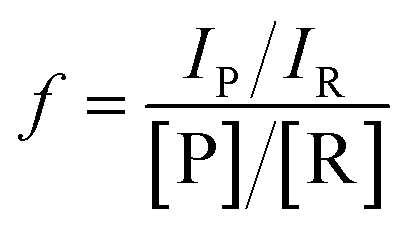DOI:
10.1039/C9SC06265C
(Edge Article)
Chem. Sci., 2020,
11, 2356-2361
High-yield gram-scale organic synthesis using accelerated microdroplet/thin film reactions with solvent recycling†
Received
10th December 2019
, Accepted 3rd January 2020
First published on 29th January 2020
Abstract
A closed system has been designed to perform microdroplet/thin film reactions with solvent recycling capabilities for gram-scale chemical synthesis. Claisen–Schmidt, Schiff base, Katritzky and Suzuki coupling reactions show acceleration factors relative to bulk of 15 to 7700 times in this droplet spray system. These values are much larger than those reported previously for the same reactions in microdroplet/thin film reaction systems. The solvent recycling mode of the new system significantly improves the reaction yield, especially for reactions with smaller reaction acceleration factors. The microdroplet/thin film reaction yield improved on recycling from 33% to 86% and from 32% to 72% for the Katritzky and Suzuki coupling reactions, respectively. The Claisen–Schmidt reaction was chosen to test the capability of this system in gram scale syntheses and rates of 3.18 g per h and an isolated yield of 87% were achieved.
Introduction
Over the past decade, a large number of mass spectrometry and fluorescence based studies have described unusual reaction behavior in small confined volumes, a topic that naturally attracts wide interest because of the close relationship to single cell biology,1–8 nanoscience9–12 and surface science.13–16 Reactions in microdroplets17–26 and thin films27–30 can be accelerated by factors of 101 to 105, and this has potential value in chemical synthesis. The partial solvation of reactants near the microdroplet–gas interface,9,15,31–33 the extremes in pH of microdroplets,9,20,22 fast solvent evaporation,29,34 special electric field and dipoles near interface10,14,35 and enhanced mass transfer36–40 make these reactions notably faster than in bulk. Although larger acceleration factors have been achieved in microdroplets,31,41 the small scale of the experiments18,29 and the failure to recycle the solvent have to be addressed to make this a practical approach to chemical synthesis. Specifically, (i) microdroplet reaction yields are restricted by the limited reaction time allowed due to the short lifetimes of microdroplets generated by electrospray,42 pneumatic spray43 and ultrasonic spray.44 (ii) The reaction scale suffers from the small volumes of the droplets (chosen because small volumes provide high acceleration factors).20,29,41,45–47 (iii) Large amounts of solvent are wasted which is undesirable from green chemistry considerations.48
To address the yield and scale issues, microdroplet collection experiments have been performed. In 2012, preparative electrospray was used to scale up the Claisen–Schmidt condensation reaction;27 the reaction mixture was electrosprayed at flow rate of 10 μL min−1 and directed into a polyethylene vessel containing glass wool to collect the product. Once the microdroplets hit the wall of the vessel or the wool fiber, they form an electroneutral thin film of reaction solution. If reaction is incomplete in the droplets it then can occur in this thin film. Although the reported reaction acceleration factors in thin films27,28,49 are not as large as in microdroplets,41 the reaction yields can be much higher due to the increased reaction time, as confirmed by dropcast thin film reactions.29 For this first 2012 preparative electrospray synthesis, the reaction scale was 35.3 mg per h (four sprayers, 90% yield).18 In 2017, deposition of uncharged microdroplets generated by sonic spray was used to fabricate a dynamic thin film for continuous synthesis.30 Again for Claisen–Schmidt reactions, the uncharged microdroplet and thin film reactions showed similar kinetics and yields to those in charged droplets, however, a larger reaction scale (120 mg per h; one sprayer, 80% yield) could be achieved due to the greater flow rate allowed in the sonic spray. Commercial pneumatic sprayers can support even greater flow rates, which means a much larger reaction scale, however, the droplet size is then too large for significant microdroplet reaction acceleration. In 2018, by choosing mesh materials with micrometer scale holes, a pneumatic sprayer was used to generate size-controlled microdroplets at a very high flow rate (8 mL min−1).43 A particular aldehyde oxidation reaction was run at a scale of 630 mg per h (one sprayer, 66% yield). Multiplexing is always a favorable strategy to achieve a larger scale. For example, the scale of two-phase microdroplet reactions13 could be increased by using multiple sprayers. Similarly, paper spray microdroplet reactions can be multiplexed by using paper with multiple tips.50
In light of this examination of earlier studies of scaled-up microdroplet synthesis, we note that although a variety of scale-up methods have been developed and larger scales have been achieved recently, the limited reaction time and lack of solvent recycling remain issues that are inadequately addressed. With a focus on these issues, we describe a new system for microdroplet synthesis with major improvements in reaction yield, product collection efficiency and solvent economy. The system can operate on the mg per h to g per h scale.
Results and discussion
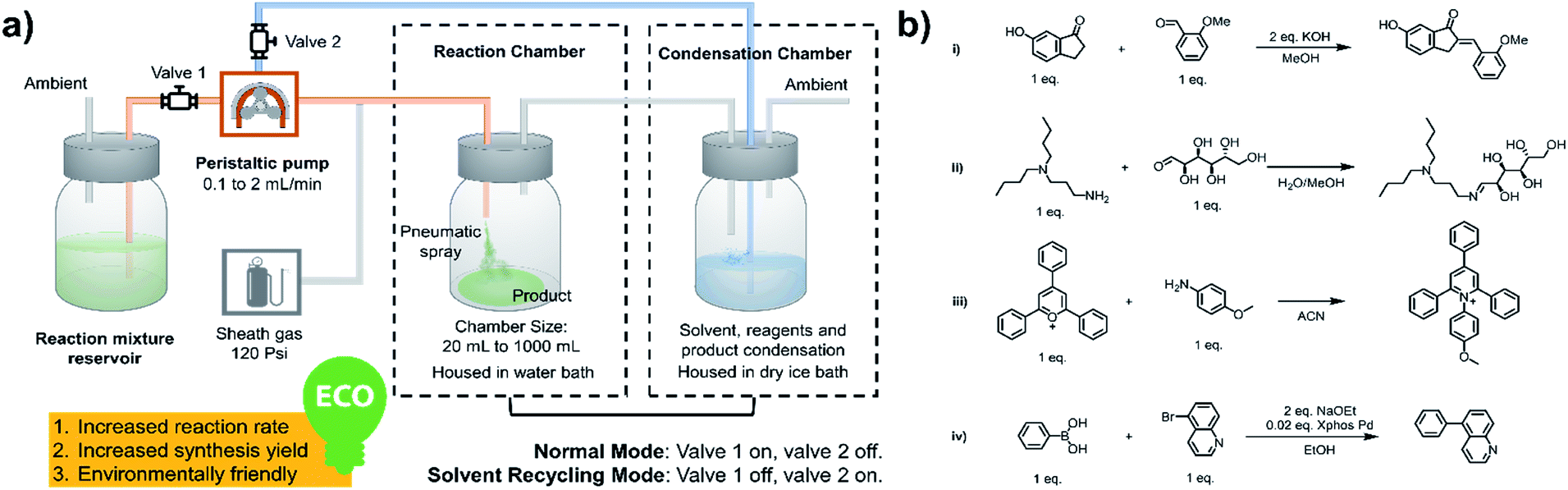
As shown in Fig. 1a, a peristaltic pump is used to introduce the reaction mixture from a reservoir, as well as to transfer the condensate back to the microdroplet reaction chamber. High-pressure nitrogen is used as a sheath gas to assist with nebulization and for protection of air/water sensitive reagents. As the synthesis begins, valve 1 is open and valve 2 is closed; the reaction mixture is transferred from the reservoir to the spray chamber to perform microdroplet and thin film reactions. The condensation chamber is used to collect the reagent- and product-containing vapors and nanodroplets. When the desired volume of reaction mixture had been transferred, valve 1 was closed and valve 2 opened, placing the system in the solvent recycling mode. During this time, reaction condensate is transferred back to the sprayer continuously to allow microdroplet and thin film reactions to proceed in the reaction chamber. This helps increase reaction time and product yield. We have performed various reactions using this new system, including typical C–C and C–N bond formation reactions, ring opening and closing reactions and coupling reactions, as listed in Fig. 1b.
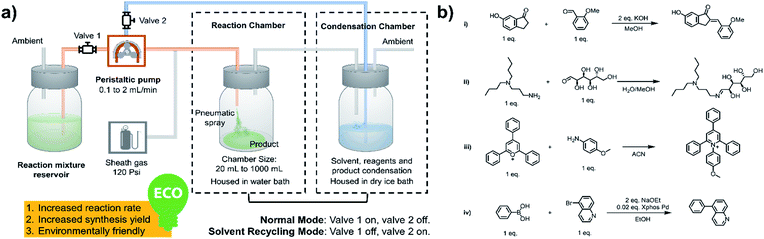 |
| | Fig. 1 (a) System for microdroplet synthesis on the mg per h to g per h scale. (b) Reactions interrogated using the new scale-up synthesis system. (b(i)) Claisen–Schmidt reaction; (b(ii)) Schiff base reaction; (b(iii)) Katritzky reaction; (b(iv)) Suzuki reaction. | |
Reaction conditions and MS measurement
All microdroplet reactions were performed at a concentration of 10 mM with equimolar reactants. For the Claisen–Schmidt reaction, 2 eq. of KOH were used to catalyze the reaction; for the Suzuki reaction, 2 eq. of EtONa and 0.02 eq. Xphos G3 were used as catalyst. The corresponding bulk reactions were run at these concentrations in 20 mL cap-sealed glass vials containing 3 mL reaction mixture and placed in an incubator and run at the desired temperatures (25 °C for Claisen–Schmidt and Katritzky reactions, 65 °C for Schiff base and Suzuki reactions). The microdroplet reactions used a flow rate of 100 μL min−1 to spray reaction mixture into the reaction chamber housed in a water bath. The concentration of reaction mixture and the chamber temperature were the same as those used for the corresponding bulk reactions. At selected times after starting a reaction, the reaction mixture (the sprayed reaction mixture plus the condensate) was quenched by dilution with quenching solution (pH adjustment of quenched solution was necessary for the Suzuki reaction; detailed information on quenching steps is given in Table S1†) prior to subsequent nanoelectrospray mass spectrometry (nESI-MS) analysis under standard non-accelerating conditions.30 By comparing the peak intensity of reactant and product ions in these mass spectra, and correcting for the difference in ionization efficiency, reaction yields were estimated from eqn (1):| | 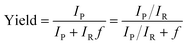 | (1) |
where IP and IR are the peak intensities of the product and reactant, respectively. The constant f reflects the difference in product and reagent ionization efficiency, defined as:| | 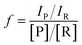 | (2) |
Constant f was measured by spiking reactant into diluted and quenched reaction mixture to measure the corresponding peak intensities in the mass spectra (see ESI†).
Microdroplet/thin film reaction without solvent recycling
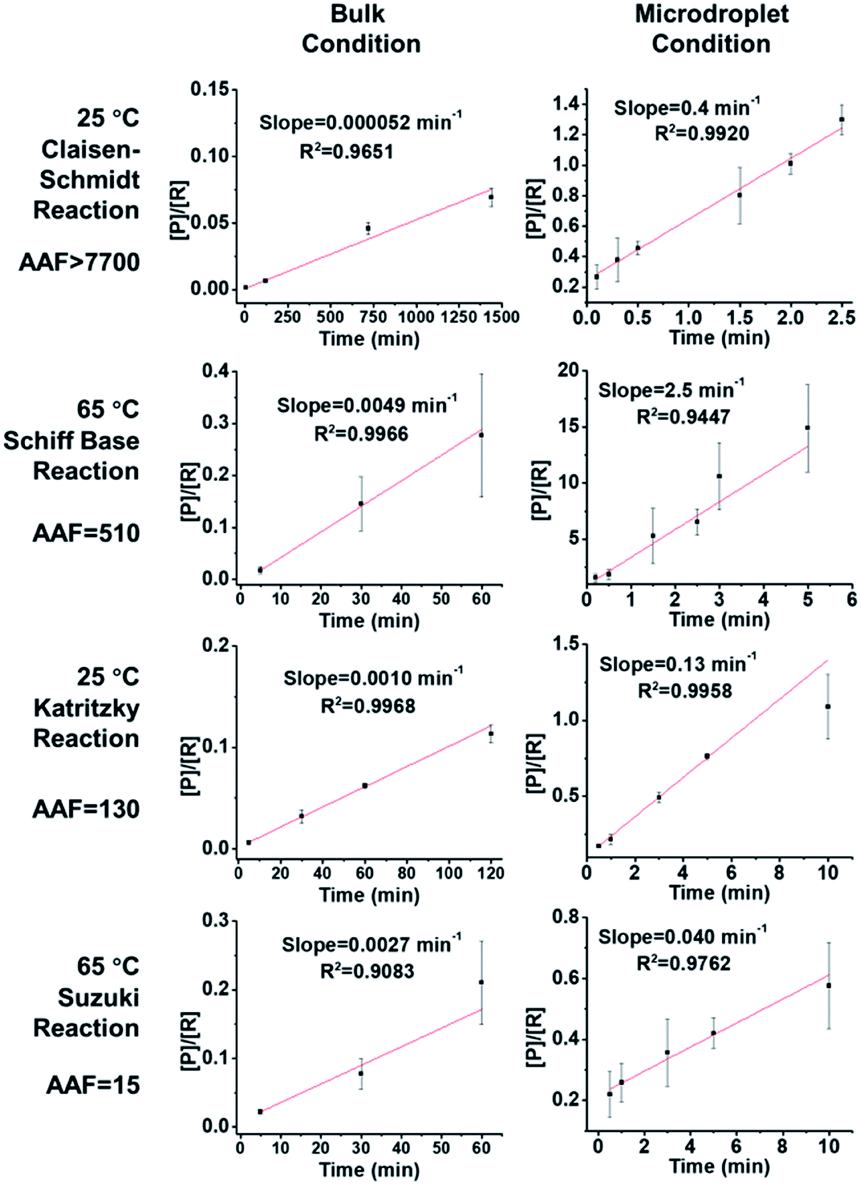
We investigated the kinetics and thermodynamics of microdroplet reactions in our system and made comparisons with bulk reactions. Organic reactions are often reversible so we investigated their reaction kinetics in the kinetic control regime, where the influence of the back reaction can be ignored. All four chosen reactions are considered as pseudo 2nd order reactions to facilitate comparison between the different reactions; note however that the order of Suzuki coupling is less certain (see the ESI† for discussion on this point). Hence the slope of the plot of [P]/[R] vs. reaction time t should be the product of reagent concentration (c0) and rate constant (k). This information can then be used to evaluate the apparent reaction acceleration factor (AAF, AAF = (c0k)droplet/(c0k)bulk) of the different chemical systems. It is noteworthy that there is a concentration effect on AAF.
As shown in Fig. 2, the slopes of the Claisen–Schmidt, Schiff base, Katritzky and Suzuki coupling reactions in bulk are 0.000052 min−1, 0.0014 min−1, 0.0049 min−1 and 0.0029 min−1, respectively; the slopes derived from the microdroplet reactor are 0.40 min−1, 0.18 min−1, 2.5 min−1 and 0.050 min−1, respectively. Taking the ratio of the slopes recorded under bulk and microdroplet conditions, and noting that both sets of experiments used the same initial concentrations, we note that the Claisen–Schmidt reaction has a very large AAF of ca. 7700, while the Schiff base and Katritzky reactions have moderate AAFs of 510 and 130, respectively, and the Suzuki coupling reaction shows a small AAF of 15 (only 15 times faster than bulk!). For the Claisen–Schmidt reaction, a model reaction often examined in microdroplet/thin film reaction acceleration studies, we note that the AAF in this new system is 7700, which is significantly higher than our previously reported value of 1000 (compared in units of [P]/[R]) in microdroplet product collection experiments.30 This improvement can be due to enhanced microdroplet collection efficiency, especially more efficient collection of the smallest droplets. It is important to emphasize that the apparent acceleration factor in these experiments, while a relative measure of rate constants, includes droplets of a range of sizes. As already noted, there is a strong inverse size effect on rate constants45 because reactions at the microdroplet/air interface are the main contributors to reaction acceleration.14–16,33,35,38,51 The closed reaction system will, no doubt, increase the collection efficiency of small sized droplets compared to ambient collection conditions (Fig. S2†) and hence increase the apparent acceleration factor measured. However, it must be acknowledged that not every reaction can be dramatically accelerated in microdroplets and that the mechanism behind this phenomenon is not completely clear.30 Empirically, it is clear that microdroplets are very effective at facilitating and accelerating bimolecular reactions, especially those involving the loss of small molecules such as condensation reactions. Moreover, solvent evaporation and the increased concentrations of acids/bases in microdroplets, as well as the super acidic/basic environment of microdroplets will facilitate reactions such as the Claisen–Schmidt reaction. These considerations help to explain acceleration in the Claisen–Schmidt, Katritzky and Schiff base reactions. However, the Suzuki coupling reaction could be different because it is a homogenous reaction catalyzed by a Pd(0) complex, wherein the reaction rate is highly dependent on the catalyst loading. The microdroplet condition does not increase the catalyst loading but the fast mass transfer in microdroplets can facilitate the renewal of catalytic sites and so increase catalyst efficiency.
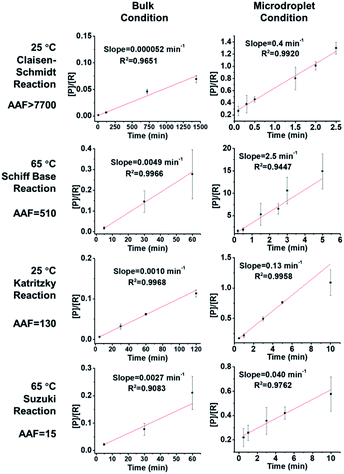 |
| | Fig. 2 Kinetics curves and apparent acceleration factors (AAF). In order to obtain measurable kinetics curves in bulk, 20 equivalent KOH had to be used as opposed to 2 equiv. in droplets. | |
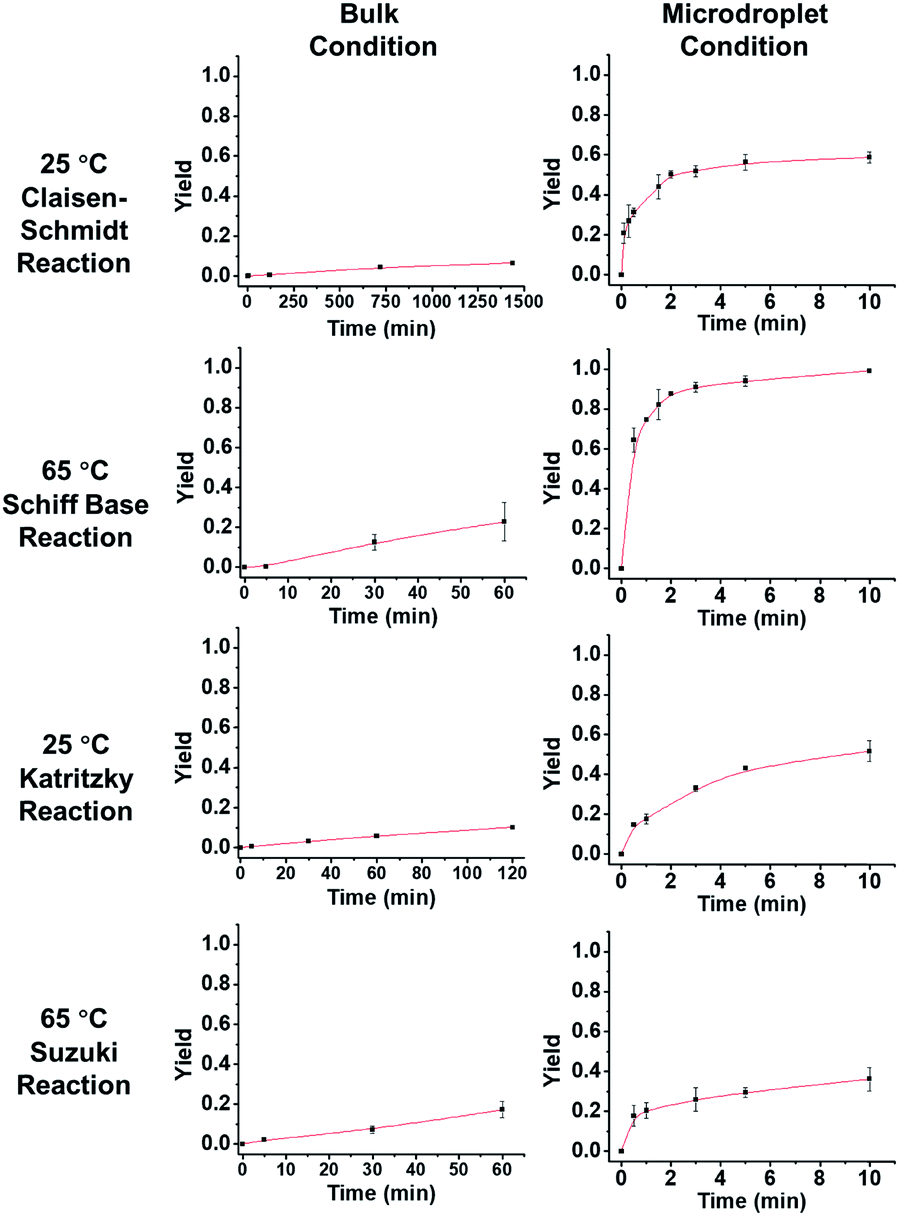
Fig. 3 describes the thermodynamics in the bulk and microdroplet reactors. The first column shows data for the bulk reactions and the second column shows those for the microdroplet reactions. For all four microdroplet reactions, there is a plateau in the plot of yield vs. reaction time that occurs within 10 min, indicating that the reactions approach equilibrium very quickly. For example, the Claisen–Schmidt and Schiff base reactions both have very large AAFs and reach a plateau (maximum yield in microdroplet reaction) within 3 min. The maximum yield after 10 min is 58% for the microdroplet/thin film Claisen–Schmidt reaction however the yield of bulk reaction is only 9%, even with 20× KOH and after 24 hour reaction. The maximum yield is 94% for the microdroplet/thin film Schiff base reaction while the yield in bulk is only 20% after 1 hour reaction. For the Katritzky and Suzuki reactions, with their medium and small AAFs, the plateau in the microdroplet reaction is not as well-defined. The yield after 10 min microdroplet reaction vs. 60 min bulk reaction is 55% vs. 14% for Katritzky reaction and 40% vs. 17% for the Suzuki reaction. Although the yield was improved in the microdroplet reaction, it is still some way from the maximum possible yield, likely due to the lack of reaction time. This is a general drawback of the continuous microdroplet reaction format, which becomes a serious problem in reactions with intrinsically small acceleration factors. In traditional microdroplet/thin film reaction format, reaction time is dependent on spray time and the later the reactants are introduced, the lower is the reaction time. The radical solution to this problem is separation of reagent introduction and solvent introduction into the system so that one can control the reaction scale and microdroplet reaction time appropriately.
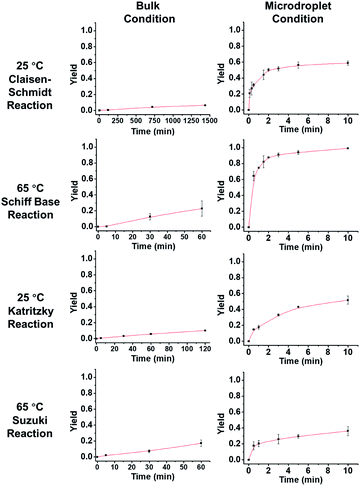 |
| | Fig. 3 Yield vs. time behavior of the four reactions under consideration. In order to obtain measurable kinetics curves in bulk, 20 equivalent KOH had to be used as opposed to 2 equiv. in droplets. | |
Comparison of microdroplet/thin film reaction with and without solvent recycling
To solve the above question, instead of separate introduction of reagents and solvent we added a solvent recycling capability to our microdroplet reaction system. We then compared the microdroplet reaction with and without the solvent recycling option. To compare these two methods, the reagent concentrations (10 mM), reaction time (10 min), reaction scale (3 mL, 30 μmol) and temperature (65 °C) were kept the same. For the simple microdroplet reaction, the flow rate was set to 0.3 mL min−1 so that 30 μmol of each reactant solution was deposited after 10 min. For the solvent recycling version of the microdroplet reaction, the flow rate was 1 mL min−1. In the first 3 min, 3 mL reaction solution (30 μmol) was deposited. Table 1 compares reaction yields with and without the solvent recycling. We see that for the Claisen–Schmidt and Schiff base reactions, two reactions with large AAFs, the yields with and without solvent recycling are similar and very high. However, for reactions with smaller AAFs, solvent recycling improved the yields dramatically, from 33% to 86% for the Katritzky reaction and 32% to 72% for the Suzuki coupling reaction. The higher yields achieved in the solvent recycling reactions are due to the increase of microdroplet reaction time. In the continuous spray microdroplet reaction format, the reaction mixture is gradually introduced to the reaction chamber to allow microdroplet reactions. The later the reaction mixture is introduced, the less microdroplet/thin film reaction time there is. However, in the solvent recycle mode, all reactants are introduced into the chamber within 3 minutes; during the other 7 min, all of the reactants are experiencing accelerated microdroplet and thin film reactions and thus a higher yield is achieved (Table 2).
Table 1 Comparison of the parameters used in normal vs. solvent recycling reactions
| Parameters |
Normal mode |
Recycling mode |
| Temperature (°C) |
65 |
65 |
| Flow rate (mL min−1) |
0.3 |
3 |
| Concentration (mM) |
10 |
10 |
| Volume transferred (mL) |
3 |
3 |
| Reaction time (min) |
10 |
10 |
| Valve 1 (reaction solution reservoir) |
Always on |
On (0 to 3 min) |
| Valve 2 (condensation chamber) |
Always off |
On (3 to 10 min) |
Table 2 Recycling/no recycling 10 min reactions
| Reaction type |
Yield (%) |
| Normal mode |
Recycling mode |
| Claisen–Schmidt reaction |
92 |
93 |
| Katritzky reaction |
33 |
86 |
| Schiff base formation |
98 |
100 |
| Suzuki coupling reaction |
32 |
72 |
Scale-up synthesis with solvent recycling
We chose the Claisen–Schmidt reaction to test the capability to perform reactions on the gram per hour scale. The photos in Fig. 4 show the instrumental setup, the reaction chamber during reaction/after reaction and the purified product acquired after the reaction. In this experiment 30 mL of the fresh mixed reaction solution (0.2 M 6-hydroxyindanone, 0.2 M 2-methoxybenzaldehyde and 0.4 M KOH) was transferred into reaction chamber at a flow rate of 2 mL min−1. After 15 min, solvent recycling was started and run for 15 min. In total, the reaction took half an hour. All of the mixture in the reaction chamber was then washed with 15 mL 2 M hydrochloride solution twice and then with 15 mL water once. The washed product was dried in an incubator at 35 °C overnight. The dried powder was weighed as 1.38 g (isolated yield of 87%). A small fraction of the powder was diluted by MeOH (with 1 mM DMAP for ionization in negative mode) to 1 mM for nESI-MS analysis. The MS is shown in Fig. S3.† Another small fraction of the product was dissolved deuterated DMSO for NMR analysis. The H-NMR and 13C-NMR spectra are shown in Fig. S4 and S5.† All the spectra indicate a very high purity of the isolated product.
 |
| | Fig. 4 Photos of (a) the scaled-up microdroplet/thin film reaction system for Claisen–Schmidt reaction, (b) reaction chamber during/after reaction and (c) purified product. | |
Conclusions
We have designed a semi-closed system for accelerated microdroplet reactions with solvent recycling capabilities. In this system, microdroplet reactions occur in a chamber housed in a water bath, which is separated from the ambient environment by a cold trap housed in a dry ice bath. The semi-closed system improves microdroplet reaction yields in three ways: (i) the reaction chamber increases the collection efficiency of small micro- or nanodroplets, enhancing the reaction acceleration factor relative to open systems; (ii) the cold trap allows carrier gas to pass into the air but captures microdroplets entrained reagents, products and solvents, and works with the peristaltic pump to recycle the condensate so improving the isolated yield in synthesis; (iii) solvents can be recycled to achieve longer microdroplet reaction times, which increases yields, especially for those reactions with small acceleration factors. This system supports flow rates ranging from 0.1 mL min−1 to 2 mL min−1, corresponding to the flexible synthetic scale of milligrams to grams per hour. We have performed the Claisen–Schmidt reaction at a scale of 3.18 g per h with subsequent purification showing an isolated yield of 86.8%. This large scale and high isolation yield emphasize the synthetic potential of microdroplet reactions. We believe this system has the potential to provide new synthesis solutions to the pharmaceutical and chemical industry although we recognize that implementation of multi-step syntheses still lies ahead.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
R. G. Cooks and Z. Wei designed the experiments and wrote the manuscript. H. Nie and Z. Wei fabricated the system and performed the experiments. L. Qiu helped in product purification and characterization. X. Chen and D. T. Holden helped with performing the reactions. The authors acknowledge support from US. Defense Advanced Research Projects Agency W911NF-16-2-0020 and National Science Foundation 1905087. H. Nie thanks the financial support from National Natural Science Foundation of China 21405006 and 21527809 and China Scholarship Council 201806015051.
Notes and references
- W. Stroberg and S. Schnell, Biophys. J., 2018, 115, 3–8 CrossRef CAS PubMed.
- X. Y. Gong, Y. Y. Zhao, S. Q. Cai, S. J. Fu, C. D. Yang, S. C. Zhang and X. R. Zhang, Anal. Chem., 2014, 86, 3809–3816 CrossRef CAS PubMed.
- X. Y. Si, X. C. Xiong, S. C. Zhang, X. Fang and X. R. Zhang, Anal. Chem., 2017, 89, 2275–2281 CrossRef CAS PubMed.
- X. C. Zhang, Q. C. Zang, H. S. Zhao, X. X. Ma, X. Y. Pan, J. X. Feng, S. C. Zhang, R. P. Zhang, Z. Abliz and X. R. Zhang, Anal. Chem., 2018, 90, 9897–9903 CrossRef CAS PubMed.
- R. M. Onjiko, S. A. Moody and P. Nemes, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6545–6550 CrossRef CAS PubMed.
- C. Lombard-Banek, S. A. Moody and P. Nemes, Angew. Chem., Int. Ed., 2016, 55, 2454–2458 CrossRef CAS.
- R. M. Onjiko, E. P. Portero, S. A. Moody and P. Nemes, Anal. Chem., 2017, 89, 7069–7076 CrossRef CAS.
- C. Lombard-Banek, S. A. Moody, M. C. Manzin and P. Nemes, Anal. Chem., 2019, 91, 4797–4805 CrossRef CAS.
- J. K. Lee, D. Samanta, H. G. Nam and R. N. Zare, Nat. Commun., 2018, 9, 1562 CrossRef PubMed.
- J. K. Lee, D. Samanta, H. G. Nam and R. N. Zare, J. Am. Chem. Soc., 2019, 141, 10585–10589 CrossRef CAS.
- A. Y. Li, Z. Baird, S. Bag, D. Sarkar, A. Prabhath, T. Pradeep and R. G. Cooks, Angew. Chem., Int. Ed., 2014, 53, 12528–12531 CAS.
- A. Y. Li, Q. J. Luo, S. J. Park and R. G. Cooks, Angew. Chem., Int. Ed., 2014, 53, 3147–3150 CrossRef CAS PubMed.
- X. Yan, H. Y. Cheng and R. N. Zare, Angew. Chem., Int. Ed., 2017, 56, 3562–3565 CrossRef CAS PubMed.
- J. K. Lee, K. L. Walker, H. S. Han, J. Kang, F. B. Prinz, R. M. Waymouth, H. G. Nam and R. N. Zare, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 19294–19298 CrossRef CAS PubMed.
- D. Kim, N. Wagner, K. Wooding, D. E. Clemmer and D. H. Russell, J. Am. Chem. Soc., 2017, 139, 2981–2988 CrossRef CAS PubMed.
- D. A. Thomas, L. T. Wang, B. Goh, E. S. Kim and J. L. Beauchamp, Anal. Chem., 2015, 87, 3336–3344 CrossRef CAS PubMed.
- M. Girod, E. Moyano, D. I. Campbell and R. G. Cooks, Chem. Sci., 2011, 2, 501–510 RSC.
- T. Muller, A. Badu-Tawiah and R. G. Cooks, Angew. Chem., Int. Ed. Engl., 2012, 51, 11832–11835 CrossRef PubMed.
- R. M. Bain, C. J. Pulliam and R. G. Cooks, Chem. Sci., 2015, 6, 397–401 RSC.
- S. Banerjee and R. N. Zare, Angew. Chem., Int. Ed., 2015, 54, 14795–14799 CrossRef CAS PubMed.
- R. M. Bain, C. J. Pulliam, F. Thery and R. G. Cooks, Angew. Chem., Int. Ed., 2016, 55, 10478–10482 CrossRef CAS PubMed.
- E. A. Crawford, C. Esen and D. A. Volmer, Anal. Chem., 2016, 88, 8396–8403 CrossRef CAS PubMed.
- J. K. Lee, H. G. Nam and R. N. Zare, Q. Rev. Biophys., 2017, 50, e2 CrossRef PubMed.
- M. I. Jacobs, J. F. Davies, L. Lee, R. D. Davis, F. Houle and K. R. Wilson, Anal. Chem., 2017, 89, 12511–12519 CrossRef CAS PubMed.
- F. A. Houle, A. A. Wiegel and K. R. Wilson, J. Phys. Chem. Lett., 2018, 9, 1053–1057 CrossRef CAS PubMed.
- C. N. Mu, J. Wang, K. M. Barraza, X. X. Zhang and J. L. Beauchamp, Angew. Chem., Int. Ed., 2019, 58, 8082–8086 CrossRef CAS PubMed.
- A. K. Badu-Tawiah, D. I. Campbell and R. G. Cooks, J. Am. Soc. Mass Spectrom., 2012, 23, 1461–1468 CrossRef CAS PubMed.
- X. Yan, R. Augusti, X. Li and R. G. Cooks, Chempluschem, 2013, 78, 1142–1148 CrossRef CAS.
- Z. W. Wei, X. C. Zhang, J. Y. Wang, S. C. Zhang, X. R. Zhang and R. G. Cooks, Chem. Sci., 2018, 9, 7779–7786 RSC.
- Z. W. Wei, M. Wleklinski, C. Ferreira and R. G. Cooks, Angew. Chem., Int. Ed., 2017, 56, 9386–9390 CrossRef CAS PubMed.
- X. Yan, R. M. Bain and R. G. Cooks, Angew. Chem., Int. Ed., 2016, 55, 12960–12972 CrossRef CAS PubMed.
- R. G. Cooks, H. Chen, M. N. Eberlin, X. Zheng and W. A. Tao, Chem. Rev., 2006, 106, 188–211 CrossRef CAS PubMed.
- A. Fallah-Araghi, K. Meguellati, J. C. Baret, A. El Harrak, T. Mangeat, M. Karplus, S. Ladame, C. M. Marques and A. D. Griffiths, Phys. Rev. Lett., 2014, 112(2), 028301 CrossRef PubMed.
- D. T. Chiu, C. F. Wilson, F. Ryttsen, A. Stromberg, C. Farre, A. Karlsson, S. Nordholm, A. Gaggar, B. P. Modi, A. Moscho, R. A. Garza-Lopez, O. Orwar and R. N. Zare, Science, 1999, 283, 1892–1895 CrossRef CAS PubMed.
- Z. P. Zhou, X. Yan, Y. H. Lai and R. N. Zare, J. Phys. Chem. Lett., 2018, 9, 2928–2932 CrossRef CAS PubMed.
- D. N. Mortensen and E. R. Williams, Anal. Chem., 2014, 86, 9315–9321 CrossRef CAS PubMed.
- R. D. Davis, M. I. Jacobs, F. A. Houle and K. R. Wilson, Anal. Chem., 2017, 89, 12494–12501 CrossRef CAS PubMed.
- S. Mondal, S. Acharya, R. Biswas, B. Bagchi and R. N. Zare, J. Chem. Phys., 2018, 148(24), 244704 CrossRef PubMed.
- D. N. Mortensen and E. R. Williams, Anal. Chem., 2015, 87, 1281–1287 CrossRef CAS PubMed.
- D. N. Mortensen and E. R. Williams, J. Am. Chem. Soc., 2016, 138, 3453–3460 CrossRef CAS PubMed.
- S. Banerjee, E. Gnanamani, X. Yan and R. N. Zare, Analyst, 2017, 142, 1399–1402 RSC.
- J. B. Fenn, M. Mann, C. K. Meng, S. F. Wong and C. M. Whitehouse, Science, 1989, 246, 64–71 CrossRef CAS PubMed.
- X. Yan, Y. H. Lai and R. N. Zare, Chem. Sci., 2018, 9, 5207–5211 RSC.
- A. Hirabayashi, M. Sakairi and H. Koizumi, Anal.
Chem., 1995, 67, 2878–2882 CrossRef CAS PubMed.
- B. M. Marsh, K. Iyer and R. G. Cooks, J. Am. Soc. Mass Spectrom., 2019, 30, 2022–2030 CrossRef CAS PubMed.
- J. K. Lee, S. Banerjee, H. G. Nam and R. N. Zare, Q. Rev. Biophys., 2015, 48, 437–444 CrossRef CAS PubMed.
- J. K. Lee, S. Kim, H. G. Nam and R. N. Zare, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 3898–3903 CrossRef CAS PubMed.
- R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC.
- Y. F. Li, X. Yan and R. G. Cooks, Angew. Chem., Int. Ed., 2016, 55, 3433–3437 CrossRef CAS PubMed.
- X. T. Zhu, W. W. Zhang, Q. Y. Lin, M. Y. Ye, L. Y. Xue, J. H. Liu, Y. C. Wang and H. Y. Cheng, ACS Sustainable Chem. Eng., 2019, 7, 6486–6491 CrossRef CAS.
- R. Augusti, H. Chen, L. S. Eberlin, M. Nefliu and R. G. Cooks, Int. J. Mass Spectrom., 2006, 253, 281–287 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc06265c |
| ‡ These authors contribute equally. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a