
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoexcited Pd(II) auxiliaries enable light-induced control in C(sp3)–H bond functionalisation†
Milena L.
Czyz
 a,
Geethika K.
Weragoda
b,
Tyra H.
Horngren
a,
Timothy U.
Connell
c,
Daniel
Gomez
c,
Richard A. J.
O'Hair
*a and
Anastasios
Polyzos
*ab
a,
Geethika K.
Weragoda
b,
Tyra H.
Horngren
a,
Timothy U.
Connell
c,
Daniel
Gomez
c,
Richard A. J.
O'Hair
*a and
Anastasios
Polyzos
*ab
aSchool of Chemistry, The University of Melbourne, Parkville 3010, Victoria, Australia. E-mail: anastasios.polyzos@unimelb.edu.au; rohair@unimelb.edu.au
bCSIRO Manufacturing, Research Way, Clayton, VIC 3168, Australia
cSchool of Science, RMIT University, Melbourne, Victoria 3000, Australia
First published on 23rd January 2020
Abstract
Herein we report the photophysical and photochemical properties of palladacycle complexes derived from 8-aminoquinoline ligands, commonly used auxiliaries in C–H activation. Spectroscopic, electrochemical and computational studies reveal that visible light irradiation induces a mixed LLCT/MLCT charge transfer providing access to synthetically relevant Pd(III)/Pd(IV) redox couples. The Pd(II) complex undergoes photoinduced electron transfer with alkyl halides generating C(sp3)–H halogenation products rather than C–C bond adducts. Online photochemical ESI-MS analysis implicates participation of a mononuclear Pd(III) species which promotes C–X bond formation via a distinct Pd(III)/Pd(IV) pathway. To demonstrate the synthetic utility, we developed a general method for inert C(sp3)–H bond bromination, chlorination and iodination with alkyl halides. This new strategy in auxiliary-directed C–H activation provides predictable and controllable access to distinct reactivity pathways proceeding via Pd(III)/Pd(IV) redox couples induced by visible light irradiation.
Introduction
Advances in homogeneous palladium catalysis have identified efficient and versatile manifolds for the selective functionalisation of C–H bonds. In contrast to the direct functionalisation of C(sp2)–H bonds in aromatic compounds,1 approaches to functionalisation of isolated alkyl C–H bonds (unactivated C(sp3)–H bonds) remain scarce.2 The challenges of inert C(sp3)–H bond functionalisation derive from their high bond strengths, and lack of selectivity arising from the indistinguishable steric and electronic properties of proximal C–H bonds in alkanes.2b,2c,3 A bidentate directing group (DG) strategy has been developed to address these selectivity and thermodynamic requirements for C(sp3)–H bond scission.4 Pioneering work by Daugulis5 demonstrated that bidentate auxiliaries lower the energy barrier for C–H bond cleavage6 and result in the formation of stable cyclometallated intermediates.The cyclometallated Pd(II) complexes undergo concerted, two-electron oxidative addition with aryl or alkyl halides under thermal conditions to forge the new C–C bond.7 A limitation of this approach is the two-electron Pd(II)/Pd(IV) redox cycle that generally imposes a high energy barrier to oxidative addition, usually mandating high temperatures, stoichiometric additives and extended reaction times.7h,8 A useful strategy to reduce this energy demand relies on the addition of a carbon-centred radical to cyclometallated Pd(II) complexes to generate a Pd(III) intermediate via a one-electron oxidative addition step.9 Recently, our research group reported that a Pd complex derived from a 5-chloro-8-aminoquinoline ligand undergoes single electron oxidation with an aryl radical to generate a Pd(III) intermediate, under synergistic photoredox and palladium catalysis.10 The overall Pd(II)/Pd(III)/Pd(IV) redox chemistry reduced the thermodynamic and kinetic barriers to direct C(sp3)–H functionalisation, allowing selective C–H arylation to proceed at room temperature with a range of functionalised arenes.
The Gevorgyan group11 recently established that visible light excitation of Pd complexes induces single electron transfer (SET) processes in the absence of exogenous photosensitisers, to engage new reactivity through a Pd(0)/Pd(I) pathway (Fig. 1a).12 This approach also improves the efficiency of Pd-catalysed cross coupling chemistry by significantly accelerating reaction rates.12a,13 Thus, we wondered whether cyclometallated Pd(II) complexes possess unexploited photochemical reactivity that promotes access to Pd(II)/Pd(III)/Pd(IV) redox couples via a direct excitation. We speculated that direct photoinduced one-electron oxidation of monomeric cyclometallated Pd(II) complex, following irradiation with visible light, may provide access to hybrid Pd(III)-radical species (Fig. 1b) following photoinduced electron transfer with an appropriate aryl or alkyl (pseudo)halide.14 The reactivity of this proposed hybrid Pd(III)-radical species remains unexplored particularly within the context of C–H activation.
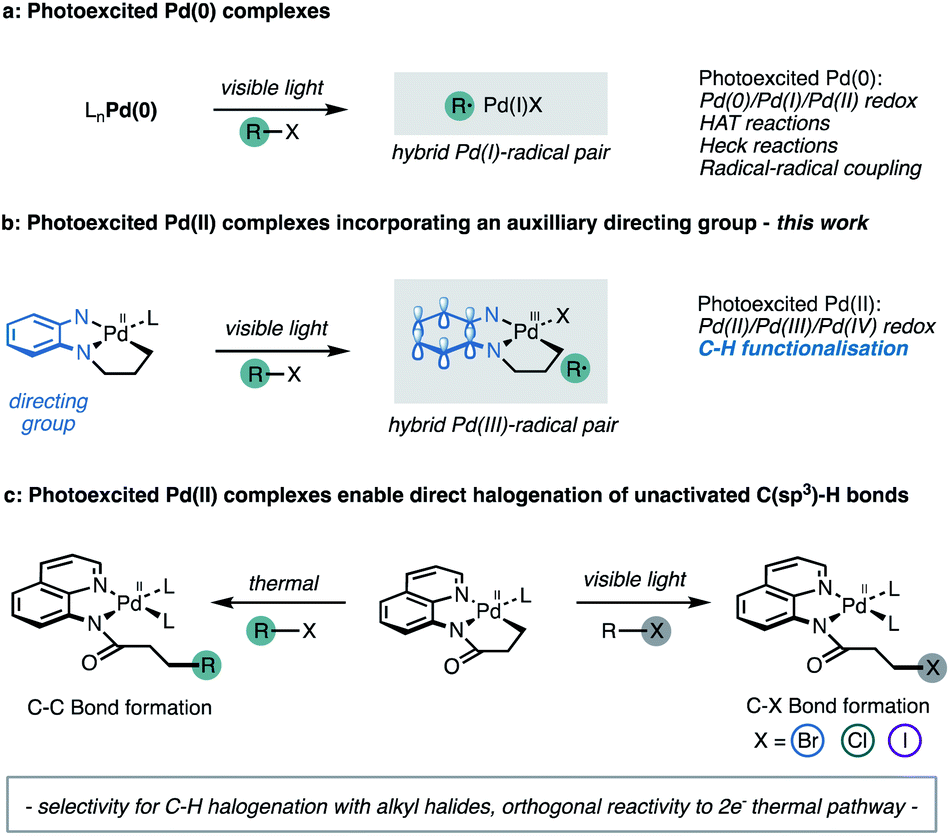 | ||
| Fig. 1 (a) Previous work. (b) Proposed hybrid Pd(III)-radical pair. (c) Reactivity control via visible light irradiation. | ||
Herein we demonstrate that cyclometallated complexes of palladium derived from the 5-chloro-8-aminoquinoline ligand possess mixed MLCT/LLCT excited states that can participate in photoinduced electron-transfer reactions. These photoactivated palladium intermediates exhibit distinct, rather than analogous reactivity relative to the conventional and thermally induced Pd(II)/Pd(IV) redox couples.5,15 Thus, these photoexcited complexes initiate electron transfer with alkyl halides leading to the halogenation of unactivated C(sp3)–H bonds (Fig. 1c). This unexpected reactivity enabled the development of synthetically relevant, and general reaction manifold for the direct halogenation of C–H bonds under mild reaction conditions.16 This offers a conceptually distinct approach towards directed C–H functionalisation, wherein the auxiliary plays a dual role of promoting selective activation of specific C–H bonds as well as absorbing visible light to allow one-electron changes to the oxidation states of the metal and ligand in the complex.
Results and discussion
Studies commenced with an investigation of the photophysical and electrochemical properties of cyclometallated Pd(II) complex derived from 5-chloro-8-aminoquinoline ligand. We selected this palladacycle because complex 1 is readily isolable, highly coloured and stable to air and moisture. X-Ray diffraction analysis confirmed that the structure is monomeric, and the metal centre has a slightly distorted square planar geometry (Fig. 2a), consistent with related Pd(II) complexes.5b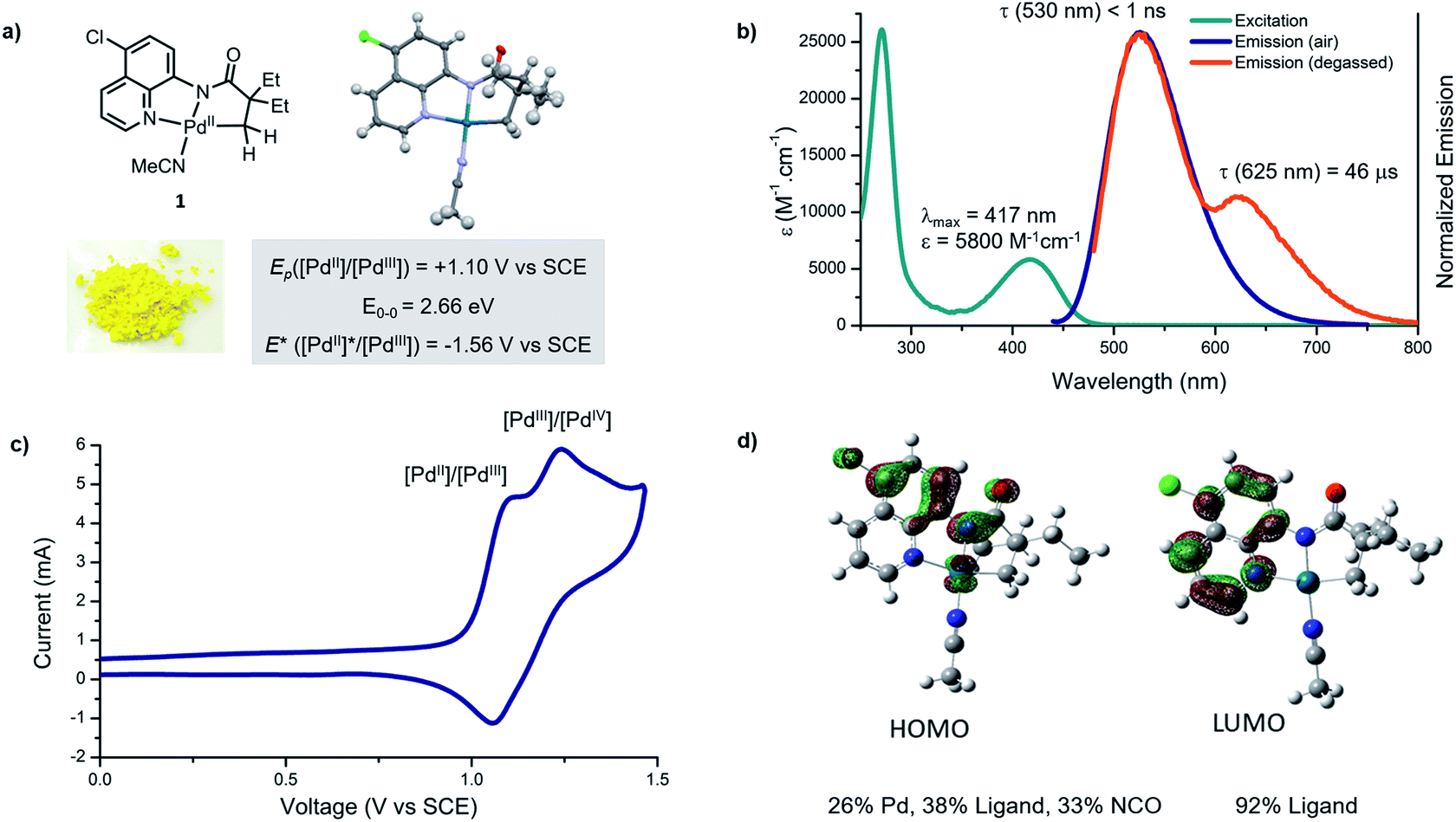 | ||
| Fig. 2 Photophysical and photochemical characteristics of palladacycle 1. (a) X-ray crystal structure and summary of redox properties in the ground and excited states; (b) absorption and emission profiles in acetonitrile (20 μM); (c) cyclic voltammogram of palladacycle 1 (1 μM) measured in degassed acetonitrile, reported relative to SCE using Fe+/0 couple as an internal standard; (d) ground state frontier molecular orbitals for the palladacycle 1 (BP86/def2-TZVP). | ||
The ultraviolet-visible absorbance spectrum of palladacycle 1 in acetonitrile (Fig. 2b) reveals two absorption features; a sharp band in the ultraviolet (λmax = 270 nm), and a broader band in the visible region (λmax = 418 nm). Based on comparison with the free ligand and other cyclometallated Pd complexes reported in the literature,17 the high energy feature likely arises from ligand-based, metal-perturbed π–π* transitions. The visible feature is assigned to mixed ligand–ligand and metal-to-ligand charge transfers, that typically give bands of medium intensity (102 to 104 M−1 cm−1) and have been observed in planar Pd(II) complexes featuring strong σ donors and low-lying, π-extended conjugated ligands.17b
Palladacycle 1 in acetonitrile is emissive at room temperature and displays a single luminescent band (λmax = 530 nm) under air (Fig. 1b). A second, less-intense and overlapping emission band (λmax = 630 nm) was observed after removal of oxygen. The excited state lifetimes of these two features are <1 ns and 46 μs, respectively. The long, triplet based emission lifetime results from the diminished spin orbit coupling of Pd relative to other metals such as Pt(II) and Ir(III), and has been observed for Pd(II) complexes with rigid metal chelating ring systems.17c,17d
Electrochemical methods were used to evaluate ground and excited state redox characteristics of palladacycle 1 (Fig. 1c). In acetonitrile, the complex features two closely overlapping quasi-reversible one electron oxidations at 1.10 and 1.24 V vs. SCE, which we attributed to Pd(II)/Pd(III) and Pd(III)/Pd(IV) redox couples, respectively. The relatively high positive potential is consistent with a monomeric structure that does not benefit from Pd–Pd interactions,18 or polydentate ligands capable of stabilising Pd(III) intermediates.19 Based on its electrochemistry and the value E0–0 = 2.66 eV (derived from the intersection of the excitation and emission profile),20 the singlet excited state reduction potential is estimated to be −1.56 V vs. SCE.
Time-dependent density functional theory (TD-DFT) calculations were performed to further probe the nature of the electronic transitions. The theoretical absorption spectra matched closely with the experimental results (Fig. S19†). The molecular orbital analysis revealed that the highest occupied molecular orbital (HOMO) receives large contributions from the π and the σ orbitals of the 8-aminoquinoline ligand (38%), and from Pd (26%) (Fig. 1d, S21 and S22†). On the other hand, the lowest unoccupied molecular orbital (LUMO) is π*-centred, indicating that the electronic transition at ca. 420 nm has mixed ligand-to-ligand (LLCT) and metal-to-ligand (MLCT) charge transfer character. Furthermore, the DFT computed 1MLCT transition in the visible manifold (420 nm, 2.81 eV) is in good agreement with the electrochemically estimated gap (2.88 eV), but is higher than the spectroscopically estimated band gap of 2.66 eV (Fig. S20†).
The reduction potential of −1.56 V vs. SCE suggests that the excited state palladacycle 1* should undergo exergonic electron transfer with activated alkyl iodides, such as perfluorobutyl iodide 2 (Ep = −1.42 V vs. SCE). Luminescence quenching experiments identified that 2 quenched both singlet and triplet emission bands of the 1* excited state (Fig. S6A†). The linear correlation of the Stern–Volmer is consistent with a purely dynamic quenching process (Fig. S6B†).20
The results of spectroscopic and electrochemical studies established that excitation of palladacycle 1 with visible light triggers a mixed ligand-to-ligand and metal-to-ligand charge transfer, followed by photoinitiated electron transfer with an activated alkyl iodide. To investigate the synthetic utility of this pathway, we probed the reactivity through combining palladacycle 1 with one equivalent of perfluorobutyl iodide 2 in acetonitrile and irradiating with 15 W blue LEDs at room temperature overnight (Table 1, entry 1). Unexpectedly, the initial experiment conditions generated the iodinated amide 3 in 54% isolated yield. Importantly, the iodinated product was not detected in the absence of irradiation at ambient temperature, or with heating at 65 °C (Table 1, entry 2–3). In both cases, cyclometallated complex 1 was recovered in quantitative yield, ruling out a 2-electron, SN2-type oxidative addition pathway. Irradiation with a wavelength beyond the absorption maximum of palladacycle 1 (530 nm, with the green LEDs) led to decreased conversion, which further supports the operation of a photon-initiated reaction (Table 1, entry 4). Furthermore, in the absence of the cyclometallated complex 1, minimal homolysis of perfluorobutyl iodide 2 under blue light irradiation was observed by 19F NMR spectroscopy, suggesting that a homolytic cleavage of perfluorobutyl iodide, followed by trapping of Pd(II) by an iodine radical, is also unlikely (Fig. S26†).21 Increasing perfluorobutyl iodide from one to two equivalents did not improve the yield of the reaction, providing further evidence that the halogenation reagent was not decomposing through direct photolysis, or unproductive radical chain propagation pathways. On the other hand, addition of one equivalent of oxone, increased the yield of 3 to 89% yield. Related oxidants, K2S2O8 and (NH4)2S2O8 were not effective (Table 1, entries 8–9), and the use of (diacetoxyiodo)benzene, a known 2-electron oxidant for C–H functionalization chemistry, led to extensive decomposition of the starting material (Table 1, entry 10). Conversely, application of a mild, one-electron oxidant, ferrocenium hexafluorophosphate, also led to a high yield of 86% (Table 1, entry 11). Silver salts were not compatible with reaction conditions and triggered formation of silver mirror within minutes of irradiation. (Table 1, entry 12) The reaction continued to furnish high yield of iodinated product 3 in the presence of molecular oxygen, suggesting reactivity is not dependent on the generation of a triplet excited state (Table 1, entry 7). Finally, we established that iodinated product 3 could not oxidise palladacycle 1 under photochemical conditions (Fig. S27†).
|
|
||||
|---|---|---|---|---|
| Entry | Light | Additive (1.0 equiv.) | Temp. [°C] | Yield [%] |
| a General conditions: palladacycle 1 (1.0 equiv.), CF3(CF2)3I (2) (1 equiv.), solvent (0.1 M in substrate), 16 h. b Yield determined by 1H NMR spectroscopy using 2,5-dimethylfuran as an internal standard. c Isolated yield. d Under air. | ||||
| 1 | 15 W blue LEDs (448 nm) | — | R.T. | 55 (54)c |
| 2 | Dark | — | R.T. | N. D. |
| 3 | Dark | — | 65 | N. D. |
| 4 | 15 W green LEDs (530 nm) | — | R.T. | 6 |
| 5 | 15 W blue LEDs (448 nm) | Run with 2 equiv. of CF3(CF2)3I | R.T. | 50 |
| 6 | 15 W blue LEDs (448 nm) | Oxone | R.T. | 89c |
| 7d | 15 W blue LEDs (448 nm) | Oxone | R.T. | 77c |
| 8 | 15 W blue LEDs (448 nm) | K2S2O8 | R.T. | 58 |
| 9 | 15 W blue LEDs (448 nm) | (NH4)2S2O8 | R.T. | 50 |
| 10 | 15 W blue LEDs (448 nm) | PhI(OAc)2 | R.T. | N. D. |
| 11 | 15 W blue LEDs (448 nm) | [FeCp2]PF6 | R.T. | 86c |
| 12 | 15 W blue LEDs (448 nm) | AgOAc | R.T. | 33 |
Further mechanistic considerations: reductive elimination
We next probed the underlying mechanism of this new C–H iodination reaction and the role of oxidants in improving the efficiency of the transformation. We posited an open-shell Pd(III) complex as the key intermediate. Both, bimetallic Pd(III) complexes with bridging acetate ligands and monomeric Pd(III) complexes, have been reported to undergo reductive elimination to form carbon–heteroatom, carbon–carbon and carbon–halogen bonds.22 However, mononuclear Pd(III) complexes have also been demonstrated to undergo instantaneous net disproportionation to generate Pd(IV) and Pd(II) species, followed by reductive elimination from Pd(IV) intermediate to form a C–C or C–X bond.22e,23In order to differentiate between these different scenarios, we turned to electrospray ionisation mass spectrometry (ESI-MS) as a means of gaining information about the key bond-forming events downstream from the initial excitation and single electron transfer events in the excited-state.24 A continuous flow photoreactor was interfaced to the ESI sources of two different mass spectrometers to enable real-time monitoring of the reaction and detection of key intermediates (Fig. 3 and S9†). To enable these studies, a charge-tag strategy was employed to enhance signal sensitivity in MS detection.25 This approach requires the installation a cationic alkylammonium group on the palladacycle, at a remote location away from the reaction site enabling signal monitoring in positive ion mode. The charge tag complex 4 was applied in the standard iodination protocol in continuous flow, which was then infused directly into the mass spectrometers. This allowed for continuous sampling of the reaction mixture over a period of 30 minutes.
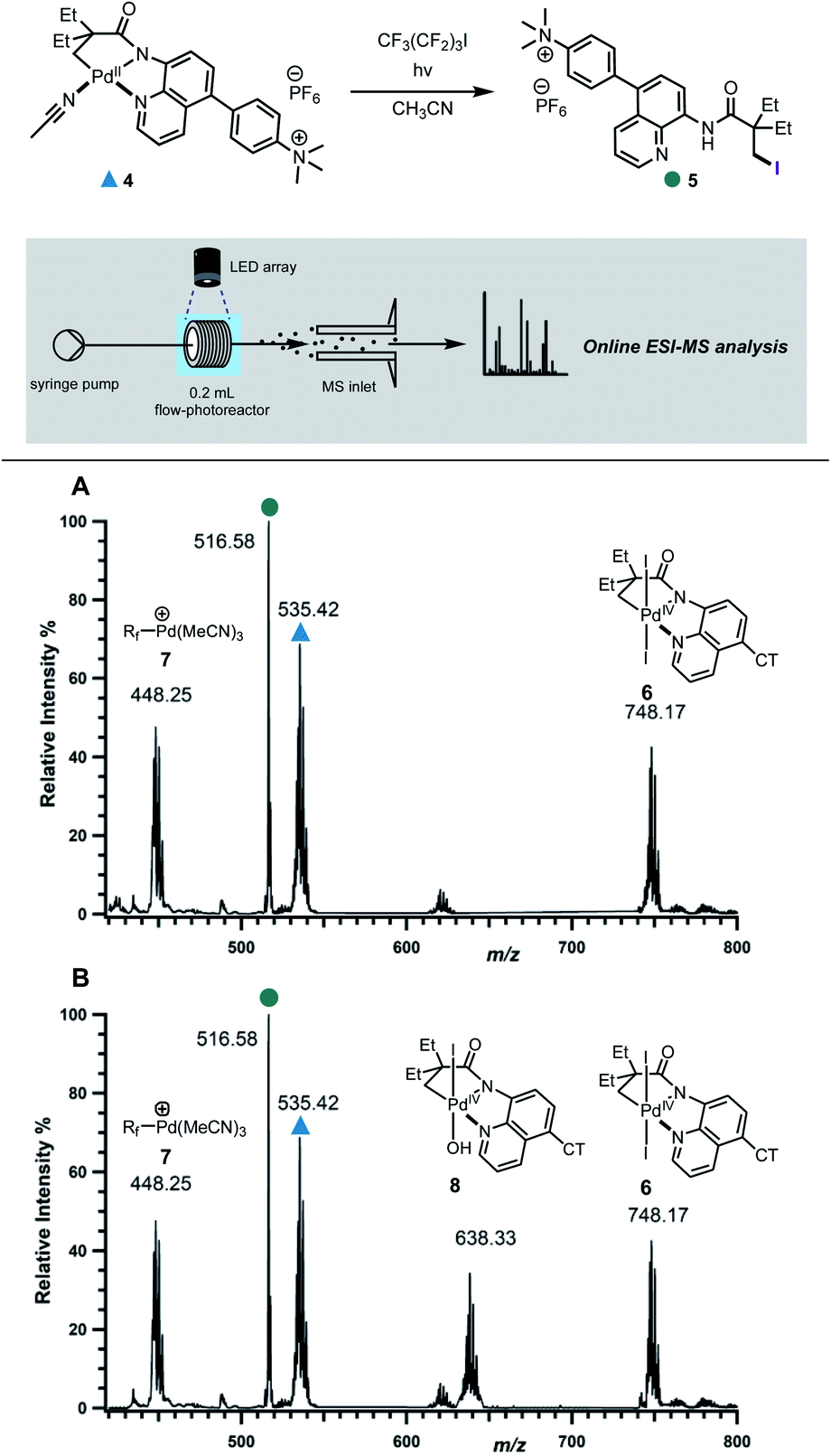 | ||
| Fig. 3 Mechanistic investigation via online photochemical ESI-MS analysis. Mass spectra of the reaction mixture in the: (A) absence of oxone taken at t = 10 min; (B) presence of oxone taken at t = 12 min. | ||
Firstly, the reaction progress was analysed in the absence of oxone. Within 6 minutes of irradiation with blue LED lamp, the precursor peak for complex 4 (m/z 535) started depleting with the concurrent formation of a new peak at m/z 748, followed by those at m/z 448 and 516 (Fig. 3A, S10 and S17†). We assigned the peak at m/z 516 to the iodinated product 5. On the other hand, the isotope pattern at m/z 748 was consistent with the formation of a new bis-iodinated palladium complex. To gain more information about its structure, collision induced dissociation (CID) was carried out. The MS/MS experiment revealed fragmentation of m/z 748 to give a new fragment at m/z 494, which corresponds to the loss of I2 (Fig. S12†). This is consistent with elimination of two iodide ions from a bis-iodinated Pd(IV) intermediate 6. Importantly, the simulated HRMS pattern for 6 matched very closely with experimental results (Fig. S14†). Furthermore, plots of relative intensity versus time are consistent with the detected Pd(IV) complex 6 being a competent intermediate in the C–H iodination reaction (Fig. S17†).
The ion at m/z 448 was found to correspond to Pd(II)-perfluorobutyl adduct 7, which results from trapping of the Pd catalyst by the perfluorobutyl radical. Interestingly, products arising from trapping of the perfluorobutyl fragment with the cyclometallated adducts were not detected, suggesting that the radical might react with palladium following the reductive elimination step.12b
We next probed the role of oxone in improving the efficiency of the reaction and its involvement in the reductive elimination step. When oxone was added as an external oxidant, a new prominent peak with palladium isotope pattern was detected at m/z 638 (Fig. 3B and S11†), consistent with Pd(IV) intermediate 8. This was further supported by the fragmentation pattern observed in the MS/MS experiment and analysis of the isotope pattern (Fig. S13 and S15†). Kinetic profiles revealed that both 8 and 6 were reactive intermediates, capable of undergoing reductive elimination to forge the new C–I bond (Fig. S18†).
Formation of Pd complex 8 in the presence of oxone can be rationalised by an inner-sphere, one-electron oxidation of Pd(III)–I intermediate with a transfer of the hydroxyl group. It should be noted that oxone is typically considered to be a two-electron oxidant, however one electron-oxidation pathways are well-established, particularly in the presence of metals.26 On the other hand, the standard 2-electron reduction potential for oxone has been reported to be E° = 1.81 V,27 which prompted further investigation into the possibility of oxone interacting with Pd(II) in the ground or excited state. Owing to a low solubility in acetonitrile, Stern–Volmer quenching experiments with oxone were deemed inconclusive; however, control experiments revealed that irradiation of complex 1 with one equivalent of oxone led to slow conversion of the starting material to hydroxylated product 9 (15% yield after 24 hours, see Table S4†). Furthermore, stirring a mixture of palladacycle, oxone and perfluorobutyl iodide at room temperature with the exclusion of light gave only 5% of the hydroxylated product 9 (Table S4†). These results suggest that ground- or excited-state oxidation of palladacycle by oxone is an unlikely mechanistic pathway for the iodination reaction.
To gain further evidence for the proposed mechanism, the reaction was analysed in the presence of ferrocenium hexafluorophosphate (Fc+), an unambiguous one electron, outer sphere oxidant. The new major intermediate was detected at m/z 331, consistent with one-electron oxidation of a transient Pd(III)–I complex through an outer sphere mechanism to give cationic Pd(IV) complex 10 (Fig. S16†). This result supports our hypothesis that intermediates 6 and 8 derive from a common precursor.
Based on the ESI-MS data, the following mechanism for the halogenation reaction was proposed (Fig. 4A). Complex 1 absorbs light in the visible region and undergoes mixed LLCT/MLCT transitions to give a highly reducing (−1.56 V vs. SCE) singlet excited state, 1*. Single electron transfer to an alkyl halide generates a transient Pd(III) intermediate and a perfluroalkyl radical. In path A, Pd(III) undergoes net disproportionation to generate Pd(IV) complex 6-Cl. For related Pd(III) complexes, disproportionation has been reported to proceed either through a homolytic cleavage of Pd(III)–ligand bond, or by direct ligand transfer between coordinatively unsaturated Pd(III) complexes.22e In path B, Pd(III) is oxidised by an added oxidant to generate Pd(IV) complex 8-Cl. Computational studies confirmed that solvent-assisted, concerted reductive elimination from both 6-Cl and 8-Cl are thermodynamically feasible, and forge the new C–I bond (Fig. 4B, S23 and S24†). Our DFT calculations also suggest that concerted C–OH reductive elimination from complex 8-Cl has a higher energy barrier (TS barrier = 35 kcal mol−1) than C–I reductive elimination (TS barrier = 9 kcal mol−1), which agrees with the observed C–H iodination selectivity under both reaction conditions (Fig. 4B).
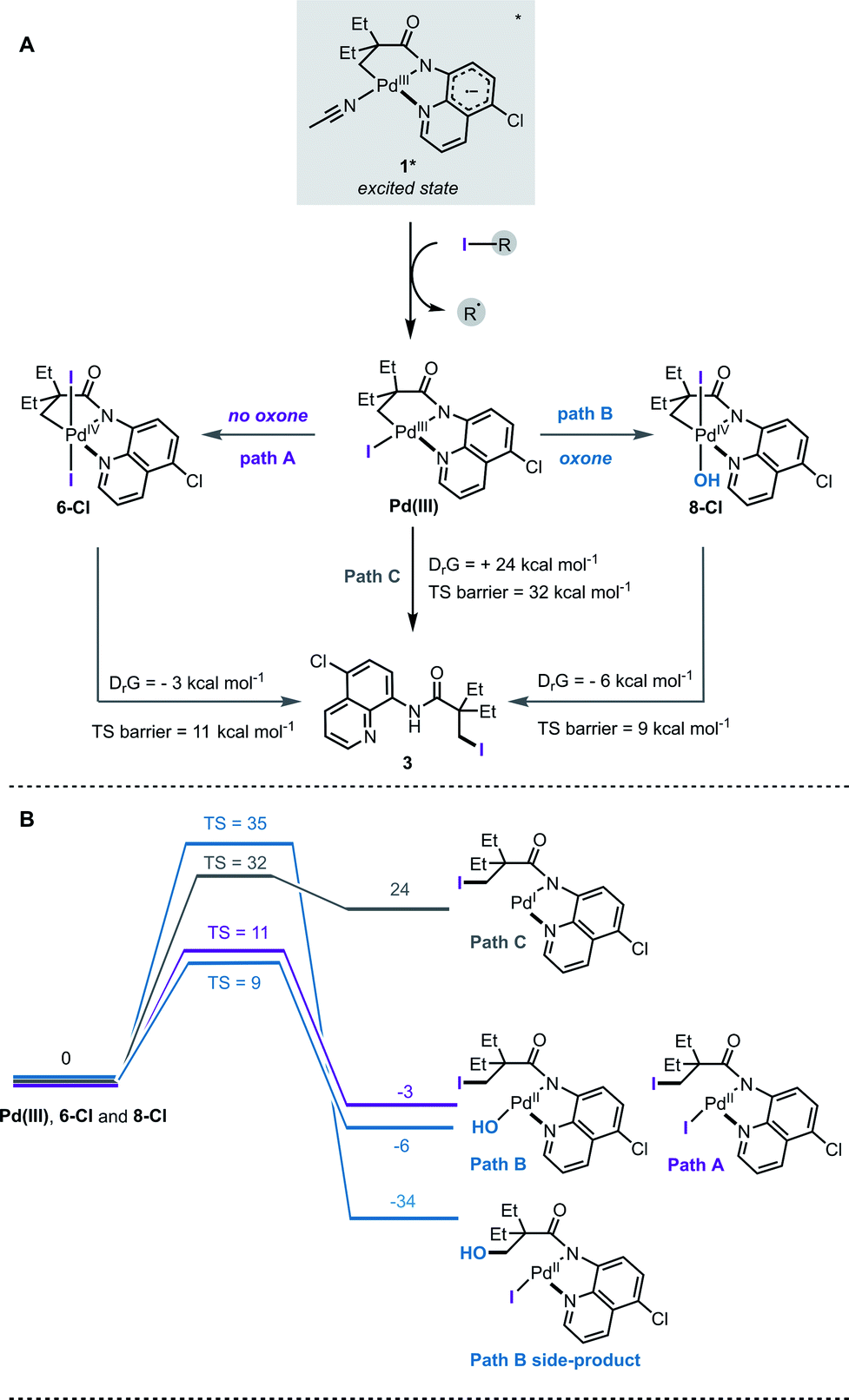 | ||
| Fig. 4 (A) Proposed mechanistic pathway in the absence of oxone (path A) and in the presence of oxone (path B), and via direct reductive elimination from Pd(III) (path C); (B) DFT computed thermodynamics of reductive elimination to form C–I bond from intermediates 6-Cl, 8-Cl and from Pd(III). All values in kcal mol−1. | ||
Observation of the Pd(IV) intermediates 6 and 8 by ESI-MS is also consistent with a relatively high energy barrier (TS = 32 kcal mol−1) for the reductive elimination from the Pd(III)–I species (path C); however we cannot exclude that all three pathways proceed simultaneously under the reaction conditions.
Reaction scope
Following investigation of the iodination mechanism, we examined the suitability of the cyclopalladated complex as a general platform for the halogenation of C(sp3)–H bonds. We hypothesised that a light-activated C–H activation pathway may provide a general route to direct halogenation (iodination, bromination and chlorination) of unactivated C(sp3)–H bonds, rather than direct alkylation that has been reported under thermal conditions.5,7i,15 Oxone was chosen as the co-oxidant for all of the reactions owing to the intense absorption of ferrocenium hexafluorophosphate in the visible region.Firstly, we synthesised a series of palladacycles by stirring the corresponding aliphatic amides with Pd(OAc)2 in acetonitrile at room temperature. Pleasingly, structural changes to the carboxylic acid moiety did not affect photoreactivity of the corresponding palladacycles (Fig. 5). The reaction was found to be highly selective for the β-methyl C(sp3)–H bonds for all substrates containing both β-methyl and β-methylene C(sp3)–H bonds, with halogenation of the β-methyl group exclusively observed (Fig. 5, products 3, 13–14, 16, 18). Longer alkyl chains (Fig. 5, products 13, 18), ether groups (Fig. 5, products 14, 17), halogens (Fig. 5, product 19), phenyl rings (Fig. 5, products 13) and saturated carbocycles (Fig. 5, product 20) and heterocycles (Fig. 5, product 17) were well tolerated. Although β-halogenation of α-halogenated substrates has been reported to be exceptionally challenging,16e α-difluoro compound was successfully iodinated under our reaction conditions to give 19 in a good yield.
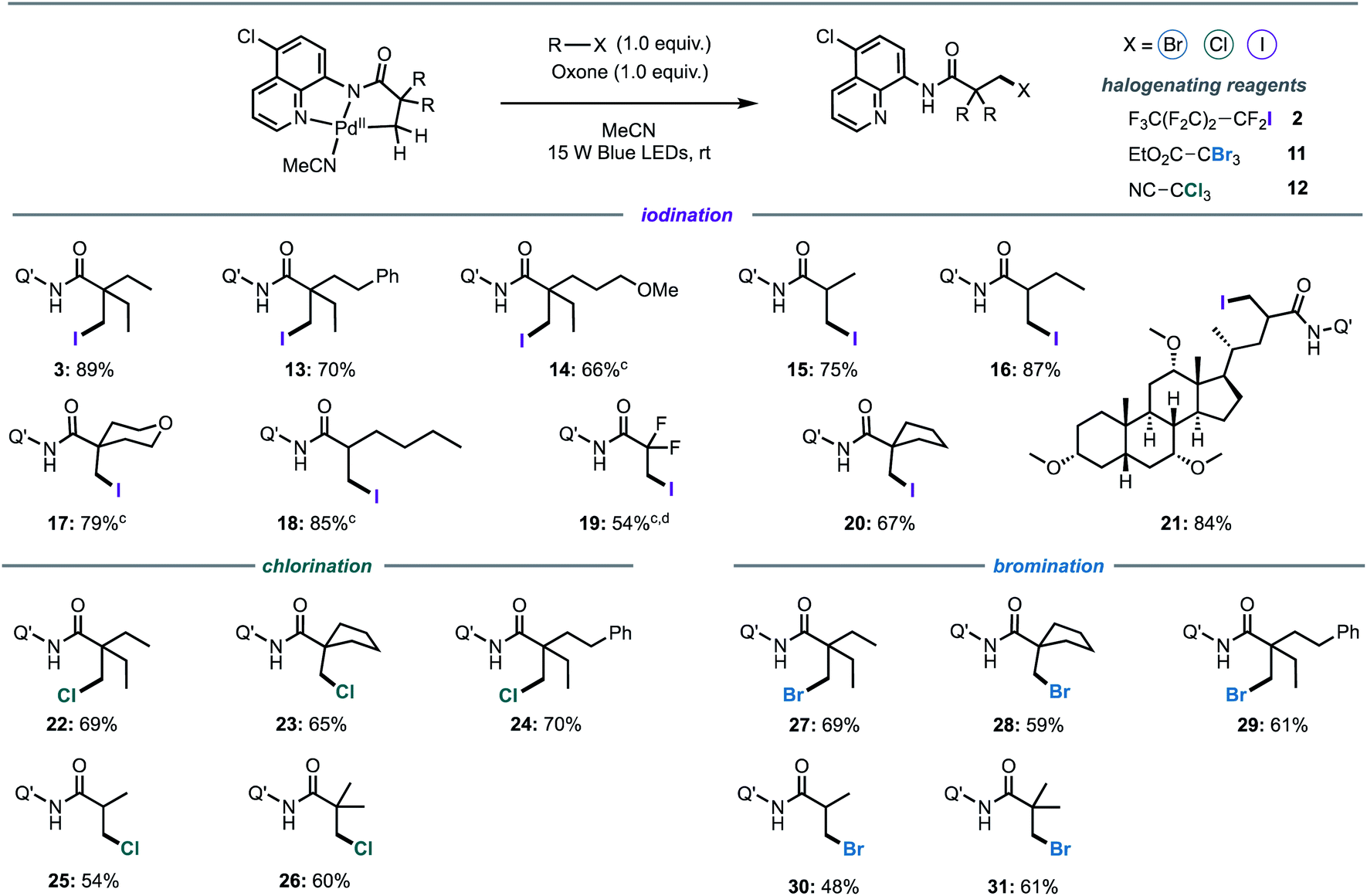 | ||
| Fig. 5 Light-promoted iodination, bromination and chlorination of aliphatic C–H bondsa,b. aGeneral conditions: R–X (1.0 equiv.) MeCN (0.1 M in palladacycle), r.t., 16 h, 15 W Blue LEDs (448 nm). bIsolated yields. Q′ = 5-chloroquinoline. cPalladacycle prepared in situ from the corresponding amide and Pd(OAc)2. dReaction run with 20 μL of DMF to improve solubility. | ||
Substrates bearing α-tertiary carbon centres are often incompatible with direct C–H halogenation due to competing α-halogenation of the more acidic proton, or elimination of the β-halogenated product.16e Pleasingly, these substrates were well tolerated furnishing the desired products in excellent yields (Fig. 5, 15–16 and 18). Competing side products were not evidenced by 1H-NMR spectroscopic analysis of reaction mixtures. Furthermore, it should be highlighted that most palladacycles derived from amides bearing α-tertiary carbon centres were not sufficiently stable to isolate, however these products could be prepared in situ from the corresponding amide and Pd(OAc)2 (see ESI for Experimental details†), suggesting that palladacycle stability does not exert a significant influence on the photophysical properties. Finally, we investigated the potential of the newly developed method for late-stage functionalisation of a complex, biologically active molecule and a cholic acid derivative was iodinated in an excellent 84% yield, and without epimerisation of the α-C–H bond (Fig. 5, product 21).
We then turned our attention to the development of bromination and chlorination protocols. A series of activated alkyl bromides and chlorides were examined under optimised conditions. It was found that ethyl tribromoacetate 11 (Ep = −0.78 V vs. SCE) and trichloroacetonitrile 12 (Ep ≅ −1.0 V vs. SCE) were the most efficient brominating and chlorinating reagents, respectively. Both bromination and chlorination reactions were compatible across a range of different substrates and functional groups (Fig. 5). Furthermore, the reaction showed excellent levels of selectivity for mono-halogenated products 26 and 31, with only traces of bis- and tris-halogenated products detected in the purified sample (see ESI for details†).
To further demonstrate synthetic utility of this methodology, we investigate possible methods for the selective removal of the auxiliary in the presence of the C–I bonds. Rao and co-workers reported that the 5-chloro-8-aminoquinoline directing group can be readily cleaved in the presence of C(sp3)–I bonds using BF3·Et2O or H2SO4 in methanol.16a Indeed, exposing iodinated product 37 to acidic conditions cleanly produced deprotected ester 37a with no evidence for the elimination or substitution of the alkyl iodide (See ESI for Experimental details†).
Catalysis
The newly discovered reactivity of the photoexcited palladacycle and elucidation of the iodination reaction mechanism paves the way for the development of C–H halogenation reaction that operates under catalytic conditions with respect to palladium. Formation of complex 7, which results from trapping of the perfluorobutyl radical by the palladium catalyst (see Fig. 3), suggested that the catalyst might be deactivated following reductive elimination. Consistent with this hypothesis, our first attempt towards a catalytic reaction gave the desired iodinated product 3 in 20% yield when amide 3a with 20 mol% Pd(TFA)2, perfluorobutyl iodide, oxone and trifluoroacetic acid in acetonitrile were irradiated with blue light. On the other hand, the use of ethyl difluoroiodoacetate as the iodination reagent provided indication of catalyst turnover, producing the desired product 3 in 30% yield under the same reaction conditions. Furthermore, the addition of mesitylene as a radical trap increased reaction yield to 46% (Scheme 1).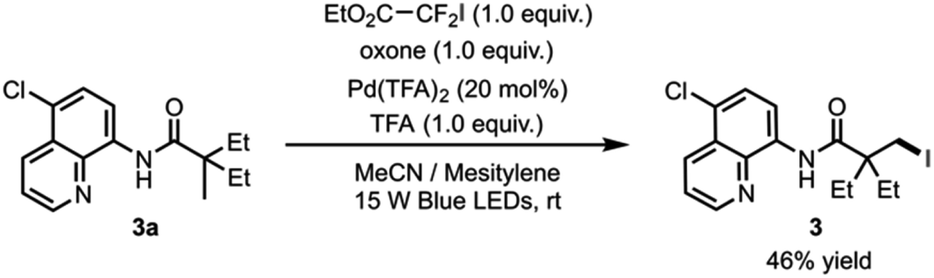 | ||
| Scheme 1 Catalytic iodination. | ||
These results suggest that the development of an efficient catalytic process is possible, if the radical by-product is effectively deactivated. Our current efforts are focussed on the design of a family of halogenation reagents compatible with the catalytic reaction conditions.
Conclusion
Here we established that cyclometallated Pd complexes derived from 5-chloro-8-aminoquinoline auxiliary, which are key intermediates for the functionalization of C(sp3)–H bonds under thermal conditions, also possess highly reactive photoexcited states that can be easily and predictably accessed via irradiation with blue light. From the excited state, the palladium complexes can participate in single electron transfer processes with organic substrates, providing access to highly reactive Pd(II)/Pd(III)/Pd(IV) redox couples. We have also established that Pd turnover is viable under light-induced conditions. These findings uncover a conceptually distinct and unconventional mechanistic paradigm for C–H activation chemistry, which should allow the design of a range of broadly useful C–H functionalisation reactions. Work is presently underway in our laboratory to identify new directing group auxiliaries to enable attenuation the photochemical properties of the resulting palladacycle intermediates, along with the development of catalytic C–H halogenation reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MLC acknowledges the ARC Industrial Transformation Training Centre Post-Doctoral Fellowship; GKW acknowledges CSIRO ResearchPlus Post-Doctoral Fellowships program; THH acknowledges the University of Melbourne, Melbourne Research Scholarship (MRS); AP acknowledges the University of Melbourne and CSIRO for the joint Establishment Grant and the ARC (IC1701000020). RAJO thanks the ARC (DP180101187). We thank the Bio21 Mass Spectrometry and Proteomics Facility for access to the Thermo OrbiTrap Fusion Lumos mass spectrometer. The authors acknowledge Prof. Jonathan White for XRD analysis, and Dr Carol Hua for advice on processing of XRD data.References
- (a) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053–1064 CrossRef CAS; (b) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef CAS PubMed; (c) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed.
- (a) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed; (b) J. C. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62–101 CrossRef CAS PubMed; (c) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754–8786 CrossRef CAS PubMed; (d) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC; (e) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC; (f) R. Jazzar, J. Hitce, A. Renaudat, J. Sofack-Kreutzer and O. Baudoin, Chem.–Eur. J., 2010, 16, 2654–2672 CrossRef CAS PubMed; (g) R. R. Karimov and J. F. Hartwig, Angew. Chem., Int. Ed., 2018, 57, 4234–4241 CrossRef CAS PubMed; (h) B. Wang, D. Qiu, Y. Zhang and J. Wang, Beilstein J. Org. Chem., 2016, 12, 796–804 CrossRef CAS PubMed.
- (a) J. F. Hartwig and M. A. Larsen, ACS Cent. Sci., 2016, 2, 281–292 CrossRef CAS PubMed; (b) J. C. K. Chu and T. Rovis, Nature, 2016, 539, 272 CrossRef; (c) M. Pfeffer and J. Spencer, in Comprehensive Organometallic Chemistry III, ed. D. M. P. Mingos and R. H. Crabtree, Elsevier, Oxford, 2007, pp. 101–166 Search PubMed.
- P. L. Alsters, P. F. Engel, M. P. Hogerheide, M. Copijn, A. L. Spek and G. van Koten, Organometallics, 1993, 12, 1831–1844 CrossRef CAS.
- (a) E. T. Nadres, G. I. F. Santos, D. Shabashov and O. Daugulis, J. Org. Chem., 2013, 78, 9689–9714 CrossRef CAS; (b) D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965–3972 CrossRef CAS.
- H. Tang, X.-R. Huang, J. Yao and H. Chen, J. Org. Chem., 2015, 80, 4672–4682 CrossRef CAS PubMed.
- (a) P. Sehnal, R. J. K. Taylor and I. J. S. Fairlamb, Chem. Rev., 2010, 110, 824–889 CrossRef CAS PubMed; (b) E. L. Weinberg, B. K. Hunter and M. C. Baird, J. Organomet. Chem., 1982, 240, 95–102 CrossRef CAS; (c) P. K. Byers, A. J. Canty, B. W. Skelton and A. H. White, J. Chem. Soc., Chem. Commun., 1986, 1722–1724 RSC; (d) B. A. Markies, M. D. Janssen, J. Boersma, G. van Koten, A. J. Canty and A. L. Spek, Recl. Trav. Chim. Pays-Bas, 1991, 110, 477–479 CrossRef CAS; (e) A. J. Canty, J. Patel, M. Pfeffer, B. W. Skelton and A. H. White, Inorg. Chim. Acta, 2002, 327, 20–25 CrossRef CAS; (f) A. J. Canty, M. C. Denney, G. van Koten, B. W. Skelton and A. H. White, Organometallics, 2004, 23, 5432–5439 CrossRef CAS; (g) J. J. Topczewski and M. S. Sanford, Chem. Sci., 2015, 6, 70–76 RSC; (h) Y. Dang, S. Qu, J. W. Nelson, H. D. Pham, Z.-X. Wang and X. Wang, J. Am. Chem. Soc., 2015, 137, 2006–2014 CrossRef CAS; (i) S.-Y. Zhang, Q. Li, G. He, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2015, 137, 531–539 CrossRef CAS; (j) A. J. Canty, M. J. G. Hettinga, J. Patel, M. Pfeffer, B. W. Skelton and A. H. White, Inorg. Chim. Acta, 2002, 338, 94–98 CrossRef CAS.
- Y. Wei, H. Tang, X. Cong, B. Rao, C. Wu and X. Zeng, Org. Lett., 2014, 16, 2248–2251 CrossRef CAS PubMed.
- (a) D. Kalyani, K. B. McMurtrey, S. R. Neufeldt and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18566–18569 CrossRef CAS PubMed; (b) S. R. Neufeldt and M. S. Sanford, Adv. Synth. Catal., 2012, 354, 3517–3522 CrossRef CAS PubMed; (c) A. Polley, K. Varalaxmi and R. Jana, ACS Omega, 2018, 3, 14503–14516 CrossRef CAS PubMed; (d) J. Jiang, W.-M. Zhang, J.-J. Dai, J. Xu and H.-J. Xu, J. Org. Chem., 2017, 82, 3622–3630 CrossRef CAS PubMed; (e) Q. Liu, X. Dong, J. Li, J. Xiao, Y. Dong and H. Liu, ACS Catal., 2015, 5, 6111–6137 CrossRef CAS; (f) M. D. Levin, S. Kim and F. D. Toste, ACS Cent. Sci., 2016, 2, 293–301 CrossRef CAS PubMed; (g) W.-M. Cheng, R. Shang, H.-Z. Yu and Y. Fu, Chem.–Eur. J., 2015, 21, 13191–13195 CrossRef CAS PubMed; (h) C. Zheng, W.-M. Cheng, H.-L. Li, R.-S. Na and R. Shang, Org. Lett., 2018, 20, 2559–2563 CrossRef CAS PubMed; (i) H.-H. Zhang, J.-J. Zhao and S. Yu, J. Am. Chem. Soc., 2018, 140, 16914–16919 CrossRef CAS PubMed; (j) T. Liu, W. Zhou and J. Wu, Org. Lett., 2017, 19, 6638–6641 CrossRef CAS PubMed.
- M. L. Czyz, D. W. Lupton and A. Polyzos, Chem.–Eur. J., 2017, 23, 14450–14453 CrossRef CAS PubMed.
- (a) M. Parasram, P. Chuentragool, D. Sarkar and V. Gevorgyan, J. Am. Chem. Soc., 2016, 138, 6340–6343 CrossRef CAS; (b) M. Parasram, P. Chuentragool, Y. Wang, Y. Shi and V. Gevorgyan, J. Am. Chem. Soc., 2017, 139, 14857–14860 CrossRef CAS; (c) D. Kurandina, M. Parasram and V. Gevorgyan, Angew. Chem., Int. Ed., 2017, 56, 14212–14216 CrossRef CAS; (d) M. Parasram and V. Gevorgyan, Chem. Soc. Rev., 2017, 46, 6227–6240 RSC.
- (a) P. Chuentragool, D. Kurandina and V. Gevorgyan, Angew. Chem., Int. Ed., 2019, 58, 2–15 CrossRef; (b) G.-Z. Wang, R. Shang, W.-M. Cheng and Y. Fu, J. Am. Chem. Soc., 2017, 139, 18307–18312 CrossRef CAS; (c) G.-Z. Wang, R. Shang and Y. Fu, Org. Lett., 2018, 20, 888–891 CrossRef CAS; (d) S. Sumino, M. Uno, H.-J. Huang, Y.-K. Wu and I. Ryu, Org. Lett., 2018, 20, 1078–1081 CrossRef CAS PubMed; (e) W.-J. Zhou, G.-M. Cao, G. Shen, X.-Y. Zhu, Y.-Y. Gui, J.-H. Ye, L. Sun, L.-L. Liao, J. Li and D.-G. Yu, Angew. Chem., Int. Ed., 2017, 56, 15683–15687 CrossRef CAS.
- R. Kancherla, K. Muralirajan, B. Maity, C. Zhu, P. E. Krach, L. Cavallo and M. Rueping, Angew. Chem., Int. Ed., 2019, 58, 3412–3416 CrossRef CAS.
- H. M. Omer and P. Liu, J. Am. Chem. Soc., 2017, 139, 9909–9920 CrossRef CAS.
- (a) K. Chen and B.-F. Shi, Angew. Chem., Int. Ed., 2014, 53, 11950–11954 CrossRef CAS; (b) S.-Y. Zhang, G. He, W. A. Nack, Y. Zhao, Q. Li and G. Chen, J. Am. Chem. Soc., 2013, 135, 2124–2127 CrossRef CAS PubMed; (c) S.-Y. Zhang, Q. Li, G. He, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2013, 135, 12135–12141 CrossRef CAS PubMed; (d) Q. Yang and S.-D. Yang, ACS Catal., 2017, 7, 5220–5224 CrossRef CAS; (e) T. Kinsinger and U. Kazmaier, Org. Lett., 2018, 20, 7726–7730 CrossRef CAS PubMed.
- (a) X. Yang, Y. Sun, T.-y. Sun and Y. Rao, Chem. Commun., 2016, 52, 6423–6426 RSC; (b) H.-Y. Xiong, D. Cahard, X. Pannecoucke and T. Besset, Eur. J. Org. Chem., 2016, 2016, 3625–3630 CrossRef CAS; (c) R. Giri, X. Chen and J.-Q. Yu, Angew. Chem., 2005, 117, 2150–2153 CrossRef; (d) R.-Y. Zhu, L.-Y. Liu and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 12394–12397 CrossRef CAS PubMed; (e) R.-Y. Zhu, T. G. Saint-Denis, Y. Shao, J. He, J. D. Sieber, C. H. Senanayake and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 5724–5727 CrossRef CAS PubMed; (f) R. K. Rit, M. R. Yadav, K. Ghosh, M. Shankar and A. K. Sahoo, Org. Lett., 2014, 16, 5258–5261 CrossRef CAS PubMed; (g) R. Giri, M. Wasa, S. P. Breazzano and J.-Q. Yu, Org. Lett., 2006, 8, 5685–5688 CrossRef CAS PubMed; (h) M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 14058–14059 CrossRef CAS PubMed; (i) X.-C. Wang, Y. Hu, S. Bonacorsi, Y. Hong, R. Burrell and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 10326–10329 CrossRef CAS PubMed.
- (a) C. S. Consorti, G. Ebeling, F. Rodembusch, V. Stefani, P. R. Livotto, F. Rominger, F. H. Quina, C. Yihwa and J. Dupont, Inorg. Chem., 2004, 43, 530–536 CrossRef CAS PubMed; (b) P.-K. Chow, W.-P. To, K.-H. Low and C.-M. Che, Chem.–Asian J., 2014, 9, 534–545 CrossRef CAS PubMed; (c) P.-K. Chow, G. Cheng, G. S. M. Tong, C. Ma, W.-M. Kwok, W.-H. Ang, C. Y.-S. Chung, C. Yang, F. Wang and C.-M. Che, Chem. Sci., 2016, 7, 6083–6098 RSC; (d) P. K. Chow, C. Ma, W.-P. To, G. S. M. Tong, S.-L. Lai, S. C. F. Kui, W.-M. Kwok and C.-M. Che, Angew. Chem., Int. Ed., 2013, 52, 11775–11779 CrossRef CAS; (e) M. Ghedini, I. Aiello, A. Crispini and M. L. Deda, Dalton Trans., 2004, 1386–1392 RSC; (f) F. S. Mancilha, L. Barloy, F. S. Rodembusch, J. Dupont and M. Pfeffer, Dalton Trans., 2011, 40, 10535–10544 RSC.
- (a) B. N. Nguyen, L. A. Adrio, T. Albrecht, A. J. P. White, M. A. Newton, M. Nachtegaal, S. J. A. Figueroa and K. K. Hii, Dalton Trans., 2015, 44, 16586–16591 RSC; (b) Y. B. Dudkina, K. V. Kholin, T. V. Gryaznova, D. R. Islamov, O. N. Kataeva, I. K. Rizvanov, A. I. Levitskaya, O. D. Fominykh, M. Y. Balakina, O. G. Sinyashin and Y. H. Budnikova, Dalton Trans., 2017, 46, 165–177 RSC.
- (a) L. M. Mirica and J. R. Khusnutdinova, Coord. Chem. Rev., 2013, 257, 299–314 CrossRef CAS; (b) J. R. Khusnutdinova, N. P. Rath and L. M. Mirica, Inorg. Chem., 2014, 53, 13112–13129 CrossRef.
- (a) L. Buzzetti, G. E. M. Crisenza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 3730–3747 CrossRef CAS PubMed; (b) D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803–5820 RSC.
- P. B. Arockiam, L. Guillemard and J. Wencel-Delord, Adv. Synth. Catal., 2017, 359, 2571–2579 CrossRef CAS.
- (a) D. C. Powers and T. Ritter, Nat. Chem., 2009, 1, 302 CrossRef CAS PubMed; (b) D. C. Powers, D. Benitez, E. Tkatchouk, W. A. Goddard and T. Ritter, J. Am. Chem. Soc., 2010, 132, 14092–14103 CrossRef CAS PubMed; (c) J. R. Khusnutdinova and L. M. Mirica, in C-H and C-X Bond Functionalization: Transition Metal Mediation, The Royal Society of Chemistry, 2013, pp. 122–158 Search PubMed; (d) F. A. Cotton, J. Gu, C. A. Murillo and D. J. Timmons, J. Am. Chem. Soc., 1998, 120, 13280–13281 CrossRef CAS; (e) F. Tang, F. Qu, J. R. Khusnutdinova, N. P. Rath and L. M. Mirica, Dalton Trans., 2012, 41, 14046–14050 RSC; (f) G. Maestri, M. Malacria and E. Derat, Chem. Commun., 2013, 49, 10424–10426 RSC.
- (a) M. P. Lanci, M. S. Remy, W. Kaminsky, J. M. Mayer and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 15618–15620 CrossRef CAS; (b) J. R. Khusnutdinova, N. P. Rath and L. M. Mirica, J. Am. Chem. Soc., 2010, 132, 7303–7305 CrossRef CAS; (c) R. Jagadeesan, G. Sabapathi, J. Madhavan and P. Venuvanalingam, Inorg. Chem., 2018, 57, 6833–6846 CrossRef CAS PubMed; (d) D. C. Powers and T. Ritter, in Higher Oxidation State Organopalladium and Platinum Chemistry, ed. A. J. Canty, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 129–156 Search PubMed; (e) F. Tang, S. V. Park, N. P. Rath and L. M. Mirica, Dalton Trans., 2018, 47, 1151–1158 RSC.
- (a) D. Schröder, Acc. Chem. Res., 2012, 45, 1521–1532 CrossRef; (b) K. L. Vikse, Z. Ahmadi and J. Scott McIndoe, Coord. Chem. Rev., 2014, 279, 96–114 CrossRef CAS; (c) C. Iacobucci, S. Reale and F. De Angelis, Angew. Chem., Int. Ed., 2016, 55, 2980–2993 CrossRef CAS.
- (a) D. M. Chisholm and J. Scott McIndoe, Dalton Trans., 2008, 3933–3945 RSC; (b) J. Limberger, B. C. Leal, A. L. Monteiro and J. Dupont, Chem. Sci., 2015, 6, 77–94 RSC.
- (a) R. C. Thompson, Inorg. Chem., 1981, 20, 3745–3748 CrossRef CAS; (b) G. Lente, J. Kalmár, Z. Baranyai, A. Kun, I. Kék, D. Bajusz, M. Takács, L. Veres and I. Fábián, Inorg. Chem., 2009, 48, 1763–1773 CrossRef CAS PubMed.
- M. Spiro, Electrochim. Acta, 1979, 24, 313–314 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1965101. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc05722f |
| This journal is © The Royal Society of Chemistry 2020 |