
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral N,N′-dioxide/Mg(OTf)2 complex-catalyzed asymmetric [2,3]-rearrangement of in situ generated ammonium salts†
Qianchi
Lin
 ,
Bowen
Hu
,
Xi
Xu
,
Shunxi
Dong
,
Xiaohua
Liu
* and
Xiaoming
Feng
*
,
Bowen
Hu
,
Xi
Xu
,
Shunxi
Dong
,
Xiaohua
Liu
* and
Xiaoming
Feng
*
Key Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, China. E-mail: liuxh@scu.edu.cn; xmfeng@scu.edu.cn
First published on 19th February 2020
Abstract
Catalytic enantioselective [2,3]-rearrangements of in situ generated ammonium ylides from glycine pyrazoleamides and allyl bromides were achieved by employing a chiral N,N′-dioxide/MgII complex as the catalyst. This protocol provided a facile and efficient synthesis route to a series of anti-α-amino acid derivatives in good yields with high stereoselectivities. Moreover, a possible catalytic cycle was proposed to illustrate the reaction process and the origin of stereoselectivity.
Introduction
[2,3]-Rearrangements have been regarded as a class of synthetically powerful organic transformations due to their inherently high efficiency.1 In particular, [2,3]-rearrangement of ammonium ylides has been extensively investigated for rapid construction of valuable nitrogen-containing molecules.2 It is highly attractive to disclose asymmetric versions of such intriguing rearrangement,3–5 but only a few examples concerning the catalytic enantioselective [2,3]-rearrangement of ammonium ylides have been reported to date.5 In 2014, Smith and co-workers demonstrated the first example of chiral isothiourea-catalyzed [2,3]-rearrangement of allylic ammonium ylides to gain optically enriched syn-configured α-amino acid derivatives (Scheme 1a).5a In 2017, they developed an elegant tandem in situ protocol utilizing Pd/chiral isothiourea relay catalysis, which provides a direct method for the synthesis of syn-α-amino acid derivatives from N,N-disubstituted glycine aryl esters and allylic phosphates (Scheme 1b).5e Recently, the group of Song reported an interesting study on an isothiourea catalyzed asymmetric [2,3]-rearrangement reaction of propargyl ammonium salts, which allows access to optically active allenyl α-amino amides.5f Despite these impressive advances, there is still room for further development. For instance, chiral isothiourea is a unique catalyst with which syn-α-amino acid derivatives were preferentially afforded in current reports.5a–e Given the wide and versatile use of α-amino acids in organic synthesis and pharmaceutical chemistry,6 it is highly desirable to search for new catalytic systems for [2,3]-rearrangement of ammonium ylides in terms of the catalyst, substrate scope and the method of ammonium ylide formation, as well as stereodivergence of products.7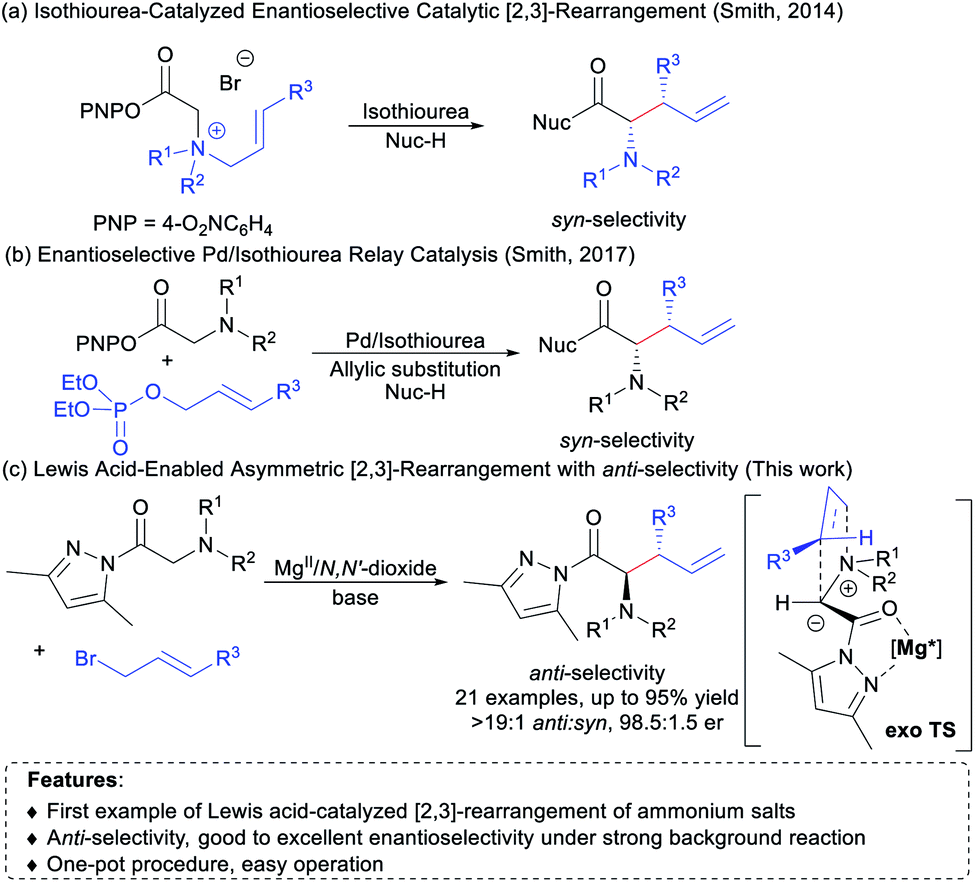 | ||
| Scheme 1 [2,3]-Rearrangements of allylic ammonium ylides. | ||
Inspired by the achievements in enantioselective [2,3]-rearrangement1–5,8 and our ongoing interest in synthesis of unnatural α-amino acid derivatives,9 we envisaged that chiral N,N′-dioxide-metal complex catalysts10 developed by our group would be potentially useful in promoting asymmetric [2,3]-rearrangement of allylic ammonium ylides upon rationally introducing pyrazoleamide groups.11 As depicted in Scheme 1c, pyrazoleamide ammonium salts generated from glycine pyrazoleamides 1 and allyl bromides 2 were selected as the precursors of [2,3]-rearrangement. It was thought that such allylic ammonium salts could be activated by bidentate coordination with the chiral N,N′-dioxide–metal complex and then subjected to deprotonation with the assistance of an external base to afford ammonium ylides, which subsequently undergo [2,3]-rearrangement to deliver non-racemic α-amino acid derivatives. There are some difficulties associated with the catalytic asymmetric [2,3]-rearrangement, such as the compatibility of all reactants with the catalyst5a,f and the background reaction in the presence of the external base.12 Herein, we wish to disclose our effort toward one-pot asymmetric [2,3]-rearrangement of in situ formed allylic ammonium ylides. Chiral N,N′-dioxide/Mg(OTf)2 (ref. 13) was found to promote the diastereo- and enantioselective rearrangement efficiently, and various anti-α-amino acid derivatives14 were readily obtained in good yields with high stereoselectivities (up to 95% yield, >19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 anti:syn and 98.5:1.5 er) from easily available glycine pyrazoleamides and allyl bromides.
:1 anti:syn and 98.5:1.5 er) from easily available glycine pyrazoleamides and allyl bromides.
Results and discussion
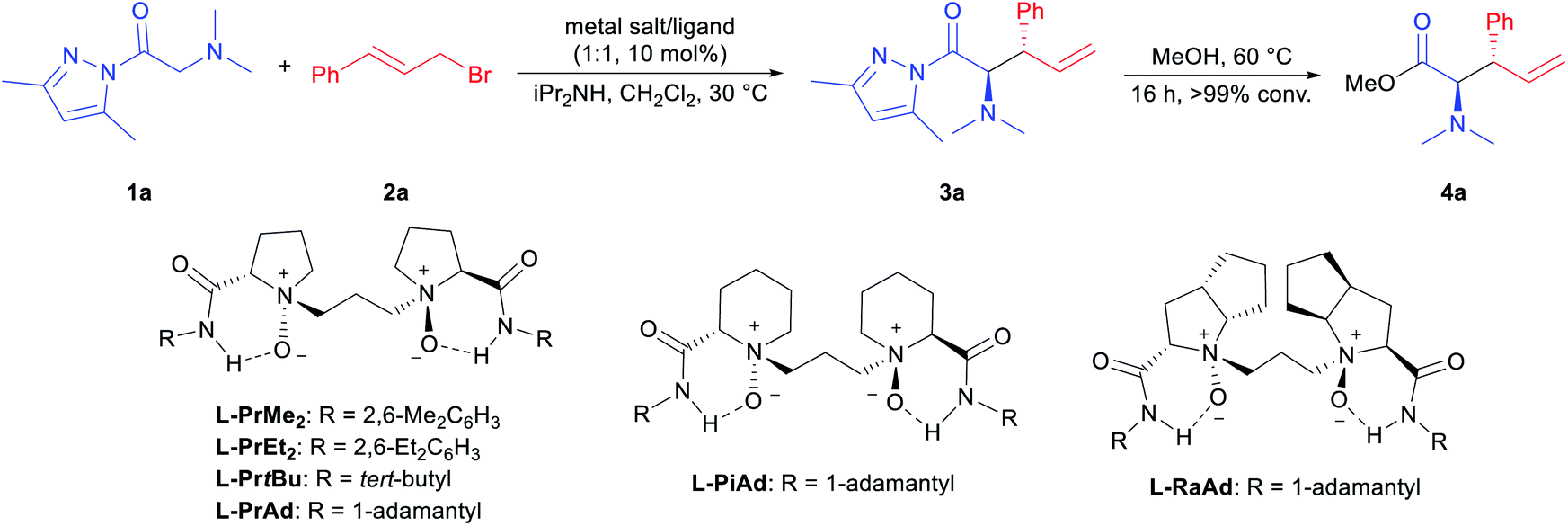
In the initial study, N,N-dimethylglycine pyrazoleamide (1a) and cinnamyl bromide (2a) were selected as the model substrates to optimize the reaction conditions. The preliminary study indicated that the tandem ammonium salt formation/[2,3]-rearrangement took place well in the presence of an external base, and the desired product 3a was isolated in 91% yield with a 1:1 anti:syn ratio by using diisopropylamine as the base (see page 9 in the ESI† for more details). This result showed the difficulty in achieving the asymmetric version of such one-pot transformation. For the purpose of determination of the er value, compound 3a was converted to 4a quantitatively with MeOH at 60 °C for further analysis. Next, different metal salts coordinated with chiral N,N′-dioxide L-PrMe2 were examined in CH2Cl2 at 30 °C (Table 1, entries 1–4). Mg(OTf)2 performed better than other metal salts, giving the desired rearranged product 3a in 91% yield, 1.8:1 anti:syn, and 67.5:32.5 er for the major diastereomer (Table 1, entry 3). The complex of Mg(NTf)2 provided a comparable result (Table 1, entry 4). Subsequently, the amide moiety in the ligand was evaluated, and it was found that sterically bulky 1-adamantyl amine derived L-PrAd afforded better stereoselectivities (Table 1, entry 7 vs. entries 5 and 6). Further investigations on the chiral backbone in the ligand showed that the L-ramipril-derived L-RaAd was superior to the L-proline-derived L-PrAd and (S)-pipecolic acid-derived L-PiAd in terms of enantioselectivity (91% yield, 3:1 anti:syn, and 81:19 er; entries 7–9). Other reaction parameters were investigated and the solvent was proven to play a significant role in the reaction. With MeCN as the solvent, the amino acid derivative 3a was produced in 99% yield with 13:1 anti:syn and 93.5:6.5 er for the major diastereomer (entry 10). The enantioselectivity could be further enhanced when the reaction was performed at decreased temperature (−20 °C)15 while a slightly lower yield was obtained (82% yield, 12:1 anti:syn, 95.5:4.5 er; entry 11). To our delight, the addition of NaBArF4 as an additive and preparation of the catalyst in EtOAc produced optimized results (entry 12; 94% yield, >19:1 anti:syn, 97:3 er). Moreover, when the product with a low anti:syn ratio was subjected to the reaction conditions, it was found that both anti:syn ratios and er values did not change. This result implied that an ultimately high anti:syn ratio was obtained during the [2,3]-rearrangement rather than epimerization of the product during the reaction (for further details, see ESI,† page 11).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Metal salt | Ligand | Yield of 3ab (%) |
anti:syn of 3ac |
er of 4ad |
|
a Unless otherwise noted, all reactions were carried out with 1a (0.10 mmol), 2a (0.10 mmol), iPr2NH (0.15 mmol) and metal salt/ligand (1:1, 10 mol%) in CH2Cl2 (1.0 mL) at 30 °C for 14 h.
b Isolated yield of 3a.
c Determined by 1H NMR.
d Determined by HPLC on a chiral stationary phase.
e Carried out in MeCN. The metal salt/ligand was pretreated in CH2Cl2.
f −20 °C for 24 h.
g NaBArF4 {NaB[3,5-(F3C)2C6H3]4} (20 mol%) was added. The metal salt/ligand/NaBArF4 was pretreated in EtOAc instead of CH2Cl2. Tf = trifluoromethanesulfonyl.
|
|||||
| 1 | Sc(OTf)3 | L-PrMe2 | 77 | 1:1 |
race/race |
| 2 | Ni(OTf)2 | L-PrMe2 | 90 | 1.1:1 |
54:46/57.5:42.5 |
| 3 | Mg(OTf)2 | L-PrMe2 | 91 | 1.8:1 |
67.5:32.5/67.5:32.5 |
| 4 | Mg(NTf)2 | L-PrMe2 | 93 | 1.6:1 |
65.5:34.5/64.5:35.5 |
| 5 | Mg(OTf)2 | L-PrEt2 | 99 | 3:1 |
61:39/80:20 |
| 6 | Mg(OTf)2 | L-PrtBu | 90 | 2:1 |
56.5:43.5/52.5:47.5 |
| 7 | Mg(OTf)2 | L-PrAd | 98 | 5:1 |
75.5:24.5/57:43 |
| 8 | Mg(OTf)2 | L-PiAd | 94 | 2:1 |
52.5:47.5/race |
| 9 | Mg(OTf)2 | L-RaAd | 91 | 3:1 |
81:19/54.5:45.5 |
| 10e | Mg(OTf)2 | L-RaAd | 99 | 13:1 |
93.5:6.5 |
| 11e,f | Mg(OTf)2 | L-RaAd | 82 | 12:1 |
95.5:4.5 |
| 12e,f,g | Mg(OTf)2 | L-RaAd | 94 | >19:1 |
97:3 |
With the optimized reaction conditions in hand, the substrate scope of [2,3]-rearrangement was screened (Table 2). Varying N-substituents in the glycine pyrazoleamides had no effect on the anti:syn ratio of the reaction (>19:1 in all cases). However, the reactivity and enantioselectivity dropped sharply with the increase of the ring size and steric hindrance of N-substituents (3a–3e). As shown in Table 2, the use of glycine pyrazoleamides bearing symmetrical N,N-dialkyl substituents, such as N,N-diethylglycine pyrazoleamide, gave the desired product 3b in 64% yield with a 95:5 er value. Cyclic N-piperidinyl and N-azepanyl substituents were also tolerated in this reaction, delivering the products 3c and 3d in moderate yields with satisfactory enantioselectivities. Lower yield (27% yield) with a good enantiomeric ratio (92:8 er) was obtained for product 3e bearing a N-heterocycle (morpholinyl) substituent. Next, the reaction of 1a with allyl bromide compounds 2 bearing different cinnamic aryl substituents was evaluated. Both the position and electronic properties of substituents had obvious effects on the reaction. Meta-substituted (E)-(3-bromopropenyl)benzenes afforded the corresponding products (3g and 3o) with better results than those with substituents at ortho- or para-positions (3f, 3h, 3n, 3p). Generally, the substrate with an electron-rich group transformed into the rearrangement product with a slightly higher anti:syn ratio than the substrate with an electron-deficient group (3h and 3p). Other para-halogen-substituted aryl rings, including 4-FC6H4, 4-BrC6H4, and 4-IC6H4 delivered the expected products (3j–3l) in 57–70% isolated yields of the major diastereomers with 90.5:9.5 to 96:4 er. The presence of the 4-F3CC6H4 substituent led to acceptable outcomes (3m, 57% yield, 4:1 anti:syn and 92:8 er). The allyl bromide compounds bearing a 2-naphthyl group and heteroaromatic ring could also perform well to yield the desired products (3q–3s) with high diastereo- and enantioselectivities. Moreover, the allyl bromide compound 2 bearing a styryl functional group was suitable for the current reaction, producing the amino acid derivative 3t in 82% yield with >19:1 anti:syn and 97:3 er. In addition, this rearrangement process of N,N-dimethylglycine pyrazoleamide 1a with cinnamyl bromide bearing a methyl on the double bond gave the amino acid derivative 3u in excellent yields (95% yield, >19:1 anti:syn and 98.5:1.5 er). Unfortunately, when cyclohexyl-substituted allyl bromide was employed, only a trace amount of the desired product (3v) was detected even with a prolonged reaction time (7 days). The absolute configuration of product 4u was determined to be (2R,3S) by X-ray crystallography analysis,16 and the others were assigned by comparing their CD spectra with that of 3u (see pages 96–105 in the ESI† for more details).
|
a All reactions were carried out with L-RaAd/Mg(OTf)2/NaBArF4 (1:1:2, 10 mol%), 1 (0.2 mmol), 2 (0.2 mmol), and iPr2NH (0.3 mmol) in MeCN (2.0 mL) at −20 °C. Isolated total yield of product 3. The anti:syn ratio was determined by 1H NMR analysis. The er value was determined by HPLC on a chiral stationary phase.
b Isolated yield of the major diastereomer 3.
c er value of product 4.
|
|---|

|
To illustrate the potential utility of the methodology, a scale-up synthesis of 3r was carried out under the optimized reaction conditions. As illustrated in Scheme 2a, 3 mmol of compound 1a reacted smoothly with equal amounts of allyl bromide 2n, furnishing the desired product 3r in 89% yield with >19:1 anti:syn and 98:2 er. Further transformations of the product 4a were conducted (Scheme 2b). Compound 4a was easily reduced to 5a in 88% yield with maintained stereoselectivities (>19:1 anti:syn, 96.5:3.5 er) by treatment with 10% Pd/C in methanol. Additionally, the reduction of 4a with LiAlH4 generated the corresponding alcohol 6a in 71% yield (>19:1 anti:syn, 97.5:2.5 er).
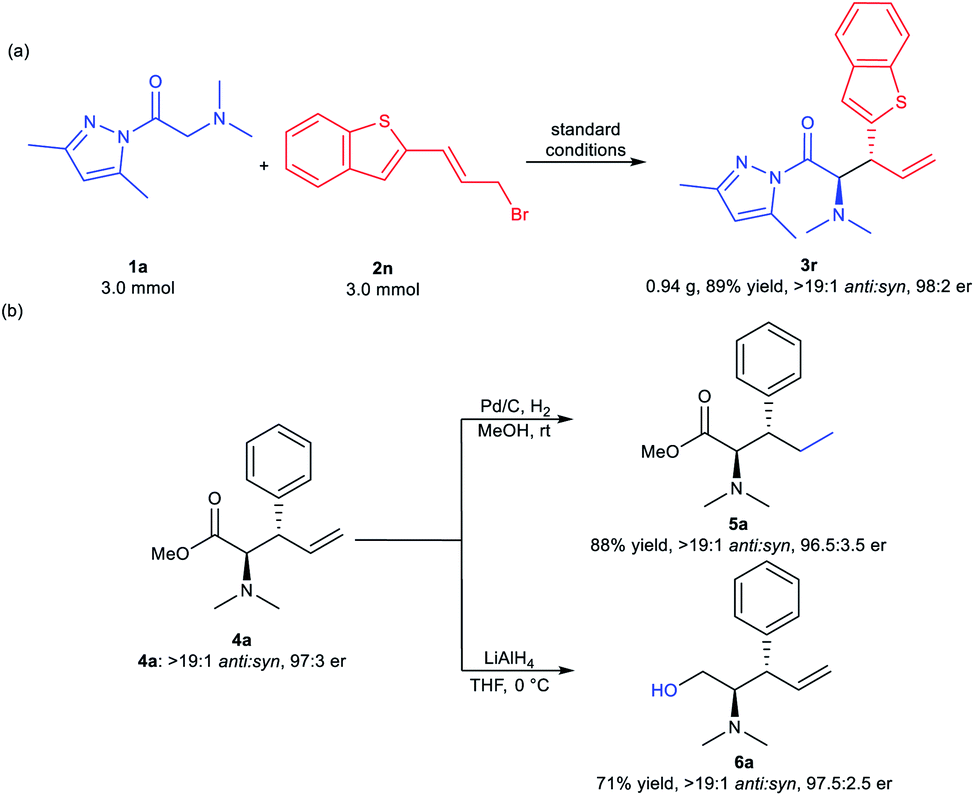 | ||
| Scheme 2 (a) Scale-up synthesis of 3r; (b) further transformation of product 4a. | ||
Based on previous work,17 the proposed catalytic cycle and a possible working mode of the enantioselective [2,3]-rearrangements of ammonium ylides are depicted in Scheme 3. Initially, the reaction of N,N-dimethylglycine pyrazoleamide (1a) and cinnamyl bromide (2a) produced the corresponding ammonium salt I which was activated by bidentate coordination with the N,N′-dioxide–metal complex and subjected to deprotonation with the assistance of iPr2NH to afford metal bonded ammonium ylide II. Due to the steric repulsion of the aryl group of the cinnamyl moiety of the substrate with the octahydrocyclopenta[b]pyrrole unit in the ligand L-RaAd as well as the pyrazoleamide unit in the exo-transition state,14 the rearrangement occurred preferentially to afford the anti-configured α-amino acid derivative (2R,3S)-3a, which was consistent with the experimental results.
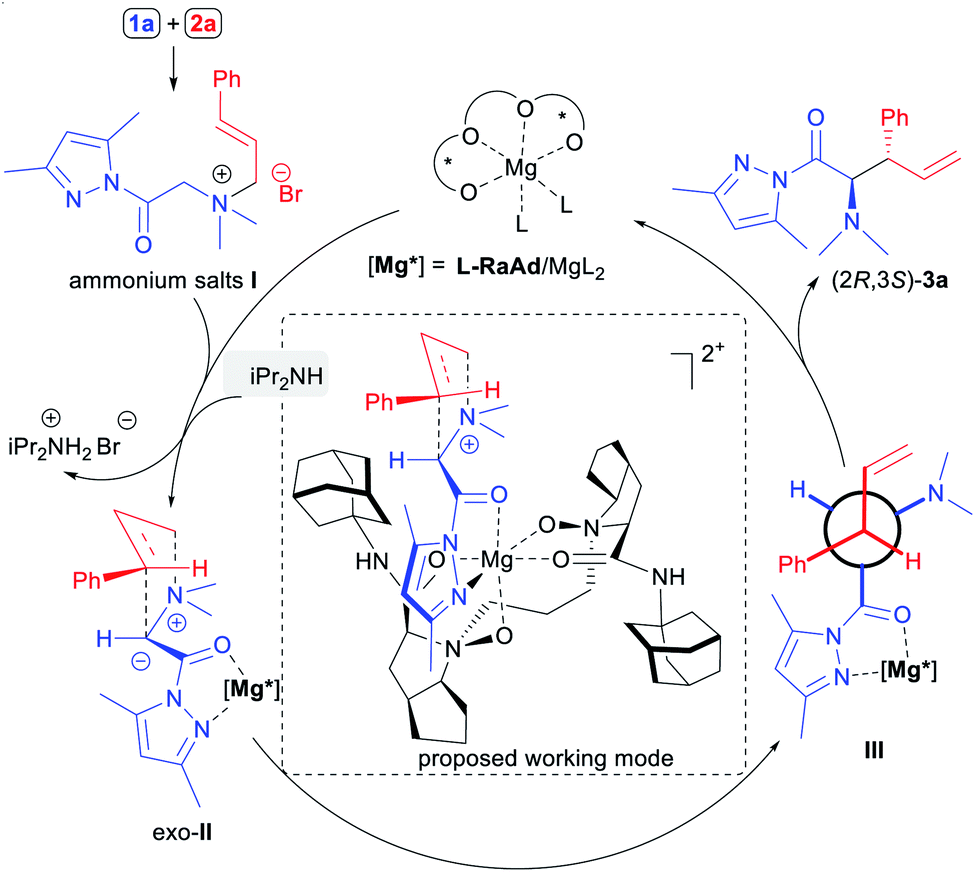 | ||
| Scheme 3 The proposed catalytic cycle and working mode. | ||
Conclusions
We have successfully developed the first Lewis acid catalyzed asymmetric [2,3]-rearrangement of quaternary ammonium ylides formed in situ from glycine pyrazoleamides and allyl bromides. The N,N′-dioxide/Mg(OTf)2 catalytic system benefited the rearrangement process efficiently, providing diverse chiral anti-α-amino acid derivatives in good yields with high stereoselectivities (up to 95% yield, >19:1 anti:syn and 98.5:1.5 er). Besides, the potential use of the current method was illustrated by gram-scale synthesis and further transformations of products. A possible catalytic cycle along with the working mode was proposed to elucidate the reaction process and chiral induction. Further investigations on other reactions enabled by chiral N,N′-dioxide–metal complex catalysts are ongoing in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate the National Natural Science Foundation of China (No. 21625205 and 21772127) for financial support.Notes and references
- For selected books and reviews on [2,3]-rearrangements, see: (a) A.-H. Li, L.-X. Dai and V. K. Aggarwal, Chem. Rev., 1997, 97, 2341 CrossRef CAS PubMed; (b) A. P. A. Arboré, D. J. Cane-Honeysett, I. Coldham and M. L. Middleton, Synlett, 2000, 2000, 236 CrossRef; (c) J. B. Sweeney, Chem. Soc. Rev., 2009, 38, 1027 RSC; (d) Nitrogen, Oxygen and Sulfur Ylide Chemistry: A Practical Approach in Chemistry, ed. J. S. Clark, Oxford University Press, NewYork, 2002 Search PubMed; (e) H. L. Bao and U. K. Tambar, [2,3]-Rearrangements of Ammonium Zwitterions, in Molecular Rearrangements in Organic Synthesis, ed. C. M. Rojas, John Wiley & Sons, Inc., Hoboken, N.J., USA, 2015, pp 459–496 Search PubMed; (f) A. C. Jones, J. A. May, R. Sarpong and B. M. Stoltz, Angew. Chem., Int. Ed., 2014, 53, 2556 CrossRef CAS PubMed; (g) T. H. West, S. S. M. Spoehrle, K. Kasten, J. E. Taylor and A. D. Smith, ACS Catal., 2015, 5, 7446 CrossRef CAS; (h) Z. Sheng, Z. K. Zhang, C. H. Chu, Y. Zhang and J. B. Wang, Tetrahedron, 2017, 73, 4011 CrossRef CAS; (i) H. Wu, Q. Wang and J. P. Zhu, Eur. J. Org. Chem., 2019, 2019, 1964 CrossRef CAS.
- For recent examples of [2,3]-rearrangement of ammonium ylides to synthesize useful natural products or active molecules, see: (a) J. A. Vanecko, H. Wan and F. G. West, Tetrahedron, 2006, 62, 1043 CrossRef CAS; (b) H. Cho, H. Jeon, J. E. Shin, S. Lee, S. Park and S. Kim, Chem.–Eur. J., 2019, 25, 2447 CrossRef CAS PubMed; (c) H. Cho, J. Jung, J. Kim, S. Park and S. Kim, Asian J. Org. Chem., 2019, 8, 1010 CrossRef CAS; (d) H. Jeon, H. Cho and S. Kim, Org. Lett., 2019, 21, 1121 CrossRef CAS PubMed.
- For selected examples of asymmetric [2,3]-rearrangement of ammonium ylides with chiral auxiliaries or substrate control, see: (a) K. Hiroi and K. Nakazawa, Chem. Lett., 1980, 1077 CrossRef CAS; (b) J. A. Workman, N. P. Garrido, J. Sançon, E. Roberts, H. P. Wessel and J. B. Sweeney, J. Am. Chem. Soc., 2005, 127, 1066 CrossRef CAS PubMed; (c) T.-S. Zhu and M.-H. Xu, Chem. Commun., 2012, 48, 7274 RSC; (d) E. Tayama, N. Naganuma, H. Iwamoto and E. Hasegawa, Chem. Commun., 2014, 50, 6860 RSC.
- For selected Lewis acid mediated asymmetric [2,3]-rearrangements of ammonium ylides, see: (a) J. Blid and P. Somfai, Tetrahedron Lett., 2003, 44, 3159 CrossRef CAS; (b) J. Blid, P. Brandt and P. Somfai, J. Org. Chem., 2004, 69, 3043 CrossRef CAS PubMed; (c) J. Blid, O. Panknin, P. Tuzina and P. Somfai, J. Org. Chem., 2007, 72, 1294 CrossRef CAS PubMed.
- (a) T. H. West, D. S. B. Daniels, A. M. Z. Slawin and A. D. Smith, J. Am. Chem. Soc., 2014, 136, 4476 CrossRef CAS PubMed; (b) T. H. West, D. M. Walden, J. E. Taylor, A. C. Brueckner, R. C. Johnston, P. H.-Y. Cheong, G. C. Lloyd-Jones and A. D. Smith, J. Am. Chem. Soc., 2017, 139, 4366 CrossRef CAS PubMed; (c) T. H. West, S. S. M. Spoehrle and A. D. Smith, Tetrahedron, 2017, 73, 4138 CrossRef CAS; (d) K. Kasten, A. M. Z. Slawin and A. D. Smith, Org. Lett., 2017, 19, 5182 CrossRef CAS PubMed; (e) S. S. M. Spoehrle, T. H. West, J. E. Taylor, A. M. Z. Slawin and A. D. Smith, J. Am. Chem. Soc., 2017, 139, 11895 CrossRef CAS PubMed; (f) L. Zhang, Z.-J. Zhang, J.-Y. Xiao and J. Song, Org. Lett., 2018, 20, 5519 CrossRef CAS PubMed.
- For selected reviews, see: (a) E. R. Jarvo and S. J. Miller, Tetrahedron, 2002, 58, 2481 CrossRef CAS; (b) G. Prabhu, N. Narendra, Basavaprabhu, V. Panduranga and V. V. Sureshbabu, RSC Adv., 2015, 5, 48331 RSC; (c) S.-M. Paek, M. Jeong, J. Jo, Y. M. Heo, Y. T. Han and H. Yun, Molecules, 2016, 21, 951 CrossRef PubMed; (d) P. Singh, S. L. K. Manda, K. Samanta and G. Panda, Tetrahedron, 2017, 73, 1911 CrossRef CAS; (e) E. D. Carpio, L. Hernández, C. Ciangherotti, V. V. Coa, L. Jiménez, V. Lubes and G. Lubes, Coord. Chem. Rev., 2018, 372, 117 CrossRef.
- For selected reviews, see: (a) Y. Ohfune and T. Shinada, Eur. J. Org. Chem., 2005, 5127 CrossRef CAS; (b) H. Vogt and S. Bräse, Org. Biomol. Chem., 2007, 5, 406 RSC; (c) K. Bera and I. N. N. Namboothiri, Asian J. Org. Chem., 2014, 3, 1234 CrossRef CAS; (d) Y. B. Wang, X. H. Song, J. Wang, H. Moriwaki, V. A. Soloshonok and H. Liu, Amino Acids, 2017, 49, 1487 CrossRef CAS PubMed.
- For selected reviews, see: (a) M. P. Doyle and D. C. Forbes, Chem. Rev., 1998, 98, 911 CrossRef CAS PubMed; (b) A. Moyano, N. El-Hamdouni and A. Atlamsani, Chem.–Eur. J., 2010, 16, 5260 CrossRef CAS PubMed. For selected examples of enantioselective [2,3]-rearrangement in our laboratory, see: (c) X. Xu, J. L. Zhang, S. X. Dong, L. L. Lin, X. B. Lin, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2018, 57, 8734 CrossRef CAS PubMed; (d) X. B. Lin, Y. Tang, W. Yang, F. Tan, L. L. Lin, X. H. Liu and X. M. Feng, J. Am. Chem. Soc., 2018, 140, 3299 CrossRef CAS PubMed; (e) X. B. Lin, W. Yang, W. K. Yang, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2019, 58, 13492 CrossRef CAS PubMed.
- (a) D. J. Shang, Y. L. Liu, X. Zhou, X. H. Liu and X. M. Feng, Chem.–Eur. J., 2009, 15, 3678 CrossRef CAS PubMed; (b) J. Yang, P. R. Ruan, W. Yang, X. M. Feng and X. H. Liu, Chem. Sci., 2019, 10, 10305 RSC.
- For reviews on N,N′-dioxide ligands, see: (a) X. H. Liu, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2011, 44, 574 CrossRef CAS PubMed; (b) X. H. Liu, L. L. Lin and X. M. Feng, Org. Chem. Front., 2014, 1, 298 RSC; (c) X. H. Liu, H. F. Zheng, Y. Xia, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2017, 50, 2621 CrossRef CAS PubMed; (d) X. H. Liu, S. X. Dong, L. L. Lin and X. M. Feng, Chin. J. Chem., 2018, 36, 791 CrossRef CAS.
- (a) Q. Yao, Z. Wang, Y. H. Zhang, X. H. Liu, L. L. Lin and X. M. Feng, J. Org. Chem., 2015, 80, 5704 CrossRef CAS PubMed; (b) Y. Zhang, Y. T. Liao, X. H. Liu, Q. Yao, Y. H. Zhou, L. L. Lin and X. M. Feng, Chem.–Eur. J., 2016, 22, 15119 CrossRef CAS PubMed; (c) G. J. Wang, Y. Tang, Y. Zhang, X. H. Liu, L. L. Lin and X. M. Feng, Chem.–Eur. J., 2017, 23, 554 CrossRef CAS PubMed.
- For a recent example, see: A. Murre, K. Erkman, S. Kaabel, I. Järving and T. Kanger, Synthesis, 2019, 51, 4183–4197 CrossRef CAS.
- For a recent review on magnesium catalysis, see: (a) D. X. Yang, L. Q. Wang, D. Li and R. Wang, Chem, 2019, 5, 1108 CrossRef CAS. For selected examples of reactions catalyzed by the N,N′-dioxide/MgII complex, see: (b) X. H. Zhao, X. H. Liu, H. J. Mei, J. Guo, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2015, 54, 4032 CrossRef CAS PubMed; (c) X. Zhong, Q. Tang, P. F. Zhou, Z. W. Zhong, S. X. Dong, X. H. Liu and X. M. Feng, Chem. Commun., 2018, 54, 10511 RSC; (d) Q. Xiong, S. X. Dong, Y. S. Chen, X. H. Liu and X. M. Feng, Nat. Commun., 2019, 10, 2116 CrossRef PubMed; (e) Q. Xiong, G. L. Li, S. X. Dong, X. H. Liu and X. M. Feng, Org. Lett., 2019, 12, 8771 CrossRef PubMed.
- Tambar and co-workers reported several fascinating examples of highly diastereoselective synthesis of anti-amino acid derivatives via palladium-catalyzed tandem allylic amination/[2,3]-rearrangement, see: (a) A. Soheili and U. K. Tambar, J. Am. Chem. Soc., 2011, 133, 12956 CrossRef CAS PubMed; (b) A. Nash, A. Soheili and U. K. Tambar, Org. Lett., 2013, 15, 4770 CrossRef CAS PubMed; (c) A. Soheili and U. K. Tambar, Org. Lett., 2013, 15, 5138 CrossRef CAS PubMed.
- Even at −20 °C, the background reaction still occurred, and the desired racemic product was isolated in 40% yield with a slightly lower anti:syn ratio (1:1.5).
- CCDC 1960932 (4u)†.
- (a) W. W. Luo, X. Yuan, L. L. Lin, P. F. Zhou, X. H. Liu and X. M. Feng, Chem. Sci., 2016, 7, 4736 RSC; (b) D. Zhang, L. L. Lin, J. Yang, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2018, 57, 12323 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: [1H, 13C{1H} and 19F{1H} NMR, HPLC spectra, and CD spectra (PDF). X-ray crystallographic data for 4u (CIF)]. CCDC 1960932. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc06342k |
| This journal is © The Royal Society of Chemistry 2020 |