
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA simple and efficient approach toward deep-red to near-infrared-emitting iridium(III) complexes for organic light-emitting diodes with external quantum efficiencies of over 10%†
Zhao
Chen‡
 *a,
Hongyang
Zhang‡
bc,
Dawei
Wen
a,
Wenhai
Wu
a,
Qingguang
Zeng
a,
Shuming
Chen
*d and
Wai-Yeung
Wong
*bc
*a,
Hongyang
Zhang‡
bc,
Dawei
Wen
a,
Wenhai
Wu
a,
Qingguang
Zeng
a,
Shuming
Chen
*d and
Wai-Yeung
Wong
*bc
aSchool of Applied Physics and Materials, Wuyi University, Jiangmen 529020, P. R. China. E-mail: chenzhao2006@163.com
bDepartment of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University (PolyU), Hung Hom, Hong Kong, P. R. China. E-mail: wai-yeung.wong@polyu.edu.hk
cPolyU Shenzhen Research Institute, Shenzhen, 518057, P. R. China
dDepartment of Electrical and Electronic Engineering, Southern University of Science and Technology, Shenzhen, 518055, P. R. China. E-mail: chen.sm@sustc.edu.cn
First published on 3rd February 2020
Abstract
While the external quantum efficiency (EQE) of iridium(III) (Ir(III)) phosphor based near-infrared organic light-emitting diodes (NIR OLEDs) has been limited to 5.7% to date, there is no significant EQE improvement for these types of OLEDs due to the lack of efficient Ir(III) emitters. Here, a convenient approach within three synthetic steps is developed to afford two novel and efficient deep-red to near-infrared (DR-NIR) emitting phosphors (CNIr and TCNIr), in which a cyano group is added into a commercial red emitter named Ir(piq)2(acac) to significantly stabilize the lowest unoccupied molecular orbitals of the newly designed Ir(III) complexes. They emit strong DR-NIR phosphorescence emissions at a wavelength of around 700 nm, with relatively high absolute quantum efficiencies of around 45% for their doped films. DR-NIR OLEDs made using CNIr and TCNIr exhibit high-efficiencies, affording peak EQEs of 10.62% and 9.59% with emission peak wavelengths of 690 and 706 nm, respectively. All these devices represent the most efficient Ir(III)-based DR-NIR OLEDs with a similar color gamut. The simplified synthesis procedure of the DR-NIR-emitting phosphors in conjunction with their excellent performance in OLEDs confirms our efficient strategy to achieve the DR-NIR-emitting Ir(III) phosphors.
Introduction
Due to their great potential for applications in photodynamic therapy, signal processing, night-vision devices and information-secured displays, near-infrared (NIR) light-emitting organic materials and their devices have aroused growing interest recently.1–4 Thermally activated delayed fluorescence (TADF) organics5–8 and transition metal based phosphors,9–11 which convert both 25% singlet and 75% triplet excited states into light emissions, are commonly used to achieve efficient NIR emitters. It is a real challenge to attain NIR organic light-emitting diodes (NIR OLEDs) with external quantum efficiencies (EQEs) of over 10% due to the lack of efficient NIR emitters.1 To date, NIR OLEDs based on TADF organics, platinum(II) (Pt(II)) and osmium(II) (Os(II)) phosphors can achieve this goal.5,7–9,11–13 In general, the TADF emitter based NIR OLEDs exhibit very low luminance values to date. For example, by using an acenaphtho[1,2-b]pyrazine-8,9-dicarbonitrile (APDC) acceptor and two diphenylamine (DPA) donor units, Liao and co-workers synthesized NIR-emitting 3,4-bis(4-(diphenylamino)phenyl)ace-naphtho[1,2-b]pyrazine-8,9-dicarbonitrile (APDC-DTPA). Its NIR-emitting device shows a high EQE of 10.19% with an emission wavelength of 693 nm but a maximum luminance of around 100 cd m−2.7 It is noted that these types of OLEDs exhibit significant efficiency roll-offs at high current densities. Meanwhile, the NIR emission of the Pt(II) phosphor is from the excited dimer and/or oligomer (excimers) due to the square-planar geometry of the Pt(II) complexes.9,11 For example, by using 2-(3-(trifluoromethyl)-1H-pyrazol-5-yl)pyridine (fppz), 4-(tert-butyl)-2-(3-(trifluoromethyl)-1H-pyrazol-5-yl)pyridine (tbfppz) and 2-(3-(trifluoromethyl)-1H-pyrazol-5-yl)pyrazine (fprpz) as the cyclometalated ligands, Chi and co-workers designed three NIR-emitting Pt(II) phosphors with an emission wavelength of over 700 nm in their neat films. The nondoped OLEDs afforded a world-record EQE of around 24% at the emission wavelength of 740 nm. However, in such a case, a nondoped emissive layer (EML) consisting of a high concentration of the Pt(II) emitter is generally used to achieve excimer based NIR OLEDs, which significantly increases the cost of these types of devices and may induce an undesired efficiency roll-off due to the self-quenching of the long-lifetime triplet excitons inside the EML.11 Moreover, current studies demonstrate that the NIR OLEDs made using Os(II) complexes, which possess a stronger metal-to-ligand charge transfer (MLCT) contribution and less intermolecular interaction due to their octahedral configuration for efficient phosphorescence emissions, exhibit an excellent performance, giving the highest EQE of 11.5% and an emission wavelength of 710 nm. Remarkably, the efficiency roll-offs for these NIR OLEDs are finely restricted.13 It seems that the Os(II) phosphors could be the best candidates for efficient and stable NIR OLEDs if the cost of the starting material dodecacarbonyltriosmium (Os3(CO)12) is low. Therefore, these problems (such as low brightness values, serious efficiency roll-offs and high cost) will restrict the potential applications of these NIR-emitting materials and their electroluminescent (EL) devices.It is well known that the phosphorescent OLEDs based on iridium(III) (Ir(III)) complexes, which have been proved to be one of the most efficient triplet emitters in the visible region, exhibit unexpectedly high efficiencies, satisfactory brightness values, and restricted efficiency roll-offs when the Ir(III) phosphors were doped into the host materials with a low consumption of emitters.14–16 Meanwhile, these types of emitters also adopt an octahedral geometry which is the same as the Os(II) complexes. Besides, the metal salt iridium chloride hydrate (IrCl3·3H3O) as the starting material for the preparation of the Ir(III) phosphor is relatively low-cost. Therefore, the use of NIR-emitting Ir(III) phosphors to achieve NIR OLEDs may overcome the drawbacks arising from TADF and Pt(II) emitters very well. However, the NIR-emitting iridium(III) (Ir(III)) phosphors have been confronted with a lot of problems, such as an insufficient strategy for the design of their chemical structures, inferior photoluminescence quantum yields (PLQYs) and extremely low EQEs of their OLEDs.17–21 By considering the medical and optical applications,22,23 development of more efficient NIR Ir(III) phosphors is urgently needed.
In general, only cyclometalated ligands with extensively conjugated rings can be used to synthesize NIR-emitting Ir(III) complexes.17–21 For example, Cao and his co-workers designed two NIR-emitting Ir(III) complexes, namely (fldpqx)2Ir(acac) and (thdpqx)2Ir(acac), by using 2,3-bis(4-(9,9-bis((methoxymethoxy)methyl)-9H-fluoren-2-yl)phenyl)-6,7-bis(9,9-diethyl-9H-fluoren-3-yl)quinoxaline and 2,3-bis(4-(9,9-bis((methoxymethoxy)methyl)-9H-fluoren-2-yl)phenyl)-6,7-bis(5-hexylthiophen-2-yl)quinoxaline as the cyclometalated ligands (Scheme 1). The PLQYs of (fldpqx)2Ir(acac) and (thdpqx)2Ir(acac) were 18.6% and 14.9%, respectively. Their NIR OLEDs fabricated using a solution process afforded a peak EQE of 5.7%.19 Later, two NIR emitting Ir(III) complexes synthesized by using a cyclometalated ligand consisting of an extensively conjugated ring (benzo[g]phthalazine) were demonstrated by Qiao and co-workers. These phosphors (fac-Ir(dtbpa)3 and fac-Ir(Ftbpa)3) exhibited low-energy emissions with emission wavelengths of over 750 nm and PLQYs of around 17.3% but their NIR OLEDs only gave EQEs of around 4.5%.20 Besides, other NIR-emitting Ir(III) phosphors listed in Scheme 1 exhibit unsatisfactory PL and electroluminescence (EL) properties. Undesirably, the use of these ligands based on the extensive conjugation, on the one hand, makes the synthesis procedure of the phosphors more challenging because cumbersome and multistep synthesis procedures are commonly needed.17–21 On the other hand, it weakens the triplet metal-to-ligand charge-transfer (3MLCT) transitions of these Ir(III) complexes, leading to unsteady triplet excited states which are readily decayed to their ground states through the nonradiative channels by virtue of the energy-gap law and hence resulting in inferior PLQYs.11,24 It is reasonable that the EQEs (the world-record value of 5.7% with an emission wavelength of 690 nm (ref. 19)) of the Ir(III)-based NIR OLEDs are far behind those of the devices made using the TADF organics or Pt(II) phosphors.7–9 To deal with these problems (vide supra), one feasible solution is designing NIR-emitting Ir(III) phosphors by using simple chemical structures, which not only simplifies their synthesis procedures but also strengthens their triplet excited states for efficient phosphorescence emissions, which can favour the development of easily prepared and high-performance NIR phosphors.25,26
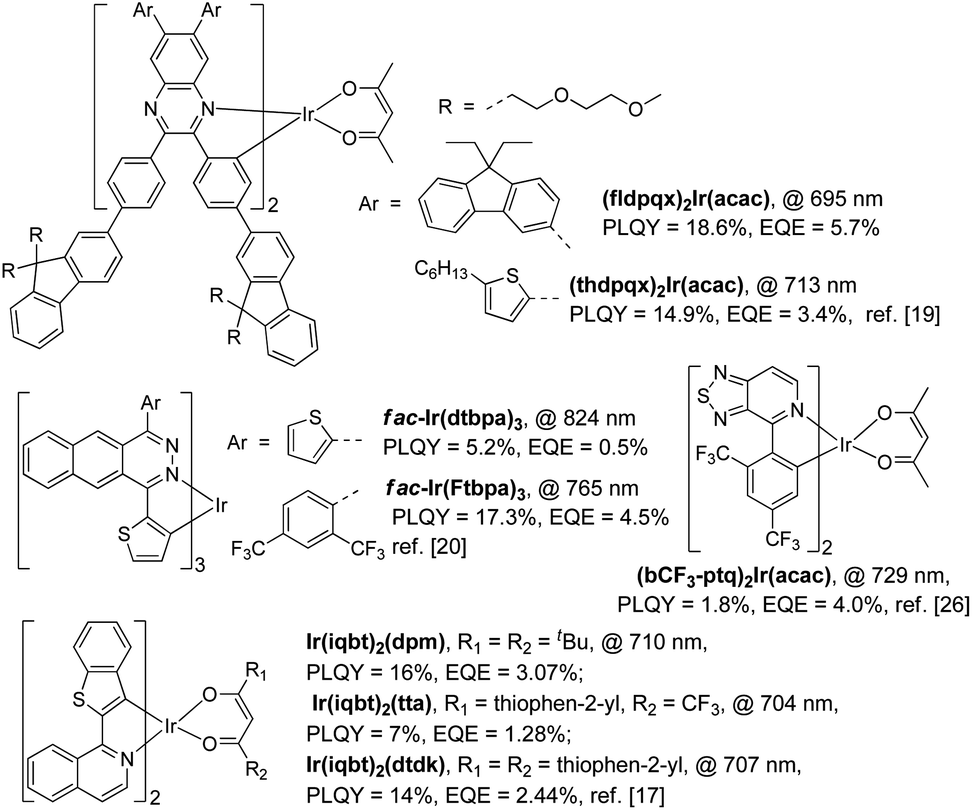 | ||
| Scheme 1 The representative near-infrared-emitting iridium(III) complexes. | ||
Herein, we demonstrate a novel and efficient strategy to design NIR Ir(III) phosphors by adding a cyano (CN) group on the isoquinoline part of a red emitter Ir(piq)2(acac) (Scheme 2), a commercial phosphor with an emission wavelength of around 625 nm.27 A strong electron-withdrawing CN group is used to decrease the LUMO of its Ir(III) complex,26,28 corresponding to a narrow emission energy gap (Eg) and a long wavelength emission. Meanwhile, in contrast to the traditional strategy to design NIR-emitting Ir(III) phosphors based on the extensive conjugation, this work aims at strengthening the triplet excited states of the NIR-emitting phosphors by using the structurally simple Ir(III) complexes.20,25 The newly designed Ir(III) phosphors, Ir(piqCN)2(acac) (CNIr) and Ir(tiqCN)2(acac) (TCNIr) (Scheme 2), exhibit the most efficient phosphorescence emissions (PLQYs ∼ 45%) in doped films with emission wavelengths of around 700 nm, corresponding to the deep-red to NIR (DR-NIR) region. It is noted that the DR-NIR OLEDs fabricated using CNIr and TCNIr afford peak EQEs of 10.62% and 9.59%, respectively, and are the most efficient Ir(III)-based DR-NIR OLEDs to date and comparable to the devices fabricated by adopting TADF, Pt(II) and Os(II) emitters.1,7,9,19,20
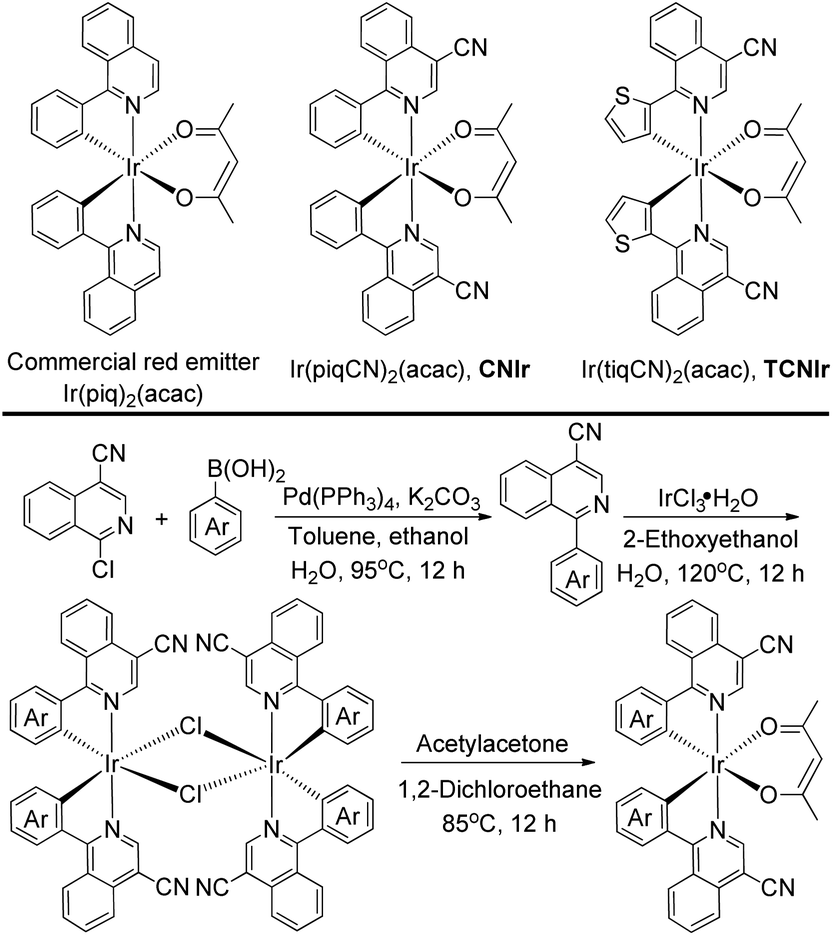 | ||
| Scheme 2 The chemical structures and synthesis route of the DR-NIR-emitting CNIr and TCNIr. | ||
Results and discussion
According to the synthesis route shown in Scheme 2, the phosphors were prepared in three steps, confirming the efficient and convenient strategy to attain the NIR-emitting Ir(III) phosphors in this work. Firstly, the cyclometalated ligands were obtained through the Suzuki coupling reaction in the presence of a palladium catalyst. Secondly, the μ-chloro-bridged dimers were synthesized through the reaction between the cyclometalated ligands and IrCl3·3H2O. Lastly, the dimers were allowed to react with acetylacetone to produce CNIr and TCNIr (see Experimental).26In tetrahydrofuran (THF) solution, CNIr and TCNIr exhibit strong absorptions with larger molar extinction coefficients (ε > 1.0 × 104 M−1 cm−1) in the wavelength region from 200 to 400 nm, which are attributed to the ligand-centered (LC) π–π* transitions (Fig. 1a). The values of ε gradually decrease in the absorption spectrum of CNIr when the wavelength is beyond 400 nm, corresponding to the MLCT transition.29,30 In contrast to CNIr, TCNIr exhibits a significantly stronger absorption (ε = 1.12 × 104 M−1 cm−1) at a wavelength of around 586 nm. It may be due to the transition state resulting from the charge transfer (CT) from electron-donating thienyl ring to the electron-withdrawing CN moiety in the cyclometalated ligand.31 Meanwhile, the emission peaks of CNIr and TCNIr are found at 696 and 708 nm in their photoluminescence spectra (Fig. 1a and Table 1), corresponding to the DR-NIR region for CNIr and the NIR regime for TCNIr. A red-shift of about 70 nm is observed by comparing the peak emission wavelength of CNIr with that of Ir(piq)2(acac) (that is at around 625 nm).27 In comparison with the PL spectra measured in the solutions at room temperature, a slight red-shift of the emission peak for CNIr (696 nm in solution and 699 nm in the film) is observed in its doped film; almost no change is found for the emission peak of the TCNIr (708 nm) doped CBP film (Fig. S1b†). Meanwhile, these peaks demonstrate a significant blue-shift (about 15 nm) when they were measured at 77 K, and are located at 681 and 694 nm for the triplet emissions of CNIr and TCNIr, respectively (Fig. S1a†). To investigate their phosphorescence performance, absolute PLQY measurements were conducted. The values are positively related to the number of photons emitted from the emitter and negatively related to the number of photons absorbed by the emitter (ESI, eqn (1)†). The absolute PLQYs of the solutions containing 10−4 M CNIr and TCNIr recorded at room temperature are 16 ± 1% and 6 ± 1% (excited at 450 nm), which are significantly inferior to those Ir(III) phosphors with the emissions in the visible region. According to the energy-gap theory, the low PLQYs of these DR-NIR phosphors are reasonable.11 In solution, it is difficult to confine the low-energy triplet excited states of the NIR phosphors, which easily decay to their ground states through the non-radiative channels. Fortunately, in their transparent doped films (15 wt% CNIr and TCNIr in the host of 4,4′-di(9H-carbazol-9-yl)-1,1′-biphenyl (CBP)), their PLQYs could reach up to 45 ± 1% and 44 ± 1%, respectively (excited at 300 nm), which are the highest values compared to those of other NIR Ir(III) phosphors (PLQY < 20%).17–21 In their doped films, CBP is used to efficiently confine the excitons and transfer them to Ir(III) phosphors for efficient phosphorescence emissions. On the other hand, the vibrations of chemical bonds in these phosphor solutions may be stronger than those in their thin films. Therefore, the PLQYs of their films are higher than those measured in their solutions.32 In the doped films, the PL decay curves are single-exponential and their lifetimes (τ) of excited states for CNIr and TCNIr at room temperature are 0.30 and 0.83 μs, respectively (Fig. S2† and Table 1). These values were significantly increased to around 19 μs when the PL decay curves for CNIr and TCNIr solutions were measured at 77 K, which corresponds to the steady triplet excited states of these phosphors.33 The calculated radiative transition rates (kr) of 1.5 × 106 and 5.3 × 105 s−1 for CNIr and TCNIr in their doped films are higher than those of most of the NIR-emitting Ir(III) phosphors. It is found that their nonradiative transition rates (knr) are much higher than kr values but they are quite smaller than the knr values of other NIR-emitting Ir(III) emitters.19,20 These mainly contribute to the high PLQYs of these newly designed phosphors17–21 and support that their triplet excited states may be subject to less nonradiative quenching compared to these Ir(III) phosphors synthesized by using extensive conjugations.11
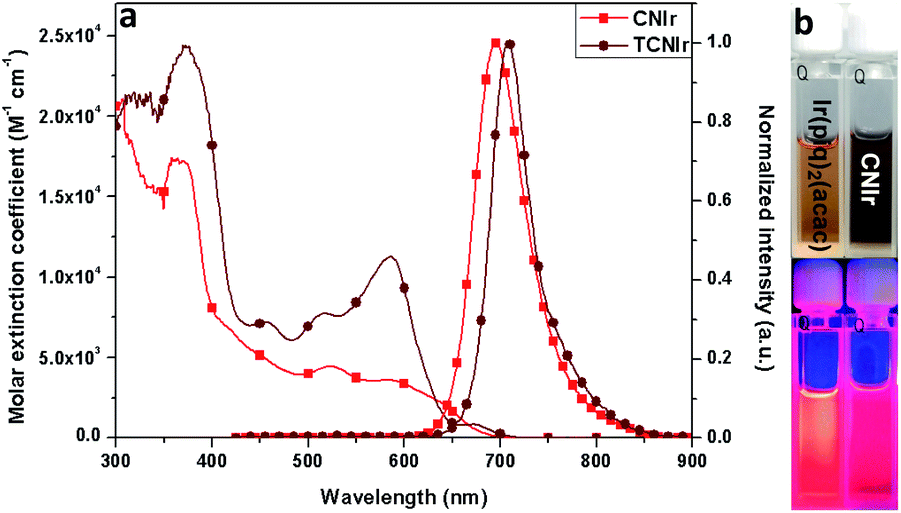 | ||
| Fig. 1 (a) The spectra of UV absorption and PL (at a concentration of 10−4 M) and (b) the images of phosphor solutions before and after UV light (λexcitation = 365 nm) radiation. | ||
| Ir | UV-absorption (ε) [nm] ([103 M−1 cm−1]) | PL [nm] | PLQY [%] | τ [μs] | k r [105 s−1] | k nr [105 s−1] | HOMO [eV] | LUMO [eV] | E T [eV] | T dec [°C] |
|---|---|---|---|---|---|---|---|---|---|---|
| a Measured in THF at room temperature with the concentration of Ir(III) phosphor at 10−4. b Measured at 77 K with the concentration of Ir(III) phosphor at 10−4 M. c Measured in films (15 wt% phosphors in CBP) at room temperature. d Calculated by using the equations of (kr + knr) = 1/τ and PLQY = kr/(kr + knr) and these values are for films. e Energy levels estimated from CV curves. f Energy levels estimated from the DFT calculation results. g The triplet energy level (ET) was estimated from the emission maxima of the PL spectra at 77 K. h Onset decomposition temperature at 5% degradation measured by thermogravimetric analysis. | ||||||||||
| CNIr | 305 (21.1), 359 (17.3), 457 (4.8), 526 (4.5), 591 (3.6), 650 (1.7) | 696a, 681b, 699c | 16 ± 1a, 45 ± 1c | 0.14a, 19.82b, 0.30c | 15.0 | 18.3 | −5.50e, −5.65f | −3.74e, −2.92f | 1.82 | 347 |
| TCNIr | 312 (21.2), 372 (24.4), 460 (7.2), 516 (7.8), 586 (11.2), 678 (0.9) | 708a, 694b, 708c | 6 ± 1a, 44 ± 1c | 0.15a, 18.74b, 0.83c | 5.3 | 6.8 | −5.53e, −5.70f | −3.81e, −2.89f | 1.79 | 273 |
As shown in Fig. 2, the LUMOs of these complexes are distributed on both N-heterocyclic isoquinoline and aromatic phenyl/thienyl rings, while the HOMOs are mainly located on the aromatic rings.21,26,34 Therefore, there is a good overlap between the HOMO and LUMO, resulting in a narrow Eg and low-energy emission. In general, the addition of the electron-withdrawing CN group will decrease the electron density on the isoquinoline part, resulting in much deeper LUMOs and HOMOs of CNIr and TCNIr. However, the influence of CN on the LUMO plays a much more significant role than that on the HOMO. Therefore, the use of CN can significantly decrease the Eg values of phosphors, changing the red-emitting Ir(piq)2(acac) to the NIR-emitting CNIr and TCNIr. The calculated HOMOs of −5.65 and −5.70 eV for CNIr and TCNIr are close to the data estimated from cyclic voltammetry (CV) curves (Fig. S3†).
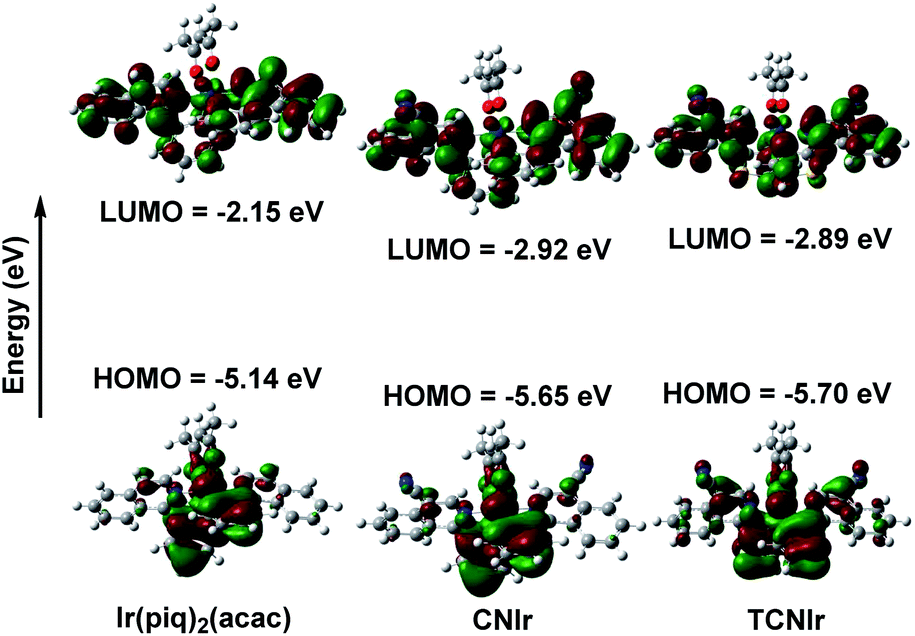 | ||
| Fig. 2 The calculated HOMO and LUMO energy levels of Ir(III) complexes. | ||
Since these phosphors have high potential for efficient NIR OLED applications, the light-emitting devices were firstly fabricated by using the architectures of ITO/HATCN (20 nm)/TAPC (40 nm)/TcTa (5 nm)/CNIr in the host (10 wt%, 30 nm)/TPBi (40 nm)/LiF (1 nm)/Al (100 nm) to investigate the performance of the OLEDs, where the host materials were TcTa (4,4′,4′′-tris(carbazol-9-yl)-triphenylamine), TPBi (1,3,5-tris(1-phenyl-1H-benzo[d]imidazol-2-yl)benzene), CBP and CDBP (9,9′-(2,2′-dimethyl-[1,1′-biphenyl]-4,4′-diyl)bis(9H-carbazole)) for D1–D4, respectively. ITO (indium tin oxide) and Al (aluminum) served as the electrodes, LiF (lithium fluoride) and HATCN (dipyrazino[2,3-f:2′,3′-h]quinoxaline-2,3,6,7,10,11-hexacarbonitrile) as the electron and hole injection layers (EIL/HIL), 4,4′-(cyclohexane-1,1-diyl)bis(N,N-di-p-tolylaniline) (TAPC) as the hole transporting layer (HTL), 5 nm TcTa as the hole conductive electron blocking layer (EBL)35 and 40 nm TPBi as the electron transporting layer (ETL). D4 shows the highest EQE of 9.89%, but the emission peak wavelength shifts to 677 nm (a blue-shift of 22 nm by comparison with the PL spectrum of CNIr) with a weak emission from the TcTa at around 380 nm (Fig. S5†).36 Therefore, the transfer of triplet excitons from CDBP (ET = 3.0 eV (ref. 37)) to CNIr (ET = 1.82 eV) in D4 is not complete, resulting in a slight exciton harvesting by TcTa (ET = 2.76 eV (ref. 36)) from CDBP. It is found that all these DR-NIR OLEDs exhibit a significant EQE roll-off under high current density, corresponding to the inferior confinement of triplet exctions or unbalanced distribution of charge carriers inside the EMLs.36–38 By considering a high EQE (8.66%) and pure emission (λEL = 683 nm) of D3 (Fig. S5†), CBP was selected as an optimal host material for further EQE improvement.
Due to the deep HOMO of CBP (HOMO = −6.0 eV, ET = 2.56 eV), another hole conductive EBL, mCP39 (1,3-di(9H-carbazol-9-yl)benzene), with a well-matched HOMO energy level, was used to fabricate D5–D8: ITO/HATCN (20 nm)/TAPC (40 nm)/mCP (5 nm)/CNIr in CBP (x wt%, 30 nm)/TPBi (40 nm)/LiF (1 nm)/Al (100 nm), where the concentrations of CNIr are 5, 10, 15 and 20 wt% for D5–D8, respectively (Fig. 3a). The use of mCP (ET > 2.9 eV) not only transfers more holes inside the EML to form a balanced recombination of holes and electrons but also efficiently confines the triplet excitons formed on CBP so as to completely transfer them into CNIr for efficient triplet emission. Therefore, all these devices display relatively high EQEs of over 10% (10.51% for D5, 10.52% for D6, 10.62% for D7 and 10.18% for D8) (Fig. 3d, S6† and Table 2), which are the champion EQEs among those of the devices fabricated using other DR-NIR-emitting Ir(III) phosphors17–21 and are comparable to those devices made based on TADF, Pt(II) and Os(II) emitters7–9 while the colour gamut is similar. The concentration of CNIr has a significant effect on the emission peak wavelength of the devices, which is gradually shifted from 681 to 693 nm when the concentration of the dopant is increased from 5 wt% to 20 wt% (Fig. 3b). In general, the increased concentration of Ir(III) phosphor inside the EML would induce the aggregation of the emitter, resulting in a reasonably red-shifted EL emission.40 The Commission Internationale de l'Eclairage (CIE) chromaticity coordinates of these devices are listed in Table 2. It was found that these data are almost located at the edge of the red region in the CIE coordinates map (Fig. 3b, inset).
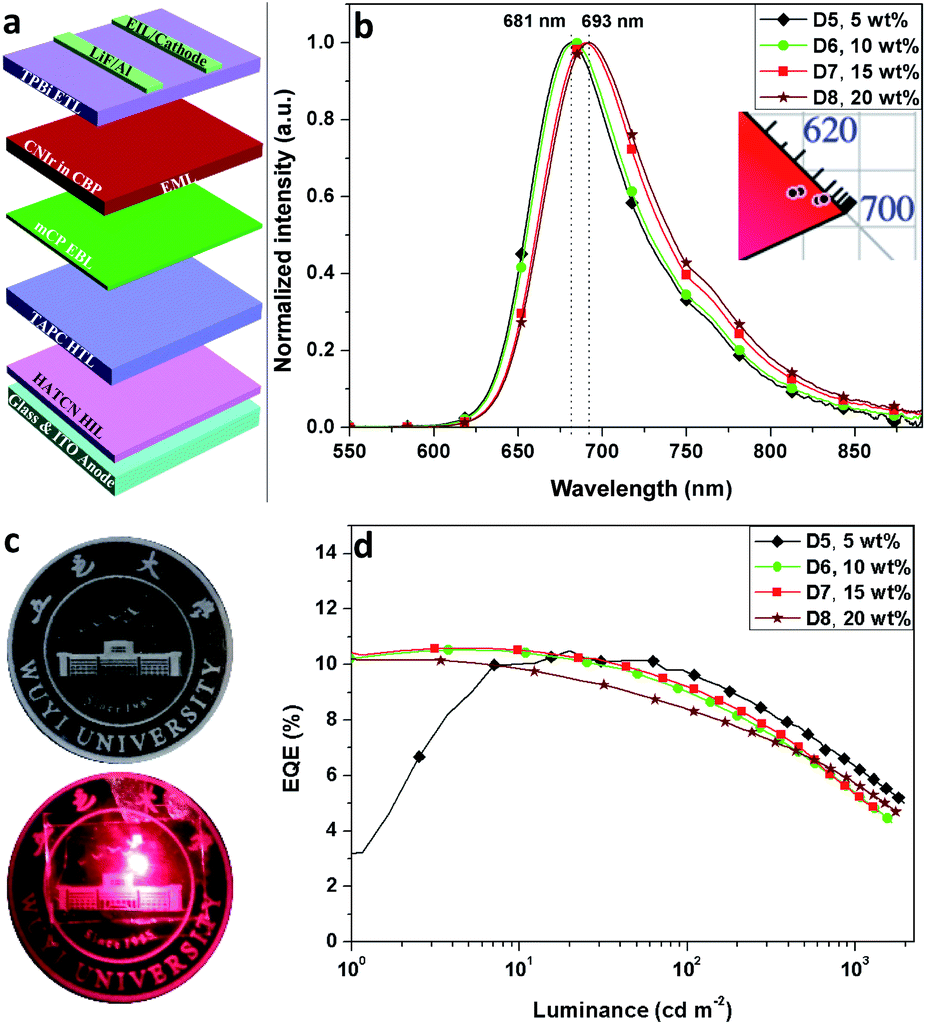 | ||
| Fig. 3 (a) Device architecture, (b) the EL spectra (inset: CIE coordinates) of D5–D8, (c) the image of emission color from D7 and (d) EQE-luminance curves of D5–D8. | ||
| Device | Emitter (x wt%) | λ EL [nm] | EQE [%] | L at 15 V [cd m−2] | CIE (x, y) | J 1/2 [mA cm−2] |
|---|---|---|---|---|---|---|
| a The peak EQE. b The EQE at the brightness values of around 500 cd m−2. c The EQE at the brightness values of around 1000 cd m−2. d The current density when the EQE was dropped to its half value. | ||||||
| D5 | CNIr (5) | 681 | 10.51a, 7.49b, 6.21c | 1967 | 0.691, 0.280 | 210 |
| D6 | CNIr (10) | 683 | 10.52a, 6.71b, 5.36c | 1665 | 0.697, 0.281 | 202 |
| D7 | CNIr (15) | 690 | 10.62a, 6.76b, 5.23c | 1367 | 0.713, 0.274 | 235 |
| D8 | CNIr (20) | 693 | 10.18a, 6.68b, 5.72c | 1746 | 0.719, 0.275 | 392 |
| D9 | TCNIr (15) | 706 | 9.59a, 1.67b | 500 | 0.707, 0.277 | 118 |
| D10 | Ir(piq)2(acac) (15) | 626 | 16.13a, 15.8b, 15.06c | 29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 230 230 |
0.680, 0.315 | 383 |
By using the same device architecture, Ir(piq)2(acac) and TCNIr (dopant concentration = 15 wt%) were also employed as the emitters to fabricate OLEDs. Although the red-emitting OLED (D10) shows a relatively high EQE of 16.13%, its emission wavelength is at around 628 nm, with a blue-shift of 63 nm compared with the device made using CNIr (Fig. 4a and Table 2). Meanwhile, the NIR-emitting TCNIr afforded its OLED (D9) an emission wavelength of 706 nm with a peak EQE of 9.59% (Fig. 4a, b and S7†). It is noted that these OLEDs made by CNIr and TCNIr represent the most efficient devices fabricated by using the NIR-emitting Ir(III) complexes (Fig. 4c).
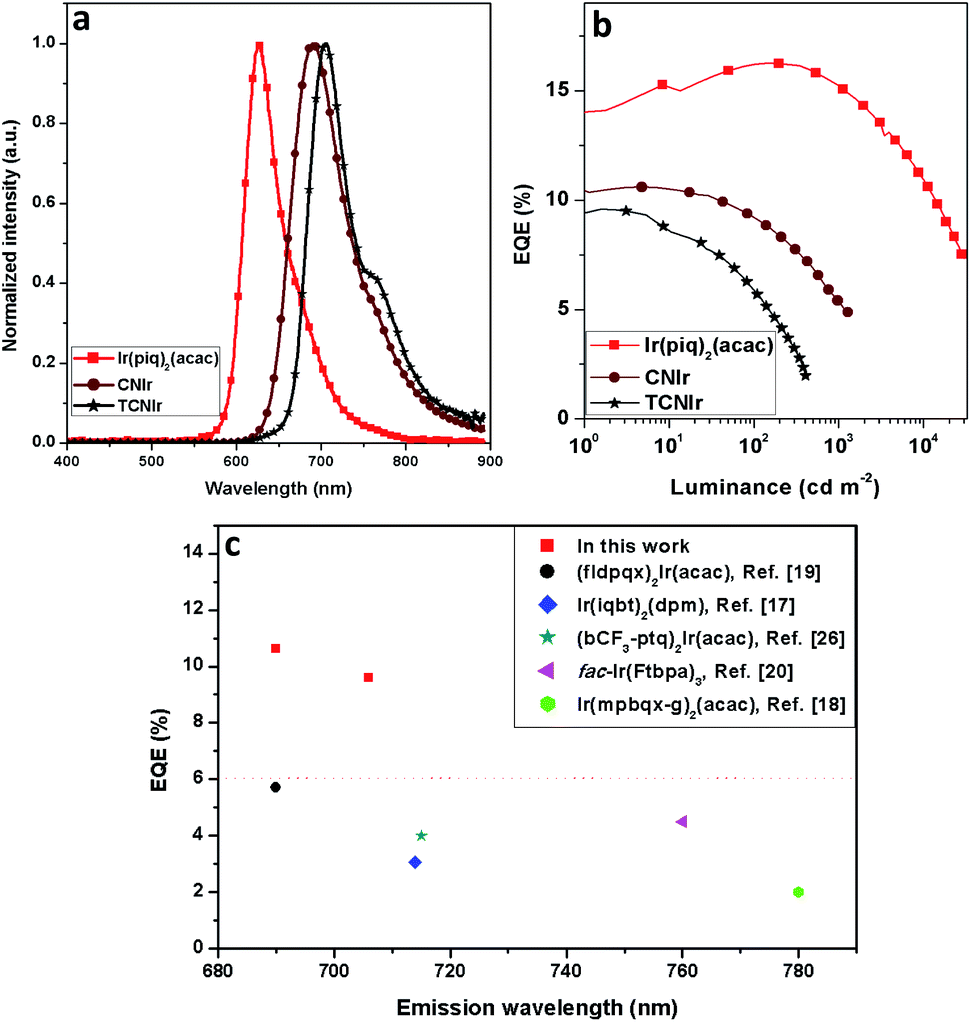 | ||
| Fig. 4 The performance of red and DR-NIR OLEDs made by Ir(piq)2(acac), CNIr and TCNIr, respectively: (a) EL spectra and (b) EQE vs. luminance curves for these devices; (c) the EQE summary of Ir(III)-based DR-NIR-emitting OLEDs with the emission peaks from 680 to 780 nm. | ||
The stability of D7 was investigated. As shown in Fig. 5a, the EL spectra exhibit an excellent stability without other impure emissions in the wavelength region from 380 to 780 nm under the gradually increased voltages from 7 to 15 V. It is noted that the EQE roll-offs of D5–D8 are finely restricted. When the peak EQEs were dropped to their half values, the current densities (J1/2) were recorded at around 210, 202, 235 and 392 mA cm−2 for D5–D8, respectively, confirming good EQE stabilities of D5–D8 when the current density was increased (Fig. 3d and Table 2). Meanwhile, the EQE of 7.90% was still maintained after measurements were performed 10 times for D7 (Fig. 5b). Moreover, it took about 17 hours (T50) for D7 to decay its initial luminance (114 cd m−2) to its half value (Fig. 5d). The operational lifetime of D7 is inferior to those of Ir(III) based green and red OLEDs. In general, the current investigation on the NIR OLEDs just focuses on the EQE improvement, but less on their stabilities. In addition, D7 gave a good EQE reproducibility and 19 out of 20 devices possessed the EQEs of over 10% (Fig. 5c). The excellent performance of these devices together with their good stabilities confirm our successful design strategy to attain efficient NIR-emitting Ir(III) phosphors. Nevertheless, for the real applications of these materials and devices, a further improvement of the performance (especially for the luminance and operational lifetime) is needed.
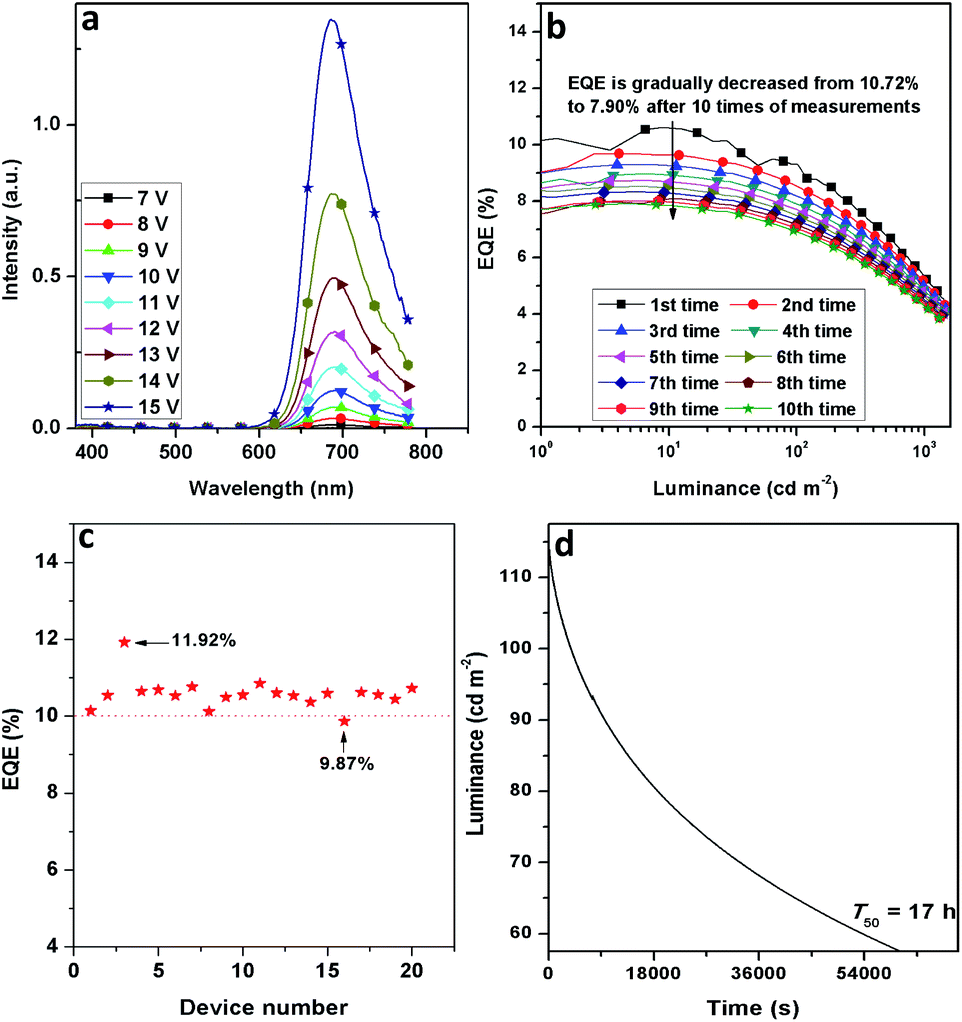 | ||
| Fig. 5 Stability of device D7: (a) EL spectra at different voltages, (b) EQE-luminance curves obtained after performing measurements 10 times, (c) EQE reproducibility and (d) operational lifetime. | ||
Experimental
General information
All reagents were used as received from commercial sources. They were weighed and handled in air. All the solvents were used directly without any treatment. 1H, 13C NMR and high resolution mass spectra (HRMS) were recorded on a Bruker AM400 spectrometer and Bruker Autoflex (Fig. S8–S13†). Chemical shifts (δ) were reported in ppm and coupling constants (J) in Hertz (Hz). The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, and m = multiplet.The synthesis of ligands and Ir(III) complexes
:ethyl acetate = 10:1, v/v) to give a white solid (243 mg, 66% yield). 1H NMR (400 MHz, CDCl3, δ): 8.98 (s, 1H), 8.25 (m, 2H), 7.92 (m, 1H), 7.71 (m, 3H), 7.57 (m, 3H); 13C NMR (100.5 MHz, CDCl3, δ): 165.0, 147.7, 138.3, 135.8, 132.7, 130.1, 129.9, 129.1, 128.8, 125.9, 124.7, 116.5, 104.9; HRMS (ESI, m/z): (M + H)+ calcd for C16H11N2, 231.0917; found: 231.0916.
:CH2Cl2 = 1:3, v/v) to provide a black solid (30 mg, 15% yield). 1H NMR (400 MHz, CDCl3, δ): 9.01 (d, J = 8.4 Hz, 2H), 8.72 (s, 2H), 8.33 (dd, J = 8 Hz, J = 0.8 Hz, 2H), 8.22 (d, J = 8.4 Hz, 2H), 7.90 (m, 4H), 6.98 (m, 2H), 6.74 (m, 2H), 6.41 (dd, J = 7.6 Hz, J = 0.8 Hz, 2H), 5.27 (s, 1H), 1.81 (s, 6H); 13C NMR (100 MHz, CDCl3, δ): 185.7, 174.3, 155.1, 146.1, 144.6, 135.7, 134.1, 133.4, 131.9, 131.1, 129.2, 128.1, 125.1, 124.6, 121.4, 115.5, 103.5, 101.3, 29.7, 28.7; MS (MALDI-TOF): calcd for C37H25IrN4O2, 750.1607; found: 750.1639.
Conclusions
In conclusion, by introducing the CN group into the commercial red phosphor Ir(piq)2(acac), the DR-NIR-emitting Ir(III) phosphor CNIr and the NIR-emitting TCNIr with very simple chemical structures can be achieved, which strengthen the triplet MLCT states to decrease the non-radiative decays of their excited states. They exhibit strong DR-NIR phosphorescence in their doped films with the PLQYs of around 45% and emission wavelengths of around 700 nm. The DR-NIR OLED fabricated using CNIr and the NIR OLED made using TCNIr exhibit excellent performance, giving the peak EQEs of 10.62% and 9.59% with the emission wavelengths of 690 and 706 nm for CNIr-based D7 and TCNIr-based D9, respectively, which are the highest among the EQEs of those devices made using Ir(III) phosphors and comparable to those measured from the NIR OLEDs based on the TADF, Pt(II) and Os(II) emitters when the color gamut is similar. It is noted that the structurally simple and newly designed DR-NIR phosphors afford the best Ir(III)-based DR-NIR OLEDs with a stable performance, confirming our efficient design strategy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Z. C. acknowledges the National Natural Science Foundation of China (No. 21901190), the Science and Technology Projects of Jiangmen (No. (2017) 307, (2017) 149, and (2018) 352), Cooperative Education Platform of Guangdong Province (No. (2016) 31) and Key Laboratory of Optoelectronic Materials and Applications in Guangdong Higher Education (No: 2017KSYS011) for the financial support. W.-Y. W. acknowledges the Hong Kong Research Grants Council (C6009-17G and PolyU 153051/17P), the National Natural Science Foundation of China (No. 51873176), Areas of Excellence Scheme of the University Grants Committee, HKSAR (AoE/P-03/08), the Hong Kong Polytechnic University (1-ZE1C) and Ms. Clarea Au (8475S) for the financial support.Notes and references
- A. Zampetti, A. Minotto and F. Cacialli, Adv. Funct. Mater., 2019, 29, 1807623 CrossRef.
- J.-X. Chen, W.-W. Tao, W.-C. Chen, Y.-F. Xiao, K. Wang, C. Cao, J. Yu, S. Li, F.-X. Geng, C. Adachi, C.-S. Lee and X.-H. Zhang, Angew. Chem., Int. Ed., 2019, 58, 14660 CrossRef PubMed.
- W.-C. Chen, B. Huang, S.-F. Ni, Y. Xiong, A. L. Rogach, Y. Wan, D. Shen, Y. Yuan, J.-X. Chen, M.-F. Lo, C. Cao, Z.-L. Zhu, Y. Wang, P. Wang, L.-S. Liao and C.-S. Lee, Adv. Funct. Mater., 2019, 29, 1903112 CrossRef.
- Y.-L. Zhang, Q. Ran, Q. Wang, Y. Liu, C. Hänisch, S. Reineke, J. Fan and L.-S. Liao, Adv. Mater., 2019, 31, 1902368 CrossRef PubMed.
- Y. Hu, Y. Yuan, Y.-L. Shi, D. Li, Z.-Q. Jiang and L.-S. Liao, Adv. Funct. Mater., 2018, 28, 1802597 CrossRef.
- D.-H. Kim, A. D'Aléo, X.-K. Chen, A. D. S. Sandanayaka, D. Yao, L. Zhao, T. Komino, E. Zaborova, G. Canard, Y. Tsuchiya, E. Choi, J. W. Wu, F. Fages, J.-L. Brédas, J.-C. Ribierre and C. Adachi, Nat. Photonics, 2018, 12, 98 CrossRef.
- Y. Yuan, Y. Hu, Y.-X. Zhang, J.-D. Lin, Y.-K. Wang, Z.-Q. Jiang, L.-S. Liao and S.-T. Lee, Adv. Funct. Mater., 2017, 27, 1700986 CrossRef.
- J. Xue, Q. Liang, R. Wang, J. Hou, W. Li, Q. Peng, Z. Shuai and J. Qiao, Adv. Mater., 2019, 31, 1808242 CrossRef PubMed.
- X. Yang, H. Guo, X. Xu, Y. Sun, G. Zhou, W. Ma and Z. Wu, Adv. Sci., 2019, 6, 1801930 CrossRef PubMed.
- K. R. Graham, Y. Yang, J. R. Sommer, A. H. Shelton, K. S. Schanze, J. Xue and J. R. Reynolds, Chem. Mater., 2011, 23, 5305 CrossRef.
- K. T. Ly, R.-W. Chen-Cheng, H.-W. Lin, Y.-J. Shiau, S.-H. Liu, P.-T. Chou, C.-S. Tsao, Y.-C. Huang and Y. Chi, Nat. Photonics, 2017, 11, 63 CrossRef.
- J.-L. Liao, Y. Chi, C.-C. Yeh, H.-C. Kao, C.-H. Chang, M. A. Fox, P. J. Low and G.-H. Lee, J. Mater. Chem. C, 2015, 3, 4910 RSC.
- Y. Yuan, J.-L. Liao, S.-F. Ni, A. K.-Y. Jen, C.-S. Lee and Y. Chi, Adv. Funct. Mater., 2019, 30, 1906738 CrossRef.
- J. Lee, H.-F. Chen, T. Batagoda, C. Coburn, P. I. Djurovich, M. E. Thompson and S. R. Forrest, Nat. Mater., 2016, 15, 92 CrossRef PubMed.
- K.-H. Kim, E. S. Ahn, J.-S. Huh, Y.-H. Kim and J.-J. Kim, Chem. Mater., 2016, 28, 7505 CrossRef.
- G.-Z. Lu, Q. Zhu, L. Liu, Z.-G. Wu, Y.-X. Zheng, L. Zhou, J.-L. Zuo and H. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 20192 CrossRef PubMed.
- S. Kesarkar, W. Mróz, M. Penconi, M. Pasini, S. Destri, M. Cazzaniga, D. Ceresoli, P. R. Mussini, C. Baldoli, U. Giovanella and A. Bossi, Angew. Chem., Int. Ed., 2016, 55, 2714 CrossRef PubMed.
- R. Tao, J. Qiao, G. Zhang, L. Duan, C. Chen, L. Wang and Y. Qiu, J. Mater. Chem. C, 2013, 1, 6446 RSC.
- X. Cao, J. Miao, M. Zhu, C. Zhong, C. Yang, H. Wu, J. Qin and Y. Cao, Chem. Mater., 2015, 27, 96 CrossRef.
- J. Xue, L. Xin, J. Hou, L. Duan, R. Wang, Y. Wei and J. Qiao, Chem. Mater., 2017, 29, 4775 CrossRef.
- M. Song, J. S. Park, Y.-S. Gel, S. Kang, J. Y. Lee, J. W. Lee and S.-H. Jin, J. Phys. Chem. C, 2012, 116, 7526 CrossRef.
- G. Qian and Z. Y. Wang, Chem.–Asian J., 2010, 5, 1006 CrossRef.
- H. Xiang, J. Cheng, X. Ma, X. Zhou and J. J. Chruma, Chem. Soc. Rev., 2013, 42, 6128 RSC.
- W. Siebrand, J. Chem. Phys., 1967, 47, 2411 CrossRef.
- C. C. Tong and K. C. Hwang, J. Phys. Chem. C, 2007, 111, 3490 CrossRef.
- Z. Chen, L. Wang, C.-L. Ho, S. Chen, S. Suramitr, A. Plucksacholatarn, N. Zhu, S. Hannongbua and W.-Y. Wong, Adv. Opt. Mater., 2018, 6, 1800824 CrossRef.
- Y.-T. Huang, T.-H. Chuang, Y.-L. Shu, Y.-C. Kuo, P.-L. Wu, C.-H. Yang and I.-W. Sun, Organometallics, 2003, 24, 6230 CrossRef.
- S. Chen, G. Tan, W.-Y. Wong and H.-S. Kwok, Adv. Funct. Mater., 2011, 21, 3785 CrossRef.
- C.-H. Hsieh, F.-I. Wu, C.-H. Fan, M.-J. Huang, K.-Y. Lu, P.-Y. Chou, Y.-H. Ou Yang, S.-H. Wu, I.-C. Chen, S.-H. Chou, K.-T. Wong and C.-H. Cheng, Chem.–Eur. J., 2011, 17, 9180 CrossRef.
- G. Li, P. Li, X. Zhuang, K. Ye, Y. Liu and Y. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 11749 CrossRef.
- N. Blouin, A. Michaud, D. Gendron, S. Wakim, E. Blair, R. Neagu-Plesu, M. Belletête, G. Durocher, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2008, 130, 732 CrossRef.
- S. Tokito, T. Lijima, Y. Suzuri, H. Kita, T. Tsuzuki and F. Sato, Appl. Phys. Lett., 2003, 83, 569 CrossRef CAS.
- H. Sasabe, J. Takamatsu, T. Motoyama, S. Watanabe, G. Wagenblast, N. Langer, O. Molt, E. Fuchs, C. Lennartz and J. Kido, Adv. Mater., 2010, 22, 5003 CrossRef CAS.
- S. Kang, J. Y. Lee, S. H. Jung, H. S. Kim, M. Y. Chae and S. H. Jin, Adv. Funct. Mater., 2009, 19, 2205 CrossRef.
- K. Udagawa, H. Sasabe, C. Cai and J. Kido, Adv. Mater., 2014, 26, 5062 CrossRef CAS.
- Y. Tao, C. Yang and J. Qi, Chem. Soc. Rev., 2011, 40, 2943 RSC.
- S. H. Kim, J. Jang and J. Y. Lee, Appl. Phys. Lett., 2007, 91, 083511 CrossRef.
- H. A. Al Attar and A. P. Monkman, Adv. Funct. Mater., 2006, 16, 2231 CrossRef CAS.
- V. Adamovich, J. Brooks, A. Tamayo, A. M. Alexander, P. I. Djurovich, B. W. D'Andrade, C. Adachi, S. R. Forrest and M. E. Thompson, New J. Chem., 2002, 26, 1171 RSC.
- H. J. Bolink, L. Cappelli, S. Cheylan, E. Coronado, R. D. Costa, N. Lardiés, M. K. Nazeeruddin and E. Ortí, J. Mater. Chem., 2007, 17, 5032 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc05492h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |