional aliquot of the matrix solution was added to dilute the sample and spotted again (repeat 3×). Spectra can be obtained in positive mode and the data were processed in mMass and Origin.
Preparation of biotinylated mOrange protein
Each protein expression plasmid (mOrange and BirA) was transformed into BL21(DE3) E. coli and starting from a single colony an overnight culture in 10 mL LB medium with 50 μg mL−1 kanamycin was prepared. The overnight culture was transferred into 1 L LB medium with 50 μg mL−1 kanamycin and incubated at 37 °C, 250 rpm until the OD600 = 0.6–0.8 and then the protein expression was induced with 1 mM IPTG. Then, the cultures were incubated at 18 °C, 250 rpm overnight and harvested the next day by centrifugation at 6000 rpm, 4 °C for 8 min (Beckman Coulter Avanti J-26S XP, JA-10 rotor). The bacteria pellet was resuspended in 20 mL buffer A supplemented with 1 mM protease inhibitor phenylmethane sulfonyl fluoride (PMSF) and 1 mM DL-dithiothreitol (DTT). The bacteria were lysed by sonication and the lysate was cleared by centrifugation at 12000 rpm (Beckman Coulter Avanti J-26S XP, JA-25.50 rotor) for 30 min, followed by filtration through a 0.45 μm filter twice. The lysate was loaded onto a 5 mL Ni2+-NTA agarose column (HisTrap™ HP, column volume 5 mL). The column was washed with 50 mL buffer C (Buffer A with 25 mM imidazole and 1 mM DTT) and the protein was eluted with 10 mL buffer B (Buffer A with 250 mM imidazole and 1 mM DTT). The purified proteins were dialyzed against 2 L buffer A with 1 mM DTT twice for at least 6 h at 4 °C.
mOrange contains a biotinylation sequence (GLNDIFEAQKIEWHE) at its N-terminal, which is recognized by the enzyme BirA.18 To prepare biotinylated mOrange, 50 μM mOrange, 5 mM MgCl2, 1 mM ATP, 1 μM BirA and 70 μM biotin were mixed in 1 mL buffer A and incubated at room temperature with gentle mixing (100 rpm) for 1 h. Then, additional 1 μM BirA and 70 μM biotin were added to the reaction mixture and incubated for 1 h. The reaction mixture was dialyzed (10 kDa molecular weight cut-off) against 2 L buffer A twice for at least 6 h at 4 °C.
Preparation and analysis of mOrange streptavidin conjugates
Typically, 1 μM S(Bio-His-Tag)1, S(Bio-His-Tag)2 or S(Bio-His-Tag)3 was mixed with 5 μM, 4 μM or 3 μM biotinylated mOrange, respectively, for at least 1 h at 4 °C. Then, 400 μL of each reaction mixture was injected onto a HiLoad™ 16/600, Superdex™ 200 pg size elution column and eluted with 150 mL buffer A at a flow rate of 1 mL min−1. The elution of different species was monitored through the absorbance at 280 nm. The molecular weight of the different species was determined based on a calibration curve established with five standard proteins (GE healthcare life science, 5 mg mL−1 thyroglobulin: MW 669 kDa, 0.3 mg mL−1 ferritin: MW 440 kDa, 4 mg mL−1 aldolase: MW 158 kDa, 3 mg mL−1 conalbumin: MW 75 kDa and 4 mg mL−1 ovalbumin: MW 44 kDa). For this, the partition coefficient (Kav) for each protein was calculated as follows:where Vo is the column void volume, Ve is the elution volume and Vc is the geometric column volume, which is equal to 120 mL for the HiLoad™ 16/600, Superdex™ 200 pg size elution column. The Vo was determined running blue dextran 2000 (400 μL, 1 mg mL−1) (GE healthcare life science) on the same column and the first eluted peak was used as the void volume. The molecular weights of the different streptavidin mOrange (O) species (S(Bio-His-Tag)1O3, S(Bio-His-Tag)2O2 and S(Bio-His-Tag)3O1) were determined based on the linear fit of the Kavversus molecular weight curve (Fig. S1†): S(Bio-His-Tag)1O3 (theo. 148.4 kDa, exp. 141.0 kDa), S(Bio-His-Tag)2O2 (theo. 118.6 kDa, exp. 108.6 kDa) and S(Bio-His-Tag)3O1 (theo. 88.8 kDa, exp. 81.3 kDa).
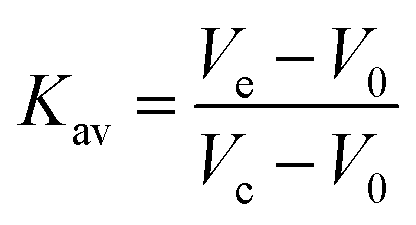
Labeling of biotinylated cell surface molecules
MDA-MB-231 cells were seeded at 3 × 104 cells per cm2 in an 8-well cell culture plate (glass bottom, ibidi) in 300 μL Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat inactivated fetal bovine serum (FBS, 10%) and 1% penicillin/streptomycin (P/S) and incubated at 37 °C, 5% CO2 overnight. The next day, the cells were washed three times using cold phosphate buffer saline supplemented with 1 mM CaCl2 and 0.1 mM MgCl2 (PBS-CM) and then the cell membrane was biotinylated using 0.25 mM sulfo-NHS-LC-biotin in PBS-CM for 30 min on ice.10 After washing the cells twice using cold PBS-CM, the cells were incubated with 100 mM glycine in PBS-CM for 2 min on ice to stop further biotinylation. The cells were washed again twice with cold PBS-CM and then incubated with 1.5 μM SA1, SA2 or SA3 in DMEM + 10% FBS + 1% P/S for 20 min on ice to label biotinylated surface molecules. The cells were washed twice with PBS, the medium was exchanged with pre-warmed DMEM + 10% FBS + 1% P/S and the cells were incubated for 0, 10 and 30 min at 37 °C in a 5% CO2 atmosphere. Then the cells were washed twice with PBS and fixed using 4% paraformaldehyde (PFA) in PBS and stained with 1 μg mL−1 TO-PRO-3. Finally, the samples were washed twice with PBS and imaged in the atto-565 and far-red channels using a confocal laser scanning microscope (Leica TCS SP8) equipped with 561 nm and 633 nm laser lines and a 63× H2O objective. For live cell imaging, the cells were treated as described above and imaged in the atto-565 channel at room temperature after adding 1.5 μM SA1 (Movie S1†), SA2 (Movie S2†) or SA3 (Movie S3†). Images were analysed by Fiji ImageJ. For the analysis, the average fluorescence intensities of single cells in the atto-565 channel were measured by encircling single cells and measuring their average intensities. 25 cells were analysed per sample and fluorescence was corrected for the background.Preparation of fluorescently labelled folic acid streptavidin conjugates and their cellular uptake
10 μM of SA1, SA2 or SA3 (A: atto-565-biotin) was mixed with 40 μM, 30 μM or 20 μM folic acid–PEG–biotin (F), respectively and incubated for 20 min at room temperature. Each reaction mixture was dialyzed (10 kDa molecular weight cut-off) against 2 L buffer A twice for at least 6 h yielding SA1F3, SA2F2 and SA3F1. The protein concentration of each conjugate and their relative fluorescence were determined by UV-Vis and fluorescence spectroscopy, respectively (Table S1†).For cellular uptake studies, MDA-MB-231 or MCF-7 cells were seeded at 5 × 104 cells per well on glass coverslips (VWR, diameter 18 mm) in 12-well cell culture plates (Greiner bio-one, F-bottom) and were cultured in RPMI-1640 medium without folic acid + 10% FBS + 1% P/S at 37 °C, 5% CO2 overnight. The next day, the cells were washed twice with PBS, 500 μL of RPMI-1640 medium without folic acid + 10% FBS + 1% P/S containing 1 μM of different streptavidin conjugates (SA1, SA2, SA3, SA1F3, SA2F2 or SA3F1) was added to each cell type and the cells were incubated at 37 °C, 5% CO2 for 4 h. Afterward, the cells were washed twice with PBS and fixed with 4% PFA in PBS for 15 min at room temperature. The cells were washed twice with PBS and were mounted on a glass slide (ROTH, 24 × 60 mm) with 40 μL Mowiol-488 containing 1 μg mL−1 DAPI. The cells were imaged in the atto-565 and DAPI channels using a confocal laser scanning microscope (Leica TCS SP8) equipped with 405 nm and 552 nm laser lines and a 63× H2O objective. Images were analysed by Fiji ImageJ. For the analysis, the average fluorescence intensities of single cells in the atto-565 channel were measured by encircling single cells and measuring their average intensities. 25 cells were analyzed per sample and fluorescence was corrected for the background. Then, fluorescence intensities measured for SA1F3, SA2F2 and SA3F1 in the cells were normalized taking into account the relative fluorescence brightness of SA1F3, SA2F2 and SA3F1 as measured in solution (Table S2†).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is part of the MaxSynBio Consortium, which is jointly funded by the Federal Ministry of Education and Research (BMBF) of Germany (FKZ 031A359L) and the Max Planck Society. We would like to thank Prof. Katharina Landfester for her support. Dongdong Xu would like to thank the Chinese Scholarship Council for his doctoral fellowship.Notes and references
- N. M. Green, Methods Enzymol., 1990, 184, 51–67 CAS.
- Y. P. Wu, C. Y. Chew, T. N. Li, T. H. Chung, E. H. Chang, C. H. Lam and K. T. Tan, Chem. Sci., 2018, 9, 770–776 RSC.
- A. Goujon, K. Strakova, N. Sakai and S. Matile, Chem. Sci., 2019, 10, 310–319 RSC.
- E. Migliorini, P. Horn, T. Haraszti, S. V. Wegner, C. Hiepen, P. Knaus, R. P. Richter and E. A. Cavalcanti-Adam, Adv. Biosyst., 2017, 1, 1600041 CrossRef.
- C. M. Dundas, D. Demonte and S. Park, Appl. Microbiol. Biotechnol., 2013, 97, 9343–9353 CrossRef CAS PubMed.
- G. Lemercier and K. Johnsson, Nat. Methods, 2006, 3, 247–248 CrossRef CAS PubMed.
- M. Howarth, D. J. F. Chinnapen, K. Gerrow, P. C. Dorrestein, M. R. Grandy, N. L. Kelleher, A. El-Husseini and A. Y. Ting, Nat. Methods, 2006, 3, 267–273 CrossRef CAS PubMed.
- M. Howarth, W. H. Liu, S. Puthenveetil, Y. Zheng, L. F. Marshall, M. M. Schmidt, K. D. Wittrup, M. G. Bawendi and A. Y. Ting, Nat. Methods, 2008, 5, 397–399 CrossRef CAS PubMed.
- I. Chamma, M. Letellier, C. Butler, B. Tessier, K. H. Lim, I. Gauthereau, D. Choquet, J. B. Sibarita, S. Park, M. Sainlos and O. Thoumine, Nat. Commun., 2016, 7, 10773 CrossRef CAS PubMed.
- J. M. Lee, J. A. Kim, T. C. Yen, I. H. Lee, B. Ahn, Y. Lee, C. L. Hsieh, H. M. Kim and Y. Jung, Angew. Chem., Int. Ed., 2016, 55, 3393–3397 CrossRef CAS PubMed.
- J. Leppiniemi, J. A. Määttä, H. Hammaren, M. Soikkeli, M. Laitaoja, J. Jänis, M. S. Kulomaa and V. P. Hytönen, PLoS One, 2011, 6, e16576 CrossRef CAS PubMed.
- V. P. Hytönen, H. R. Nordlund, J. Hörhä, T. K. Nyholm, D. E. Hyre, T. Kulomaa, E. J. Porkka, A. T. Marttila, P. S. Stayton, O. H. Laitinen and M. S. Kulomaa, Proteins: Struct., Funct., Bioinf., 2005, 61, 597–607 CrossRef PubMed.
- S. L. Kuan, D. Y. Ng, Y. Wu, C. Fortsch, H. Barth, M. Doroshenko, K. Koynov, C. Meier and T. Weil, J. Am. Chem. Soc., 2013, 135, 17254–17257 CrossRef CAS PubMed.
- R. Mittal and M. P. Bruchez, Bioconjugate Chem., 2011, 22, 362–368 CrossRef CAS PubMed.
- H. J. Gruber, G. Kada, M. Marek and K. Kaiser, BBA, Biochim. Biophys. Acta, Gen. Subj., 1998, 1381, 203–212 CrossRef CAS.
- G. O. Reznik, S. Vajda, T. Sano and C. R. Cantor, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 13525–13530 CrossRef CAS PubMed.
- V. Gaberc-Porekar and V. Menart, Chem. Eng. Technol., 2005, 28, 1306–1314 CrossRef CAS.
- M. Howarth and A. Y. Ting, Nat. Protoc., 2008, 3, 534–545 CrossRef CAS PubMed.
- Y. Ma, M. Sadoqi and J. Shao, Int. J. Pharm., 2012, 436, 25–31 CrossRef CAS PubMed.
- Y. H. Park, S. Y. Park and I. In, J. Ind. Eng. Chem., 2015, 30, 190–196 CrossRef CAS.
- Y. Chen, W. B. Cao, J. L. Zhou, B. Pidhatika, B. Xiong, L. Huang, Q. Tian, Y. W. Shu, W. J. Wen, I. M. Hsing and H. K. Wu, ACS Appl. Mater. Interfaces, 2015, 7, 2919–2930 CrossRef CAS PubMed.
- G. V. Dubacheva, C. Araya-Callis, A. G. Volbeda, M. Fairhead, J. Codee, M. Howarth and R. P. Richter, J. Am. Chem. Soc., 2017, 139, 4157–4167 CrossRef CAS PubMed.
- M. Fairhead, G. Veggiani, M. Lever, J. Yan, D. Mesner, C. V. Robinson, O. Dushek, P. A. van der Merwe and M. Howarth, J. Am. Chem. Soc., 2014, 136, 12355–12363 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc01589j |
| This journal is © The Royal Society of Chemistry 2020 |