
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sulfatase-cleavable linkers for antibody-drug conjugates†
Jonathan D.
Bargh
 a,
Stephen J.
Walsh
ab,
Albert
Isidro-Llobet
c,
Soleilmane
Omarjee
b,
Jason S.
Carroll
b and
David R.
Spring
*a
a,
Stephen J.
Walsh
ab,
Albert
Isidro-Llobet
c,
Soleilmane
Omarjee
b,
Jason S.
Carroll
b and
David R.
Spring
*a
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: spring@ch.cam.ac.uk
bCancer Research UK Cambridge Institute, University of Cambridge, Robinson Way, Cambridge, CB2 0RE, UK
cGSK, Gunnels Wood Road, Stevenage, SG1 2NY, UK
First published on 27th January 2020
Abstract
Antibody-drug conjugates (ADCs) are a class of targeted drug delivery agents combining the cell-selectivity of monoclonal antibodies (mAbs) and the cytotoxicity of small molecules. These two components are joined by a covalent linker, whose nature is critical to the efficacy and safety of the ADC. Enzyme-cleavable dipeptidic linkers have emerged as a particularly effective ADC linker type due to their ability to selectively release the payload in the lysosomes of target cells. However, these linkers have a number of drawbacks, including instability in rodent plasma and their inherently high hydrophobicity. Here we show that arylsulfate-containing ADC linkers are cleaved by lysosomal sulfatase enzymes to tracelessly release their payload, while circumventing the instability problems associated with dipeptide-linkers. When incorporated with trastuzumab and the highly potent monomethyl auristatin E (MMAE) payload, the arylsulfate-containing ADC 2 and ADC 3 were more cytotoxic than the non-cleavable ADC 4 against HER2-positive cells, while maintaining selectivity over HER2-negative cells. We propose that the stability, solubility and synthetic tractability of our arylsulfate linkers make them an attractive new motif for cleavable ADC linkers, with clear benefits over the widely used dipeptidic linkers.
Introduction
Antibody-drug conjugates (ADCs) are now established as an important class of therapeutics for the treatment of cancer. There are currently seven FDA-approved ADCs, with at least 60 more in clinical development.1 The success of ADCs arises from the combination of the exquisite cell-selectivity of monoclonal antibodies and the cytotoxicity of small molecule chemotherapies. However, for the mAb and drug to exert their maximum therapeutic potential, the covalent linker between the two groups must exhibit the following properties: (1) high plasma stability; the long circulatory lifetimes (t1/2 > 1 week) of ADCs places stringent stability requirements on the linker to avoid off-target payload release. (2) High aqueous solubility to aid bioconjugation of lipophilic payloads and avoid antibody aggregation. (3) Release of a potent cytotoxin from the antibody in the target cell.2The majority of ADCs employ antibodies that target internalising antigens, overexpressed on the surface of certain cancer cells. Upon antibody-antigen binding at the target cell, the ADC is endocytosed and trafficked to the highly hydrolytic lysosomal compartments, where the antibody is broken down into its constituent amino acids.3 In the case of a non-cleavable linker, the active intracellular cytotoxin is released as the linker-payload with the terminal amino acid from the mAb still attached.4 Conversely, a cleavable linker is designed to release the unmodified drug at this point, or upon subsequent entry to the cytosol.
Cleavable linkers are generally preferred to non-cleavable linkers in ADC research for a number of reasons.5 First, traceless drug release allows the unmodified payload to perform its intracellular function without an unwanted linker appendage, thereby maximising cytotoxicity. Second, the unmodified payload can be uncharged, allowing it to diffuse into ‘bystander’ tumour cells that may not be expressing the target antigen. This ‘bystander effect’ provides an important mechanism for eradicating tumours with heterogeneous antigen expression.6
Enzyme-cleavable linkers are the most widely used group of ADC linkers. Other cleavable linkers, such as reducible disulfides or acid-sensitive motifs have been developed but their stability in human plasma is generally inferior.7,8 The benefits of enzyme-cleavable linkers include their ability to selectively induce drug release at target cells rather than in circulation. Thus far, the only clinically explored enzyme-cleavable linkers are peptides, sensitive to cleavage by lysosomal cathepsin proteases.9–13 Linkers containing Val–Cit or Val–Ala sequences are most widely employed, due to their high stability in human plasma and efficient drug release within the lysosomes of target cells (Fig. 1a). A self-immolative para-aminobenzoyl carbamate (PABC) spacer is also required, to ensure the cathepsin-mediated cleavage is unimpeded by the payload.
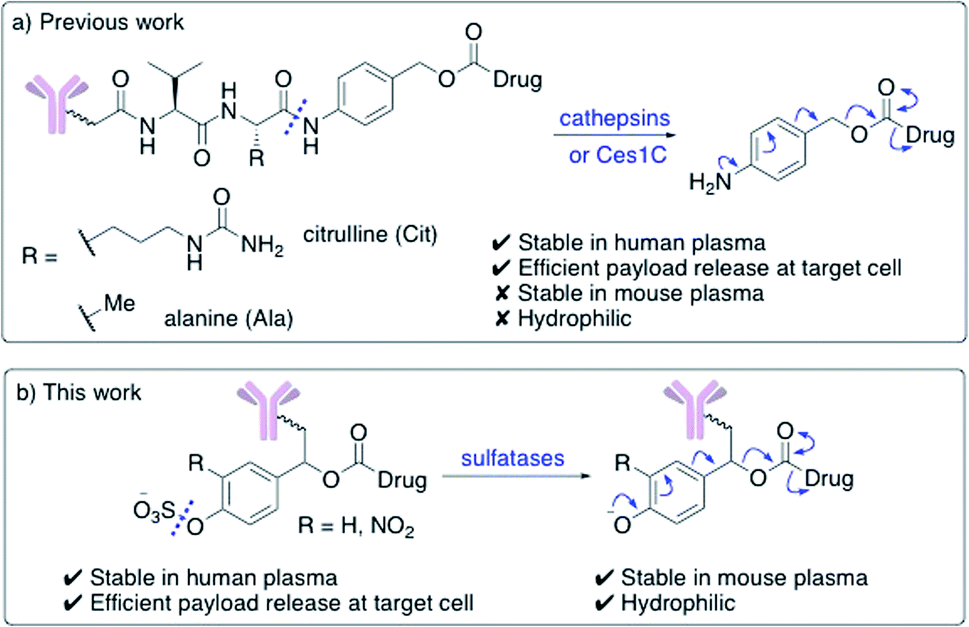 | ||
| Fig. 1 (a) Previously developed dipeptide linkers, cleaved by lysosomal cathepsins and mouse plasma enzyme Ces1C; (b) arylsulfate linkers for use in ADCs are cleaved by lysosomal sulfatases. | ||
Unfortunately, dipeptidic linkers feature a number of major drawbacks. First, they are unstable in rodent blood, due to their susceptibility to the Ces1C hydrolase enzyme in rodent plasma.7,14,15 This hydrolysis causes premature payload release in vivo and limits the accuracy of preclinical efficacy and safety studies. Second, these linkers are hydrophobic, thereby hindering bioconjugation of lipophilic payloads and causing high levels of antibody aggregation upon conjugation.16,17 Despite these shortcomings, Val–Cit and Val–Ala dipeptides continue to be widely used in ADC research, routinely alongside monomethyl auristatin E (MMAE) or pyrrolobenzodiazepine- (PBD) dimer payloads respectively.
Other non-peptidic linkers have also been developed for ADCs. Linkers sensitive to the lysosomal β-glucuronidase and β-galactosidase enzymes have been described, which appear to address the problems associated with linker-payload hydrophobicity and rodent-plasma instability.11,18 However, their stereochemical complexity and possible overreliance on one specific lysosomal enzyme may explain their lack of development in clinically tested ADCs. Additionally, pyrophosphatase-cleavable motifs have also been described, for use with alcohol-linked glucocorticoid payloads.19,20
Novel linkers employing different modes of action are required to expand the ADC toolbox, given the therapeutic importance of the linker and the shortcomings of current cleavable linker technology.5 Herein, we describe the development of novel sulfatase-cleavable linkers for ADCs (Fig. 1b). Initial linker studies, facilitated by their simple synthesis, reveal the highly soluble arylsulfate linkers are stable in both human and mouse plasma. In addition, a series of arylsulfate-ADCs employing a trastuzumab antibody and MMAE as a payload were prepared and their cytotoxicity assessed against HER2-positive and HER2-negative cells.
Results and discussion
Design of linkers
Effective enzyme-cleavable ADC linkers must be highly stable in circulatory conditions but labile upon entry to the lysosomes of target cells, efficiently releasing their payloads. Sulfatases therefore offer an opportunity for selective payload release due to their high activity within the lysosomes and low activity in human and rodent plasma.21 A number of different sulfatases reside in the lysosome, catalysing the hydrolysis of alkylsulfate esters to alcohols.22 Although selective towards their natural substrates, they also each display arylsulfatase activity. Furthermore, sulfatases are overexpressed in a number of different cancer types, thereby offering the possibility of additional selectivity for arylsulfate-containing ADCs towards tumours.23 It was anticipated that the charged sulfates would be amenable to bioconjugation in aqueous media and their synthesis simplified by previously reported protecting group strategies.24,25 Accordingly, arylsulfate ADC linkers would potentially display significant advantages over dipeptide-based linkers.Arylsulfate linker motifs were designed such that, upon hydrolysis, a 4-alkoxybenzyl carbamate would be revealed, primed for spontaneous 1,6-elimination of an amine-linked payload (Fig. 1b). To link to the antibody, we were first inspired by β-glucuronidase-cleavable linkers, which are linked to the mAb from an amide ortho- to the enzyme cleavable group (Fig. 2, blue route).18 It was also postulated that arylsulfates linked to the antibody by branching from the benzyl position would be less hindered at the cleavable sulfate, potentially improving sensitivity towards sulfatases (Fig. 2, red route). For our preliminary stability and release studies, we employed the fluorescent 7-amino-4-methyl coumarin (AMC) group as a model payload. AMC is widely used within enzymatic probes, as fluorescence is only observed upon release of the amino-coumarin from an amide or carbamate precursor.26 Therefore, through the use of fluorimetry, payload release at physiologically relevant concentrations could be measured in enzyme solutions and plasma. Model linkers 7 and 12 incorporate the two antibody-attachment designs, with AMC as their payload (Fig. 3).
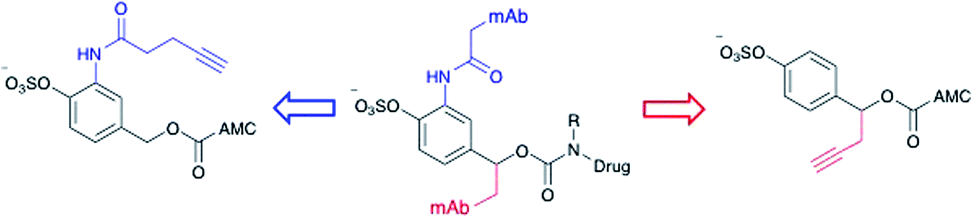 | ||
| Fig. 2 Design of linker-AMC model compounds. It was envisaged that the linker-payloads could be joined to the antibody through an ortho-amide (blue route) or a benzyl-alkyl (red route). | ||
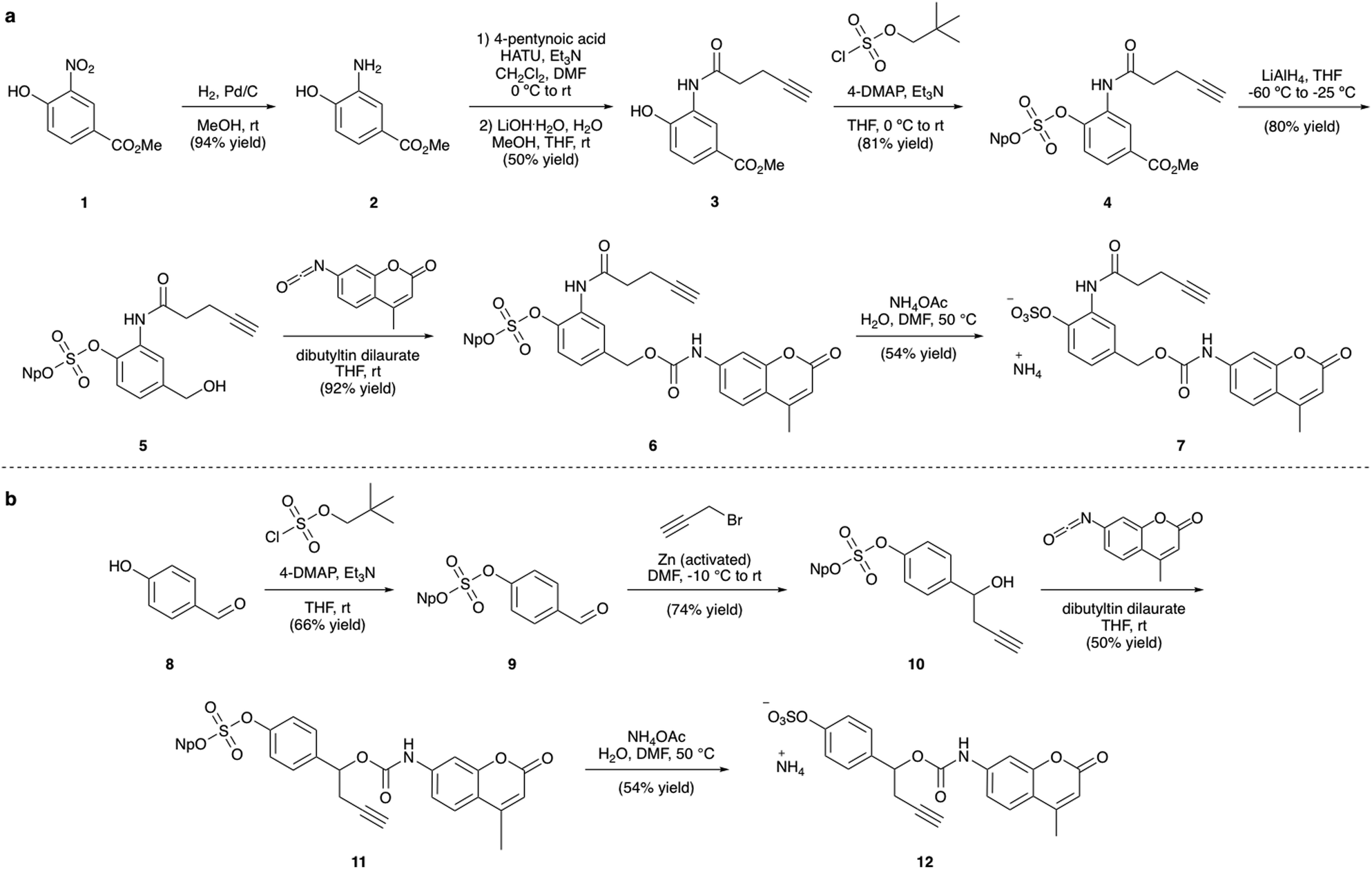 | ||
| Fig. 3 (a) Synthesis of linker-AMC 7 and (b) synthesis of linker-AMC 12. Np = neopentyl. | ||
Synthesis of linker-coumarins
The synthesis of the arylsulfate linkers required a sulfate protecting group strategy to limit the number of steps requiring purification of charged molecules. The neopentyl moiety was chosen as an appropriate protecting group due to its stability, facile installation and functional group compatibility.25 Although harsh deprotection conditions are typically required, a procedure described by Simpson et al. employing aqueous NH4OAc appeared to offer a mild alternative.27 Synthesis of linker-AMC 7 was achieved in six steps from nitroarene 1 (Fig. 3a). First, 1 was hydrogenated to yield aniline 2, which was then modified via HATU-mediated amide coupling to yield alkyne 3. Next, installation of the neopentyl-protected sulfate was achieved by reaction with neopentyl sulfurochloridate. The resulting methyl ester 4 was then selectively reduced with LiAlH4 before reaction with AMC-isocyanate in the presence of a dibutyltin dilaurate catalyst afforded carbamate 6. Gratifyingly, deprotection of the neopentyl sulfate with aqueous NH4OAc occurred smoothly to afford linker-AMC 7 in good overall yield. Linker-AMC 12 was synthesised via an analogous synthetic route (Fig. 3b). First, the neopentyl sulfate was introduced as before, with subsequent Barbier reaction with propargyl bromide and zinc powder affording alcohol 10. Finally, reaction with AMC-isocyanate was followed by neopentyl deprotection to generate linker-AMC 12 with moderate to good yields throughout.Stability and release studies
With constructs 7 and 12 in hand, the lability of the linkers under conditions representative of the lysosomal compartments and blood plasma was evaluated (Fig. 4a). First, to approximate the lysosomal environment, the susceptibility of the arylsulfate linkers 7 and 12 to sulfatase cleavage was determined using sulfatase from Helix pomatia (EC 3.1.6.1). This enzyme has been used previously to approximate sulfate hydrolysis, given its general sulfatase activity.28 The arylsulfates were incubated with the sulfatase and the fluorescence of the released AMC payload was measured over 8 h (Fig. 4b). The resulting increase in fluorescence confirmed that both linkers are substrates for sulfatases and the subsequent 1,6-elimination occurs as expected. Although both linkers were hydrolysed by the sulfatase, the enzymolysis rate of benzyl-linked 12 (t1/2 = 24 min) was dramatically superior to that of ortho-amide linked 7 (t1/2 > 12 h) under the same conditions. These results suggest an ADC linker based on 12 would release its payload much more efficiently in the lysosome than 7, increasing the cytotoxicity of the ADC.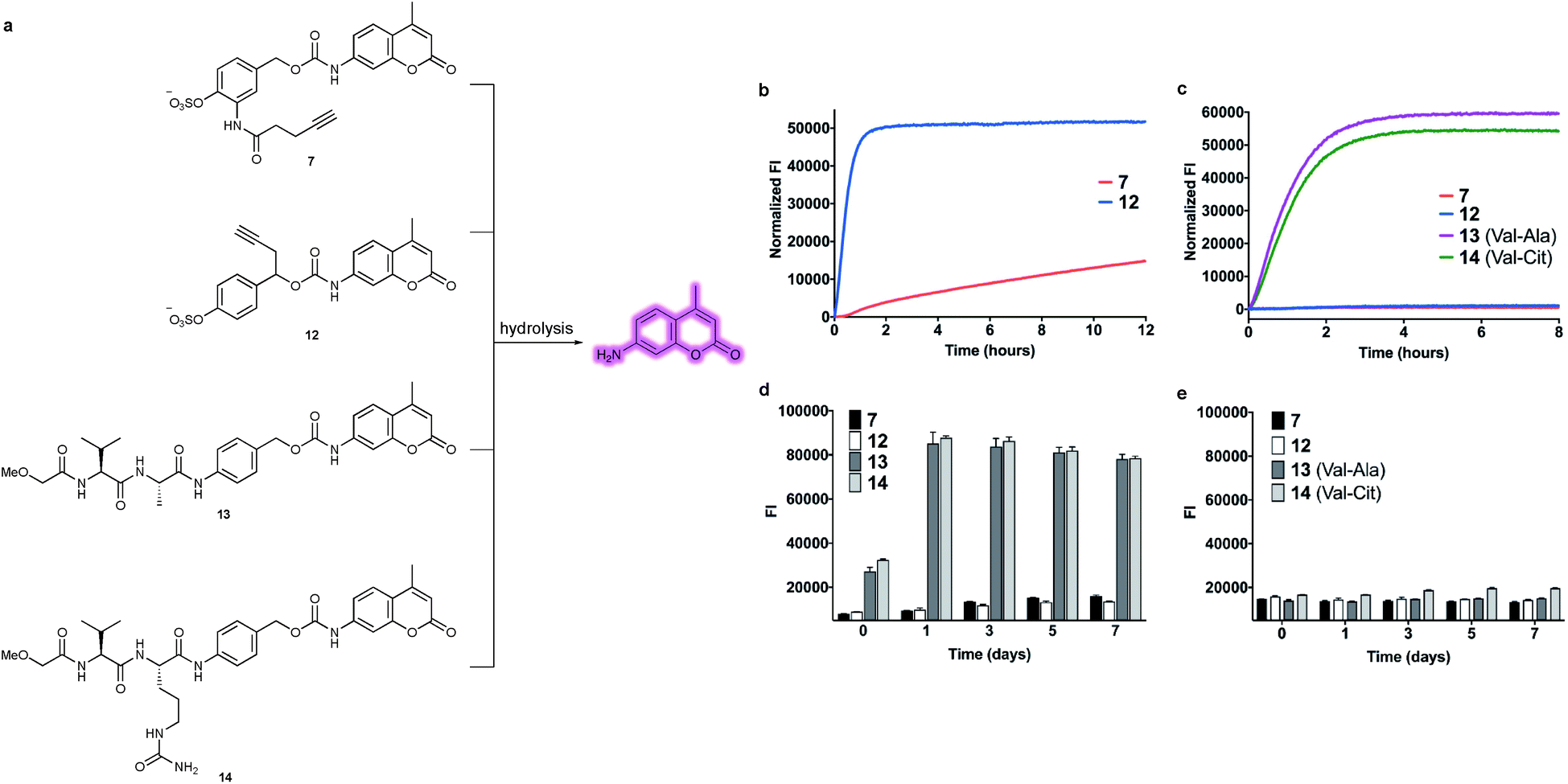 | ||
| Fig. 4 (a) Structures of model linker-AMC constructs including arylsulfates 7 and 12 and Val–Ala 13 and Val–Cit 14. Upon hydrolysis and subsequent 1,6-elimination, the AMC payload is released and fluoresces. (b) Comparison of enzymolysis rates of 7versus12 when incubated with sulfatase from Helix pomatia. (c) Stability comparison of arylsulfates 7 and 12versus dipeptides 13 and 14 in mouse plasma over 8 hours. (d) Stability comparison in mouse plasma over 7 days. The t = 0 reading was taken after adding plasma to all the samples, by which time 13 and 14 were already partially hydrolysed. (e) Stability comparison in human plasma over 7 days. | ||
To investigate the enzymatic nature of the hydrolysis, sulfates 7 and 12 were incubated with the sulfatase in the presence of phenyl sulfamate, a known sulfatase inhibitor (Fig. S3†).29 Pleasingly, no fluorescence was observed under these conditions, confirming that enzymolysis is required for release. Furthermore, incubation of 12 at pH 7.4 and pH 9 demonstrated the acidic optimum pH of the sulfatase hydrolysis, with the cleavage rate decreasing with increasing pH (Fig. S4†). Finally, to affirm its susceptibility towards human lysosomal sulfatases, 12 was incubated with recombinant human lysosomal arylsulfatase A (ARSA) and arylsulfatase B (ARSB), upon which, fluorophore release was observed in both cases (Fig. S5†).
The plasma stability of arylsulfate-AMC constructs 7 and 12 was then investigated, first by incubation in mouse plasma (Fig. 4c). Under these conditions, the sulfate linkers 7 and 12 were highly stable, with no significant increase in fluorescence. Contrastingly, dipeptides 13 and 14 were rapidly hydrolysed in mouse plasma, with half-lives of less than one hour. The exceptional stability of 7 and 12 was further confirmed by incubation in both human and mouse plasma over seven days, with minimal hydrolysis observed (Fig. 4d and e). Moreover, sulfates 7 and 12 were shown to be stable in the presence of an intracellular nucleophile (glutathione) (Fig. S6†).
Design of linker-MMAE constructs
Having confirmed the favourable properties of arylsulfates 7 and 12, the linkers were elaborated to include an antibody attachment group and a cytotoxic payload, for biological evaluation of the proceeding ADCs. The divinylpyrimidine (DVP) group has been reported by our group as an effective method for creating stable, homogeneous ADCs with an average drug-antibody ratio (DAR) of four.30 We therefore included this functional handle, as well as the cytotoxic MMAE payload for the full linker-payloads. Linker-payloads 15 and 16a are derived from the model compounds 7 and 12 respectively, and were expected to exhibit similar plasma stability and sensitivity towards sulfatases. Linker-payload 16b is an analogue of 16a with an added electron-withdrawing NO2 group on the arylsulfate, included because of the increased activity of sulfatases towards electron-poor arylsulfates.31 Non-cleavable analogue 17 and Val–Ala-containing linker payload 18 were also synthesised so that their biological properties could be compared to the arylsulfates in vitro (structures in Fig. 5, full synthetic details described in the ESI†).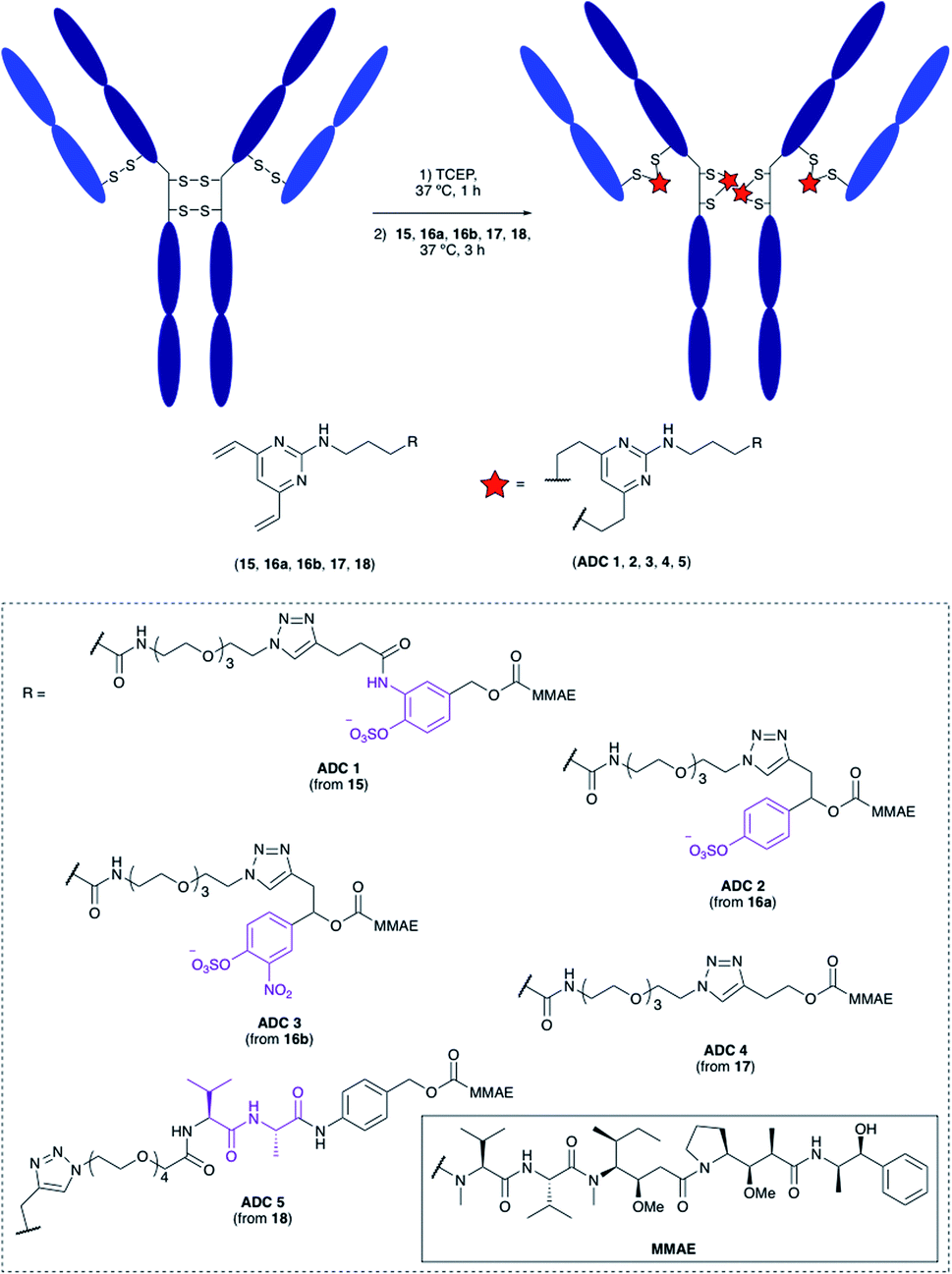 | ||
| Fig. 5 Bioconjugation of linker-payloads 15, 16a, 16b, 17 and 18 to trastuzumab to afford ADCs 1–5. | ||
Bioconjugation
The linker-payloads were conjugated to trastuzumab, an FDA-approved IgG mAb which targets the HER2 receptor, an internalising antigen that is overexpressed in around 20% of breast cancers.32 Our earlier studies confirmed the excellent retention of antibody binding affinity and internalisation of DVP-derived trastuzumab ADCs, as well as the high plasma stability of the cysteine-DVP linkage.30 First, the four interchain disulfides were reduced by TCEP for 1 h to reveal eight reactive cysteine residues (Fig. 5). Then linker-payloads 15, 16a, 16b, 17 and 18 were incubated for 3 h to react with the cysteine residues, rebridging them to form five ADCs (ADCs 1–5). Efficient conversion (>95%) to the desired conjugates was confirmed by LC-MS and SDS-PAGE analysis (Fig. S1 and S2†).In vitro cytotoxicity
Having successfully synthesised the cleavable and non-cleavable ADCs (ADCs 1–5), their cytotoxicity against HER2-positive (BT474) and HER2-negative (MCF7) cells was evaluated (Fig. 6). As expected, the cathepsin-cleavable ADC 5 demonstrated increased cytotoxicity (IC50 = 92 pM) towards the HER2-positive BT474 cells compared with the non-cleavable ADC 4 (IC50 = 609 pM). This six-fold increase in potency can be explained by the traceless release of MMAE, compared with the modified MMAE-metabolite released from non-cleavable ADC 4, whose tubulin-binding activity is expected to be lower. Gratifyingly, arylsulfate-containing ADC 2 and ADC 3 compared similarly with the dipeptidic ADC 5; both were also 5–10 times more cytotoxic than non-cleavable ADC 4 (Fig. 6a). The similar performance of these arylsulfate- and dipeptide-ADCs suggests that the arylsulfate linkers are also being cleaved in the cells following ADC internalisation. Contrastingly, ADC 1 was non-toxic across both HER2-positive cell lines, demonstrating that ortho-amide containing arylsulfate linkers are unsuitable for ADCs. The complete lack of cytotoxicity indicates that the in vitro sulfatase cleavage of ADC 1 is occurring at an insufficient rate and the maintained presence of the anionic sulfate group adjacent to the auristatin payload inhibits its tubulin-binding ability.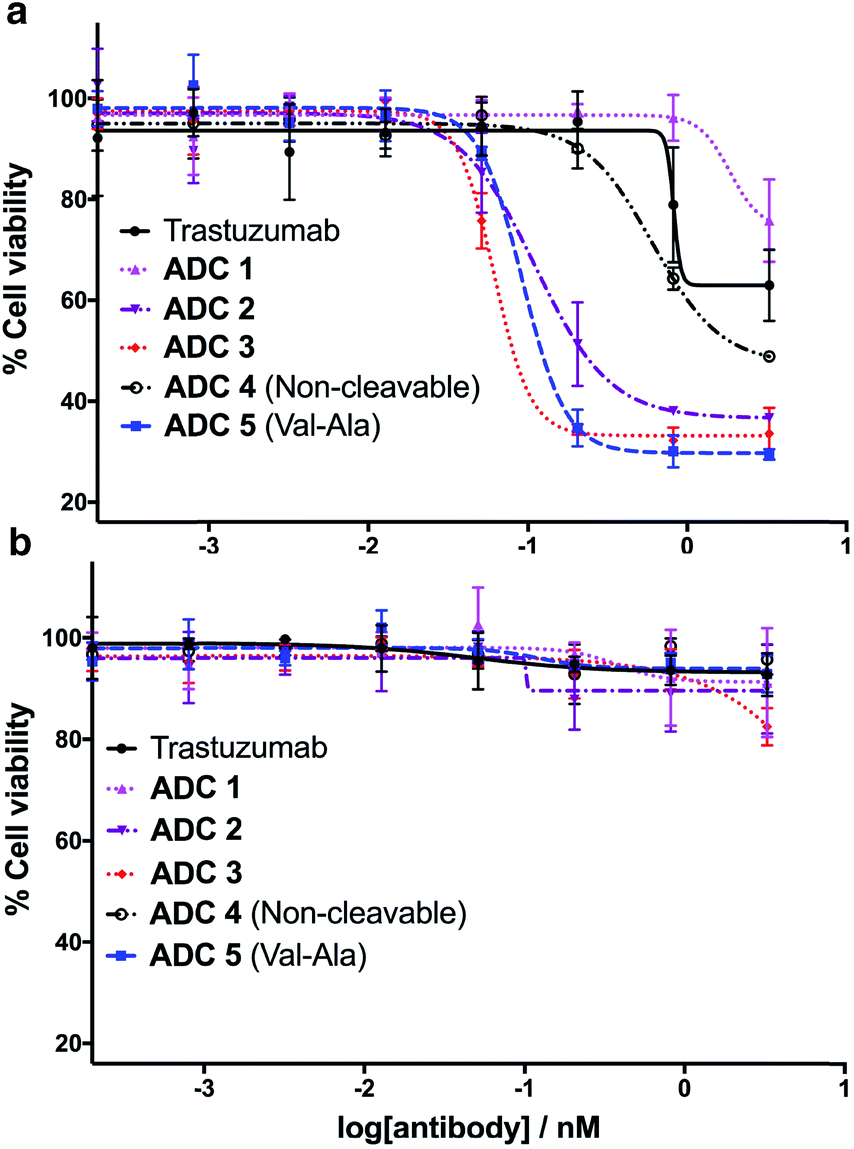 | ||
| Fig. 6 In vitro biological evaluation of ADCs 1–5 in (a) HER2-positive BT474 cells and (b) HER2-negative MCF7 cells. Viability data shows the mean of three independent experiments and error bars represent standard error of the mean. For BT474 cells, IC50 values are as follows: ADC 1 = N/A; ADC 2 = 111 pM; ADC 3 = 61 pM; ADC 4 = 609 pM; ADC 5 = 92 pM. | ||
All five ADCs were non-toxic towards the HER2-negative MCF7 cells up to 3 nM, validating the stability of the linkers in media and the retained functionality of the mAb to bind and internalise with the HER2 antigen (Fig. 6b). Furthermore, trastuzumab's lack of cytotoxicity against HER2-positive BT474 cells confirms that the MMAE payload is critical to the cell-killing ability of the ADCs. The results of our in vitro evaluations were confirmed by testing the ADCs in additional HER2-positive (SKBR3) and HER2-negative (T47D) cell lines, where similar trends were observed (Fig. S7 and Table S1†).
Conclusions
In conclusion, we have demonstrated the suitability of arylsulfates as cleavable linkers for ADCs. Model studies of the linkers with a fluorometric AMC payload validate their susceptibility towards sulfatase enzymes to tracelessly release amine-linked payloads, as well as their vastly superior mouse plasma stability compared to dipeptidic linkers. Upon conjugation to trastuzumab with an MMAE payload, all five ADCs retained selectivity towards HER2-positive cells. The arylsulfate-containing ADC 2 and ADC 3 were significantly more potent against HER2-positive cells than the non-cleavable ADC 4, suggesting the highly potent MMAE payload is being released inside the cells. The dramatic difference in cytotoxicity between ADC 1 and ADCs 2 and 3 reflects the difference in enzymolysis rates of 7 and 12, suggesting that rapid payload release rates are crucial to the cell-killing capacity of the arylsulfate-ADCs. An appropriate choice of aryl-substitution is therefore vital to the cytotoxicity of sulfatase-cleavable linkers.We envision that the arylsulfate linkers should be applicable to other lipophilic payloads, given their solubility and synthetic viability. Furthermore, the ubiquity of lysosomal sulfatases should enable pairing of the arylsulfate linkers with alternative antibodies for the treatment of a range of cancer types.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Dr Marko Hyvönen for allowing us access to the Pherastar FS plate reader and Teodors Pantelejevs for his guidance with its operation. J. D. B. acknowledges an iCASE studentship from GlaxoSmithKline/EPSRC. D. R. S. acknowledges support from the Engineering and Physical Sciences Research Council (EP/P020291/1) and Royal Society (Wolfson Research Merit Award). The Spring lab acknowledges general lab support from the EPSRC, BBSRC, MRC and Royal Society.References
- V. Chudasama, Drug Discovery Today: Technol., 2018, 30, 1–2 CrossRef.
- J. Lu, F. Jiang, A. Lu and G. Zhang, Int. J. Mol. Sci., 2016, 17, 561 CrossRef PubMed.
- R. V. J. Chari, M. L. Miller and W. C. Widdison, Angew. Chem., Int. Ed., 2014, 53, 3796–3827 CrossRef CAS PubMed.
- J. M. Lambert and R. V. J. Chari, J. Med. Chem., 2014, 57, 6949–6964 CrossRef CAS PubMed.
- J. D. Bargh, A. Isidro-Llobet, J. S. Parker and D. R. Spring, Chem. Soc. Rev., 2019, 48, 4361–4374 RSC.
- Y. V. Kovtun, C. A. Audette, Y. Ye, H. Xie, M. F. Ruberti, S. J. Phinney, B. A. Leece, T. Chittenden, W. A. Blättler and V. S. Goldmacher, Cancer Res., 2006, 66, 3214–3221 CrossRef CAS PubMed.
- S. O. Doronina, B. E. Toki, M. Y. Torgov, B. a Mendelsohn, C. G. Cerveny, D. F. Chace, R. L. DeBlanc, R. P. Gearing, T. D. Bovee, C. B. Siegall, J. A. Francisco, A. F. Wahl, D. L. Meyer and P. D. Senter, Nat. Biotechnol., 2003, 21, 778–784 CrossRef CAS PubMed.
- G. D. Lewis Phillips, G. Li, D. L. Dugger, L. M. Crocker, K. L. Parsons, E. Mai, W. A. Blättler, J. M. Lambert, R. V. J. Chari, R. J. Lutz, W. L. T. Wong, F. S. Jacobson, H. Koeppen, R. H. Schwall, S. R. Kenkare-Mitra, S. D. Spencer and M. X. Sliwkowski, Cancer Res., 2008, 68, 9280–9290 CrossRef CAS PubMed.
- G. M. Dubowchik and R. A. Firestone, Bioorg. Med. Chem. Lett., 1998, 8, 3341–3346 CrossRef CAS PubMed.
- G. M. Dubowchik, K. Mosure, J. O. Knipe and R. A. Firestone, Bioorg. Med. Chem. Lett., 1998, 8, 3347–3352 CrossRef CAS PubMed.
- S. Kolodych, C. Michel, S. Delacroix, O. Koniev, A. Ehkirch, J. Eberova, S. Cianférani, B. Renoux, W. Krezel, P. Poinot, C. D. Muller, S. Papot and A. Wagner, Eur. J. Med. Chem., 2017, 142, 376–382 CrossRef CAS PubMed.
- Y. Ogitani, T. Aida, K. Hagihara, J. Yamaguchi, C. Ishii, N. Harada, M. Soma, H. Okamoto, M. Oitate, S. Arakawa, T. Hirai, R. Atsumi, T. Nakada, I. Hayakawa, Y. Abe and T. Agatsuma, Clin. Cancer Res., 2016, 22, 5097–5108 CrossRef CAS PubMed.
- A. C. Tiberghien, J. N. Levy, L. A. Masterson, N. V. Patel, L. R. Adams, S. Corbett, D. G. Williams, J. A. Hartley and P. W. Howard, ACS Med. Chem. Lett., 2016, 7, 983–987 CrossRef CAS PubMed.
- S. Cazzamalli, A. Dal Corso and D. Neri, J. Controlled Release, 2017, 246, 39–45 CrossRef PubMed.
- M. Dorywalska, R. Dushin, L. Moine, S. E. Farias, D. Zhou, T. Navaratnam, V. Lui, A. Hasa-Moreno, M. G. Casas, T.-T. Tran, K. Delaria, S.-H. Liu, D. Foletti, C. J. O'Donnell, J. Pons, D. L. Shelton, A. Rajpal and P. Strop, Mol. Cancer Ther., 2016, 15, 958–970 CrossRef CAS PubMed.
- Y. Wang, S. Fan, W. Zhong, X. Zhou and S. Li, Int. J. Mol. Sci., 2017, 18, 1860 CrossRef PubMed.
- P. J. Burke, P. D. Senter, D. W. Meyer, J. B. Miyamoto, M. Anderson, B. E. Toki, G. Manikumar, M. C. Wani, D. J. Kroll and S. C. Jeffrey, Bioconjugate Chem., 2009, 20, 1242–1250 CrossRef CAS PubMed.
- S. C. Jeffrey, J. B. Andreyka, S. X. Bernhardt, K. M. Kissler, T. Kline, J. S. Lenox, R. F. Moser, M. T. Nguyen, N. M. Okeley, I. J. Stone, X. Zhang and P. D. Senter, Bioconjugate Chem., 2006, 17, 831–840 CrossRef CAS PubMed.
- J. C. Kern, M. Cancilla, D. Dooney, K. Kwasnjuk, R. Zhang, M. Beaumont, I. Figueroa, S. C. Hsieh, L. Liang, D. Tomazela, J. Zhang, P. E. Brandish, A. Palmieri, P. Stivers, M. Cheng, G. Feng, P. Geda, S. Shah, A. Beck, D. Bresson, J. Firdos, D. Gately, N. Knudsen, A. Manibusan, P. G. Schultz, Y. Sun and R. M. Garbaccio, J. Am. Chem. Soc., 2016, 138, 1430–1445 CrossRef CAS PubMed.
- P. E. Brandish, A. Palmieri, S. Antonenko, M. Beaumont, L. Benso, M. Cancilla, M. Cheng, L. Fayadat-Dilman, G. Feng, I. Figueroa, J. Firdos, R. Garbaccio, L. Garvin-Queen, D. Gately, P. Geda, C. Haines, S. Hseih, D. Hodges, J. Kern, N. Knudsen, K. Kwasnjuk, L. Liang, H. Ma, A. Manibusan, P. L. Miller, L. Y. Moy, Y. Qu, S. Shah, J. S. Shin, P. Stivers, Y. Sun, D. Tomazela, H. C. Woo, D. Zaller, S. Zhang, Y. Zhang and M. Zielstorff, Bioconjugate Chem., 2018, 29, 2357–2369 CrossRef CAS PubMed.
- S. R. Hanson, M. D. Best and C. H. Wong, Angew. Chem., Int. Ed., 2004, 43, 5736–5763 CrossRef CAS PubMed.
- B. A. Schröder, C. Wrocklage, A. Hasilik and P. Saftig, Proteomics, 2010, 10, 4053–4076 CrossRef PubMed.
- N. Albin, L. Massaad, C. Toussaint, M. C. Mathieu, J. Morizet, O. Parise, A. Gouyette and G. G. Chabot, Cancer Res., 1993, 53, 3541–3546 CAS.
- Y. Liu, I. F. F. Lien, S. Ruttgaizer, P. Dove and S. D. Taylor, Org. Lett., 2004, 6, 209–212 CrossRef CAS PubMed.
- L. S. Simpson and T. S. Widlanski, J. Am. Chem. Soc., 2006, 128, 1605–1610 CrossRef CAS PubMed.
- C. R. Drake, D. C. Miller and E. F. Jones, Curr. Org. Synth., 2011, 8, 498–520 CrossRef CAS PubMed.
- L. S. Simpson, J. Z. Zhu, T. S. Widlanski and M. J. Stone, Chem. Biol., 2009, 16, 153–161 CrossRef CAS PubMed.
- K. M. Weir, T. D. Sutherland, I. Horne, R. J. Russell and J. G. Oakeshott, Appl. Environ. Microbiol., 2006, 72, 3524–3530 CrossRef CAS PubMed.
- S. R. Hanson, L. J. Whalen and C. H. Wong, Bioorg. Med. Chem., 2006, 14, 8386–8395 CrossRef CAS PubMed.
- S. J. Walsh, S. Omarjee, W. R. J. D. Galloway, T. T. L. Kwan, H. F. Sore, J. S. Parker, M. Hyvönen, J. S. Carroll and D. R. Spring, Chem. Sci., 2019, 10, 694–700 RSC.
- K. S. Dodgson, G. F. White and J. W. Fitzgerald, Sulfatases of microbial origin, CRC Press, Boca Raton, 2018, vol. 2 Search PubMed.
- I. L. Andrulis, S. B. Bull, M. E. Blackstein, D. Sutherland, C. Mak, S. Sidlofsky, K. P. Pritzker, R. W. Hartwick, W. Hanna, L. Lickley, R. Wilkinson, A. Qizilbash, U. Ambus, M. Lipa, H. Weizel, A. Katz, M. Baida, S. Mariz, G. Stoik, P. Dacamara, D. Strongitharm, W. Geddie and D. McCready, J. Clin. Oncol., 1998, 16, 1340–1349 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc06410a |
| This journal is © The Royal Society of Chemistry 2020 |