
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bright red aggregation-induced emission nanoparticles for multifunctional applications in cancer therapy†
Liping
Zhang
a,
Weilong
Che
a,
Zhiyu
Yang
b,
Xingman
Liu
a,
Shi
Liu
 b,
Zhigang
Xie
*b,
Dongxia
Zhu
*a,
Zhongmin
Su
a,
Ben Zhong
Tang
*c and
Martin R.
Bryce
*d
b,
Zhigang
Xie
*b,
Dongxia
Zhu
*a,
Zhongmin
Su
a,
Ben Zhong
Tang
*c and
Martin R.
Bryce
*d
aKey Laboratory of Nanobiosensing and Nanobioanalysis at Universities of Jilin Province, Department of Chemistry, Northeast Normal University, 5268 Renmin Street, Changchun, Jilin Province 130024, P. R. China. E-mail: zhudx047@nenu.edu.cn; zmsu@nenu.edu.cn
bState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry Chinese Academy of Sciences, Changchun 130022, P. R. China. E-mail: xiez@ciac.ac.cn
cState Key Laboratory of Molecular Neuroscience Institute for Advanced Study Institute of Molecular Functional Materials, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China. E-mail: tangbenz@ust.hk
dDepartment of Chemistry, Durham University, Durham DH1 3LE, UK. E-mail: m.r.bryce@durham.ac.uk
First published on 29th January 2020
Abstract
Developing multifunctional photosensitizers (PSs) is needed to effectively simplify cancer treatment, but it remains a big challenge. Here, two red-emitting AIE-active, donor–acceptor (D–A) PSs with small ΔEST and their AIE nanoparticles, are rationally designed and synthesized. The PS1 NPs exhibit bright red-emission with high quantum yield, appropriate 1O2 generation ability and good biocompatibility. More importantly, PS1 NPs can strongly light up the cytoplasm by gently shaking the cells for only 5 s at room temperature, indicating ultrafast staining and mild incubation conditions. In vitro and in vivo cell tracing demonstrate that PS1 NPs can track cells over 14 days, and effectively inhibit tumor growth upon irradiation. To the best of our knowledge, this work is the first example of a PS that integrates image-guided PDT, ultrafast staining and long-term tracing functions, demonstrating the “all-in-one” concept which offers great advantages for potential clinical applications.
Introduction
Image-guided photodynamic therapy (PDT) has recently gained increasing attention. This technique simultaneously achieves real-time molecular diagnosis and concurrent light-triggered therapy, and has surpassed traditional surgery, such as radiotherapy and chemotherapy.1–8 Noninvasive fluorescence imaging has emerged as a very powerful tool for visualizing clinical diagnostics and for biological research.9–14 Unfortunately, discontinuous and short-term cellular tracing is unable to provide continuous real-time dynamic information, thereby limiting the insights into a variety of complex biological processes.15–21 Long incubation times and harsh incubation conditions are critical barriers for cellular fluorescence imaging in clinical applications.22–25 Cancer treatments mainly include three essential steps: ultrafast staining, therapy, and long-term tracing. The former two play important roles for the discovery and excision of tumors, and the last is to further monitor the metastasis of residual tumor.15,21 The current scope of photosensitizers (PSs) is far from ideal, and until now, only a few PSs can achieve partial procedures, whereas there are no reports on simultaneous multiple applications, namely: (i) image-guided PDT, (ii) ultrafast staining and (iii) long-term tracking. Is it possible to design a single “all-in-one” PS that embraces all the above functions at the same time?PSs with efficient generation of singlet oxygen (1O2) and bright red-emission are crucial to realize imaging-guided PDT.26 An effective strategy to improve the efficiency of 1O2 generation is to accelerate the intersystem crossing (ISC) and to reduce ΔEST (the energy gap between S1 and T1 states).27–31 A small ΔEST is obtained by constructing a conjugated donor–acceptor (D–A) structure, which is beneficial for strong intramolecular charge transfer (ICT) with efficient separation of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), leading to red-shifted emission.32–35 Red emission is important because of its minimized autofluorescence interference, increased penetration depth, and less damage to tissue.36–38 However, traditional organic molecules tend to form aggregates in aqueous media, for example by π–π stacking, which directly leads to reduced 1O2 production, non-radiative pathways and quenched fluorescence.27,39–41
To overcome this challenge, the concept of aggregation-induced emission (AIE) has been exploited, notably by Tang's group.42–45 A series of AIEgen-based PSs have been reported to improve both the fluorescence intensity and the 1O2 production ability in the aggregated state, due to the restriction of intramolecular motions (RIMs) which lead to energy dissipation.46–54 Recently, our group obtained three red-emitting AIEgen nanoparticles (NPs) with higher brightness, enhanced 1O2 generation, and better biocompatibility compared to the pure iridium(III) complexes.55 These precedents make AIE NPs with purely organic D–A units particularly suitable as smart PSs to successfully obtain an “all-in-one” (image-guided PDT, ultrafast staining and long-term tracing) system.
In this contribution new PSs are designed and easily synthesized in high yield via a three-bladed propeller-like triphenylamine (TPA) as the strong donor and the dicyanovinyl (DC) groups as the acceptor, in which TPA also acts as a rotor to realize AIE.56 The two red-emitting AIE D–A and D–A–D molecules are PS1 and PS2 (Fig. 1A) and their corresponding polymer-encapsulated NPs are PS1 NPs and PS2 NPs. PS1 NPs possess bright red-emission, a large Stokes shift, appropriate 1O2 generation ability, good biocompatibility and excellent image-guided PDT activity. More importantly, PS1 NPs achieve ultrafast staining of cells in only 5 s at room temperature, with excellent long-term imaging of more than 14 days in vitro and in vivo.
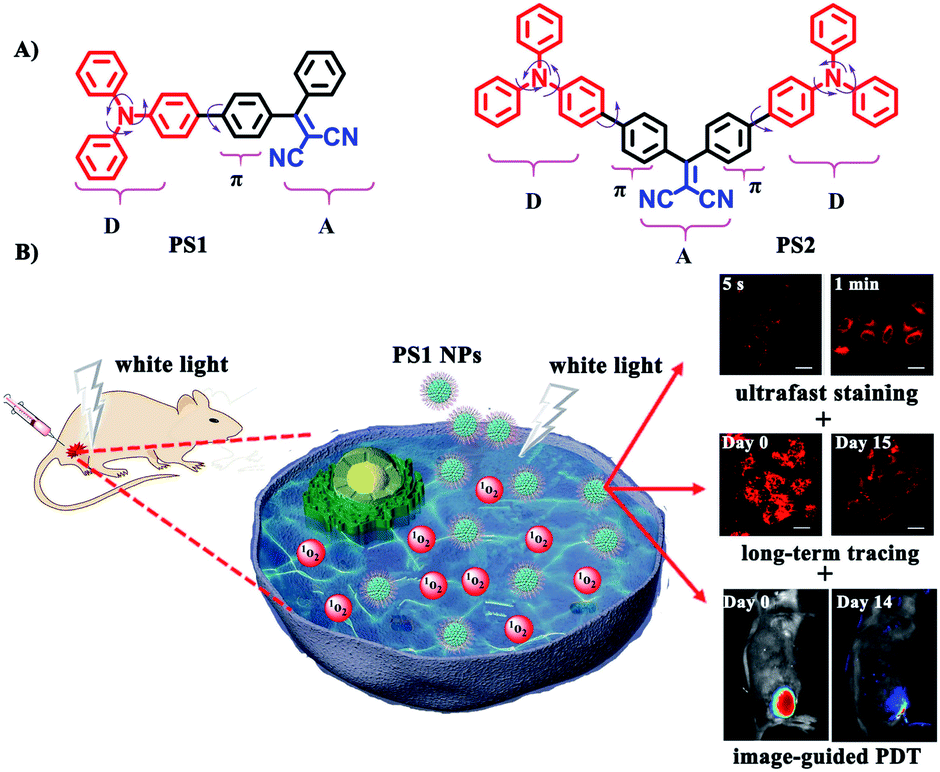 | ||
| Fig. 1 (A) Structural formulas of PS1 and PS2. (B) Schematic illustration of PS1 NPs as PSs for “all-in-one” PDT. | ||
Results and discussion
To gain an insight into the molecular properties of PS1 and PS2, time-dependent density theory (TD-DFT) calculations were performed (Fig. 2A). The HOMOs are mainly localized on TPA units, whereas the LUMOs are located on DC units, with significant HOMO–LUMO separation. The energy gap between the HOMO and LUMO of PS1 and PS2 is 2.449 and 2.425 eV, respectively. These relatively narrow band gaps induce the red-shifted absorption. Furthermore, the additional triphenylamine unit in PS2 results in a slight reduction of ΔEST (PS2 0.204 eV; PS1 0.229 eV), owing to a smaller spatial overlap of the Frontier orbitals. The small ΔEST values of PS1 and PS2 are beneficial for ISC, which will facilitate highly efficient 1O2 generation.29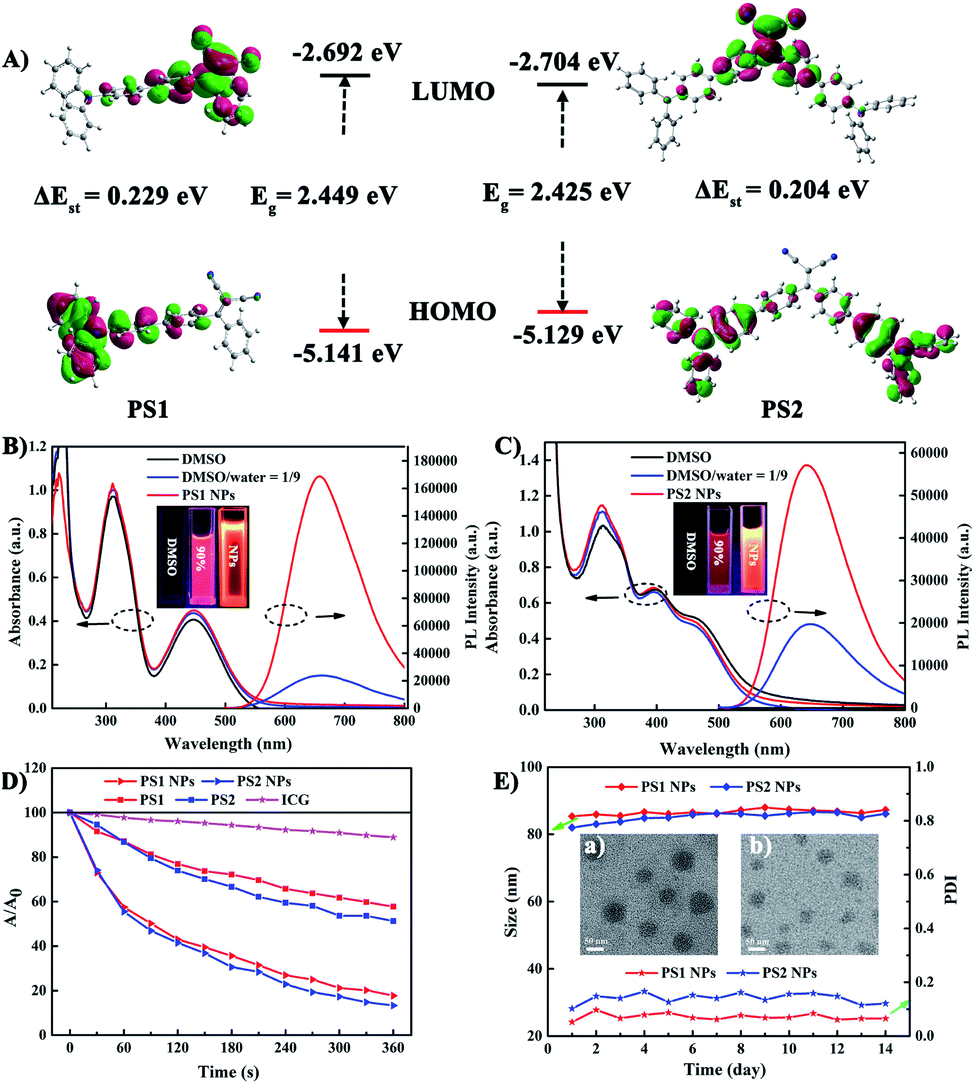 | ||
| Fig. 2 (A) HOMO and LUMO distributions of PS1 and PS2 based on TD-DFT. UV-vis and emission spectroscopies of (B) PS1 and (C) PS2 in DMSO, DMSO/water (v/v) = 1/9, and the corresponding PS NPs in water (λex = 488 nm), insert: the fluorescent image of PSs and their NPs under 365 nm UV light. (D) The decomposition rates of ICG with different PSs under white irradiation (20 mW cm−2). (E) Size measurement of different PSs in 14 days, insert: the TEM images of (a) PS1 NPs and (b) PS2 NPs. | ||
The photophysical properties of the PSs were investigated by absorption and photoluminescence (PL) spectroscopy. In Fig. 2B and C the absorption bands at about 210–315 nm are assigned to the π–π* transition of the conjugated backbone. The bands located in the 380–600 nm region belong to the intense ICT process between the D–A units. The absorption band covers most of the visible light range, which is beneficial for matching the white light spectrum.28 In Fig. S9 (ESI†), both PS1 and PS2 show negligible fluorescence in DMSO, because the rotational motions of the triphenylamine moieties increase the dissipation of energy. However, the PL intensities of PS1 and PS2 gradually increase upon raising the water content, when intramolecular rotation is restricted by the formation of aggregates, revealing a typical AIE effect. Compared with PS1 (λmax 660 nm) and PS2 (645 nm), their NPs show similar emission peaks but with significantly enhanced intensities. The PL intensities of PS1 NPs and PS2 NPs are 6.9 and 2.86 times higher than those of PS1 and PS2, respectively, with PS1 NPs 2.9 times higher than PS2 NPs in the same conditions. The fluorescence spectra of these AIE NPs extend into the near-infrared region, and they exhibit a large Stokes shift which can eliminate interference from the background. These factors are favorable for bioimaging. The fluorescence quantum yields (QYs) of PS1 NPs and PS2 NPs in water are 40% and 32%, respectively, which are much higher than PS1 and PS2 (16% and 13%, respectively) in DMSO-water mixtures. Compared with tetraphenylethylene (TPE),41 in the present work TPA with stronger electron donating ability promoted a red-shift in the absorption and emission of the PSs. This shift is beneficial for efficient biological applications, such as image-guided PDT, ultrafast staining and long-term tracking. These results confirm that the AIE NPs possess favourable photophysical properties.
Photosensitized 1O2 generation plays an important role in PDT.57 To evaluate the 1O2 generation ability of PS1, PS2 and their NPs, indocyanine green (ICG) was used as an indicator. No obvious changes of absorbance were observed in three control groups: (i) ICG + irradiation, (ii) PSs + irradiation, and (iii) ICG + PSs without irradiation (ESI, Fig. S14–S16†), verifying that all the PSs show good photostability.58 As expected, upon irradiation of ICG (5 μg mL−1) solutions in the presence of PS1, PS2 and their NPs (30 μg mL−1), the absorption peak of ICG at 790 nm gradually decreases in intensity (Fig. 2D; ESI, Fig. S17†), indicating that the PSs possess 1O2 generation ability. In Fig. S18 (ESI†) all the PSs follow a first-order 1O2 generation behavior. The slopes can be ordered as PS1 (1.45 × 10−3) < PS2 (1.88 × 10−3) < PS1 NPs (4.36 × 10−3) < PS2 NPs (5.32 × 10−3). The slopes of the kinetic decays of PS1 NPs and PS2 NPs are 3.01 and 2.83 times higher than for PS1 and PS2, respectively. A steeper slope represents a quicker decay rate of ICG and increased ability to generate 1O2. The 1O2 quantum yields of PS1, PS2, PS1 NPs and PS2 NPs are 38%, 43%, 67% and 76% with methylene blue (MB) as the reference (ΦΔ = 52% in MeCN). Overall, these NPs generate 1O2 very effectively and they possess great potential as PSs for PDT applications.
In addition, the morphology, size and stability of these NPs are important premises for biomedical applications. PS1 NPs and PS2 NPs are spherical and possess uniform sizes of 52 nm and 46 nm, respectively (Fig. 2E, insert). The corresponding hydrodynamic sizes are 82 nm and 79 nm, respectively (ESI, Fig. S19†). In addition, the diameters of PS1 NPs and PS2 NPs are stable within 14 days in water (Fig. 2E). Meanwhile, the fluorescence and absorbance intensity of PS1 NPs and PS2 NPs both retain more than 80% and 98%, respectively, of their initial values after 7 days (ESI, Fig. S11 and S12†), indicating that these NPs have excellent optical stability. The combination of spherical morphology, suitable size, superior physical stability and excellent photostability suggest that these NPs are suitable for the ensuing biological applications.
In vitro cytotoxicity of all the PSs toward HeLa cells was measured by MTT colorimetric assay.59 The cells retained >95% viability after incubation with PS1 NPs and PS2 NPs for 24 h (Fig. 3A and B) indicating good cytocompatibility. Under white-light irradiation PS1 NPs and PS2 NPs have dose-dependent cytotoxicity. The half-maximal inhibitory concentration (IC50) is in the sequence PS1 NPs (5.72 μg mL−1) > PS2 NPs (4.31 μg mL−1). In contrast, PS1 and PS2 exhibit some dark cytotoxicity and low phototoxicity (ESI, Fig. S20†). The same results were observed in A549 cells and MDA-MB-231 cells (ESI, Fig. S21†). Intracellular 1O2 generation of PS2 NPs was further investigated by using DCFH-DA as indicator. In Fig. S23 (ESI†) after irradiation the green emission from PS2 NPs is evidently brighter than that from PS2, indicating the enhanced 1O2 generation ability of the nanoparticle formulation. Moreover, the anticancer efficiencies of PS1, PS2 and their NPs were examined by live/dead staining techniques (ESI, Fig. S22†). Under light irradiation, negligible green fluorescence has been observed which indicates that the cancer cells are completely killed. The proportion of dead cells in the group of PS1 NPs and PS2 NPs under irradiation was much higher than with PS1 and PS2, which agrees with the above MTT results.
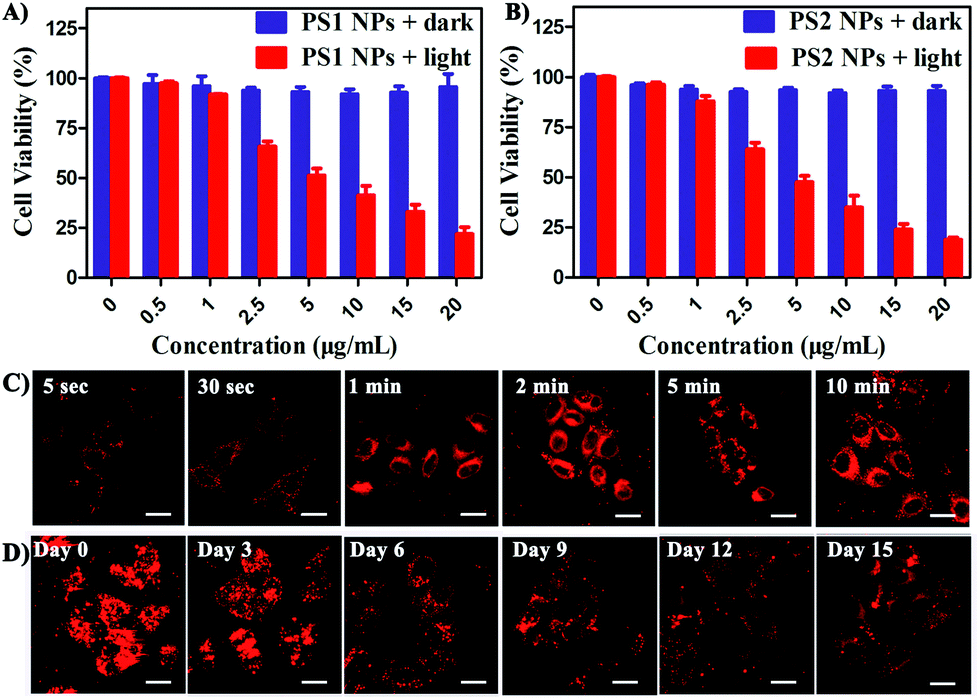 | ||
| Fig. 3 Viability of HeLa cells treated with (A) PS1 NPs and (B) PS2 NPs with or without white light (20 mW cm−2, 60 min). (C) CLSM images of HeLa cells after incubation with PS1 NPs (3 μg mL−1) for different times. (D) Long-term cell tracing images of the PS1 NPs (20 μg mL−1) at 37 °C for 6 h and then subcultured for a different number of days. Scale bar = 20 μm for all images. | ||
The bright fluorescence emission, large Stokes shift, favorable stability, and good cytocompatibility of PS1 NPs and PS2 NPs inspired us to conduct further investigation in their intracellular imaging behavior. HeLa cells were incubated with PS1 NPs and PS2 NPs (3 μg mL−1) for different times as depicted in Fig. 3C and S28 (ESI†). Bright red emission was observed from the cells treated with PS1 NPs, whereas negligible emission was observed with PS2 NPs. The PS1 NPs are more suitable for ultrafast staining, because as described above that PS1 NPs possess brighter red emission and higher fluorescence quantum yield (40%) than PS2 NPs. The cytoplasm was brightly emissive after simply shaking the cell incubate with PS1 NPs for 5 s at room temperature, demonstrating its ultrafast staining on the timescale of a few seconds. More strikingly, the cellular interior was lit up by incubation with 0.5 μg mL−1 of PS1 NPs (ESI, Fig. S24†). These results strongly validate that the PS1 NPs could be applied for ultrafast staining.
The applications of the PS1 NPs and PS2 NPs for in vitro long-term cellular tracing were investigated. As shown in Fig. 3D and S31 (ESI†), the internal cells were stained by PS1 NPs and PS2 NPs with bright red fluorescence in their initial stages which gradually decreased with time. Obviously, the cell tracking ability of PS1 NPs is better than that of PS2 NPs, which could be attributed to the brighter red emission of PS1 NPs than PS2 NPs. Negligible change in cell morphology was observed from day 1 to day 15 during long-term cell tracing. This indicates that there are no cytotoxicity effects for long-term cell tracing with PS1 and PS2 NPs. Under the same conditions, flow cytometry results also demonstrate that both PS1NPs and PS2 NPs have a tendency to accumulate in HeLa cells after 15 days incubation (ESI, Fig. S33†). Therefore, the PS1 NPs could perform as a promising long-term tracing probe.
Motivated by the excellent performance in vitro, PS1 NPs were further evaluated in vivo with U14 tumor-bearing mice. In Fig. 4A, obvious fluorescent signals were obtained from the site injected with PS1 NPs, indicating good in vivo imaging. With increasing time, the fluorescence intensity gradually decreased, but still retained 42% of the initial intensity after 14 days (Fig. 4B), indicating the excellent in vivo long-term tracing ability of PS1 NPs. After PDT treatment, faint fluorescence could be observed, suggesting high PDT efficacy of PS1 NPs. Simultaneously, in Fig. 4C, in the control groups, the relative tumor volumes increased by 10–13 times after 14 days, implying that treatment only by irradiation or by PS1 NPs, has negligible influence on tumor growth. As expected, tumor volume in the experimental group showed a significant reduction, confirming the good anticancer effect of PS1 NPs under white-light irradiation. Negligible changes of body weight were observed after various treatments (ESI, Fig. S34D†) indicating the low systemic toxicity of PS1 NPs. The biosafety of PS1 NPs was further evaluated by hematoxylin and eosin (H&E) staining of major organs. No pathological changes were found in the major organs of the four groups. These results reveal that PS1 NPs are excellent candidates for in vivo long-term tracing and image-guided PDT.
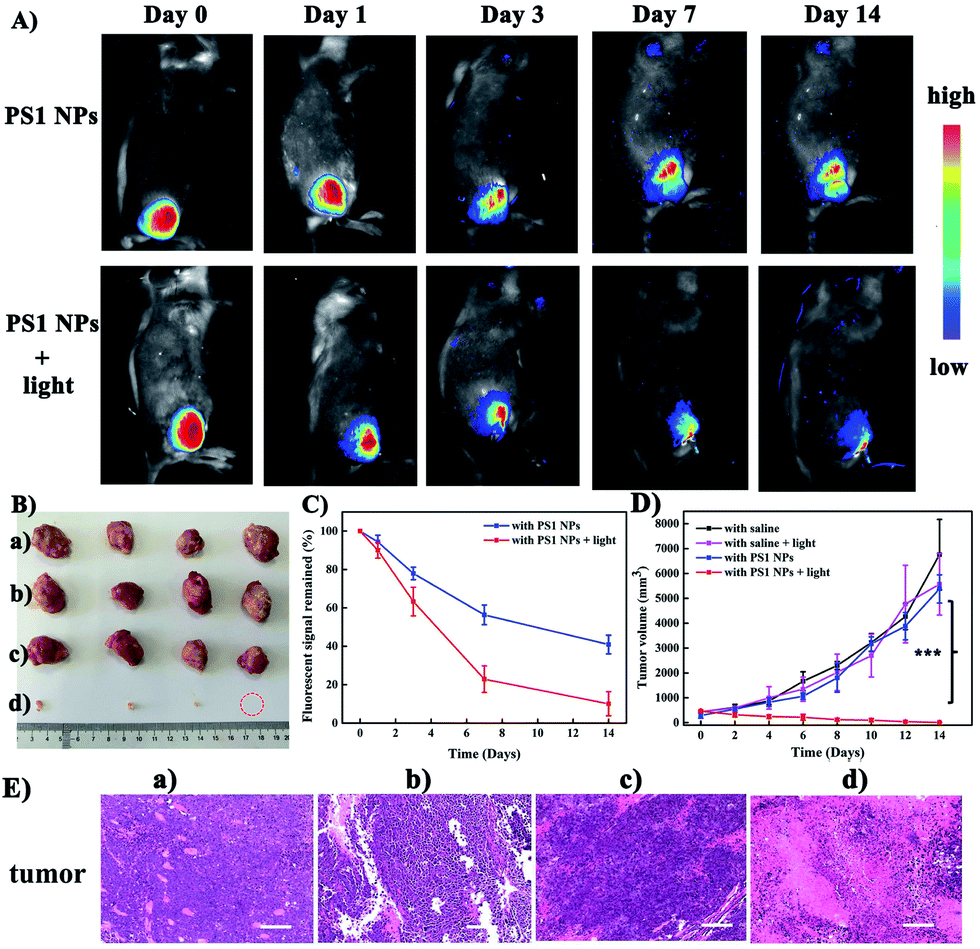 | ||
| Fig. 4 (A) Time-dependent in vivo fluorescence images of U14 tumor-bearing mice after intratumoral injection with PS1 NPs (100 μg mL−1, 100 μL) or with PS1 NPs + white light (200 mW cm−2, 20 min). (B) Harvested tumors from various groups treated. (C) Fluorescence intensity of U14 tumor-bearing mice in different mice groups. (D) Tumor volume measurement for different treatments of mice (***P < 0.001, n = 4 per group, PDT vs. other groups). (E) H&E staining images of tumor slices from each groups. Scale bars: 100 μm. (a) With saline, (b) with saline and light, (c) with PS1 NPs, (d) with PS1 NPs and light (100 μg mL−1, 100 μL), white light (200 mW cm−2, 20 min). | ||
Conclusions
In summary, we have successfully constructed for the first time a single PS to simultaneously achieve image-guided PDT, ultrafast staining and long-term tracing. Two red-emitting AIE-active D–A organic molecules and their NPs, were rationally designed and synthesized. The small ΔEST increases the intersystem crossing (ISC) process, leading to effective 1O2 generation for the two PSs. The construction of nanoparticles further improves the fluorescence intensity and the 1O2 production ability. Especially the PS1 NPs are ideal for biological applications owing to the following advantages: bright red emission, large Stokes shift, high fluorescence quantum yield (40%), appropriate 1O2 generation ability, good biocompatibility, and excellent image-guided PDT activity. More importantly, PS1 NPs can stain cells in only 5 s at room temperature, and they display excellent long-term imaging for more than 14 days in vitro and in vivo. This work successfully achieves a smart AIE PS with image-guided PDT, ultrafast staining and long-term tracing functions. This prototype example of “all-in-one” PS NPs should stimulate the design and development of new multifunctional PSs for potential clinical applications.Ethical statement
All animal studies were performed in strict accordance with the NIH guidelines for the care and use of laboratory animals (NIH Publication No. 85-23 Rev. 1985) and were approved by the guidelines of the Committee on Animal Use and Care of the Chinese Academy of Sciences.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was funded by NSFC (No. 51473028), the Key Scientific and Technological Project of Jilin Province (20160307016GX, 20190701010GH), the Development and Reform Commission of Jilin Province (20160058). The Project was supported by Open Research Fund of State Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences. M. R. B. thanks EPSRC grant EL/L02621X/1 for funding.Notes and references
- F. Hu, D. Mao, Kenry, X. L. Cai, W. B. Wu, D. L. Kong and B. Liu, Angew. Chem., Int. Ed., 2018, 57, 10182–10186 CrossRef CAS PubMed.
- Z. L. Dong, L. Z. Feng, Y. Hao, M. C. Chen, M. Gao, Y. Chao, H. Zhao, W. W. Zhu, J. J. Liu, C. Liang, Q. Zhang and Z. Liu, J. Am. Chem. Soc., 2018, 140, 2165–2178 CrossRef CAS PubMed.
- P. Huang, X. Qian, Y. Chen, L. Yu, H. Lin, L. Wang, Y. Zhu and J. Shi, J. Am. Chem. Soc., 2017, 139, 1275–1284 CrossRef CAS PubMed.
- B. Yu, H. Wei, Q. He, C. A. Ferreira, C. J. Kutyreff, D. Ni, Z. T. Rosenkrans, L. Cheng, F. Yu, J. W. Engle, X. Lan and W. Cai, Angew. Chem., Int. Ed., 2018, 57, 218–222 CrossRef CAS PubMed.
- X. Zhen, J. Zhang, J. Huang, C. Xie, Q. Miao and K. Pu, Angew. Chem., Int. Ed., 2018, 57, 7804–7808 CrossRef CAS PubMed.
- X. Zheng, L. Wang, M. Liu, P. Lei, F. Liu and Z. Xie, Chem. Mater., 2018, 30, 6867–6876 CrossRef CAS.
- X. Zheng, L. Wang, S. Liu, W. Zhang, F. Liu and Z. Xie, Adv. Funct. Mater., 2018, 28, 1706507 CrossRef.
- T. Zhang, Y. Li, Z. Zheng, R. Ye, Y. Zhang, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2019, 141, 5612–5616 CrossRef CAS PubMed.
- A. Goujon, A. Colom, K. Straková, V. Mercier, D. Mahecic, S. Manley, N. Sakai, A. Roux and S. Matile, J. Am. Chem. Soc., 2019, 141, 3380–3384 CrossRef CAS PubMed.
- D. Ni, E. B. Ehlerding and W. Cai, Angew. Chem., Int. Ed., 2019, 58, 2570–2579 CrossRef CAS PubMed.
- Z. Gao, A. J. Thompson, J. C. Paulson and S. G. Withers, Angew. Chem., 2018, 130, 13726–13729 CrossRef.
- J. Zhang, B. Xu, W. Tian and Z. Xie, Chem. Sci., 2018, 9, 2620–2627 RSC.
- G. K. Park, J. H. Lee, A. Levitz, G. El Fakhri, N. S. Hwang, M. Henary and H. S. Choi, Adv. Mater., 2019, 31, 1806216 CrossRef PubMed.
- X. Luo, B. Xue, G. Feng, J. Zhang, B. Lin, P. Zeng, H. Li, H. Yi, X. L. Zhang, H. Zhu and Z. Nie, J. Am. Chem. Soc., 2019, 141, 5182–5191 CrossRef CAS PubMed.
- N. Alifu, A. Zebibula, J. Qi, H. Zhang, C. Sun, X. Yu, D. Xue, J. W. Y. Lam, G. Li, J. Qian and B. Z. Tang, ACS Nano, 2018, 12, 11282–11293 CrossRef CAS PubMed.
- X. Ji, F. Peng, Y. Zhong, Y. Su, X. Jiang, C. Song, L. Yang, B. Chu, S. T. Lee and Y. He, Adv. Mater., 2015, 27, 1029–1034 CrossRef CAS PubMed.
- J. Zhang, M. Zheng, F. Zhang, B. Xu, W. Tian and Z. Xie, Chem. Mater., 2016, 28, 8825–8833 CrossRef CAS.
- L. Shi, X. Gao, W. Yuan, L. Xu, H. Deng, C. Wu, J. Yang, X. Jin, C. Zhang and X. Zhu, Small, 2018, 14, 1800223 CrossRef PubMed.
- L. Wyld, R. A. Audisio and G. J. Poston, Nat. Rev. Clin. Oncol., 2014, 12, 115 CrossRef PubMed.
- C. S. Ke, C. C. Fang, J. Y. Yan, P. J. Tseng, J. R. Pyle, C. P. Chen, S. Y. Lin, J. Chen, X. Zhang and Y. H. Chan, ACS Nano, 2017, 11, 3166–3177 CrossRef CAS PubMed.
- G. Driessens, B. Beck, A. Caauwe, B. D. Simons and C. Blanpain, Nature, 2012, 488, 527–530 CrossRef CAS PubMed.
- A. Nicol, R. T. K. Kwok, C. Chen, W. Zhao, M. Chen, J. Qu and B. Z. Tang, J. Am. Chem. Soc., 2017, 139, 14792–14799 CrossRef CAS PubMed.
- W. Che, L. Zhang, Y. Li, D. Zhu, Z. Xie, G. Li, P. Zhang, Z. Su, C. Dou and B. Z. Tang, Anal. Chem., 2019, 91, 3467–3474 CrossRef CAS PubMed.
- D. Wang, H. Su, R. T. K. Kwok, X. Hu, H. Zou, Q. Luo, M. M. S. Lee, W. Xu, J. W. Y. Lam and B. Z. Tang, Chem. Sci., 2018, 9, 3685–3693 RSC.
- H. Shi, J. Liu, J. Geng, B. Z. Tang and B. Liu, J. Am. Chem. Soc., 2012, 134, 9569–9572 CrossRef CAS PubMed.
- F. Hu, S. Xu and B. Liu, Adv. Mater., 2018, 30, 1801350 CrossRef PubMed.
- S. Liu, H. Zhang, Y. Li, J. Liu, L. Du, M. Chen, R. T. K. Kwok, J. W. Y. Lam, D. L. Phillips and B. Z. Tang, Angew. Chem., 2018, 130, 15409–15413 CrossRef.
- X. Gu, X. Zhang, H. Ma, S. Jia, P. Zhang, Y. Zhao, Q. Liu, J. Wang, X. Zheng, J. W. Y. Lam, D. Ding and B. Z. Tang, Adv. Mater., 2018, 30, 1801065 CrossRef PubMed.
- X. Miao, W. Hu, T. He, H. Tao, Q. Wang, R. Chen, L. Jin, H. Zhao, X. Lu, Q. Fan and W. Huang, Chem. Sci., 2019, 10, 3096–3102 RSC.
- B. M. Luby, C. D. Walsh and G. Zheng, Angew. Chem., Int. Ed., 2019, 58, 2558–2569 CrossRef CAS PubMed.
- C. Ji, Q. Gao, X. Dong, W. Yin, Z. Gu, Z. Gan, Y. Zhao and M. Yin, Angew. Chem., Int. Ed., 2018, 57, 11384–11388 CrossRef CAS PubMed.
- W. Wu, D. Mao, F. Hu, S. Xu, C. Chen, C. J. Zhang, X. Cheng, Y. Yuan, D. Ding, D. Kong and B. Liu, Adv. Mater., 2017, 29, 1700548 CrossRef PubMed.
- W. Wu, S. Xu, G. Qi, H. Zhu, F. Hu, Z. Liu, D. Zhang and B. Liu, Angew. Chem., 2019, 131, 3094–3098 CrossRef.
- C. Li, R. Duan, B. Liang, G. Han, S. Wang, K. Ye, Y. Liu, Y. Yi and Y. Wang, Angew. Chem., Int. Ed., 2017, 56, 11525–11529 CrossRef CAS PubMed.
- W. Zeng, H. Y. Lai, W. K. Lee, M. Jiao, Y. J. Shiu, C. Zhong, S. Gong, T. Zhou, G. Xie, M. Sarma, K. T. Wong, C. C. Wu and C. L. Yang, Adv. Mater., 2018, 30, 1704961 CrossRef PubMed.
- Z. Zheng, T. Zhang, H. Liu, Y. Chen, R. T. K. Kwok, C. Ma, P. Zhang, H. H. Y. Sung, I. D. Williams, J. W. Y. Lam, K. S. Wong and B. Z. Tang, ACS Nano, 2018, 12, 8145–8159 CrossRef CAS PubMed.
- D. Wang, M. M. S. Lee, G. Shan, R. T. K. Kwok, J. W. Y. Lam, H. Su, Y. Cai and B. Z. Tang, Adv. Mater., 2018, 30, 1802105 CrossRef PubMed.
- D. Cheng, J. Peng, Y. Lv, D. Su, D. Liu, M. Chen, L. Yuan and X. Zhang, J. Am. Chem. Soc., 2019, 141, 6352–6361 CrossRef CAS PubMed.
- H. Q. Peng, B. Liu, P. Wei, P. Zhang, H. Zhang, J. Zhang, K. Li, Y. Li, Y. Cheng, J. W. Y. Lam, W. Zhang, C. S. Lee and B. Z. Tang, ACS Nano, 2019, 13, 839–846 CrossRef CAS PubMed.
- M. Li, Y. Gao, Y. Yuan, Y. Wu, Z. Song, B. Z. Tang, B. Liu and Q. C. Zheng, ACS Nano, 2017, 11, 3922–3932 CrossRef CAS PubMed.
- B. Gu, W. Wu, G. Xu, G. Feng, F. Yin, P. H. J. Chong, J. Qu, K. T. Yong and B. Liu, Adv. Mater., 2017, 29, 1701076 CrossRef PubMed.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 18, 1740–1741 RSC.
- Y. Chen, W. Zhang, Z. Zhao, Y. Cai, J. Gong, R. T. K. Kwok, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams and B. Z. Tang, Angew. Chem., Int. Ed., 2018, 57, 5011–5015 CrossRef CAS PubMed.
- C. Zhou, W. Xu, P. Zhang, M. Jiang, Y. Chen, R. T. K. Kwok, M. M. S. Lee, G. Shan, R. Qi, X. Zhou, J. W. Y. Lam, S. Wang and B. Z. Tang, Adv. Funct. Mater., 2019, 29, 1805986 CrossRef.
- S. Liu, X. Zhou, H. Zhang, H. Ou, J. W. Y. Lam, Y. Liu, L. Shi, D. Ding and B. Z. Tang, J. Am. Chem. Soc., 2019, 141, 5359–5368 CrossRef CAS PubMed.
- C. Teh, P. N. Manghnani, G. N. H. Boon, T. Y. De Cheng, W. T. Lim, E. H. Lim, B. T. Chua and B. Liu, Adv. Funct. Mater., 2019, 29, 1901226 CrossRef.
- J. S. Ni, P. Zhang, T. Jiang, Y. Chen, H. Su, D. Wang, Z. Q. Yu, R. T. K. Kwok, Z. Zhao, J. W. Y. Lam and B. Z. Tang, Adv. Mater., 2018, 30, 1805220 CrossRef PubMed.
- X. Cai, D. Mao, C. Wang, D. Kong, X. Cheng and B. Liu, Angew. Chem., Int. Ed., 2018, 57, 16396–16400 CrossRef CAS PubMed.
- D. Wang, H. Su, R. T. K. Kwok, G. Shan, A. C. S. Leung, M. M. S. Lee, H. H. Y. Sung, I. D. Williams, J. W. Y. Lam and B. Z. Tang, Adv. Funct. Mater., 2017, 27, 1704039 CrossRef.
- R. Y. Zhang, Y. K. Duan and B. Liu, Nanoscale, 2019, 11, 19241–19250 RSC.
- M. M. S. Lee, L. Zheng, B. R. Yu, W. H. Xu, R. T. K. Kwok, J. W. Y. Lam, F. J. Xu, D. Wang and B. Z. Tang, Mater. Chem. Front., 2019, 3, 1454–1461 RSC.
- M. M. Kang, C. C. Zhou, S. M. Wu, B. R. Yu, Z. J. Zhang, N. Song, M. M. S. Lee, W. H. Xu, F. J. Xu, D. Wang, L. Wang and B. Z. Tang, J. Am. Chem. Soc., 2019, 141, 16781–16789 CrossRef CAS PubMed.
- D. Wang, M. M. S. Lee, W. H. Xu, G. G. Shan, X. Y. Zheng, R. T. K. Kwok, J. W. Y. Lam, X. L. Hu and B. Z. Tang, Angew. Chem., Int. Ed., 2019, 58, 5628–5632 CrossRef CAS PubMed.
- D. Wang and B. Z. Tang, Acc. Chem. Res., 2019, 52, 2559–2570 CrossRef CAS PubMed.
- L. Zhang, Y. Li, W. Che, D. Zhu, G. Li, Z. Xie, N. Song, S. Liu, B. Z. Tang, X. Liu, Z. Su and M. R. Bryce, Adv. Sci., 2019, 6, 1802050 CrossRef PubMed.
- S. Wang, X. Yan, Z. Cheng, H. Zhang, Y. Liu and Y. Wang, Angew. Chem., Int. Ed., 2015, 54, 13068–13072 CrossRef CAS PubMed.
- Q. Pei, X. Hu, X. Zheng, S. Liu, Y. Li, X. Jing and Z. Xie, ACS Nano, 2018, 12, 1630–1641 CrossRef CAS PubMed.
- X. Zheng, L. Wang, Q. Pei, S. He, S. Liu and Z. Xie, Chem. Mater., 2017, 29, 2374–2381 CrossRef CAS.
- D. Gerlier and N. Thomasset, J. Immunol. Methods, 1986, 94, 57–63 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental details and methods, supplementary figures and tables of data. See DOI: 10.1039/c9sc06310b |
| This journal is © The Royal Society of Chemistry 2020 |