
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntibody-recruiting protein-catalyzed capture agents to combat antibiotic-resistant bacteria†
Matthew N.
Idso
 a,
Ajay Suresh
Akhade
a,
Mario L.
Arrieta-Ortiz
a,
Bert T.
Lai
b,
Vivek
Srinivas
a,
James P.
Hopkins
Jr.
a,
Ana Oliveira
Gomes
a,
Naeha
Subramanian
a,
Nitin
Baliga
a and
James R.
Heath
*a
a,
Ajay Suresh
Akhade
a,
Mario L.
Arrieta-Ortiz
a,
Bert T.
Lai
b,
Vivek
Srinivas
a,
James P.
Hopkins
Jr.
a,
Ana Oliveira
Gomes
a,
Naeha
Subramanian
a,
Nitin
Baliga
a and
James R.
Heath
*a
aInstitute for Systems Biology, 401 Terry Ave North, Seattle, 98109, USA. E-mail: jim.heath@isbscience.org
bIndi Molecular, Inc., 6162 Bristol Parkway, Culver City, CA 90230, USA
First published on 12th February 2020
Abstract
Antibiotic resistant infections are projected to cause over 10 million deaths by 2050, yet the development of new antibiotics has slowed. This points to an urgent need for methodologies for the rapid development of antibiotics against emerging drug resistant pathogens. We report on a generalizable combined computational and synthetic approach, called antibody-recruiting protein-catalyzed capture agents (AR-PCCs), to address this challenge. We applied the combinatorial protein catalyzed capture agent (PCC) technology to identify macrocyclic peptide ligands against highly conserved surface protein epitopes of carbapenem-resistant Klebsiella pneumoniae, an opportunistic Gram-negative pathogen with drug resistant strains. Multi-omic data combined with bioinformatic analyses identified epitopes of the highly expressed MrkA surface protein of K. pneumoniae for targeting in PCC screens. The top-performing ligand exhibited high-affinity (EC50 ∼50 nM) to full-length MrkA, and selectively bound to MrkA-expressing K. pneumoniae, but not to other pathogenic bacterial species. AR-PCCs that bear a hapten moiety promoted antibody recruitment to K. pneumoniae, leading to enhanced phagocytosis and phagocytic killing by macrophages. The rapid development of this highly targeted antibiotic implies that the integrated computational and synthetic toolkit described here can be used for the accelerated production of antibiotics against drug resistant bacteria.
Introduction
The emergence of antibiotic-resistant bacteria represents a major threat to human health,1–4 with both increasing mortality rates and costs of care.5–8 One example is carbapenem-resistant Klebsiella pneumoniae (K. pneumoniae) strains that harbor resistance against many or all antibiotics1,6,9,10 and frequently cause hospital-acquired infections11 with high mortality rates.12 Compounding the general problem of antibiotic resistance are challenges in developing and approving new antibiotics.13 This has spurred research into understanding resistance mechanisms,14,15 host defenses,16–18 diagnostics19 and antibiotic generation.20,21 Ultimately, without a rapid and perhaps general method to develop new targeted antibiotics, therapies might relapse towards those of the pre-antibiotic era.4,13Immunomodulation by synthetic molecules is an emerging therapeutic strategy to combat bacterial and fungal infections,22–25 as well as other human diseases including cancer.26,27 The guiding principle is to employ an immunogenic agent to elicit a targeted immune response against a particular pathogen. The archetypical immune agent is an antibody and, while many therapeutic antibodies neutralize pathogens by direct interaction, a few operate by enhancing immune responses against the antibody target.22,28 Examples include antibodies against the Ebola virus,29Staphylococcus aureus,30Pseudomonas aeruginosa,28,31 and K. pneumoniae.32,33 While effective, antibodies can be challenging to produce and globally distribute at scale.34,35 Alternative compelling strategies include employing synthetic molecules that bind bacterial surface proteins36,37 or peptidoglycans,36,38 and present haptens so as to recruit the native immune system to promote bacterial clearance. Other synthetic approaches include metabolically incorporating non-native haptens into bacterial surface components.39,40 While inexpensive and scalable, these technologies can be challenging to adapt to different pathogens, or they can be non-selective, raising concerns about deleterious off-target effects.
Guided by these approaches, we envisioned a new class of highly targeted antibiotics called antibody-recruiting protein-catalyzed capture agents (AR-PCCs) that could be rapidly developed against a specified drug-resistant bacterium (Fig. 1). AR-PCCs consist of two molecular motifs. The first is a macrocyclic polypeptide ligand (the protein-catalyzed capture agent, or PCC) developed against a designated epitope of a specific bacterial surface protein. The second is an antibody-recruiting (AR) label on the PCC that promotes phagocytosis of the pathogen by innate immune cells (Fig. 1). To generate the AR-PCC ligand, we employed the recently reviewed,41 all synthetic epitope-targeted PCC method,42–45 coupled with a bioinformatics approach to identify epitopes for targeting. By targeting highly exposed epitopes of the type 3 Fimbrial Shaft (MrkA) surface protein of K. pneumoniae, we developed a small macrocyclic peptide binder in a single generation screen. That binder exhibits high affinity for the MrkA protein, high selectivity for carbapenem-resistant K. pneumoniae, and, when labeled with the AR tag, promotes macrophage-mediated phagocytosis of the pathogen. This work demonstrates that AR-PCCs can be used to target multi-drug resistant K. pneumoniae, and that the basic technology might provide a route towards drugging “undruggable” pathogenic bacteria.
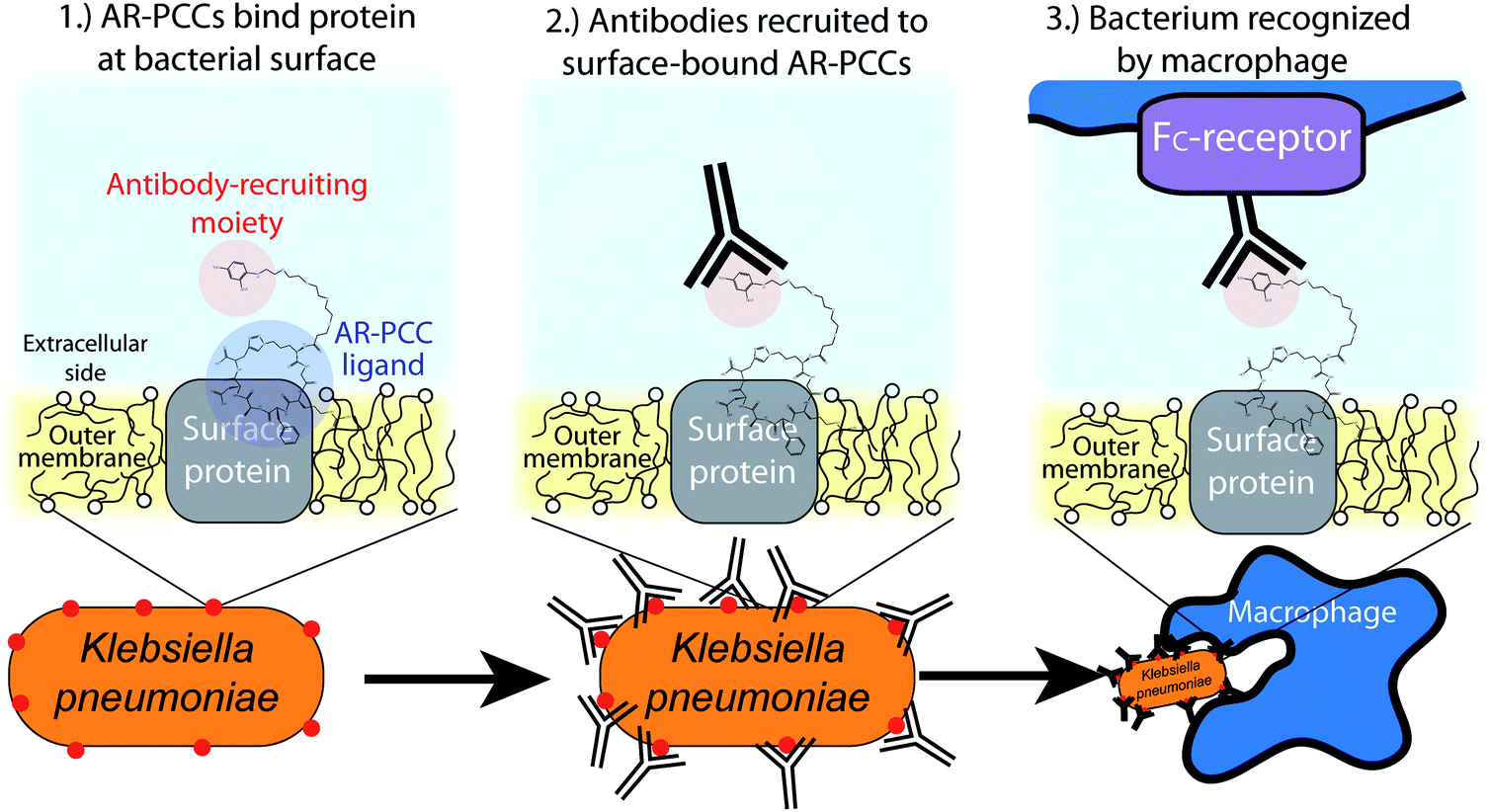 | ||
| Fig. 1 Schematic depicting the mode-of-action of AR-PCCs against antibiotic-resistant Klebsiella pneumoniae. AR-PCC molecules (1) bind to a surface protein on a K. pneumoniae bacterium, then (2) recruit antibodies that opsonize the bacterium, which leads to (3) recognition and phagocytosis of the bacterium by immune cells (e.g., macrophages). | ||
Results and discussion
Multi-omic analyses to select a target protein on K. pneumoniae
Here we describe the algorithm used to identify protein targets, and epitopes on those targets, for drugging K. pneumoniae using AR-PCCs. Traditional drugging strategies tend to rely on disrupting the function of, for example, an enzyme by competing for occupancy within a strategic hydrophobic binding pocket. Our requirements are very different. Instead, favorable aspects of target proteins are high expression levels on only the pathogen of interest, plus localization of that protein to the outer membrane or extracellular space of the pathogen. Further, once such a target protein is identified, there are additional considerations regarding which epitopes of that protein present the greatest opportunities for exploiting AR-PCCs.The flow diagram in Fig. 2A delineates the strategy for identifying the major fimbrial subunit, MrkA, as an ideal target protein for K. pneumoniae. Protein expression levels can vary across environments and growth phases, so we analyzed the transcriptional data reported by Guilhen et al.46 to identify proteins with consistently high transcript levels across three major life phases of K. pneumoniae: exponential phase, stationary phase, and biofilms (including detached cells) (see Materials and Methods section). Briefly, we focused on the top 10% of highly-expressed genes across these life phases (515 genes out of 5146 genes in the transcriptomics dataset). Subsequent cross-referencing of these 515 genes with proteomics-derived information about the localization of K. pneumoniae proteins47–49 elucidated 13 highly expressed genes that encode proteins localized to the outer membrane or extracellular space. A literature search (see Materials and Methods section for specific references) then narrowed the selection by prioritizing essential virulence- and pathogenicity-related genes as well as protein orientation in the outer membrane, to five proteins: (i) FhuA, a siderophore, (ii) Lpp, a lipoprotein, (iii) Pal, a peptidoglycan-associated lipoprotein, (iv) NlpD, a lipoprotein, (v) MrkA, a subunit of the type 3 fimbriae. Ultimately, MrkA was chosen as a target due to its key roles in infection and persistence,50 its presence in the majority of sequenced K. pneumoniae strains,51–53 and, critically, its location in fimbrial rods. These rods are large extracellular structures (0.5–2 μm long, 4-to-5 nm in diameter)54,55 that are each comprised of up to 1000s of MrkA copies.56
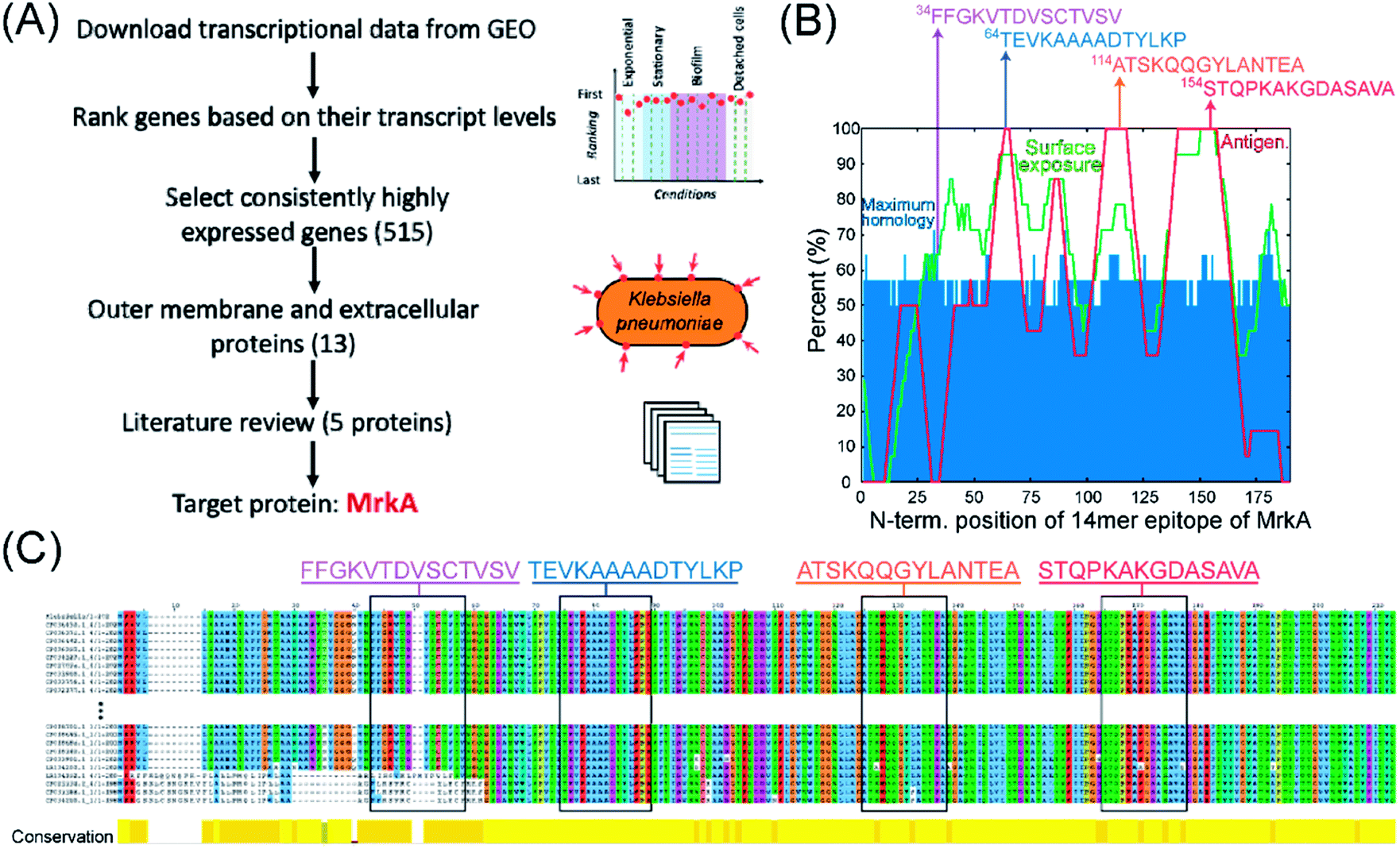 | ||
| Fig. 2 Strategy for selecting protein and epitope targets on K. pneumoniae bacteria for AR-PCCs. (A) Flow diagram illustrating the multi-omic-driven approach for selecting a protein target: analysis of transcriptomic and proteomic data identified highly expressed surface proteins of K. pneumoniae. Among those proteins, MrkA was targeted due to its association with virulence and extracellular localization (literature review). (B) Plot of predicted surface-exposure (“Surface exposure”), maximum homology with the human proteome (“Maximum homology”), and antigenicity (“Antigen.”) for all 14-residue epitopes of MrkA, as derived from bioinformatics tools (NetSurfP-2.0,57 BepiPred 2.0,58 and BLAST, using parameters defined in ref. 59). Arrows indicate the locations of four 14-residue epitopes with high-exposure and low-homology that were selected as targets. The sequences of these epitopes are shown above the plot. (C) Alignment analyses of MrkA protein sequences from 380 K. pneumoniae strains in which target epitopes are shown in rectangles. Multiple sequence alignment was performed using Fast Fourier Transform (MAFFT) and visualized in Jalview.60,61 Only the first and last 10 sequences of the total 380 sequences are shown here. | ||
The selection of target epitopes on MrkA is illustrated in Fig. 2B. Selection of surface exposed epitopes would be aided by protein structure, but no published structures exist for MrkA. As a surrogate, we employed bioinformatics tools to survey MrkA for epitopes with high surface exposure and low homology with the human proteome. B-cell antigenicity was also mapped, but wasn't a critical selection consideration.
The surface exposure, homology, and antigenicity of all 14-residue epitopes on the MrkA sequence were predicted and superimposed in Fig. 2B. Surface-exposure and antigenicity were calculated by averaging the predicted values assigned to each residue in the full-length MrkA sequence (by NetSurfP-2.0 or BepiPred-2.0 or)57,58 over the entire 14-residue epitope, while homology was predicted by comparing each 14-residue epitope with the human proteome using BlastP 2.0 with parameters defined in the heuristic string method by Berglund et al.59 This homology search yielded many partial matches per MrkA epitope, and the “Maximum homology” value plotted in Fig. 2B represents the percentage overlap with the best match. Predicted and averaged values from these analyses are tabulated in Table S1.† The plot in Fig. 2B reveals several regions containing epitopes of high (>60%) predicted surface exposure (Fig. 2B, green line) and relatively low (∼50–55%) maximum homology to the human proteome (Fig. 2B, blue bars). Note that epitopes in surface-exposed regions range in predicted antigenicity from 0 to 100%.
Four epitopes, indicated by arrows in Fig. 2B, were selected based on their predicted high surface exposures (>60%), limited maximum homology (57–71%), and broad range of antigenicity values. The sequences of these candidate epitopes are shown in Fig. 2B. Importantly, the MrkA protein and candidate epitopes are highly conserved among K. pneumoniae strains (Fig. 2C). In fact, the MrkA sequence (with 202 amino acids) is 95% conserved across 380 analyzed K. pneumoniae strains.60,61
Epitope-targeted PCC ligands against the MrkA protein
PCC ligands against the four selected epitopes were identified from a combinatorial library of macrocyclic peptides by using the epitope-targeted PCC method.42–45 This method exploits non-catalyzed click chemistry via an in situ click screen. For the screen, an alkyne-presenting, one-bead one-compound (OBOC) combinatorial library of approximately 1 M peptide macrocycles with a 5-residue variable region is screened against synthetic variants of the epitopes (SynEps). Each SynEp is designed with a biotin assay handle and a strategically incorporated azide click handle (ESI Fig. S1 and S2† contain the structures and characterization data for all MrkA SynEps).42 The concept behind the screen is that select OBOC library elements will bind to a SynEp in just the right orientation so as to promote the azide-acetylene click reaction, thus covalently linking the SynEp to the bead. This product can be detected using the biotin assay handle on the SynEp, coupled with enzymatic amplification, to add color to the hit bead. Prior to screening SynEps, the library is cleared of beads that bound the detection antibody, streptavidin-alkaline phosphatase (SAv-AP). Hit beads are separated, and the hit candidate peptides are cleaved and sequenced using tandem mass spectrometry.For this work, we performed a single screen of the OBOC library against all four target MrkA SynEps. The screen yielded 26 hits (Fig. S3, ESI†) that were sequenced, scaled up, and tested for binding to full-length recombinant MrkA in solution by a single-point sandwich enzyme-linked immunosorbent assay (ELISA) (Fig. S4, ESI†). The top-performing ligand was cy(LLFFF) (structure in Fig. 3A inset), where “cy” represents azidolysine-propargylglycine cyclization and the letters represent single-letter amino acid codes. This ligand exhibited an EC50 value of 50 nM to full-length recombinant MrkA protein (Fig. 3A). Additional ligands, but with lower affinities to MrkA, were also discovered, with sequences cy(TTFFF), cy(YRHLG) and cy(GVHRL). Based upon chemical homology, cy(TTFFF) likely binds the same epitope as cy(LLFFF), but cy(YRHLG) and cy(GVHRL) presumably bind a different one.
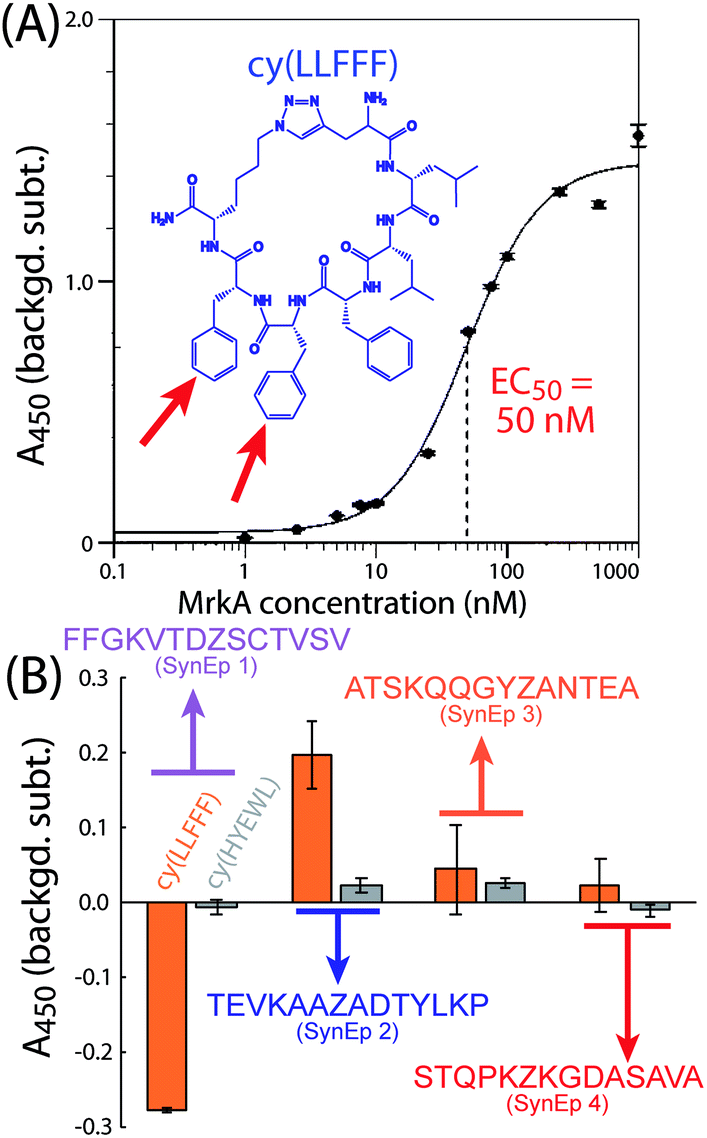 | ||
| Fig. 3 Binding of the top-performing PCC ligand cy(LLFFF) to protein and epitope targets. Sandwich enzyme-linked immunosorbent assays (ELISAs) reveal that the lead AR-PCC ligand cy(LLFFF) (A) binds to full-length MrkA protein with an EC50 value of 50 nM and (B) selectively binds synthetic epitope (SynEp) 2 versus other targeted epitopes (sequences given). The cy(HYEWL) ligand is a negative control. All measurements were performed in triplicate, and signals were background-corrected by subtracting the absorbance from otherwise identically prepared wells, except with biotin-peg6 conjugated. Error bars reflect standard deviations. Each PCC and SynEp used here had an N-terminal Biotin-peg5 group. | ||
We next identified the particular epitope target to which cy(LLFFF) binds. We performed a sandwich ELISA in which the biotinylated SynEps were immobilized. The lead ligand cy(LLFFF), conjugated to a 2,4-dinitrophenyl (DNP) moiety, was titrated at a 500 nM concentration, and anti-DNP was used as a detection antibody. The results in Fig. 3B show the highest signals from SynEp 2 (TEVKAAZADTYLKP). A background level for this assay was established using a biotinylated 6-mer polyethylene glycol (biotin-PEG6). Negative signals observed for the case of SynEp 1 (FFGKVTDZSCTVSV) indicate that this SynEp blocks non-specific binding of cy(LLFFF) or anti-DNP more than the biotin-peg6 control. As the native epitope for SynEp1 (FFGKVTDVSCTVSV) has a GRAVY score of 1.01, while the native versions of the other SynEps range between −0.9 and −0.28, the negative ELISA signals may arise from the relatively high hydrophobicity of SynEp 1. The blocking may also result from the reactivity of the cysteine residue in SynEp 1. Overall, these findings suggest that cy(LLFFF) binds the epitope TEVKAAAADTYLKP in native MrkA. This epitope is 100% conserved across the 380 K. pneumoniae isolates analyzed (Fig. 2C), suggesting that cy(LLFFF) would bind the entire cohort of these K. pneumoniae strains.
An alanine scan was performed to establish which residues of cy(LLFFF) contribute most to MrkA binding. For this experiment, a sandwich ELISA is used to quantify the affinity of several cy(LLFFF) analogues, in which one residue is substituted with an alanine, towards full-length MrkA protein. The ELISA results in Fig. 4 show lower signal for every alanine-substituted cy(LLFFF) analogue compared to unmodified cy(LLFFF), establishing that the native compound has the highest affinity. Modestly lower binding is observed for cy(ALFFF), cy(LAFFF), and cy(LLAFF), but binding is substantially reduced for analogues with alanine substitutions at the C-terminal diphenylalanine motif. Thus, these two C-terminal phenylalanine residues (Fig. 3A, red arrows in inset) play critical roles in MrkA binding, and are targets for modifications to improve affinity. Another important detail of these alanine scan results is a strong dependence of MrkA binding on the residue position, independent of residue type. For example, cy(LLAFF) and cy(LLFFA) have very different binding affinities, despite having the same phenylalanine-to-alanine substitution. This indicates that MrkA-binding is sequence dependent, suggesting selectivity of cy(LLFFF) for MrkA at the TEVKAAAADTYLKP epitope.
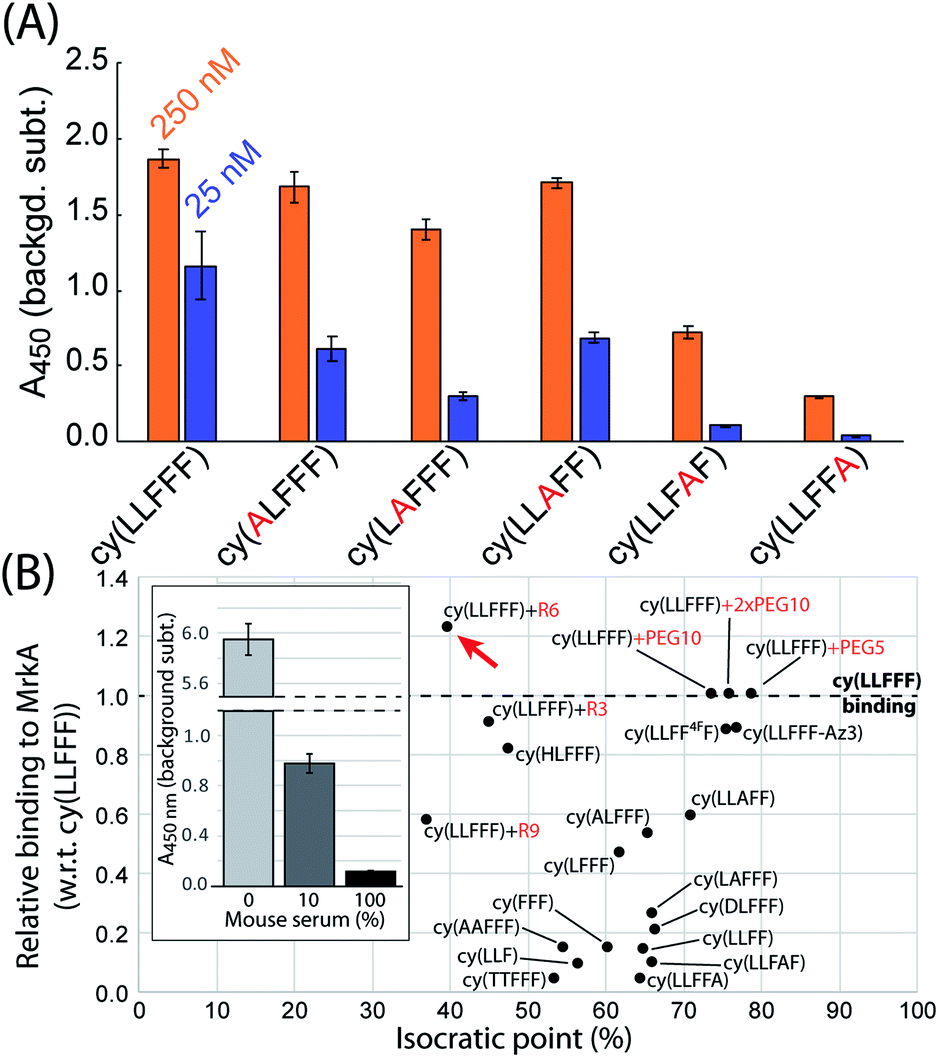 | ||
| Fig. 4 Understanding and optimizing the cy(LLFFF) ligand through strategic chemical modifications. (A) Sandwich ELISA results of an alanine scan of cy(LLFFF), which evaluates the affinity of alanine-substituted cy(LLFFF) analogues to full-length MrkA protein. (B) Plot of the isocratic point versus affinity to MrkA protein, relative to cy(LLFFF), for various cy(LLFFF) analogues in which residues were either substituted or removed, or an N-terminal peptide tag was appended. Modifications generally reduced isocratic point but also reduced target binding, with the exception of an N-terminal hexa-arginine tag (“cy(LLFFF)+R6”). This compound also bound MrkA protein in solutions of 10 and 100% mouse sera, albeit at lower levels versus in 0% serum (inset). The relative affinities plotted in (B) are the ratio of ELISA signals yielded by the analogue and those produced by a cy(LLFFF) reference on the same ELISA plate. All ELISA data are background corrected to signal from an otherwise identical measurement except with biotin-peg6 conjugated instead of the PCC. Error bars are standard deviations. Each PCC used here had an N-terminal Biotin-peg group. | ||
Synthetic modifications to optimize AR-PCC pharmacokinetics and avidity
The efficacy of AR-PCCs in vivo will not only depend on target binding, but also pharmacokinetic (PK) properties, of which clearance pathway is a critical parameter. Separate mouse studies in our labs indicate that PCCs with isocratic points (IPs) above 35% predominantly clear via the liver (i.e., hepatic clearance), while more hydrophilic compounds with lower isocratic points clear by the kidneys (i.e., renal clearance).62 Thus, the highly hydrophobic cy(LLFFF) ligand would likely exhibit hepatic clearance. To afford greater control over PK properties, we explored synthetic modifications to improve the hydrophilicity of cy(LLFFF), to favor renal clearance, while retaining the desired avidity characteristics. Modifications include single- and double-residue substitutions, residue removal, and the addition of non-ionic PEG and charged poly-arginine tags (Ri, where i represents the number of arginine). Results of this optimization are shown in Fig. 4B, which plots IP versus the affinity of the compound to MrkA, relative to cy(LLFFF). The same results are tabulated in Table S2.†Residue substitutions and removals categorically reduced affinity to MrkA, indicating that the structure of cy(LLFFF) compound is somewhat optimized to bind the TEVKAAADTYLKP epitope. These residue substitutions and removals provided up to 30% reductions in IP versus cy(LLFFF). By comparison, the addition of polyarginine tags reduced the IP to 40% without a substantial loss in MrkA avidity (three leftmost data points in Fig. 4B). In fact, cy(LLFFF) with a hexa-arginine tag, “cy(LLFFF)+R6”, shows a 22% stronger affinity to MrkA over cy(LLFFF) and had an isocratic point near 40%, as indicated by the red arrow in Fig. 4B. This represents an improvement of ∼35% versus the IP of unmodified cy(LLFFF). An additional sandwich ELISA conducted with MrkA protein at 250 nM in solutions of 10 and 100% mouse sera demonstrate that cy(LLFFF)+R6 binds MrkA protein in the context of sera (Fig. 4B inset), making this molecule promising for in vivo translation.
AR-PCC binding to multidrug resistant Klebsiella pneumoniae surfaces
While cy(LLFFF) exhibits high affinity towards recombinant full-length MrkA protein in solution, the structure of MrkA is likely much different in native fimbriae, in which MrkA proteins oligomerize into a helix-like assembly.55,63 The affinity of cy(LLFFF) towards MrkA in the fimbriae of K. pneumoniae was interrogated using whole-cell ELISAs (Fig. 5A). In this assay, bacteria are exposed to biotinylated PCC ligands, then to SAv-horseradish peroxidase (SAv-HRP), and then developed to produce a colorimetric signal that correlates with SAv-HRP binding.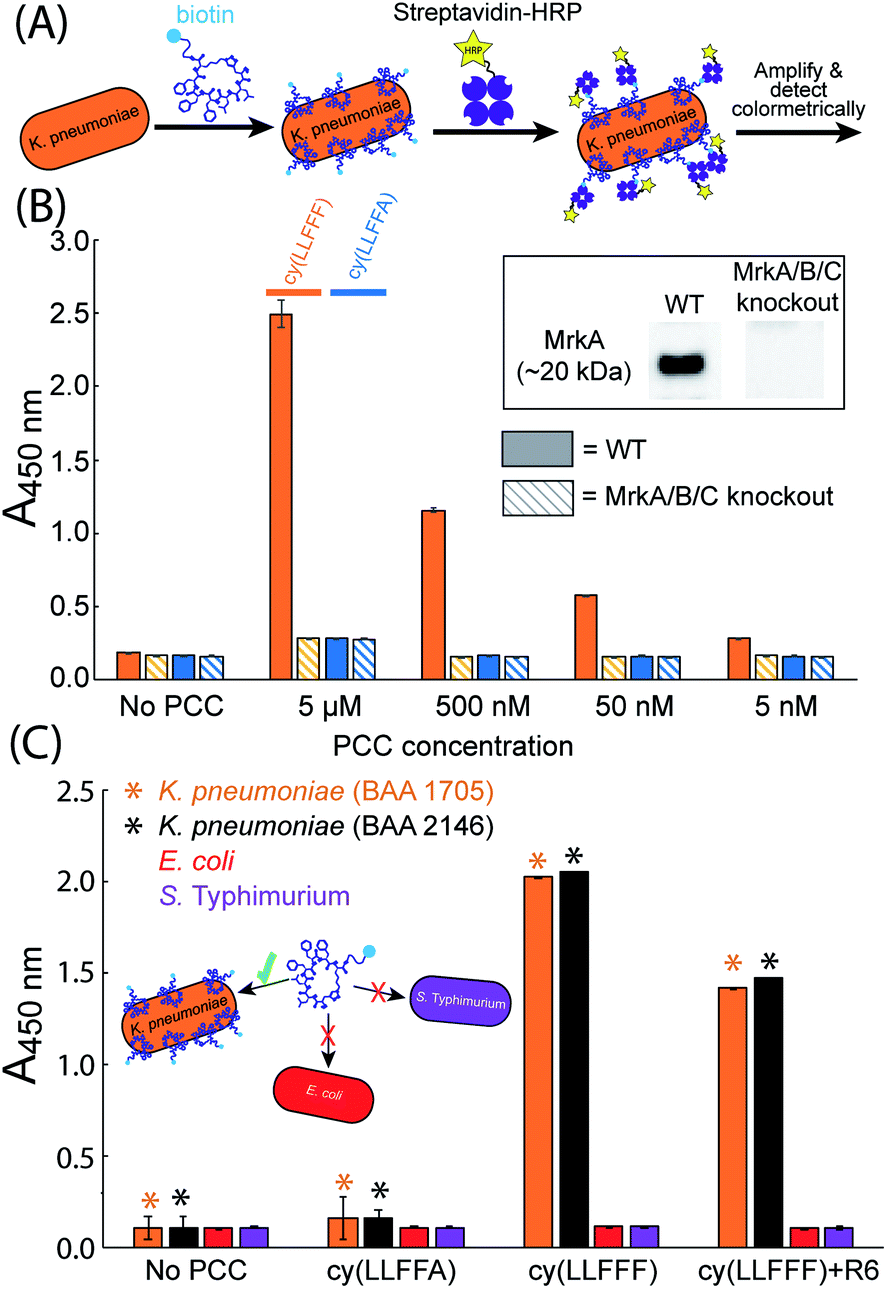 | ||
| Fig. 5 Binding specificity of cy(LLFFF) towards MrkA-expressing drug-resistant K. pneumoniae cells. (A) Schematic illustration of a whole-cell ELISA affinity test in which K. pneumoniae was incubated with PCCs conjugated with biotin, opsonized with streptavidin-HRP, and then detected colorimetrically. (B) Whole-cell ELISA results show that biotinylated cy(LLFFF) binds wild type MrkA-expressing KPPR1 cells (solid orange bars), but not MrkA-knockout KPPR1 (dashed orange bars) cells that do not produce MrkA. Much lower binding is observed from the highly homologous control compound cy(LLFFA) (solid and dashed blue bars). Western blots of wild type and MrkA-knockout KPPR1 cells are shown in the inset. (C) Cell-binding ELISA results show that biotinylated cy(LLFFF) at 5 μM binds two strains of MrkA-expressing K. pneumoniae cells with excellent selectivity versus E. coli and S. Typhimurium. By comparison a biotinylated control ligand cy(LLFFA) shows no or minimal binding above baseline. Asterisks (*) indicate ELISA data for target K. pneumoniae strains, and error bars reflect standard deviations. | ||
Whole-cell ELISA assays were conducted on the wild type version of the clinically-relevant strain of K. pneumoniae, KPPR1 (ATCC 43816). Wild type KPPR1 cells were cultured in G-CAA media to promote fimbrial expression,54 and MrkA expression in KPPR1 was confirmed by Western blot (Fig. 5B inset, full blot in Fig. S5†). The ELISA results in Fig. 5B (solid orange bars) show strong signals from wild type KPPR1 cells exposed to cy(LLFFF) at concentrations ranging from 5 μM down to 5 nM. By comparison, KPPR1 cells treated with a control compound cy(LLFFA), which differs from cy(LLFFF) by a single phenyl moiety, produce much lower intensities (Fig. 5B, solid blue bars) that are comparable to those of untreated KPPR1 cells (“No PCC”, Fig. 5B). These data not only demonstrate that cy(LLFFF) binds MrkA-expressing cells, but also reveal a high sensitivity of cy(LLFFF) structure on cell binding, establishing a strong structure–activity relationship for this PCC.
The specificity of cy(LLFFF) towards MrkA in particular was further interrogated by affinity tests to KPPR1 cells in which the gene encoding MrkA was knocked out (along with mrkB and mrkC).64 Western blot analysis confirmed that this modified KPPR1 strain does not produce MrkA (Fig. 5B inset, full blot in Fig. S5†). Knockout KPPR1 cells treated with cy(LLFFF) and control cy(LLFFA) yielded ELISA intensities comparable to baseline (Fig. 5B, dashed bars). This clearly establishes that cy(LLFFF) binds MrkA on K. pneumoniae cells. The specificity of cy(LLFFF) towards MrkA is further borne out by separate whole-cell ELISAs performed with K. pneumoniae cells (strain BAA 1705) cultured in minimal media containing 1% glycerol and 0.3% casamino acids (G-CAA media, which elicits high MrkA expression, or Lysogeny Broth (LB), which yields almost undetectable quantities of MrkA (Fig. S6 and S7†). ELISA signals were only observed for K. pneumoniae cells (strain BAA 1705) cultured in G-CAA media (Fig. S7A†), when treated with cy(LLFFF) at concentrations of μM or lower. However, when higher 50 μM PCC concentrations were used, both cy(LLFFF) and control cy(LLFFA) bound K. pneumoniae cells regardless of MrkA-expression level (Fig. S7B†), suggesting non-selective hydrophobic binding at this high concentration.
The MrkA-specific binding of cy(LLFFF) anticipates selectivity towards MrkA-expressing K. pneumoniae versus bacteria devoid of MrkA. To test this, whole-cell ELISAs were performed on MrkA-producing multidrug-resistant K. pneumoniae (strains BAA 1705 & 2146) and the bacteria Escherichia coli (E. coli) and Salmonella enterica serovar Typhimurium (S. Typhimurium) that do not express MrkA, as confirmed by Western blot (Fig. S6†). Whole-cell ELISA data in Fig. 5C show strong signals from the two strains of K. pneumoniae (BAA 1705 and BAA 2146) but only weak signals from E. coli or S. Typhimurium cells. The same bacterial strains treated instead with the control compound, biotinylated cy(LLFFA), produced low signals that were comparable to secondary antibody controls (“No PCC”, Fig. 5C), establishing low binding. Strong binding of cy(LLFFF) towards K. pneumoniae cells demonstrate excellent binding selectivity of cy(LLFFF) towards MrkA-expressing K. pneumoniae cells versus non-target bacteria. The compound cy(LLFFF)+R6, which was chemically optimized for increased hydrophilicity versus cy(LLFFF), exhibited similarly high selectivity towards MrkA-expressing cells (Fig. 5C). Importantly, though not explicitly tested here, E. coli and S. Typhimurium are known to express fimbria under growth conditions similar to those used here,65,66 suggesting selectivity of cy(LLFFF) to type 3 fimbriae of K. pneumoniae. Moreover, salt aggregation tests reveal that the cell surfaces of the K. pneumoniae strains used here have similar or lower hydrophobicities than E. coli or S. Typhimurium (Fig. S8 and S9†), indicating that binding selectivity cannot be explained simply by differences in surface hydrophobicity across these bacteria.
AR-PCC-driven opsonization of K. pneumoniae
We next sought to use cy(LLFFF) to promote opsonization by appending an antibody-recruiting (AR) handle, the hapten 2,4-dinitrophenyl (DNP), to form an AR-PCC. DNP is a hapten that is bound by 1% of endogenous human antibodies,67 and has been employed in several immune-recruiting therapeutics to recruit antibodies to various pathogens26,39,40 and cancer cells.68,69 We hypothesized that, once the AR-PCC binds to the K. pneumoniae surface, the AR handle should recruit antibodies to the pathogen. PCCs were tagged with a DNP group via conjugation of a DNP-modified lysine residue, in which a DNP moiety is covalently attached at the terminal sidechain amine. Both DNP-conjugated cy(LLFFF) and cy(HNGPT) PCCs, referred to respectively as cy(LLFFF)-DNP and cy(HNGPT)-DNP, showed absorbances at 360 nm and 420 nm (Fig. S10†) that are characteristic of DNP-modified lysine40 and indicate successful labeling of the PCC.Flow cytometry was used to determine whether the cy(LLFFF)-DNP AR-PCC promoted opsonization of resistant K. pneumoniae by anti-DNP antibodies. K. pneumoniae cells (strain BAA 2146) were first treated with cy(LLFFF)-DNP and then Alexafluor 488 fluorophore-labeled anti-DNP antibodies (Fig. 6A), so that fluorescence served as a proxy for opsonization. The cytometry data in Fig. 6B show weak fluorescence (predominantly <0.2 × 101 intensity) from K. pneumoniae cells that were either unstained or incubated with fluorescent anti-DNP only, and slightly higher fluorescence (predominantly <1.0 × 101 intensity) was observed for cells exposed to control AR-PCC cy(HNGPT)-DNP at 50 μM plus anti-DNP, indicating slight cross-reactivity. Fluorescence counts for all these samples, however, are substantially less than for cells treated with much lower concentrations (1 or 5 μM) of cy(LLFFF)-DNP plus fluorescent anti-DNP (Fig. 6B, light and dark green, respectively). This demonstrates that the cy(LLFFF)-DNP AR-PCC promotes the opsonization of K. pneumoniae cells with anti-DNP. The same behaviors were observed on a separate resistant K. pneumoniae strain, BAA 1705, (Fig. S11†) and by using whole-cell ELISA methods that were sensitive to anti-DNP binding (Fig. S12†). Paired with the recruitment of SAv-HRP to cells by biotin-cy(LLFFF) (Fig. 5B and C), these results lend to the versatility of AR-PCCs to recruit diverse and specified biomolecules to bacterial surfaces.
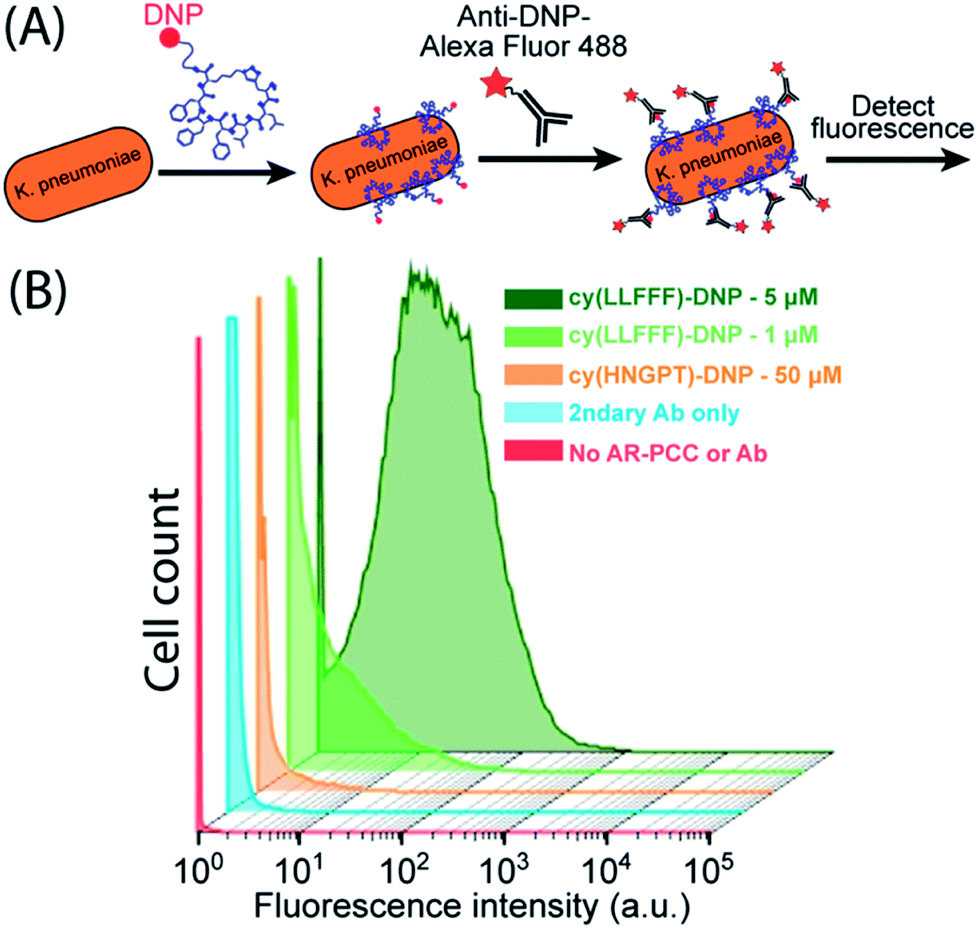 | ||
Fig. 6 Flow cytometry assay to evaluate the capacity of AR-PCCs to recruit antibodies to K. pneumoniae cell surfaces. (A) Schematic of the antibody recruitment assay in which K. pneumoniae cells are incubated with AR-PCCs that bear DNP groups, exposed to fluorescent anti-DNP antibodies, and then subjected to flow cytometry. (B) Flow cytometry results for K. pneumoniae cells (strain BAA 2146) opsonized by DNP-conjugated PCCs and fluorescently-tagged anti-DNP antibodies. Cells exposed to cy(LLFFF)-DNP, at either 1 or 5 μM concentrations, plus secondary antibody (light and dark green) showed much higher fluorescence than cells incubated with 50 μM of the control cy(HGNPT)-DNP plus secondary antibody (orange) and secondary antibody only (teal). Untreated cells showed the lowest fluorescence (red). Cytometry measurements were gated for single cells, and each distribution comprises >18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells. 000 cells. | ||
AR-PCC-driven phagocytosis and opsonophagocytic killing (OPK) of K. pneumoniae
Given that the cy(LLFFF)-DNP AR-PCC recruits antibodies to K. pneumoniae surfaces, we posited that K. pneumoniae opsonized in this manner would be more susceptible to phagocytosis by macrophages. AR-PCC-driven phagocytosis and OPK were quantified by a gentamicin protection assay, as depicted in Fig. 7A. For this assay, K. pneumoniae cells (strain BAA 1705) were treated with cy(LLFFF)-DNP, opsonized by anti-DNP antibodies, and then incubated with murine bone-marrow-derived macrophages (BMDMs). BMDMs were subsequently exposed to a solution containing gentamicin, to which K. pneumoniae strain BAA 1705 is susceptible, to kill any extracellular bacteria. Phagocytosed bacteria remain viable inside BMDMs for at least 1 h, yet are rendered inviable after 24 h by OPK (Fig. S13†). Macrophages harvested at 1 h and 24 h postinfection were lysed and plated to generate bacterial colonies, which were enumerated to quantify phagocytosis and OPK.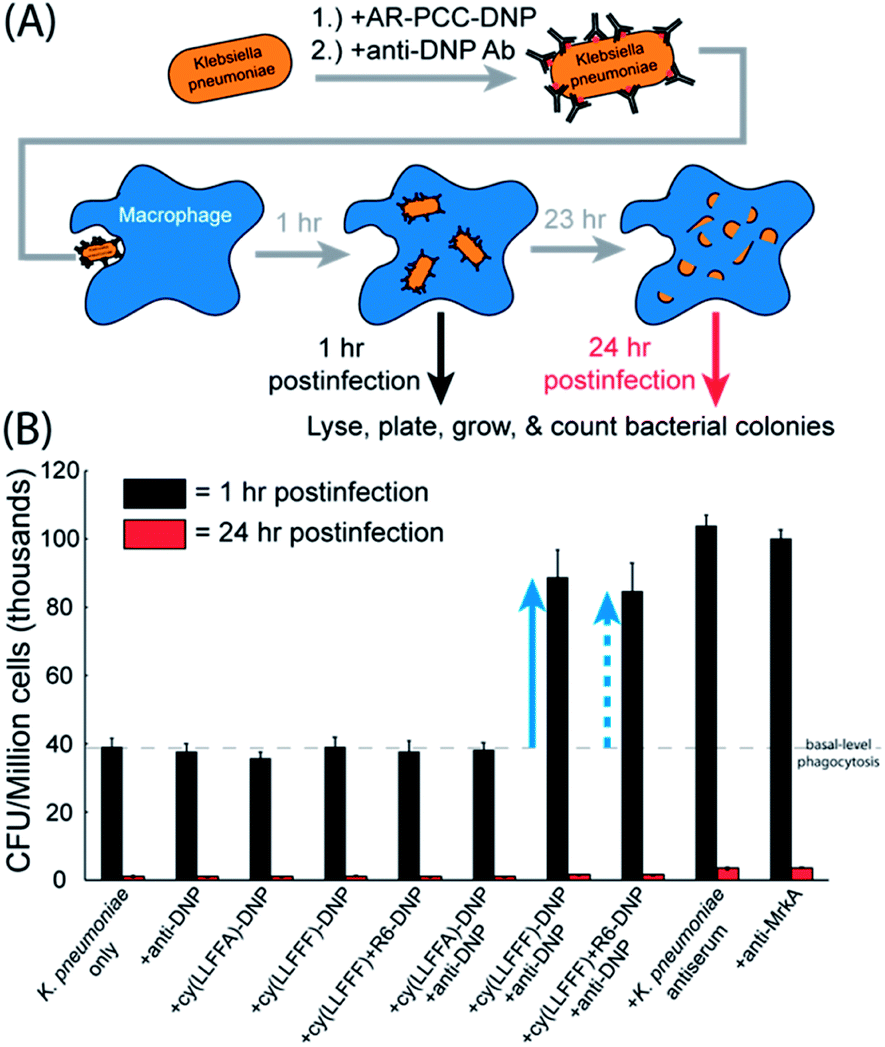 | ||
| Fig. 7 Gentamicin protection assay that evaluates AR-PCC-driven phagocytosis and opsonophagocytic killing (OPK) of K. pneumoniae cells by murine bone-marrow-derived macrophages. (A) Flow diagram depicting the gentamicin protection assay. Briefly, K. pneumoniae cells (strain BAA 1705) were opsonized by AR-PCCs and anti-DNP antibodies and then incubated with macrophages for 1 or 24 h. Macrophages were lysed, and the lysate was plated to obtain bacterial colonies that were subsequently counted. (B) Plot of colony counts for various samples subjected to the gentamicin protection assay. At 1 h, all controls (the six leftmost samples, black bars) showed basal-level CFU counts at around 39000 CFU, while K. pneumoniae cells treated with lead AR-PCC cy(LLFFF)-DNP or cy(LLFFF)+R6-DNP plus anti-DNP antibody produced much higher 89000 and 85 000 CFU respectively (blue arrows), establishing that both AR-PCC treatments promote phagocytosis of K. pneumoniae cells by macrophages. This level of phagocytosis is comparable to that promoted by K. pneumoniae antiserum and anti-MrkA antibody (at dilutions stated in the Materials and Methods section), which yielded approximately 102000 CFU. Almost no CFU were produced from any samples harvested at 24 h (red bars), indicating that AR-PCCs did not interfere with OPK activity. All AR-PCCs were used at a 5 μM concentration, measurements were conducted in triplicate, and error bars reflect standard deviations. | ||
Gentamicin protection assays were carried out using the cy(LLFFF)-DNP AR-PCC on a strain of K. pneumoniae (BAA 1705) that harbored high antimicrobial resistance, including all tested carbapenems, but is susceptible to gentamicin. As shown in Fig. 7B, BMDMs exposed to untreated K. pneumoniae cells for 1 h (“K. pneumoniae only”, black bar) yielded counts of ∼39000 CFU per million cells, establishing a basal level of phagocytosis (dashed line in Fig. 7B). Similar signals were also observed in all control samples (Fig. 7B, leftmost six samples, black bars) harvested at the 1 h time point. The controls were untreated bacteria or bacteria treated either with anti-DNP antibody only, cy(LLFFF)-DNP only, cy(LLFFA)-DNP only, cy(LLFFF)+R6-DNP only or cy(LLFFA)-DNP plus anti-DNP. None of these treatments elicited phagocytosis above the basal level. Much greater counts of 89000 CFU (indicated by the solid blue arrow, Fig. 7B) were observed from samples in which K. pneumoniae was treated with cy(LLFFF)-DNP plus anti-DNP, which compared well with the phagocytosis-promoting performances of the K. pneumoniae antiserum and the anti-MrkA antibody (Fig. 7B, rightmost two samples). When tagged with DNP, the optimized compound cy(LLFFF)+R6 also enhanced phagocytosis, as shown by the dashed blue arrow in Fig. 7B. In fact, the phagocytosis-promoting activity of DNP-tagged cy(LLFFF)-R6 was comparable to that of cy(LLFFF)-DNP. Statistical analyses (T-tests comparing phagocytosed CFUs after one hour of treatment) indicated that cy(LLFFF)-DNP plus anti-DNP promoted phagocytosis of K. pneumoniae above the basal level of the controls (p-value <0.01, Fig. S14, ESI†). Nonetheless, the level of phagocytosis induced by cy(LLFFF)-DNP plus anti-DNP was about 13% lower than that induced by K. pneumoniae antiserum and anti-MrkA treatment (positive controls: p <0.001). At 24 h of incubation, the cy(LLFFF)-DNP plus anti-DNP sample (and all other samples) showed little to no cell counts (Fig. 7B, red bars) indicating near complete OPK of phagocytosed K. pneumoniae bacteria. Thus, AR-PCCs demonstrably enhance the OPK of a highly resistant K. pneumoniae bacterium, presumably by engaging FC-receptor-mediated phagocytosis pathways.
Conclusions
We demonstrated, in vitro, a new concept for targeted antibiotics called antibody-recruiting protein-catalyzed capture agents (AR-PCCs). AR-PCCs were designed to exhibit specific in vitro antimicrobial activities against highly-resistant Klebsiella pneumoniae bacteria. AR-PCC molecules comprise of two molecular motifs: a peptide ligand that binds a specific surface protein epitope on the pathogen, and a hapten that recruits antibodies. Combined multi-omic data and bioinformatic analyses provided an algorithm for selecting a highly abundant surface protein and epitopes on K. pneumoniae as targets for AR-PCCs. A single-generation protein-catalyzed capture agent combinatorial screen was then used to rapidly identify macrocyclic peptide ligands to the chosen epitope targets. The lead AR-PCC ligand, cy(LLFFF), exhibited strong binding to full-length MrkA (EC50 ∼ 50 nM) and one of the highly conserved target epitopes, as well as high binding specificity towards MrkA-expressing K. pneumoniae versus other MrkA-deficient bacterial species. Further, the lead AR-PCC ligand conjugated with 2,4-dinitrophenyl (DNP) moieties recruited anti-DNP antibodies to the K. pneumoniae surface, which led to increased levels of phagocytosis by macrophages and ultimately greater opsonophagocytic killing. Chemical modifications to the cy(LLFFF) suggest that the macrocycle scaffold can be optimized for desired in vivo PK characteristics. Such mouse model experiments are currently underway. While K. pneumoniae served as the target in this study, the approaches used here should be adaptable to other antibiotic resistant extracellular pathogens. This versatility also makes feasible the development of cocktails of AR-PCCs that simultaneously target several conserved surface epitopes on a single pathogen, to facilitate complete clearance of bacterial populations that exhibit heterogenous surface protein expression. Overall, AR-PCCs are a promising all-synthetic molecular platform that can be rapidly designed, built and deployed against resistant microbes.Materials and Methods
Gene expression analysis
Transcriptional profiles (normalized read counts) of Klebsiella pneumoniae strain CH1034 in stationary phase, exponential phase, and biofilms (7 h, 13 h and detached cells) generated by Guilhen and collaborators46 were downloaded from the GEO database (accession number: GSE71754).70 The downloaded dataset included 5146 genes and triplicates for each condition. Although additional transcriptomes are publicly available for K. pneumoniae, we restricted our analysis to a single dataset (with key stages of K. pneumoniae life cycle) due to: (i) the high number of K. pneumoniae strains studied by different research groups. This means available transcriptomic data involve multiple strains and have been collected on multiple platforms. (ii) The large size of K. pneumoniae pangenome51 makes it possible that transcript levels of any given gene may be present in only some of the available transcriptional profiles. Inspired by the superior performance of rank-combined predictions that integrate multiple network inference methods/predictions (with respect to single ones71,72), we ranked all genes in each replicate based on their normalized read counts. Then, we computed the average ranking position of each gene along the 15 replicates. This approach identified genes with consistently high transcript levels along the sampled conditions and, for downstream analyses, we focused on the set of genes that were in the top 10% of the ranking (515 genes).Identification of highly expressed genes encoding outer membrane or extracellular proteins
We mined available literature to identify K. pneumoniae proteins that localize in the outer membrane or extracellular space.47–49 We found 54 proteins that localize to the regions of interest. Then, we evaluated the overlap between this set of proteins and the group of 515 highly expressed genes defined in the “Gene Expression Analysis” section. To compare the two sets, we first downloaded the genome annotation of Klebsiella pneumoniae strain CH1034 from the NCBI website in May 2018. We used the genome annotation to convert the locus names used as gene ID in the analyzed transcriptomics data to standard gene names (e.g. CH1034_10002 corresponds to phnV). Finally, we manually reviewed available information54,56,73–80 about genes present in both sets to select a target, prioritizing gene targets that yielded virulence-associated proteins that were either membrane-spanning or oriented on the extracellular side of the outer membrane.MrkA sequence alignment
First we performed a Blastn (megablast) in the NCBI BLAST website using as query the nucleotide sequence of mrkA in the K. pneumoniae ATCC BAA-1705 strain. We restricted the search to the K. pneumoniae taxa and set the maximum number of allowed target sequences to 500. Other parameters were kept as default. We downloaded all hits (402). Then, we removed any hit from plasmid sequences or duplicated sequences. Nucleotide hit sequences were then translated using EMBOSS Transeq.60 Finally, MrkA protein sequences were aligned using MAFFT and visualized in Jalview.60,61Bioinformatics analysis
Predictions for surface exposure and surface antigenicity were performed using NetsurfP 2.0 (ref. 57) and Bepipred2.0 (ref. 58) software (using default parameters), with the primary sequence of MrkA as an input, which assign either a value of 1 or 0 to each residue. The resulting prediction values were then averaged over 14-residue epitopes, converted to a percent value, and plotted. The uniqueness of each 14-residue epitope of MrkA among all epitopes in the human proteome was determined by performing a BlastP 2.0 search with the parameters defined in the heuristic string method by Berglund et al.59 Many matches were obtained for each epitope, and the match with greatest homology with the MrkA epitope was extracted, the corresponding homology converted to a percent value, and plotted as “Maximum homology.” All results from these bioinformatics analyses are tabulated in Table S1.†Peptide synthesis and purification
All peptides were synthesized by using standard Fmoc solid-phase peptide synthesis procedures, using either Rink Amide resin (Aapptech, RRZ005), Sieber Amide resin (Aapptech, RST001), or Tentagel S NH2 resin (Rapp Polymere, S30902). N-methylpyrrolidone (NMP, Alfa Aesar, 43894) was used as a solvent for all synthesis procedures, except the coupling of biotin (AK Scientific, C820) which used a 50/50 mixture of dimethylsulfoxide (DMSO, Fisher, D128-4) and NMP for solubility. All standard Fmoc-protected amino acids were purchased from ChemPep. Fmoc deprotection was achieved with 20% piperidine (Alfa Aesar, A12442) in NMP, and coupling reactions employed O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU, Chem-Impex Int'l Inc., 12881) as an activator and N-diisopropylethylamine (DIEA, Alfa Aesar, A11801) as the base. An Aapptech Titan 357 instrument was used to couple all Fmoc-protected standard amino acids and non-natural residues with click handles, i.e. propargylglycine (Pra, ChemPep, 180710) and azidolysine (Az4, ChemPep, 101227). Fmoc–NH–peg5-CH2CH2OH (ChemPep, 280110), Fmoc–NH–peg10-CH2CH2OH (ChemPep, 280113), and Fmoc-Lys(DNP)-OH (ApexBio, A7926) were coupled overnight with molar excesses of 2, 2, and 6 for the Fmoc-protected compound, HATU, and DIEA, respectively (excesses are with respect to the reactive groups on bead surface). The Cu-catalyzed azide–alkyne click reaction was conducted by incubating beads overnight with shaking in a solution of 1.5× molar excess of Cu(I) (Millipore Sigma, 818311) and 5× molar excess of L-ascorbic acid (Sigma, A0278) in 20% piperidine in NMP (excesses are with respect to the reactive groups on resin surface). The resin was then washed 5 × 1 minute with 5–8 mL of NMP, after which extra copper was removed by incubating beads with shaking for 5 minutes in a solution of 5 v/v% DIEA, 5 w/v% Sodium diethyldithiocarbamate trihydrate (Chem-Impex) in NMP. The resin is washed extensively with NMP (3× without shaking, 5 × 5 min or more with shaking) until the bead color turned white to light yellow and remained constant. After synthesis was complete, the resin was dried in dichloromethane (DCM, Acros Organics, 40692-0040) for at least 15 min on a vacuum manifold. Peptides were cleaved from dried resin by mixing with 10 mL solution of Trifluoroacetic acid (TFA, Alfa Aesar, L06374):Triethylsilane (TES, Millipore Sigma, 230197) Millipore Water (H2O) at volumetric ratios of 95:2.5:2.5, respectively, and vigorously stirring for 2 h. Cleavage under these acidic conditions also removed all acid-labile sidechain protecting groups. The resulting solution was added to 40 mL of diethyl ether (Acros Organics, 615080-0040) and stored overnight at −20 °C to precipitate the peptide product. The product was then pelleted by centrifugation, dried in air, and then resuspended in an aqueous solution of 30% acetonitrile (Fisher, A955-4) (aq.) with 0.1% of either TFA or formic acid (Fisher, A117-50) before purification by liquid-chromatography-mass spectrometry.
PCC compounds were purified on a Waters Autopurification system, which isolated compounds based on MS peaks corresponding to protonated [M + 1H]+ and [M + 2H]2+, and/or sodium adducts [M + Na]+ and [M + 2Na]2+. A representative HPLC chromatogram from the Waters Autopurification system with an analytical HPLC run of the corresponding product is shown in Fig. S15.† The isocratic points of PCCs were determined based on the elution of the compound from a C18 prep-scale column. Synthetic epitopes were either purified on the Waters Autopurification System, or by semi-preparative HPLC and then using matrix-assisted laser desorption/ionization mass spectroscopy to identify fractions with the desired m/z ratio. The resulting peptides were lyophilized, the yield determined by mass difference or UV-visible absorbance, and resuspended at a concentration of up to 10 mM peptide in DMSO. Peptides were stored at −80 °C before thawing for each use.
Standard Fmoc-protected synthesis and split-and-mix procedures were used to synthesize a one-bead one-compound library on TentaGel S NH2 beads with the structure NH2-Pra(80%)/Gly(20%)-X1X2X3X4X5-Az4-M-Resin, where Xi indicates one of 16 natural amino acids (excluding Methionine, Cysteine, Glutamine and Isoleucine). The coupling solution for the N-terminal residue included 80% Fmoc-propargylglycine-OH and 20% Fmoc-glycine-OH. The resulting library was clicked under copper-catalyzed conditions as described above to yield on each bead ∼80% of the cyclic product cy(Pra-X1X2X3X4X5-Az4) and 20% of the linear product Gly-X1X2X3X4X5-Az4, which facilitates identification by tandem mass spectroscopy. A final Pra was coupled onto the library to enable PCC combinatorial screening. The library was then incubated with a solution containing volumetric ratios of 95:2.5:2.5 of TFA:H2O:TES under vigorous stirring for 2 h to remove acid-labile sidechain protecting groups, and the washed 3 × 5 min in H2O, NMP, methanol (Fisher, A454-1), and then DCM.
Combinatorial PCC screening
Combinatorial in situ click screens were performed as described previously.44 Briefly, 500 mg (representing ∼1.4 copies of approximately 1000000 different compounds) of a combinatorial one-bead one-compound libraries was incubated overnight with shaking in TBS buffer (25 mM Tris HCl, 150 mM NaCl, pH 7.6). A preclear to remove beads that bound SAv-AP was performed as follows. Unless otherwise noted, all steps were performed at room temperature and all incubation and washing steps were conducted with 4 mL of the stated solution with shaking. Beads were blocked overnight by incubation in blocking buffer (TBS buffer with 1% BSA and 0.05% Tween-20, pH 7.6), rinsed with blocking buffer 3 × 1 min, incubated for 1 h with 1:10000 streptavidin-Alkaline phosphatase (SAv-AP) (Thermofisher Scientific, SA1008) in 5 mL of blocking buffer, and then washed with the following: 3 × 5 min in TBS, 3 × 5 min in 0.1 M glycine (pH 2.8), 3 × 5 min in TBS buffer, 3 × 5 min of alkaline phosphatase (AP) buffer (100 mM Tris–HCl, 150 mM NaCl, 1 mM MgCl2, pH 9.0). The beads were then split and transferred into two separate Petri dishes by using AP buffer, such that there was a total of 16 mL of AP buffer per dish. Separately, 10 mL of BCIP/NBT development buffer was prepared from the Promega BCIP/NBT Color Development substrate kit (Promega, S3771) by adding 66 μL of the NBT solution to 10 mL of AP buffer, mixing by hand, then adding 33 μL of the BCIP solution followed by vortexing. Four mL of the BCIP/NBT development buffer was added to each plate and then each plate was gently swirled for 30 seconds to ensure the development buffer was well-mixed. The reaction was quenched after 25 minutes by adding 4 mL of 5.0 N HCl (aq.) to each plate and swirling to homogenize. The beads were transferred to a new SPPS tube by using Millipore water and were then washed copiously with Millipore water (10× without shaking, then 10 × 1 minute with shaking). The beads were resuspended in 0.05 N HCl (aq.), returned to a Petri dish, and the purple beads removed by using a 10 μL pipette.
After all of the purple beads were removed, the library was incubated overnight in NMP to decolorize the residual purple coloration on the remaining beads. The library was then washed 5 × 1 minute in Millipore water and incubated overnight. The in situ click screen was then performed with the MrkA SynEps. The library was collected and rinsed in TBS buffer (3 × 5 min), incubated with a TBS buffer containing 20 μM of each purified MrkA SynEp for 6 h. The library was then subjected to the following treatments: washed with TBS buffer (10 × 1 min), incubated for 1 h in 7.5 M Guanidine-Hydrochloride (pH 2.0), washed with TBS (10 × 1 min), incubated for 2 h with blocking buffer, washed with blocking buffer (3 × 1 min), and incubated for 1 h with 1:10000 SAv-AP in 5 mL of blocking buffer. The library was then washed as follows: 3 × 5 min in TBS, 3 × 5 min in 0.1 M glycine (pH 2.8), 3 × 5 min in TBS buffer, 3 × 5 min of AP buffer. The library was then split into two Petri dishes and developed as described above. The reaction was quenched by the addition of 4 mL of 5.0 N HCl (aq.), then the beads were transferred to a new SPPS tube, washed copiously with Millipore water (10× without shaking, then 10 × 1 minute with shaking), resuspended in 0.05 N HCl, and then returned to Petri dishes. Dark and medium-dark colored beads were picked as hits and collected into Corning Costar Spin-X centrifuge tube filters (Sigma-Aldrich, CLS8170). Hit beads were then rinsed 10 × 30 s (7000 RPM, tabletop centrifuge), decolorized by overnight incubation in NMP, rinsed in Millipore water (10 × 30 s), resuspended in TBS, and stored at 4 °C.
Individual beads were transferred to wells in a 96 well plate and subjected to cyanogen-bromide cleavage, prepared, and sequenced as described previously.44,81
In vitro ELISA assays
ELISA assays for MrkA-binding and epitope selectivity were performed on clear NeutrAvidin Coated High Capacity Plates (ThermoFisher, 15507). The buffer used for all washes and to dissolve all compounds was TBST + 0.1% BSA (TBS + 0.05% Tween 20 + 0.1% BSA, pH 7.3) and, unless otherwise stated, steps were conducted at room temperature. Each wash involved a brief 20 s agitation, and all conjugation, blocking, and incubation steps were performed with gentle agitation over the entire stated time period. The general procedure for an ELISA assay was: wash 3 × 200 μL per well, conjugate with 2 μM of biotinylated compound for 2 h at room temperature (100 μL per well), wash 3 × 200 μL per well, blocked overnight in 5 wt% milk at 4 °C, wash 3 × 200 μL per well with wash buffer, incubate with titrated compound (e.g., MrkA or PCC) for 1 h, 3 × 200 μL per well, incubate with primary antibody (either or) for 1 h (100 μL per well), wash 3 × 200 μL per well, incubate with 1:2000 anti-rabbit secondary antibody-horseradish peroxidase conjugate (Cell signaling Technologies, 7074S) for 1 h, 3 × 200 μL per well, develop with the Microwell Peroxidase Substrate System (2 C) (SeraCare, 5120-0047) using 100 μL per well for 1–40 minutes, and quench using 1 M H2SO4 (aq.) at 100 μL per well. For MrkA binding assays, biotinylated PCCs were conjugated to the well surface, recombinant MrkA with a N-terminal 6xHis-SUMO-tag (MyBiosource, MBS1248970) was titrated at the desired concentration, and the primary antibody was His-tag antibody, pAb, Rabbit (Genscript, A00174) at a 1:5000 dilution. For epitope selectivity assays, biotinylated SynEps were conjugated, DNP-conjugated AR-PCCs were titrated at a desired concentration, and the primary antibody was anti-DNP antibody produced in Rabbit (Sigma-Aldrich, D9656) at a 1:8000 dilution. The PCCs and SynEps used for plate-based ELISA assays had a peg5 linker between the N-terminus of the peptide and the tag, which was either biotin or DNP-modified lysine. For binding tests in the context of mouse sera, the “titrated compound” described in the steps above was dispersed in a solution containing the desired concentration of mouse serum.
Cell culture
Bacteria were grown overnight from glycerol stocks by inoculation into either minimal media containing 1% glycerol and 0.3% casamino acids (G-CAA) or Lysogeny Broth (LB) with shaking at 37 °C. G-CAA media was used to promote the expression of MrkA, while LB was used as a control medium for binding tests that involved K. pneumoniae that do not produce MrkA. An immortalised mouse bone marrow derived macrophage cell line (iBMDM, a kind gift from Dr Eicke Latz) was used in this study. Cells were maintained in RPMI containing 10% serum at 37 °C in a humidified atmosphere with 5% CO2. K. pneumoniae strains BAA 1705 and BAA 2146 were obtained from ATCC, while wild type and MrkA/B/C knockout versions of K. pneumoniae ATCC 43816 were kindly provided by Prof. Matthew Wargo.Salt aggregation tests
Bacteria were cultured overnight in G-CAA media, washed once in 0.02 M phosphate buffer (0.01 M Na2HPO4, 0.01 M NaH2PO4, pH 8), and then resuspended to an O.D.600 of 0.95. The bacterial solutions were arrayed onto a single glass slide in 10 μL spots, and then each spot was mixed with an equal volume of phosphate buffer containing (NH4)2SO4. The glass slide was gently agitated for 2 m. Images were recorded at 10 m and 30 m following agitation, and image acquisition of all the spots took less than 1 m. Measurements were conducted on both live bacteria and heat-killed bacteria that were treated for 10 minutes at 90 °C.Whole-cell ELISAs
Detection of the binding of biotinylated PCCs to bacterial surface was performed as follows. 108 bacteria from a culture grown overnight were used for each test. Bacteria were incubated with PBS containing 1% BSA (PBS-BSA) for 1 h at 37 °C, washed once with PBS, and then incubated with various concentrations of the biotinylated PCC in PBS-BSA for 1 h at 37 °C. After washing thrice with PBS to remove unbound PCCs, bacteria were incubated with Streptavidin-HRP (1:1000) for 1 h at 37 °C. Bacteria were washed thoroughly and the TMB reagent was added until visible coloration was observed. The reaction was quenched using 2 N H2SO4 and absorbance was measured at 450 nm. The biotinylated PCCs used for cell-based ELISA assays had a peg5 linker between the N-terminus of the PCC and the biotin tag.
The protocol for detecting anti-DNP recruitment to bacterial cell surfaces is as follows. Bacteria were incubated with the desired concentration of DNP-conjugated PCCs in PBS-BSA for 1 h at 37 °C. Residual PCCs were washed off by using PBS-BSA and bacteria were incubated with anti-DNP antibody (1:1000) for 1 h at 37 °C. After washing, bacteria were incubated with HRP conjugated secondary antibody (Bio-Rad) at a dilution of 1:10000 for 1 h at 37 °C. Unbound antibody was washed off by using PBS-BSA and the cells were then developed by using TMB reagent. The reaction was quenched using 2 N H2SO4 and absorbance was measured at 450 nm. Anti-MrkA antibody was procured from Biorbyt (orb51318) and used at a concentration of 5 μg mL−1, and K. pneumoniae antiserum was obtained from abcam (ab20947) and used at a concentration of 5 μg mL−1. The DNP-conjugated PCCs used for anti-DNP recruitment assays had a peg5-peg5 linker between the N-terminus of the PCC and the DNP-modified lysine.
Opsonophagocytic killing (OPK) assays
Klebsiella pneumoniae BAA1705 was treated with DNP-conjugated PCCs followed by incubation with anti-DNP antibody, as described above in the Cell-Based ELISAs section. These opsonized bacteria were then used to infect BMDMs at a multiplicity of infection of 50 for 0.5 h at 37 °C. Bacteria were then washed by using RPMI 1640 medium and BMDMs were left in cell culture medium containing gentamicin (100 μg ml−1). After 1 h and 24 h, BMDMs were washed to remove gentamicin and intracellular bacteria were harvested by lysing BMDMs in RPMI medium containing 0.2% Triton X 100. Bacterial CFUs were enumerated by plating onto LB agar. Bacterial counts at 1 h indicated the degree of opsonization while those at 24 h served as a measure of microbicidal activity of macrophages. For these measurements, anti-MrkA antibody and K. pneumoniae antiserum were used at dilutions of 5 μg mL−1 each. The DNP-conjugated PCCs used for the phagocytosis and OPK assays had a peg5-peg5 linker between the N-terminus of the PCC and the DNP-modified lysine.Flow cytometry
Cytometry measurements were used to quantify AR-PCC-driven opsonization. For these measurements, K. pneumoniae cells were cultured in G-CAA medium overnight, washed, incubated with DNP-conjugated PCC at a desired concentration, washed, incubated with anti-DNP antibody conjugated with Alexafluor488, washed, then resuspended in PBS media. The samples were stored at 4 °C for 2 d before performing cytometry measurements. The cytometer was calibrated by using Rainbow fluorescent beads (3.5 μm diameter) from BD (559123), which aided identification of single K. pneumoniae cells in subsequent measurements. Samples were excited with 488 nm light and the fluorescence emission at 530 nm was measured. Sample with no Alexaflour488 stain was used as fluorescent minus one (FMO) control for gating of DNP+ population. FACS data was analyzed on FlowJo v10 software, and the resulting histograms each include fluorescence data from >18000 cells for strain BAA 2146 (Fig. 6B) and >4000 cells for strain BAA 1705 (Fig. S11, ESI†). PCCs used for flow cytometry assays had a peg5-peg5 linker between the N-terminus of the PCC and the DNP tag.
Conflicts of interest
J.R.H is a co-founder and B.T.L. is an employee of Indi Molecular Inc., which seeks to commercialize PCC technology.Acknowledgements
We would like to thank Professor Matthew Wargo for kindly providing the KPPR1 strains used for this study. Support for this research was provided by the 2018 ISB Innovator Award Program, the Washington State CARE Foundation, and the Institute for Collaborative Biotechnologies 6.2 program (J.R.H., #W911NF-09-D-0001). The content of the information does not necessarily reflect the position or the policy of the U.S. government, and no official endorsement should be inferred. Funding was also provided by the National Institutes of Health through grants R21AI138258 and R01AI128215, and also through the National Science Foundation grant DBI-1565166. A.S.A. was supported by an American Association of Immunologists (AAI) Careers in Immunology Fellowship.Notes and references
- L. S. Tzouvelekis, A. Markogiannakis, M. Psichogiou, P. T. Tassios and G. L. Daikos, Carbapenemases in Klebsiella pneumoniae and Other Enterobacteriaceae: an Evolving Crisis of Global Dimensions, Clin. Microbiol. Rev., 2012, 25, 682–707 CrossRef CAS PubMed.
- B. Spellberg, R. Guidos, D. Gilbert, J. Bradley, H. W. Boucher and W. M. Scheld, et al., The Epidemic of Antibiotic-Resistant Infections: A Call to Action for the Medical Community from the Infectious Diseases Society of America, Clin. Infect. Dis., 2008, 46, 155–164 CrossRef PubMed.
- World Health Organization, Antimicrobial resistance: Global Report on Surveillance, 2014 Search PubMed.
- R. J. Fair and Y. Tor, Antibiotics and Bacterial Resistance in the 21st Century, Perspect. Med. Chem., 2014, 6, 25–64 Search PubMed.
- K. Bush, P. Courvalin, G. Dantas, J. Davies, B. Eisenstein and P. Huovinen, et al., Tackling Antibiotic Resistance, Nat. Rev. Microbiol., 2011, 9, 894–896 CrossRef CAS PubMed.
- Centers for Disease Control and Prevention (US), Antibiotic Resistance Threats in the United States, 2019, 2019 Search PubMed.
- C. L. Ventola, The Antibiotic Resistance Crisis Part 1: Causes and Threats, Pharmacol. Ther., 2015, 40, 277–283 Search PubMed.
- L. S. J. Roope, R. D. Smith, K. B. Pouwels, J. Buchanan, L. Abel and P. Eibich, et al., The Challenge of Antimicrobial Resistance: What Economies Can Contribute, Science, 2019, 364, 41 CrossRef PubMed.
- R. M. Humphries, T. Kelesidis, J. D. Bard, K. W. Ward, D. Bhattacharya and M. A. Lewinski, Successful Treatment of Pan Resistant Klebsiella Pneumoniae Pneumonia and Bacteraemia with a Combination of High Dose Tigecycline and Colistin, J. Med. Microbiol., 2010, 59, 1383–1386 CrossRef PubMed.
- E. Tacconelli, E. Carrara, A. Savoldi, S. Harbarth, M. Mendelson and D. L. Monnet, et al., Discovery, Research, and Development of New Antibiotics: the WHO Priority List of Antibiotic Resistant Bacteria and Tuberculosis, Lancet Infect. Dis., 2018, 18, 318–327 CrossRef PubMed.
- J. D. D. Pitout, P. Nordmann and L. Poirel, Carbapenemase-Producing Klebsiella pneumoniae, a Key Pathogen Set for Global Nosocomial Dominance, Antimicrob. Agents Chemother., 2015, 59, 5873–5884 CrossRef CAS PubMed.
- L. S. Munoz-Price, L. Poirel, R. A. Bonomo, M. J. Schwaber, G. L. Daikos and M. Cormican, et al., Clinical Epidemiology of the Global Expansion of Klebsiella Pneumoniae Carbapenemases, Lancet Infect. Dis., 2013, 13, 785–796 CrossRef.
- E. D. Brown and G. D. Wright, Antibacterial Drug Discovery in the Resistance Era, Nature, 2016, 529, 336–343 CrossRef CAS PubMed.
- M. Lee, N. A. Pinto, C. Y. Kim, S. Yang, R. D'Souza and D. Yong, et al., Network Integrative Genomic and Transcriptomic Analysis of Carbapenem-Resistant Klebsiella pneumoniae Strains Identifies Genes for Antibiotic Resistance and Virulence, mSystems, 2019, 4, e00202–19 CrossRef PubMed.
- E. S. Kavvas, E. Catoiu, N. Mih, J. T. Yurkovich, Y. Seif and N. Dillon, et al., Machine Learning and Structural Analysis of Mycobacterium tuberculosis Pan-genome Identifies Genetic Signatures of Antibiotic Resistance, Nat. Commun., 2018, 9, 4306 CrossRef PubMed.
- F. Dotiwala, S. S. Santara, A. A. Binker-Cosen, B. Li, S. Chandrasekaran and J. Lieberman, Granzyme B Disrupts Central Metabolism and Protein Synthesis in Bacteria to Promote an Immune Cell Death Program, Cell, 2017, 171, 1125–1137 CrossRef CAS PubMed.
- M. R. Berry, R. J. Mathews, J. R. Ferdinand, C. Jing, K. W. Loudon and E. Wlodek, et al., Renal Sodium Gradient Orchestrates a Dynamic Antibacterial Defense Zone, Cell, 170(5), 860–874 CrossRef CAS PubMed.
- X. Wen, X. Xu, W. Sun, K. Chen, M. Pan and J. M. Wang, et al., G-Protein-Coupled Formyl Peptide Receptors Play a Dual Role in Neutrophil Chemotaxis and Bacterial Phagocytosis, Mol. Biol. Cell, 2019, 30, 346–356 CrossRef CAS PubMed.
- L. Barnes, D. M. Heithoff, S. P. Mahan, G. N. Fox, A. Zambrano and J. Choe, et al., Smartphone-Based Pathogen Diagnosis in Urinary Sepsis Patients, EBioMedicine, 2018, 36, 73–82 CrossRef PubMed.
- K. A. McCarthy, M. A. Kelly, K. Li, S. Cambray, A. S. Hosseini and T. van Opijnen, et al., Phage Display of Dynamic Covalent Binding Motifs Enables Facile Development of Targeted Antibiotics, J. Am. Chem. Soc., 2018, 140, 6137–6145 CrossRef CAS PubMed.
- L. Czaplewski, R. Bax, M. Clokie, M. Dawson, H. Fairhead and V. A. Fischetti, et al., Alternatives to Antibiotics – a Pipeline Portfolio Review, Lancet Infect. Dis., 2016, 16, 239–251 CrossRef CAS PubMed.
- L. L. Lu, T. J. Suscovich, S. M. Fortune and G. Alter, Beyond Binding: Antibody Effector Functions in Infectious Diseases, Nat. Rev. Immunol., 2018, 18, 46–61 CrossRef CAS PubMed.
- R. E. W. Hancock, A. Nijnik and D. J. Philpott, Modulating Immunity as a Therapy for Bacterial Infections, Nat. Rev. Microbiol., 2012, 10, 243–254 CrossRef CAS PubMed.
- P. J. McEnaney, C. G. Parker, A. X. Zhang and D. A. Spiegel, Antibody-Recruiting Molecules: An Emerging Paradigm for Engaging Immune Function in Treating Human Disease, ACS Chem. Biol., 2012, 7, 1139–1151 CrossRef CAS PubMed.
- M. S. Feigman, S. Kim, S. E. Pidgeon, Y. Yu, G. M. Ongwae and D. S. Patel, et al., Synthetic Immunotherapeutics Against Gram-negative Pathogens, Cell Chem. Biol., 2018, 25(10), 1185–1194 CrossRef CAS PubMed.
- E. Chirkin, V. Muthusamy, P. Mann, T. Roemer, P. G. Nantermet and D. A. Spiegel, Neutralization of Pathogenic Fungi with Small-Molecule Immunotherapeutics, Angew. Chem., Int. Ed., 2017, 56, 13036–13040 CrossRef CAS PubMed.
- R. P. Murelli, A. X. Zhang, J. Michel, W. L. Jorgensen and D. A. Spiegel, Chemical Control over Immune Recognition: A Class of Antibody-Recruiting Small Molecules That Target Prostate Cancer, J. Am. Chem. Soc., 2009, 131, 17090–17092 CrossRef CAS PubMed.
- A. DiGiandomenico, A. E. Keller, C. Gao, G. J. Rainey, P. Warrener and M. M. Camara, et al., A Multifunctional Bispecific Antibody Protects against Pseudomonas aeruginosa, Sci. Transl. Med., 2014, 6, 262ra155 CrossRef PubMed.
- E. O. Saphire, S. L. Schendel, B. M. Gunn, J. C. Milligan and G. Alter, Antibody-Mediated Protection Against Ebola Virus, Nat. Immunol., 2018, 19, 1169–1178 CrossRef CAS PubMed.
- U. Lorenz, B. Lorenz, T. Schmitter, K. Streker, C. Erck and J. Wehland, et al., Functional Antibodies Targeting IsaA of Staphylococcus aureus Augment Host Immune Response and Open New Perspectives for Antibacterial Therapy, Antimicrob. Agents Chemother., 2011, 55(1), 165–173 CrossRef CAS PubMed.
- A. DiGiandomenico, P. Warrener, M. Hamilton, S. Guillard, P. Ravn and R. Minter, et al., Identification of Broadly Protective Human Antibodies to Pseudomonas Aeruginosa Exopolysaccharide Psl by Phenotypic Screening, J. Exp. Med., 2012, 209(7), 1273–1287 CrossRef CAS PubMed.
- Q. Wang, C.-S. Chang, M. Pennini, M. Pelletier, S. Rajan and J. Zha, et al., Target-Agnostic Identification of Functional Monoclonal Antibodies Against Klebsiella pneumoniae Multimeric MrkA Fimbrial Subunit, J. Infect. Dis., 2016, 213, 1800–1808 CrossRef CAS PubMed.
- Q. Wang, Y. Chen, R. Cvitkovic, M. E. Pennini, C. S. Chang and M. Pelletier, et al., Anti-MrkA Monoclonal Antibodies Reveal Distinct Structural and Antigenic Features of MrkA, PLoS One, 2017, 12, e017059 Search PubMed.
- E. Sparrow, M. Friede, M. Sheikh and S. Torvaldsen, Therapeutic Antibodies for Infectious Diseases, Bull. W. H. O., 2017, 95, 235–237 CrossRef PubMed.
- E. Pelfrene, M. Mura, A. C. Sanches and M. Cavaleri, Monoclonal Antibodies as Anti-Infective Products: a Promising Future?, Clin. Microbiol. Infect., 2019, 25, 60–64 CrossRef CAS.
- M. J. Sabulski, S. E. Pidgeon and M. M. Pires, Immuno-Targeting of Staphylococcus aureus via Surface Remodeling Complexes, Chem. Sci., 2017, 8, 6804–6809 RSC.
- C. R. Bertozzi and M. D. Bednarski, A Receptor-Mediated Immune Response Using Synthetic Glycoconjugates, J. Am. Chem. Soc., 1992, 114, 5543–5546 CrossRef CAS.
- V. M. Krishnamurthy, L. J. Quinton, L. A. Estroff, S. J. Metallo, J. M. Isaacs and J. P. Mizgerd, et al., Promotion of Opsonization by Antibodies and Phagocytosis of Gram-Positive Bacteria by a Bifunctional Polyacrylamide, Biomaterials, 2006, 27, 3663–3674 CAS.
- J. M. Fura, S. E. Pidgeon, M. Birabaharan and M. M. Pires, Dipeptide-Based Metabolic Labeling of Bacterial Cells for Endogenous Antibody Recruitment, ACS Infect. Dis., 2016, 2, 302–309 CrossRef CAS.
- J. M. Fura, M. J. Sabulski and M. M. Pires, D-Amino Acid Mediated Recruitment of Endogenous Antibodies to Bacterial Surfaces, ACS Chem. Biol., 2014, 9, 1480–1489 CrossRef CAS PubMed.
- H. D. Agnew, M. B. Coppock, M. N. Idso, B. T. Lai, J. Liang and A. M. McCarthy-Torrens, et al., Protein-Catalyzed Capture Agents, Chem. Rev., 2019, 119, 9950–9970 CrossRef CAS PubMed.
- S. Das, A. Nag, J. Liang, D. N. Bunck, A. Umeda and B. Farrow, et al., A General Synthetic Approach for Designing Epitope Targeted Macrocyclic Peptide Ligands, Angew. Chem., Int. Ed., 2015, 54, 13219–13224 CrossRef CAS.
- D. N. Bunck, B. Atsavapranee, A. K. Museth, D. Vandervelde and J. R. Heath, Modulating the Folding Landscape of Superoxide Dismutase 1 with Targeted Molecular Binders, Angew. Chem., 2018, 130, 6320–6323 CrossRef.
- B. T. Lai, J. A. Wilson, J. M. Loredo, S. M. Pitram, N. A. LaBerge and J. R. Heath, et al., Epitope Targeted Macrocyclic Peptide Ligand with Picomolar Cooperative Binding to Interleukin-17F, Chem.–Eur. J., 2018, 24, 3760–3767 CrossRef CAS PubMed.
- A. M. McCarthy, J. Kim, A. K. Museth, R. K. Henning, J. E. Heath and E. Winson, et al., Allosteric Inhibitor of KRas Identified Using a Barcoded Assay Microchip Platform, Anal. Chem., 2018, 90, 8824–8830 CrossRef CAS PubMed.
- C. Guilhen, N. Charbonnel, N. Parisot, N. Gueguen, A. Iltis and C. Forestier, et al., Transcriptional Profiling of Klebsiella pneumoniae Defines Signatures for Planktonic, Sessile and Biofilm-Dispersed Cells, BMC Genomics, 2016, 17, 237 CrossRef PubMed.
- B. K. Cahill, K. W. Seeley, D. Gutel and T. N. Ellis, Klebsiella pneumoniae O Antigen Loss Alters the Outer Membrane Protein Composition and the Selective Packaging of Proteins into Secreted Outer Membrane Vesicles, Microbiol. Res., 2015, 180, 1–10 CrossRef CAS PubMed.
- A. J. Brinkworth, C. H. Hammer, L. R. Olano, S. D. Kobayashi, L. Chen and B. N. Kreiswirth, et al., Identification of Outer Membrane and Exoproteins of Carbapenem-Resistant Multilocus Sequence Type 258 Klebsiella pneumoniae, PLoS One, 2015, 10, e0123219 CrossRef PubMed.
- Y. Liu, H. Wang, X. Sun, H. Yang, Y. Wang and W. Song, Study on Mechanisms of Colonization of Nitrogen-Fixing PGPB, Klebsiella pneumoniae NG14 on the Root Surface of Rice and the Formation of Biofilm, Curr. Microbiol., 2011, 62, 1113–1122 CrossRef CAS PubMed.
- M. K. Paczosa and J. Mecsas, Klebsiella pneumoniae: Going on the Offense with a Strong Defense, Microbiol. Mol. Biol. Rev., 2016, 80, 629–661 CrossRef CAS PubMed.
- K. E. Holt, H. Wertheim, R. N. Zadoks, S. Baker, C. A. Whitehouse and D. Dance, et al., Genomic Analysis of Diversity, Population Structure, Virulence, and Antimicrobial Resistance in Klebsiella pneumoniae, an Urgent Threat to Public Health, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E3574–E3581 CrossRef CAS PubMed.
- R. M. Martin and M. A. Bachman, Colonization, Infection, and the Accessory Genome of Klebsiella pneumoniae, Front. Cell. Infect. Microbiol., 2018, 8, 4 CrossRef PubMed.
- M. D. Alcántar-Curiel, D. Blackburn, Z. Saldaña, C. Gayosso-Vásquez, N. Iovine and M. A. De la Cruz, et al., Multi-Functional Analysis of Klebsiella Pneumoniae Fimbrial Types in Adherence and Biofilm Formation, Virulence, 2013, 4, 129–138 CrossRef PubMed.
- G. F. Gerlach, B. L. Allen and S. Clegg, Molecular Characterization of the Type 3 (MR/K) Fimbriae of Klebsiella Pneumoniae, J. Bacteriol., 1988, 170, 3547–3553 CrossRef CAS PubMed.
- F.-J. Chen, C.-H. Chan, Y.-J. Huang, K.-L. Liu, H.-L. Peng and H.-Y. Chang, et al., Structural and Mechanical Properties of Klebsiella pneumoniae Type 3 Fimbriae, J. Bacteriol., 2011, 193(7), 1718–1725 CrossRef CAS PubMed.
- M. D. Alcántar-Curiel, C. A. Ledezma-Escalante, M. D. Jarillo-Quijada, C. Gayosso-Vázquez, R. Morfín-Otero and E. Rodríguez-Noriega, et al., Association of Antibiotic Resistance, Cell Adherence, and Biofilm Production with the Endemicity of Nosocomial Klebsiella pneumoniae, BioMed Res. Int., 2018, 2018, 7012958 Search PubMed.
- M. S. Klausen, M. C. Jespersen, H. Nielsen, K. K. Jensen, V. I. Jurtz and C. K. Sønderby, et al., NetSurfP-2.0: Improved Prediction of Protein Structural Features by Integrated Deep Learning, Proteins, 2019, 87, 520–527 CrossRef CAS PubMed.
- M. C. Jespersen, B. Peters, M. Nielsen and P. Marcatili, BepiPred-2.0: Improving Sequence-Based B-Cell Epitope Prediction Using Conformational Epitopes, Nucleic Acids Res., 2017, 45, W24–W29 CrossRef CAS PubMed.
- L. Berglund, J. Andrade, J. Odeberg and M. Uhlén, The Epitope Space of the Human Proteome, Protein Sci., 2008, 17, 606–613 CrossRef CAS.
- F. Mádiera, Y. M. Park, J. Lee, N. Buso, T. Gur and N. Madhusoodanan, et al., The EMBL-EBI Search and Sequence Analysis Tools APIs in 2019, Nucleic Acids Res., 2019, 47, W636–W641 CrossRef PubMed.
- A. M. Waterhouse, J. B. Procter, D. M. A. Martin, M. Clamp and G. J. Barton, Jalview Version 2—a Multiple Sequence Alignment Editor and Analysis Workbench, Bioinformatics, 2009, 25, 1189–1191 CrossRef CAS PubMed.
- Private communication with Indi Molecular Inc.
- F. G. Sauer, H. Remaut, S. J. Hultgren and G. Waksman, Fiber Assembly by the Chaperone-Usher Pathway, Biochim. Biophys. Acta, 2004, 1694, 259–267 CrossRef CAS PubMed.
- G. G. Willsey, S. Ventrone, K. C. Schutz, A. M. Wallace, J. W. Ribis, B. T. Suratt and M. J. Wargo, Pulmonary Surfactant Promotes Virulence Gene Expression and Biofilm Formation in Klebsiella Pneumoniae, Infect. Immun., 2018, 86(7), e00135-18 CrossRef.
- A. Humphries, S. DeRidder and A. J. Bäumler, Salmonella enterica Serotype Typhimurium Fimbrial Proteins Serve as Antigens during Infection of Mice, Infect. Immun., 2005, 73, 5329–5338 CrossRef CAS.
- C. Blumer, A. Kleefeld, D. Lehnen, M. Heintz, U. Dobrindt and G. Nagy, et al., Regulation of Type 1 Fimbriae Synthesis and Biofilm Formation by the Transcriptional Regulator LrhA of Escherichia coli, Microbiology, 2005, 151, 3287–3298 CrossRef CAS.
- K. Karjalaninen and O. Mäkelä, Concentrations of Three Hapten-Binding Immunoglobulins in Pooled Normal Human Serum, Eur. J. Immunol., 1976, 6, 88–93 CrossRef.
- A. F. Rullo, K. J. Fitzgerald, V. Muthusamy, M. Liu, C. Yuan and M. Huang, et al., Re-Engineering the Immune Response to Metastatic Cancer: Antibody-Recruiting Small Molecules Targeting the Urokinase Receptor, Angew. Chem., 2016, 128, 3706–3710 CrossRef.
- Y. Lu, F. You, I. Vlahov, E. Westrick, M. Fan and P. S. Low, et al., Folate-Targeted Dinitrophenyl Hapten Immunotherapy: Effect of Linker Chemistry on Antitumor Activity and Allergic Potential, Mol. Pharm., 2007, 4(5), 695–706 CrossRef CAS PubMed.
- T. Barrett, S. E. Wilhite, P. Ledoux, C. Evangelista, I. F. Kim and M. Tomashevsky, et al., NCBI GEO: Archive for Functional Genomics Data Sets—Update, Nucleic Acids Res., 2013, 41, D991–D995 CrossRef CAS PubMed.
- D. Marbach, J. C. Costello, R. Küffner, N. M. Vega, R. J. Prill and D. M. Camacho, et al., Wisdom of Crowds for Robust Gene Network Interference, Nat. Methods, 2012, 9(8), 796–804 CrossRef CAS.
- M. L. Arrieta-Ortiz, C. Hafemeister, A. R. Bate, T. Chu, A. Greenfield and B. Shuster, et al., An Experimentally Supported Model of the Bacillus subtilis Global Transcriptional Regulatory Network, Mol. Syst. Biol., 2015, 11, 839 CrossRef PubMed.
- C. E. Cowles, Y. Li, M. F. Semmelhack, I. M. Cristea and T. J. Silhavy, The Free and Bound Forms of Lpp Occupy Distinct Subcellular Locations in Escerichia coli, Mol. Microbiol., 2011, 79, 1168–1181 CrossRef CAS.
- M. M. Wilson and H. D. Bernstein, Surface-Exposed Lipoproteins: An Emerging Secretion Phenomenon in Gram-Negative Bacteria, Trends Microbiol., 2016, 24, 198–208 CrossRef CAS.
- J. C. Lee, E. J. Lee, J. H. Lee, S. H. Jun, C. W. Choi and S. I. Kim, et al., Klebsiella Pneumoniae Secretes Outer Membrane Vesicles that Induce the Innate Immune Response, FEBS Microbiol Lett, 2012, 331, 17–24 CAS.
- Y.-T. Huang, P.-F. Hsieh, J.-Y. Liu, Y.-J. Pan, M.-C. Wu and T.-L. Lin, et al., Klebsiella pneumoniae Peptidoglycan-Associated Lipoprotein and Murein Lipoprotein Contribute to Serum Resistance, Antiphagocytosis, and Proinflammatory Cytokine Stimulation, J. Infect. Dis., 2013, 208, 1580–1589 CrossRef PubMed.
- A. D. Ferguson, V. Braun, H.-P. Fiedler, J. W. Coulton, K. Diederichs and W. Welte, Crystal Structure of the Antibiotic Albomycin in Complex with the Outer Membrane Transporter FhuA, Protein Sci., 2000, 9, 956–963 CrossRef CAS PubMed.
- J. Thoma, P. Bosshart, M. Pfreundschuh and D. J. Müller, Out but Not In: The Large Transmembrane Beta-Barrel Protein FhuA Unfolds but Cannot Refold via Beta-Hairpins, Structure, 2012, 20, 2185–2190 CrossRef CAS PubMed.
- M.-J. Tsang, A. A. Yakhnina and T. G. Bernhardt, NlpD Links Cell Wall Remodeling and Outer Membrane Invagination during Cytokinesis in Escherichia coli, PLoS Genet., 2017, 13, e1006888 CrossRef PubMed.
- T. A. Van Laar, T. Chen, T. You and K. P. Leung, Sublethal Concentrations of Carbapenems Alter Cell Morphology and Genomic Expression of Klebsiella pneumoniae Biofilms, Antimicrob. Agents Chemother., 2015, 59, 1707–1717 CrossRef PubMed.
- S. S. Lee, J. Lim, J. Cha, S. Tan and J. R. Heath, Rapid Microwave-Assisted CNBr Cleavage of Bead-Bound Peptides, J. Comb. Chem., 2008, 10, 807–809 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04842a |
| This journal is © The Royal Society of Chemistry 2020 |