
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ monitoring of mechanochemical synthesis of calcium urea phosphate fertilizer cocrystal reveals highly effective water-based autocatalysis†
Patrick A.
Julien
 a,
Luzia S.
Germann
b,
Hatem M.
Titi
a,
Martin
Etter
c,
Robert E.
Dinnebier
b,
Lohit
Sharma
d,
Jonas
Baltrusaitis
*d and
Tomislav
Friščić
*a
a,
Luzia S.
Germann
b,
Hatem M.
Titi
a,
Martin
Etter
c,
Robert E.
Dinnebier
b,
Lohit
Sharma
d,
Jonas
Baltrusaitis
*d and
Tomislav
Friščić
*a
aDepartment of Chemistry, McGill University, Montreal, QC H3A 0B8, Canada. E-mail: tomislav.friscic@mcgill.ca
bMax Planck Institute for Solid State Research, Heisenbergstraße 1, 70569 Stuttgart, Germany
cDeutsches Elektronen-Synchrotron (DESY), Notkestraße 85, 22607 Hamburg, Germany
dDepartment of Chemical and Biomolecular Engineering, Lehigh University, B336 Iacocca Hall, 111 Research Drive, Bethlehem, Pennsylvania 18015, USA. E-mail: job314@lehigh.edu
First published on 13th February 2020
Abstract
Using the mechanosynthesis of the calcium urea phosphate fertilizer cocrystal as a model, we provide a quantitative investigation of chemical autocatalysis in a mechanochemical reaction. The application of in situ Raman spectroscopy and synchrotron X-ray powder diffraction to monitor the reaction of urea phosphate and either calcium hydroxide or carbonate enabled the first quantitative and in situ study of a mechanochemical system in which one of the products of a chemical reaction (water) mediates the rate of transformation and underpins positive feedback kinetics. The herein observed autocatalysis by water generated in the reaction enables reaction acceleration at amounts that are up to 3 orders of magnitude smaller than in a typical liquid-assisted mechanochemical reaction.
The need for more sustainable agriculture and agrochemicals has driven the search for novel and efficient fertilizers, which minimize the negative environmental impacts of runoff and eutrophication.1,2 Recently, co-crystallization has emerged as a powerful approach to develop new solid forms of fertilizers, which could maximize absorption by crops while minimizing runoff, which negatively impacts the environment and human health via eutrophication.3–5 Urea phosphate is one of the most prevalent fertilizer components for the delivery of critical plant nutrients nitrogen and phosphorus, with the added benefit of reducing ammonia losses by enzymatic degradation.6 The loss of ammonia from urea fertilizers is further reduced by the presence of calcium, another key nutrient for plants, leading to improved formulations for simultaneous delivery of nitrogen, phosphorus and calcium.7–9 An example is calcium urea phosphate Ca[CO(NH2)2]4(H2PO4)2 (1), an example of an agrochemical ionic cocrystal.10,11 The cocrystal 1 is highly desirable as a fertilizer due to its stability, reduction in ammonia loss, while providing three critical nutrients and good moisture stability. Mechanochemical reactions, conducted by milling, grinding or extrusion, have emerged as an efficient and rapid route to synthesize a wide range of molecules and materials, from pharmaceutical cocrystals to metal–organic frameworks.12 Mechanochemistry has been shown to enable synthesis independent of the relative solubilities of reactants, providing an excellent opportunity to form agrochemical cocrystals that are typically based on inexpensive but poorly soluble ionic starting materials.12 Mechanochemistry enables rapid reactions, such as the green synthesis of calcium urea phosphate, from very poorly soluble inorganic precursors, such as calcium carbonate or calcium hydroxide,4,13,14 producing innocuous by-products of water and CO2 (Fig. 1).
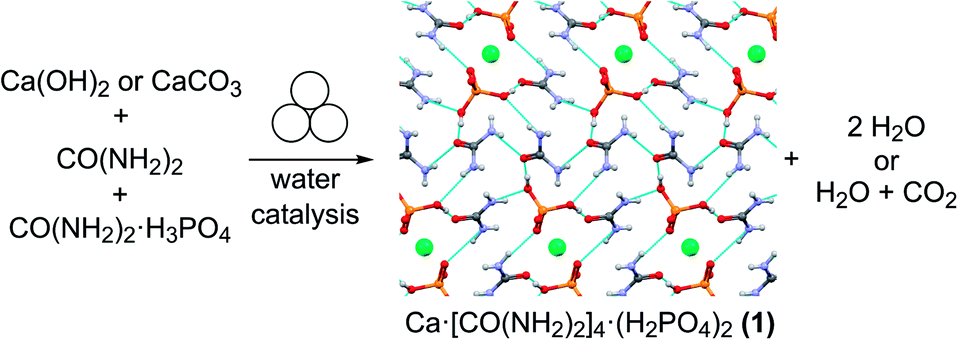 | ||
| Fig. 1 Mechanochemical synthesis of the agrochemical cocrystal 1 from urea, urea phosphate and either calcium hydroxide or carbonate. Calcium ions are shown in green, and hydrogen bonding interactions are displayed in light blue for 1 (CSD code URECAP10). | ||
Here, we investigate and compare the mechanisms of mechanochemical formation of the agrochemical cocrystal 1 by a neutralization reaction from either Ca(OH)2 or CaCO3, using the recently developed techniques for monitoring mechanochemical reactions in real time using Raman spectroscopy and synchrotron X-ray powder diffraction (XRPD).15,16 The in situ measurements reveal the appearance of sigmoidal reaction kinetics, which is herein shown to result from an autocatalytic feedback mechanism17 involving water released in the reaction. These results provide the first insight into factors controlling the mechanochemical formation of agrochemical cocrystals and, to the best of our knowledge, they also represent the first quantitative and in situ analysis of chemical autocatalysis caused by a product of a mechanochemical reaction.
Real-time monitoring by synchrotron X-ray diffraction was conducted using a previously reported design16 at the PETRA III beamline P02.1 at Deutsches Elektronen-Synchrotron (DESY), with λ = 0.207 Å, in poly(methyl methacrylate) (PMMA) milling jars. Real-time Raman spectroscopy monitoring18 was done in jars made of sapphire, as the signal from PMMA jars significantly obscured the signal for the formation of 1 (see ESI†). Milder conditions employed for synchrotron studies (smaller balls, PMMA plastic jar) resulted in a slower reaction.
Real-time Raman spectroscopy monitoring (Fig. 2) of the reaction involving CaCO3, urea, and urea phosphate in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2 respective stoichiometric ratios revealed the appearance of 1 after ca. 5 minutes milling. Despite considerable overlap of Raman bands of reactants and product, formation of 1 was clearly observable by the appearance of the characteristic band at 1083 cm−1. The reaction achieves quantitative conversion to 1 within 30 minutes, as determined by XRPD, Fourier-transform infrared attenuated total reflectance (FTIR-ATR) measurements and thermogravimetric analysis (TGA) after milling.
:2:2 respective stoichiometric ratios revealed the appearance of 1 after ca. 5 minutes milling. Despite considerable overlap of Raman bands of reactants and product, formation of 1 was clearly observable by the appearance of the characteristic band at 1083 cm−1. The reaction achieves quantitative conversion to 1 within 30 minutes, as determined by XRPD, Fourier-transform infrared attenuated total reflectance (FTIR-ATR) measurements and thermogravimetric analysis (TGA) after milling.
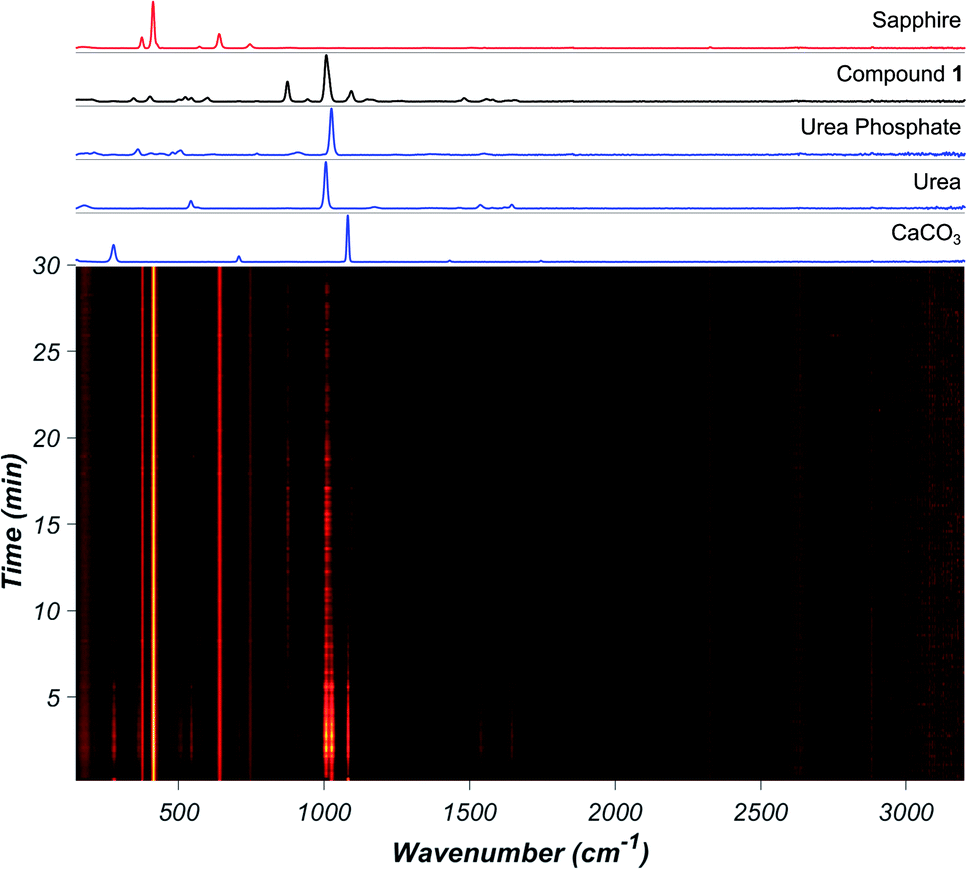 | ||
| Fig. 2 (Bottom) In situ Raman spectra of the neat milling reaction between CaCO3, urea, and urea phosphate. Brighter colour denotes higher intensity. (Top) Raman spectra of reactants (blue), the product (black), and background from the sapphire milling jar (red) with individual spectra of reactants and product above the 2D plot. | ||
In contrast, using Ca(OH)2 instead of CaCO3 revealed a dramatic acceleration in reaction rate, with near-quantitative conversion achieved in 5 minutes (see ESI†). The results of in situ Raman spectroscopy monitoring were validated by monitoring the reaction progress using in situ synchrotron XRPD, which again revealed much faster progress for the reaction involving Ca(OH)2 (Fig. 3).
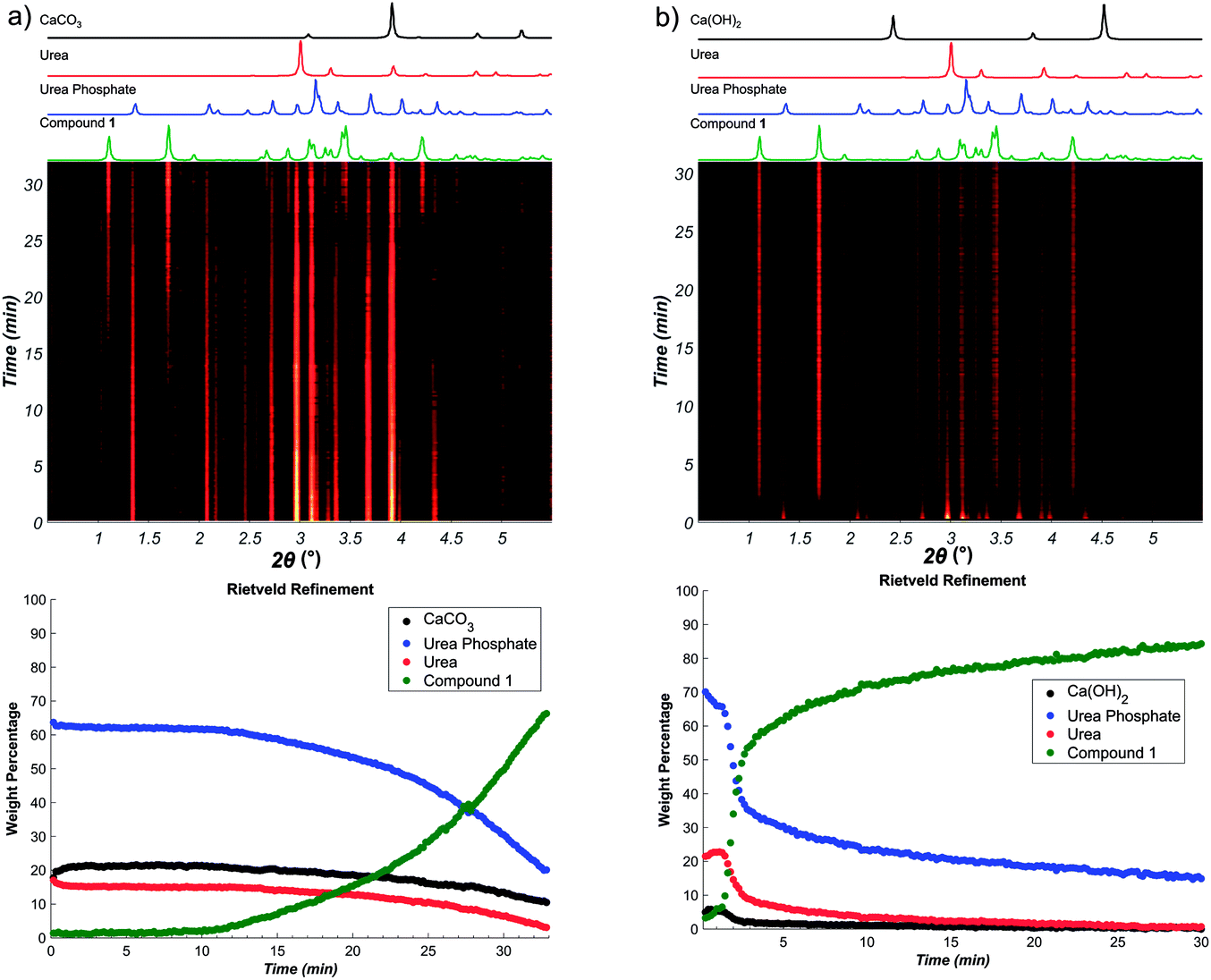 | ||
| Fig. 3 Real-time XRPD monitoring of the formation of 1: (a) time-resolved XRPD patterns of the reaction using CaCO3, with corresponding calculated patterns for reactants and product shown above the plot and the reaction profile obtained by sequential Rietveld refinement of the in situ XRPD data shown below; (b) time-resolved XRPD patterns of the reaction using Ca(OH)2, with corresponding calculated patterns for reactants and product shown above the plot and the reaction profile obtained by sequential Rietveld refinement of the in situ XRPD data shown below. | ||
Reaction profiles from in situ Raman spectroscopy (Fig. 4a) reveal sigmoidal behaviour for the reaction involving CaCO3 with 50% conversion achieved after ca. 10 minutes. In contrast, the reaction with Ca(OH)2 proceeded much faster, with a very short induction period as seen from Rietveld analysis of in situ XRPD data. Such dramatic difference in reaction rates is even more notable considering that the commercially used Ca(OH)2 contained ca. 13% CaCO3 by weight (9.6 mol%, as established by TGA) (see ESI†). The two reactions also exhibited visually observable differences in rheology during the reaction, with the reaction involving Ca(OH)2 proceeding in a moist paste, but the reaction with CaCO3 consistently being a free-flowing powder (see ESI†).
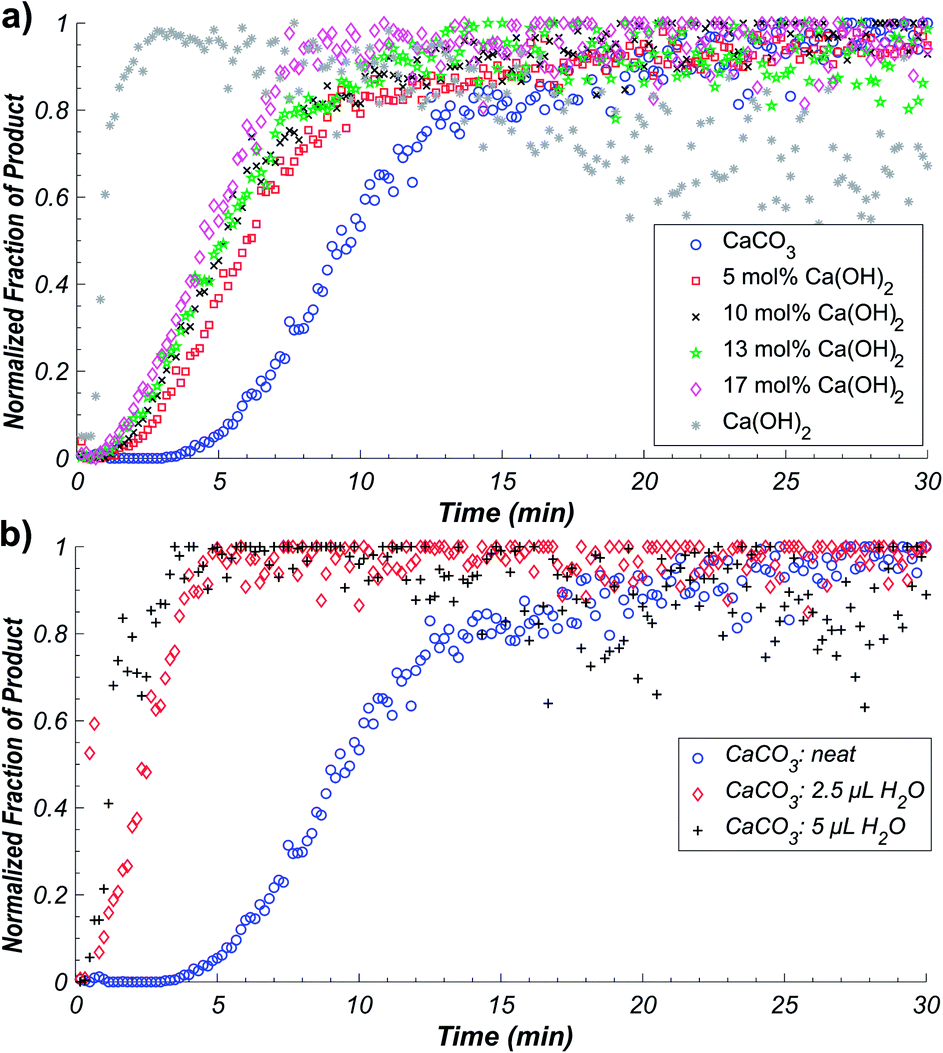 | ||
| Fig. 4 (a) Monitoring the relative rates of formation of 1 from CaCO3 (blue) and Ca(OH)2 (grey), as well as various mixtures. (b) The addition of small volumes of water to the synthesis of 1 from CaCO3, results in a significant increase in reaction rate as observed by in situ Raman spectroscopy. In each case the total weight of solid reactants was approximately 180 mg. | ||
As the reaction releases water, and TGA of the calcium precursors revealed that CaCO3 does not contain any moisture, and Ca(OH)2 might contain less than 2% by weight of absorbed water, we speculated that the progress of synthesis might be affected by the water that is formed by the neutralization reactions. As sigmoidal reaction kinetics are typically associated with autocatalysis,19 wherein a reaction product facilitates conversion of remaining reactants, we envisaged a process in which the formation of water molecules creates a more mobile environment that could facilitate mechanochemical reactivity. Indeed, previous studies have noted that a liquid phase released from a hydrated starting material can enhance the formation of hydrogen-bonded cocrystals or coordination polymers.20–25 However, there has not yet been a real-time, quantitative investigation of how a product formed in a chemical reaction, which involves the rearrangement of covalent bonds, could exhibit autocatalytic behaviour in mechanochemistry.
We explored this possibility by conducting further in situ Raman kinetic studies, but on reactions in the presence of small amounts of water added to reaction mixtures (2.5 μL water for η = 0.014 μL mg−1 and 5 μL water for η = 0.029 μL mg−1, where η corresponds to the ratio of added liquid volume to reactant weight) involving CaCO3, urea, and urea phosphate. As anticipated, the addition of water resulted in a clear reaction acceleration and shortening of the induction period of the reaction (Fig. 4b). Importantly, changing the amount of water had a significant effect on reaction kinetics, a test previously employed by the James group to evaluate autocatalytic behaviour, which confirmed autocatalysis by water generated in the reaction.17 The herein observed catalytic effect is remarkable, as the amount of added water corresponds to very low η values: one to two orders of magnitude lower than those accessible in typical liquid-assisted grinding (LAG) reactions, where η is typically between 0.1–2 μL mg−1, and reactivity enhancement is sometimes explained by physical presence of a liquid.26–28 In order to further confirm that the observed sigmoidal kinetics is due to autocatalysis by water as a product of chemical reaction, rather than due to effects of nucleation and crystal growth of 1,‡ we also performed reaction based on CaCO3 reactant that were seeded with pre-synthesized 1. The addition of different amounts of 1 to the reaction mixture was found to exhibit no significant effect on the measured reaction kinetics, which is further consistent with the sigmoidal behaviour being a result of water-based autocatalysis (see ESI†).‡
The observed high sensitivity of mechanochemical reaction kinetics to deliberate additions of minute amounts of water is consistent with the proposed self-acceleration of reaction progress due to released water. This was further validated by probing the kinetics of reactions in which a fraction of the CaCO3 reactant was replaced by an equivalent amount of Ca(OH)2. Real-time monitoring by Raman spectroscopy of mechanochemical reactions with 5, 10, 13, and 17 mol% of CaCO3 replaced by Ca(OH)2 reveals immediate and significant improvement in reaction rate and shortening of the induction period (Fig. 4a). The Ca(OH)2 reactant is expected to generate twice the amount of water by-product compared to CaCO3, effectively acting as solid source of catalytic amounts of water. Real-time monitoring of the reactions permitted us to plot the time required for 50% conversion with respect to water added as a liquid, or in the form of Ca(OH)2, revealing a non-linear relationship consistent with autocatalysis (Fig. 5a).
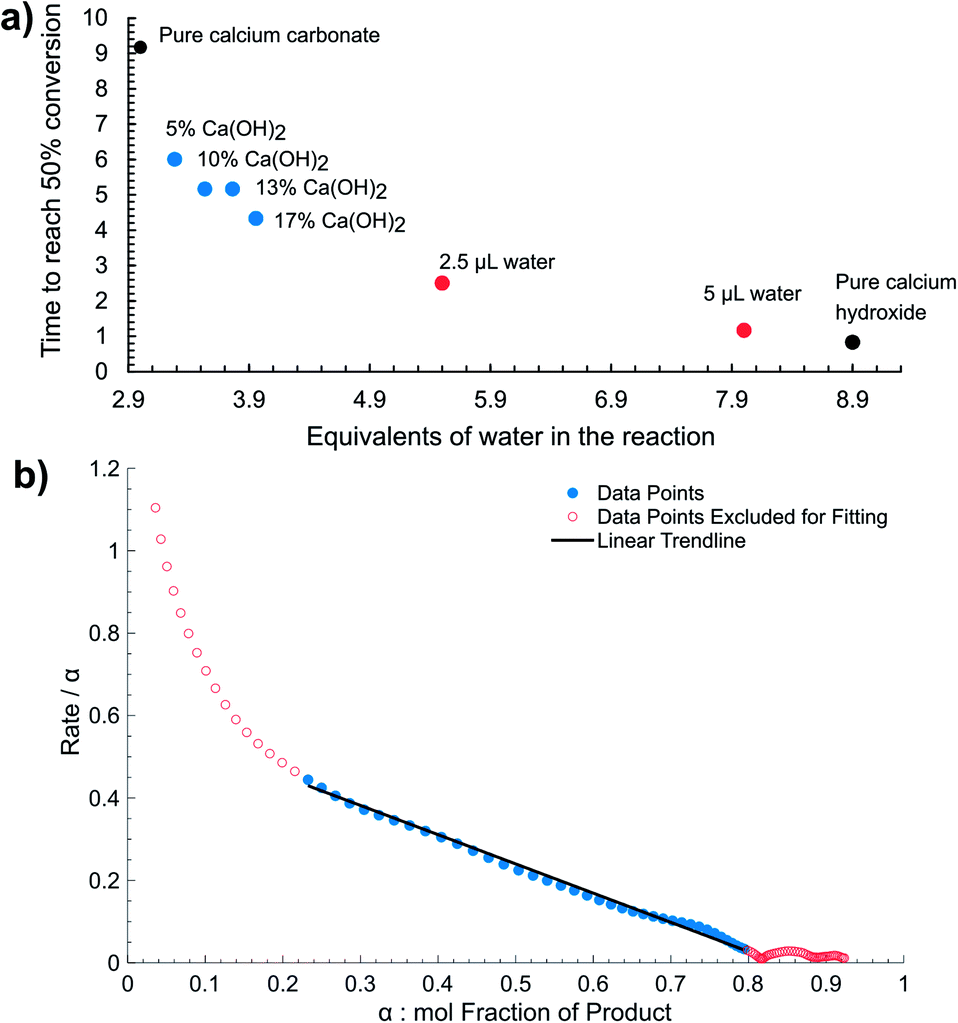 | ||
| Fig. 5 (a) The reaction rate measured as the time required to reach 50% conversion as a function of the total equivalents (relative to calcium) of water present and/or generated in the system. (b) Reaction rate (dα/dt)/α vs. α, where α is the mol fraction of 1 as determined by in situ Raman spectroscopy for the synthesis using CaCO3 reactant. Linear behaviour is observed between α = 0.2 and 0.8, consistent with autocatalysis due to a reaction product. Fitting parameters are reported in the ESI.† | ||
Autocatalytic behaviour was validated by analysing in situ data using an autocatalysis model for solid–state reactions, with respect to mol fraction of generated water (α):29
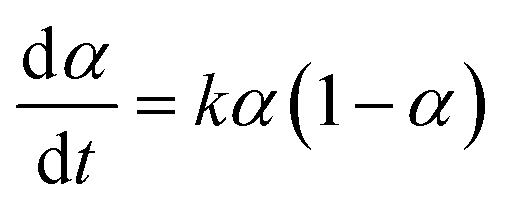 | (1) |
While the amount of water in the reaction is not readily measured by Raman spectroscopy, in the reaction of CaCO3 it should be equivalent to the amount of product formed. In the α range from 0.2 to 0.8, when the molar fraction of the product in the reaction is between 20% and 80%, eqn (1) yields a linear plot (Fig. 5b), consistent with water-mediated autocatalysis.30 The inability to obtain a linear fit during early and late phases of the reaction might be due to poor diffusion or mixing issues when the amount of product or reactants is low. It is remarkable that autocatalytic behavior can be observed at α ≈ 0.2, which would correspond to ca. 1.2 μL of water generated in the reaction, and an approximate η of 0.007 μL mg−1. Such η value is nearly three orders of magnitude lower than in a typical LAG reaction and, considering that the water is generated directly in the reaction, indicates that the observed reaction enhancement is an example of chemical autocatalysis rather than liquid-assisted26–28 mechanochemistry.
The herein presented autocatalytic behaviour is central for the ability to rapidly synthesize 1 without large amounts of water. The ability to accelerate synthesis by using water amounts that are minuscule even for LAG mechanochemistry is particularly important due to the high aqueous solubility of this material, which is beneficial for fertilizer use but hinders its preparation from solution. Once mechanochemically made, 1 exhibits properties highly desirable in a fertilizer. Thermal analysis reveals a high thermal stability for 1, which releases urea at 124 °C, a higher temperature compared to urea phosphate which decomposes at 116 °C (see ESI†). Moisture stability of 1 was compared to that of urea by dynamic vapour sorption (DVS) across the relative humidity (RH) range from 0–95%, at 25 °C. The measurements confirm the ability of 1 to interact with water more strongly, while maintaining a more modest water absorption profile compared to urea (Fig. 6). For urea, increasing RH led to negligible water uptake until the deliquescence point at 74% RH, with further increases in humidity leading to continuous hygroscopic growth of the aqueous droplet. On decreasing RH, the droplet decreased in size, below 74% RH became supersaturated with respect to urea, and below 50% RH formed an effloresced particle. Cocrystal 1 exhibited a deliquescence phase transition at 65% RH, but no distinct efflorescence point, as evidenced by a continuous hysteresis down to low humidity values (<20% RH) values. The hysteresis and presence of absorbed water in 1 over a wide range of RH values suggests a continuous transition of the bound-to-free water, potentially due to strong hydrogen bonding to hydrophilic, polar groups of 1.31 While such behaviour is beneficial for fertilizer use, it also clearly illustrates the synthetic benefit of the herein demonstrated autocatalytic effect, as it enables access to 1 as a well-defined and dry (by TGA) solid, using only traces of water.
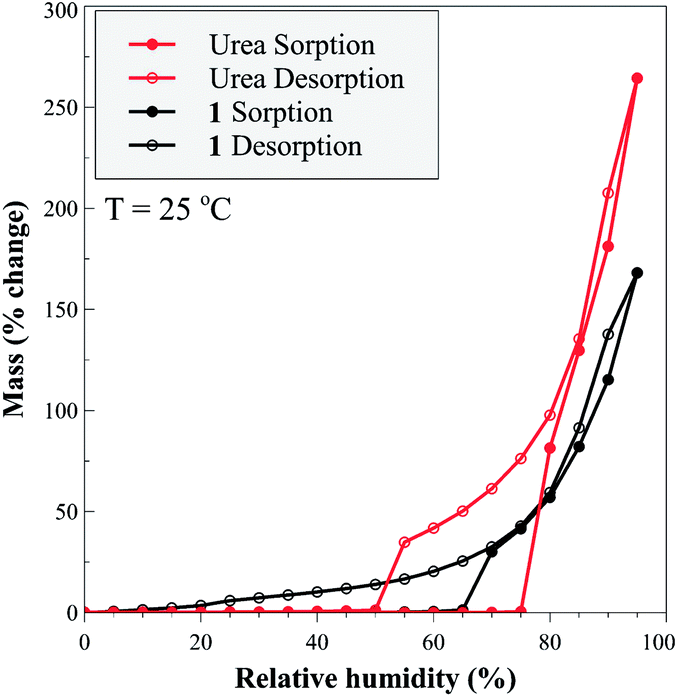 | ||
| Fig. 6 Adsorption/desorption branches of RH on urea and 1. | ||
In conclusion, we used in situ X-ray diffraction and spectroscopy to provide direct and quantitative evidence of chemical autocatalysis in a mechanochemical reaction. The presented results further our understanding of fundamental principles and mechanisms of mechanochemistry, an area that has recently attracted significant interest.32–42 While chemical autocatalysis is not the only potential reason for appearance of sigmoidal kinetics in a mechanochemical reaction,‡ this study provides direct evidence that such a model is relevant for mechanochemical reactions and, in principle, may be broadly applicable to water-forming acid–base milling reactions. The enhancement of mechanochemical reactions in liquid-assisted grinding can sometimes be explained by a purely physical effects of dissolution and having a liquid phase present.26–28 Herein, however, reaction acceleration is observed due to water directly generated by the reaction, at amounts that are up to three orders of magnitude lower than in a typical liquid-assisted grinding system, resulting in an in situ observed autocatalytic reaction profile. These observations highlight the herein explored reaction as an example of chemical autocatalysis in the solid state, which enables reaction acceleration at amounts of water significantly smaller than in liquid-assisted mechanochemistry.
The efficient autocatalysis observed herein is also of practical value, as it enables the synthesis of a highly hygroscopic fertilizer cocrystal material using only minuscule amounts of water, even for standards of liquid-assisted mechanochemistry. This demonstrates how water-based autocatalysis can be used to greatly simplify the preparation of ionic cocrystals and other valuable material whose synthesis in solution is made cumbersome by high affinity to water and tendency to form supersaturated solutions.
Conflicts of interest
T. F. is a co-founder of the company Form-Tech Scientific, Inc. that manufactures some of the equipment used in this study.Acknowledgements
We acknowledge support from DESY, and Form-Tech Scientific, Inc. T. F. acknowledges support of NSERC Discovery Grant (RGPIN-2017-06467), Discovery Accelerator (RGPAS 507837-17), and E. W. R. Steacie Memorial Fellowship (SMFSU 507347-17). J. B. and L. S. acknowledge support from USA National Science Foundation (NSF), Grant CHE 1710120.Notes and references
- J. Baltrusaitis, ACS Sustainable Chem. Eng., 2017, 5, 9527 CrossRef CAS.
- L. Casali, L. Mazzei, O. Shemchuk, K. Honer, F. Grepioni, S. Ciurli, D. Braga and J. Baltrusaitis, Chem. Commun., 2018, 54, 7637–7640 RSC.
- K. Finlay, A. Patoine, D. B. Donald, M. J. Bogard and P. R. Leavitt, Limnol. Oceanogr., 2010, 55, 1213–1230 CrossRef CAS.
- K. Honer, E. Kalfaoglu, C. Pico, J. McCann and J. Baltrusaitis, ACS Sustainable Chem. Eng., 2017, 5, 8546–8550 CrossRef CAS.
- B. Sandhu, A. S. Sinha, J. Desper and C. B. Aakeröy, Chem. Commun., 2018, 54, 4657–4660 RSC.
- J. M. Bremner and L. A. Douglas, Soil Sci. Soc. Am. J., 1971, 35, 575–578 CrossRef CAS.
- L. B. Fenn, J. E. Matocha and E. Wu, Soil Sci. Soc. Am. J., 1981, 45, 883–886 CrossRef CAS.
- L. B. Fenn, G. Tatum and G. Horst, Fert. Res., 1990, 21, 125–131 CrossRef CAS.
- J. M. Stumpe, P. L. G. Vlek and W. L. Lindsay, Soil Sci. Soc. Am. J., 1984, 48, 921–927 CrossRef CAS.
- J. P. Chen and K. Isa, J. Mass Spectrom. Soc. Jpn., 1998, 46, 299–303 CrossRef CAS.
- A. W. Frazier, J. R. Lehr and J. P. Smith, J. Agric. Food Chem., 1967, 15, 345–347 CrossRef CAS.
- (a) S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC; (b) D. Braga, L. Maini and F. Grepioni, Chem. Soc. Rev., 2013, 42, 7638–7648 RSC.
- K. Honer, C. Pico and J. Baltrusaitis, ACS Sustainable Chem. Eng., 2018, 6, 4680–4687 CrossRef CAS.
- L. Sharma, D. Kiani, K. Honer and J. Baltrusaitis, ACS Sustainable Chem. Eng., 2019, 7, 6802–6812 CrossRef CAS.
- D. Gracin, V. Štrukil, T. Friščić, I. Halasz and K. Užarević, Angew. Chem., Int. Ed., 2014, 53, 6193–6197 CrossRef CAS PubMed.
- I. Halasz, S. A. J. Kimber, P. J. Beldon, A. M. Belenguer, F. Adams, V. Honkimäki, R. C. Nightingale, R. E. Dinnebier and T. Friščić, Nat. Protoc., 2013, 8, 1718 CrossRef PubMed.
- B. P. Hutchings, D. E. Crawford, L. Gao, P. Hu and S. L. James, Angew. Chem., Int. Ed., 2017, 56, 15252–15256 CrossRef CAS PubMed.
- P. A. Julien, I. Malvestiti and T. Friščić, Beilstein J. Org. Chem., 2017, 13, 2160–2168 CrossRef CAS PubMed.
- R. Plasson, A. Brandenburg, L. Jullien and H. Bersini, J. Phys. Chem. A, 2011, 115, 8073–8085 CrossRef CAS PubMed.
- I. A. Tumanov, A. A. L. Michalchuk, A. A. Politov, E. V. Boldyreva and V. V. Boldyrev, CrystEngComm, 2017, 19, 2830–2835 RSC.
- S. Karki, T. Friščić, W. Jones and W. D. S. Motherwell, Mol. Pharm., 2007, 4, 347–354 CrossRef CAS PubMed.
- K. Užarević, V. Štrukil, C. Mottillo, P. A. Julien, A. Puškarić, T. Friščić and I. Halasz, Cryst. Growth Des., 2016, 16, 2342–2347 CrossRef.
- X. Fang, M. Yao, L. Guo, Y. Xu, W. Zhou, M. Zhuo, C. Shi, L. Liu, L. Wang, X. Li and W. Chen, ACS Sustainable Chem. Eng., 2017, 5, 10735–10743 CrossRef CAS.
- S. Lukin, T. Stolar, M. Tireli, M. V. Blanco, D. Babić, T. Friščić, K. Užarević and I. Halasz, Chem.–Eur. J., 2017, 23, 13941–13949 CrossRef CAS PubMed.
- H. Kulla, S. Greiser, S. Benemann, K. Rademann and F. Emmerling, Molecules, 2016, 21, 917 CrossRef PubMed.
- T. Friščić, S. L. Childs, S. A. A. Rizvi and W. Jones, CrystEngComm, 2009, 11, 418–426 RSC.
- G. A. Bowmaker, Chem. Commun., 2013, 49, 334–348 RSC.
- D. Hasa and W. Jones, Adv. Drug Delivery Rev., 2017, 117, 147–161 CrossRef CAS PubMed.
- E. G. Prout and F. C. Tompkins, Trans. Faraday Soc., 1944, 40, 488–498 RSC.
- F. Mata-Perez and J. F. Perez-Benito, J. Chem. Educ., 1987, 64, 925 CrossRef CAS.
- S. Basu, U. S. Shivhare and A. S. Mujumdar, Drying Technol., 2006, 24, 917–930 CrossRef.
- J. Andersen and J. Mack, Angew. Chem., 2018, 130, 13246–13249 CrossRef.
- J. M. Andersen and J. Mack, Chem. Sci., 2017, 8, 5447–5453 RSC.
- A. A. L. Michalchuk, I. A. Tumanov and E. V. Boldyreva, CrystEngComm, 2013, 15, 6403–6412 RSC.
- H. Kulla, F. Fischer, S. Benemann, K. Rademann and F. Emmerling, CrystEngComm, 2017, 19, 3902–3907 RSC.
- N. Bouvart, R.-M. Palix, S. G. Arkhipov, I. A. Tumanov, A. A. L. Michalchuk and E. V. Boldyreva, CrystEngComm, 2018, 20, 1797–1803 RSC.
- K. Užarević, N. Ferdelji, T. Mrla, P. A. Julien, B. Halasz, T. Friščić and I. Halasz, Chem. Sci., 2018, 9, 2525–2532 RSC.
- A. A. L. Michalchuk, I. A. Tumanov and E. V. Boldyreva, CrystEngComm, 2019, 21, 2174–2179 RSC.
- H. Kulla, A. A. L. Michalchuk and F. Emmerling, Chem. Commun., 2019, 55, 9793–9796 RSC.
- H. Kulla, S. Haferkamp, I. Akhmetova, M. Röllig, C. Maierhofer, K. Rademann and F. Emmerling, Angew. Chem., Int. Ed., 2018, 57, 5930–5933 CrossRef CAS PubMed.
- E. Colacino, M. Carta, G. Pia, A. Porcheddu, P. C. Ricci and F. Delogu, ACS Omega, 2018, 3, 9196–9209 CrossRef CAS PubMed.
- A. Stolle, Technical Implications of Organic Syntheses in Ball Mills, in Ball Milling Towards Green Synthesis: Applications, Projects, Challenges, ed. A. Stolle and B. Ranu, RSC Publishing, 2014, pp. 241–276 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc06224f |
| ‡ Sigmoidal profile of product formation for solvent-free (including mechanochemical) reactions can be also caused by kinetics related to product nucleation and crystal growth, and such phenomena (for example polymorph transformation or nucleation of metal–organic frameworks) have been observed in situ by both synchrotron X-ray diffraction, as well as by Raman spectroscopy, see: (a) G. Thomas and H. Dehbi, Mater. Chem. Phys., 1986, 15, 1–13; (b) T. Friščić, I. Halasz, P. J. Beldon, A. M. Belenguer, F. Adams, S. A. J. Kimber, V. Honkimäki and R. E. Dinnebier, Nat. Chem., 2013, 5, 66 and ref. 24. |
| This journal is © The Royal Society of Chemistry 2020 |