
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual Lewis site creation for activation of methanol on Fe3O4(111) thin films†
Fang
Xu
 a,
Wei
Chen
b,
Constantin A.
Walenta
a,
Christopher R.
O'Connor
a and
Cynthia M.
Friend
*ac
a,
Wei
Chen
b,
Constantin A.
Walenta
a,
Christopher R.
O'Connor
a and
Cynthia M.
Friend
*ac
aDepartment of Chemistry and Chemical Biology, Harvard University, Cambridge, MA 02138, USA. E-mail: friend@fas.harvard.edu
bDepartment of Physics, Harvard University, Cambridge, Massachusetts 02138, USA
cJohn A. Paulson School of Engineering and Applied Sciences, Harvard University, Cambridge, Massachusetts 02138, USA
First published on 24th January 2020
Abstract
Despite a wide application in heterogeneous catalysis, the surface termination of Fe3O4(111) remains controversial. Herein, a surface with both Lewis acid and base sites is created through formation of an Fe3O4(111) film on α-Fe2O3(0001). The dual functionality is generated from a locally nonuniform surface layer of O adatoms and Fetet1 sites. This reactive layer is reproducibly formed even in oxygen-free environments because of the high mobility of ions in the underlying α-Fe2O3(0001). The atomic structure of the Fe3O4(111) surface was identified by scanning tunneling microscopy (STM) and density functional theory (DFT) using the registry of the overlayers with the surface and the distinct electronic structure of oxygen adatom (Oad) and uncovered lattice Fetet1. The surface is dominated by the interface of Oad and Fetet1, a Lewis acid–base pair, which favors methanol dissociation at room temperature to form methoxy. Methoxy is further oxidized to yield formaldehyde at 700 K in temperature programmed reaction spectra, corresponding to an approximate activation barrier of 179 kJ mol−1. The surface termination of Fe3O4(111) is fully recovered by rapid heating to 720 K in vacuum, demonstrating the high mobility of ions in this material. The work establishes a clear fundamental understanding of a unique magnetite surface and provides insights into the origin of selective oxidation of alcohols on magnetite-terminated catalysts.
Introduction
The termination of metal oxide surfaces is critical for their functionality and tuning their chemical behaviour. For example, when ceria is reduced by CO, the (110) termination has the lowest oxygen vacancy formation energy of the three low Miller index terminations.1,2 Another example is that only {100} terminated Cu2O nanocrystals exhibit activity for the photodegradation of methyl orange, but not {111} or {110} surfaces.3 These termination-dependent phenomena are related to the local geometry, the bonding, and the chemical identity on the catalyst surface at an atomic level.Magnetite (Fe3O4), which is low-cost and abundant, is a promising material for a wide range of applications,4–6 including catalytic processes for mitigating environmental toxins.7 The reducible material contains a spinel structure containing both Fe2+ (octahedral coordinated) and Fe3+ (tetrahedral and octahedral coordinated). Fe3O4(111) is the most thermodynamically favoured termination of magnetite,8 forming repeating layers of –Fetet1–O1–Feoct1–O2–Fetet2–Feoct2–Fetet1– (subscripts refer to a tetrahedral, octahedral coordination, or an oxygen layer).9 All iron-containing layers share a similar hexagonal unit cell with a spacing of ∼0.6 nm.
The surface termination of Fe3O4 is important because it affects local electronic structure and therefore also local chemical reactivity. Depending on the surface termination, either Lewis acid sites (electron acceptors) or Lewis base sites (electron donors) may be present. Coordinatively unsaturated Fe cation sites, for example, FeB sites on Fe3O4(001), will act as a strong Lewis acid that leads to water dissociation.10 Anions on metal oxide surfaces are electron donors, i.e. Lewis bases, which can play a role in bond dissociation and recombination, facilitating elementary steps in a reaction on the surface.
The determination of the structure of Fe3O4(111), and therefore, understanding the chemical functionality of the surface, is challenging and remains highly debated.11–17 Very similar unit cells with various possible terminations and multiple domains may coexist.12,18 There are three predominant preparation methods for Fe3O4(111) surfaces: (1) preparation of bulk Fe3O4(111);19–22 (2) growth of thin films of Fe3O4(111) on zero-valent8,23–25 and nonreducible surfaces,26,27 especially Pt;28–30 and (3) formation of thin films via reduction of α-Fe2O3(0001).17,31 The surface termination is different for the various preparations. Of these, bulk Fe3O4(111) prepared in vacuum possesses the most varieties of surface terminations. Tetrahedrally coordinated iron, Fetet1, with a hexagonal pattern, co-exists with octahedrally coordinated iron, Feoct2,20,32 signified by the honeycomb structure which is often related to oxygen-poor conditions. Lattice oxygen, O, terminated bulk Fe3O4(111) is also observed.33 Additionally, oxygen adatoms have been proposed14,32,34 based on STM and chemical activity. The terminations of Fe3O4(111) thin films on Pt are predominantly assigned as Fetet1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 23,28,35 based on scanning tunnelling microscopy (STM),23,28,35 dynamical low energy electron diffraction (LEED) intensity analysis,23,35 infrared reflection-absorption spectroscopy (IRRAS),28 and density functional theory (DFT).28 Thin films of Fe3O4(111) formed from reduction of Fe2O3(0001) are generally thought to be terminated by Fetet1;9,31,36,37 however, the surface structures often co-exist with O terminated patches with a unique hexagonal periodicity of 0.3 nm,31,36,37 presumably due to the specific preparation conditions. Alternative preparation conditions lead to a surface terminated in the “biphase” structure in all of the three preparation methods,9,19,24,38,39 identified by STM, LEED and IRRAS. The “biphase” is a mixed oxide variously ascribed to FeO/Fe3O4 or Fe3O4/Fe2O3.13,40
23,28,35 based on scanning tunnelling microscopy (STM),23,28,35 dynamical low energy electron diffraction (LEED) intensity analysis,23,35 infrared reflection-absorption spectroscopy (IRRAS),28 and density functional theory (DFT).28 Thin films of Fe3O4(111) formed from reduction of Fe2O3(0001) are generally thought to be terminated by Fetet1;9,31,36,37 however, the surface structures often co-exist with O terminated patches with a unique hexagonal periodicity of 0.3 nm,31,36,37 presumably due to the specific preparation conditions. Alternative preparation conditions lead to a surface terminated in the “biphase” structure in all of the three preparation methods,9,19,24,38,39 identified by STM, LEED and IRRAS. The “biphase” is a mixed oxide variously ascribed to FeO/Fe3O4 or Fe3O4/Fe2O3.13,40
Herein, evidence is presented for a surface termination of Fe3O4(111) films that has dual functionality, containing both Lewis acid and base sites. The surface studied here is terminated by a mixture of tetrahedral Fetet1 and O adatoms, based on the registry of the overlayers with the surface and the differentiation of empty and filled states on different regions of the surface using STM and DFT. The surface is prepared by sputtering and annealing an α-Fe2O3(0001) single crystal in vacuum. The oxygen adatom layer was robust, even after 40+ preparation cycles in an oxygen-free environment, due to the high mobility of ions in the iron oxides. We further show that O adatoms and nearby uncovered Fetet1 are Lewis acid–base pairs that actively dissociate methanol at room temperature. The results present insight into understanding the surface chemistry of magnetite films and reduced hematite catalysts.
Methods
Sample preparation
A natural α-Fe2O3(0001) single crystal was purchased from SurfaceNet GmbH. The sample was prepared using consecutive cycles of 15 min Ar+ sputtering and 20 min annealing at 980 K in UHV until a clear LEED pattern of Fe3O4(111) was detected.Scanning tunnelling microscopy experiments
The STM experiments were carried out in a commercial Omicron VT Beam Deflection AFM/STM housed in an ultrahigh vacuum chamber equipped with LEED and direct dosing tube described in previous work.41 All images were scanned at room temperature using a commercially-cut Pt/Ir tip. Pressure of methanol vapor was read by an ion gauge placed far away from the sample. The actual methanol pressure above the sample surface during in situ experiments was estimated to be similar to the reading from the ion gauge, as a result of tip shading effect and direct dosing effect.41 All STM images were processed using SPIP 6.0.2, Scanning Probe Image Processor Software by Image Metrology.X-ray photoelectron spectroscopy experiments
XPS experiments were conducted using a commercial SPECS instrument at the Center for Functional Nanomaterials at Brookhaven National Laboratory. The UHV XPS system consisted of an analysis chamber (base pressure ∼1 × 10−9 Torr) and a preparation chamber (base pressure ∼5 × 10−10 Torr). The X-ray source uses monochromatized Al Kα radiation. The sample was irradiated under an angle of 43° and the photoelectrons were measured with normal emission using a hemispherical analyser at a pass-energy of 50 eV. Calibration was completed by setting the Ag 3d5/2 peak to 368.2 eV, the peak obtained using a reference Ag(111) crystal. Intensity was adjusted by normalizing the signal-noise ratio in baselines.Temperature programmed reaction spectroscopy experiments
TPRS experiments were carried out in a UHV setup with a base pressure of <8 × 10−11 Torr described before.42 Deuterated methanol (CD3OH, Sigma Aldrich, 99.8 atom% D) was purified by freeze-pump-thaw cycles and was introduced to the surface with a needle doser at 130 K. The TPRS experiments were carried out with a heating rate of 1 K s−1 and the resulting spectra were corrected for fragmentation pattern contributions.Density functional theory calculations
DFT calculations were performed using VASP43 with the projector-augmented wave potentials. The GGA-PBE44 exchange-correlation functional was used. The DFT-TS method45 was used to include the van der Waals correction. The on-site Coulomb repulsion of Fe 3d electrons was treated by DFT + U46 approach where Ueff is equal to 4.0 eV. The kinetic energy cut-off of the plane-wave basis sets was 550 eV. The optimized lattice constant of Fe3O4 was 8.440 Å, close to the experimental value of 8.396 Å. The Fetet1-terminated (111) surface was modelled by a slab structure, including 17 atomic layers and a more than 12 Å vacuum region along the z direction. A Gamma-centred 5 × 5 × 1 k-point mesh47 was utilized to sample the Brillouin zone of the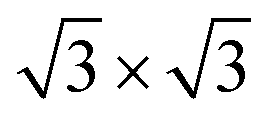 supercell (10.34 Å along the two in-plane lattice vectors). The ground-state ferrimagnetic phase48 was used for the spin configuration.
supercell (10.34 Å along the two in-plane lattice vectors). The ground-state ferrimagnetic phase48 was used for the spin configuration.
Different adsorption sites of O on the surface were calculated to find the most stable configuration. During structural relaxation, the central 5 layers of the slab were constrained to their bulk positions, and the remaining atoms were fully relaxed to a force threshold of 0.01 eV Å−1. The adsorption energy of an oxygen adatom is calculated as 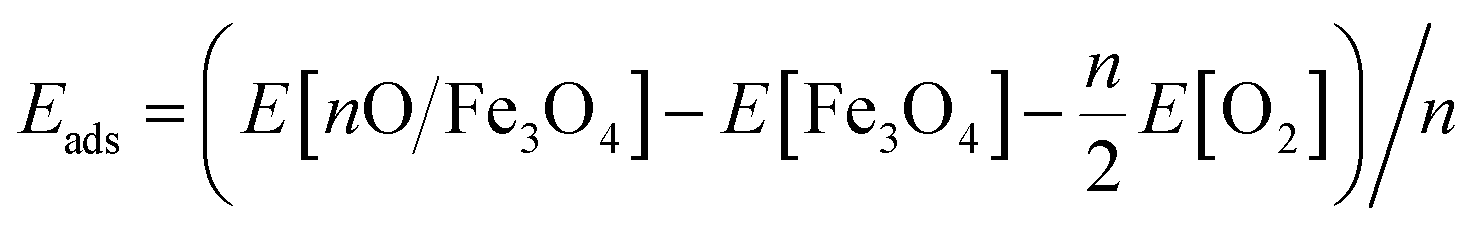 , where n is the number of O adatoms in a unit cell. The STM images at ±1.5 eV tunnelling bias were simulated using the electronic states with energies ranging from the Fermi level (EF) to ±1.5 eV. The atomic structures were visualized by QuteMol, and the simulated STM data were analysed using p4vasp with further smooth and colouring in SPIP 6.0.2.
, where n is the number of O adatoms in a unit cell. The STM images at ±1.5 eV tunnelling bias were simulated using the electronic states with energies ranging from the Fermi level (EF) to ±1.5 eV. The atomic structures were visualized by QuteMol, and the simulated STM data were analysed using p4vasp with further smooth and colouring in SPIP 6.0.2.
Results and discussion
Identity of oxygen adatoms on Fe3O4(111)/α-Fe2O3(0001)
A Fe3O4(111) film forms on top of α-Fe2O3(0001) after repeated cycles of sputtering and annealing in UHV and is evident by the transformation of the LEED patterns from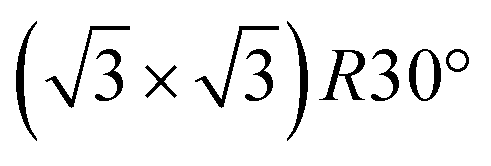 , characteristic of α-Fe2O3(0001), to a Fe3O4(111) p(2 × 2) pattern (Fig. S1†).49,50 The top layers of the surface are reduced by sputtering but subsequently re-oxidized by annealing in vacuum to 980 K for 20 minutes according to X-ray photoelectron spectroscopy results (Fig. S2†). The increase in O:Fe ratio during annealing is attributed to diffusion of iron cations and interstitials into the bulk, as the cation diffusion in the inverse spinel structure is rapid and the oxygen lattice is rigid.51–54
, characteristic of α-Fe2O3(0001), to a Fe3O4(111) p(2 × 2) pattern (Fig. S1†).49,50 The top layers of the surface are reduced by sputtering but subsequently re-oxidized by annealing in vacuum to 980 K for 20 minutes according to X-ray photoelectron spectroscopy results (Fig. S2†). The increase in O:Fe ratio during annealing is attributed to diffusion of iron cations and interstitials into the bulk, as the cation diffusion in the inverse spinel structure is rapid and the oxygen lattice is rigid.51–54
The surface termination of Fe3O4(111)/α-Fe2O3(0001) contains an unsaturated close-packed hexagonal pattern, which is assigned as an oxygen adatom layer (Fig. 1). The hexagonal pattern, which is reproducible and covers most of terraces across the surface, has a spacing of ∼0.6 nm along the close-packed directions and occupies a-top positions, templated by the substrate (Fig. 1A). The apparent height between the same feature on the two adjacent terraces is ∼0.48 nm, corresponding to a repeating distance of layers within Fe3O4(111). The a-top stacked position indicates that a new layer, other than those from bulk, is formed. The lateral periodicity of 0.6 nm rules out the termination of lattice layer O1, and the absence of honeycomb structure under all scanning conditions rules out the Feoct2 termination.
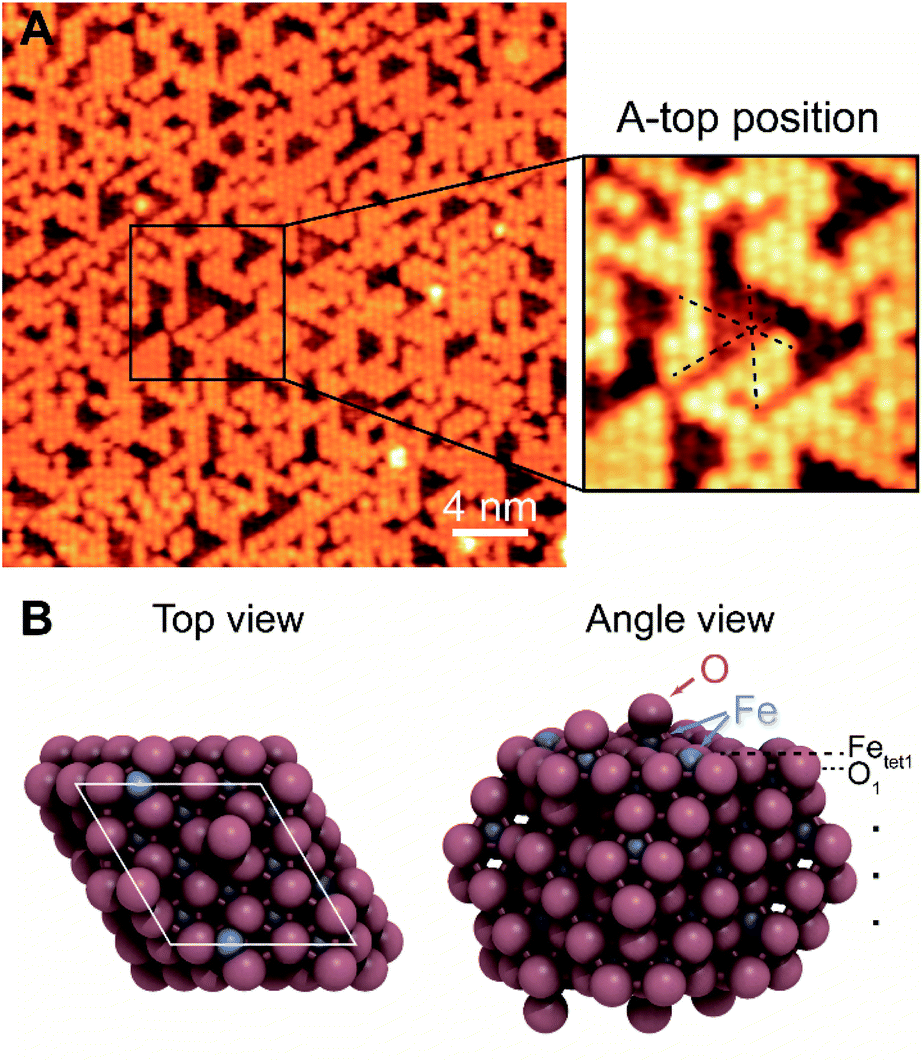 | ||
| Fig. 1 The atomic-scale structure of Fe3O4(111)/α-Fe2O3(0001) is determined using: (A) an STM image of an oxygen adatom terminated Fe3O4(111) film, and (B) the corresponding DFT slab models with O adsorption coverage of 0.67 ML. The insert in (A) is a magnified image with close-packed directions of substrate indicated in black dashed lines. Scanning conditions: −1.5 V, 0.02 nA. | ||
The surface in this work has a different topography and electronic structure compared to previous studies of Fe3O4(111).28,36,55 Although the surface prepared herein (Fig. 1) may appear qualitatively similar to images in the literature assigned to a termination by Fetet1 co-existing with lattice O that has a 0.3 nm spacing,36,56 it is clearly different, as discussed in more detail below. In this work, the top layer occupies an a-top position and is only visible in filled states in STM, different from the Fetet1 layer that contains both filled and empty states36 (Fig. 2A and S3†). The top layer in this work is not related to adsorbates from background,20 as the coverage does not increase with longer time exposure in UHV. Thus, the surface termination of the Fe3O4(111) film formed from reduction of oxygen-rich bulk Fe2O3(0001) is intrinsically different from the Fe3O4(111) terminations described in previous literature, most likely due to different preparation conditions.
Considering the re-oxidation during annealing, the surface structure is ascribed to a mixture of oxygen adatoms on the Fetet1 substrate and exposed Fetet1 sites. Under the preparation conditions used here, the coverage of the oxygen adatoms measured by STM is 0.5–0.7 ML, typically at ∼0.64 ML (1 monolayer, ML, is defined as an equivalent number of Fetet1 atoms in a primitive unit cell). The Oad atoms are templated by Fetet1, forming patches that strictly follow the Fetet1 geometry at high coverages. Different annealing temperatures (720–1010 K) in UHV do not lead to a change in the Oad structure although the population of O adatoms may vary. Oxidation in 5 × 10−7 mbar in O2 during annealing followed by a cleaning cycle results in formation of clusters, possibly oxide islands, and a slightly higher Oad coverage. Some features at the edge of close-packed O adatom patches are mobile at room temperature (Movie, M-1, in the ESI†), possibly due to surface hydroxyls or reversible dissociative adsorption of trace background water vapor.57,58
The proposed structure is validated by DFT calculations on 0.67 ML oxygen adatoms covered Fetet1 terminated surface which show that the a-top position is the most stable adsorption site among others (Fig. 1B). Other oxygen adsorption sites considered have adsorption energies weaker by at least 1.48 eV per O adatom (Fig. S4†). The adsorption energy of the a-top oxygen (Fig. 1B) is 0.24 eV per O adatom. This value becomes negative when using gas-phase O2 as a reference and considering its correction in the DFT-calculated energy (there are two different binding energies often used in the literature for O2 (0.448 or 0.6859 eV) leading to a binding energy of O of either −0.16 or −0.44 eV, respectively). The calculational results are consistent with prior theoretical work that both exposed Fetet1 and O-adsorbed Fetet1 are stable terminations.48 The calculated bond length of Oad-Fetet1 is 1.62 Å (Fig. 1B), which is substantially shorter than the lattice Fetet1-O bond length of 1.90 Å, and is comparable with the length of a Fe![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond on ferryl oxygen terminated α-Fe2O3(0001) of 1.58 Å.60 Here, the Oad is not bound to a local α-Fe2O3(0001), as measured periodicity suggests a Fe3O4(111) surface. Thus, the Oad is strongly bonded to the lattice Fetet1 on a Fe3O4(111) surface.
O bond on ferryl oxygen terminated α-Fe2O3(0001) of 1.58 Å.60 Here, the Oad is not bound to a local α-Fe2O3(0001), as measured periodicity suggests a Fe3O4(111) surface. Thus, the Oad is strongly bonded to the lattice Fetet1 on a Fe3O4(111) surface.
The assignment of oxygen adatoms is further confirmed by agreement between STM measurements and DFT calculations, including the bias dependence of the images. Similar bias-dependent features that are visible in filled states (negative bias) were reported previously; however, they are either ascribed as lattice oxygen layers with a 0.3 nm periodicity36 or as isolated OH or H adsorbates.20 In this work, the filled state image of oxygen adatoms are bright under a scanning voltage of −1.5 V (sample biased), whereas the inverted contrast was observed when switching the scanning voltage to +1.5 V (Fig. 2A, and a larger scale comparison in Fig. S3†). The contrast corresponds to a change in density of states, which is calculated using DFT. Based on models in Fig. 1B, the projected density of states (PDOS) of oxygen adatoms (Oad) and surface iron atoms (Fetet1) that were not covered by oxygen adatoms were calculated and found to be well isolated (Fig. 2C): Oad occupies two bands at about −0.5 V and −1.5 V, none of which was occupied by Fetet1; Fetet1 occupies three bands at about +0.5 V, +1.0 V and +1.7 V, neither of which was occupied by Oad. The distribution of PDOS can be used as fingerprint for element identity. The separated PDOS leads to a contrast change of Oad and Fetet1 in simulated STM images (Fig. 2B), confirming the generality for the observed surface property by STM (Fig. 2A). Further, the calculated PDOS of Oad coverages of 0, 0.67, 0.75, and 1 ML suggests an Oad coverage dependency, reinforcing the ability of oxygen adatoms to change the surface electronic properties (Fig. S5†).
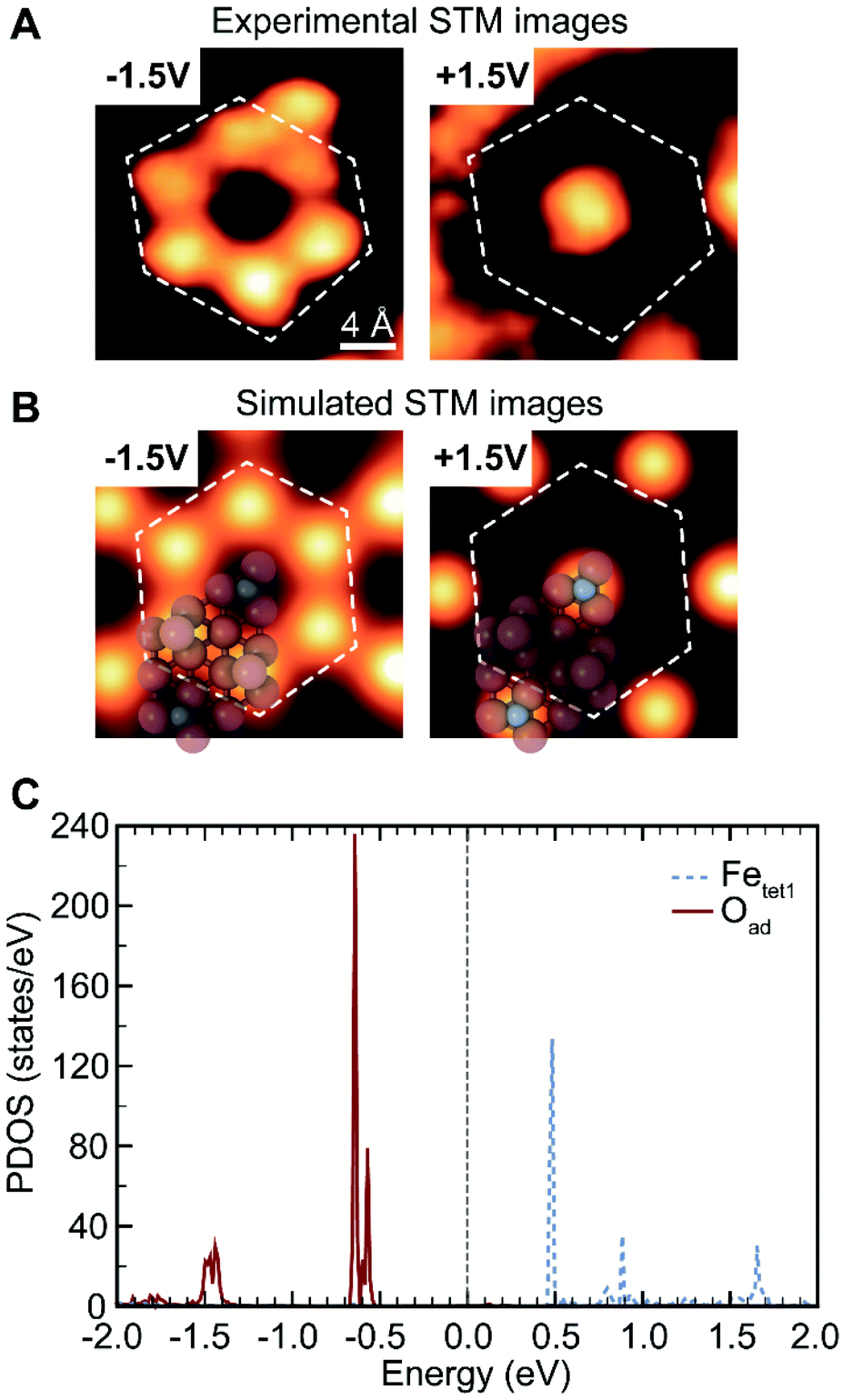 | ||
| Fig. 2 Identification of oxygen adatoms and uncovered Fetet1 by their distinct electronic states. (A) Experimental STM images showing a bias-dependent contrast change. (B) Simulated STM images based on DFT calculations. (C) DFT calculated site-projected density of states (PDOS) of oxygen adatoms (Oad) and uncovered Fetet1 atoms on the top layer. The PDOS includes both spin up and spin down components (i.e. not spin-resolved). The Fermi level is shifted to 0.0 eV. Experimental tunnelling current was 0.25 nA; probing bias was indicated in each panel. The regions encompassed by the dashed lines on the DFT model are a visual guide. | ||
The active Oad and Fetet1 interface for methanol dissociation at room temperature
Selective oxidation of methanol to formaldehyde occurs on the as-prepared Fe3O4(111)/α-Fe2O3(0001) with methoxy as an intermediate, indicated by temperature programmed reaction (Fig. 3). No other product, including CO2, is detected. Deuterated methanol, CD3OH, was used to differentiate O–H and C–H bonds. Desorption of a surface-bound layer of CD3OH was observed in a peak at 250 K, accompanied by H2O evolution from disproportionation reaction of hydroxyls and/or original water impurities in deuterated methanol. Carbon–deuterium bond cleavage starts at 600 K and peaks at 700 K, leading to the formation of formaldehyde, CD2O, methanol-d3, and deuterated water (HDO and D2O). This elementary step corresponds to an approximate activation barrier of 179 kJ mol−1 calculated assuming first-order kinetics,61 and a pre-exponential factor of 1012 s−1.62 At this temperature, trace amounts of surface hydroxyl OH contribute to a desorption peak of H2O in the high temperature oxidation feature. These data show that methoxy (CD3O) is formed at lower temperature in competition with methanol desorption; ultimately yielding CD2O and CD3OH at high temperature. Similar reactivity was previously reported on Fe3O4(111).22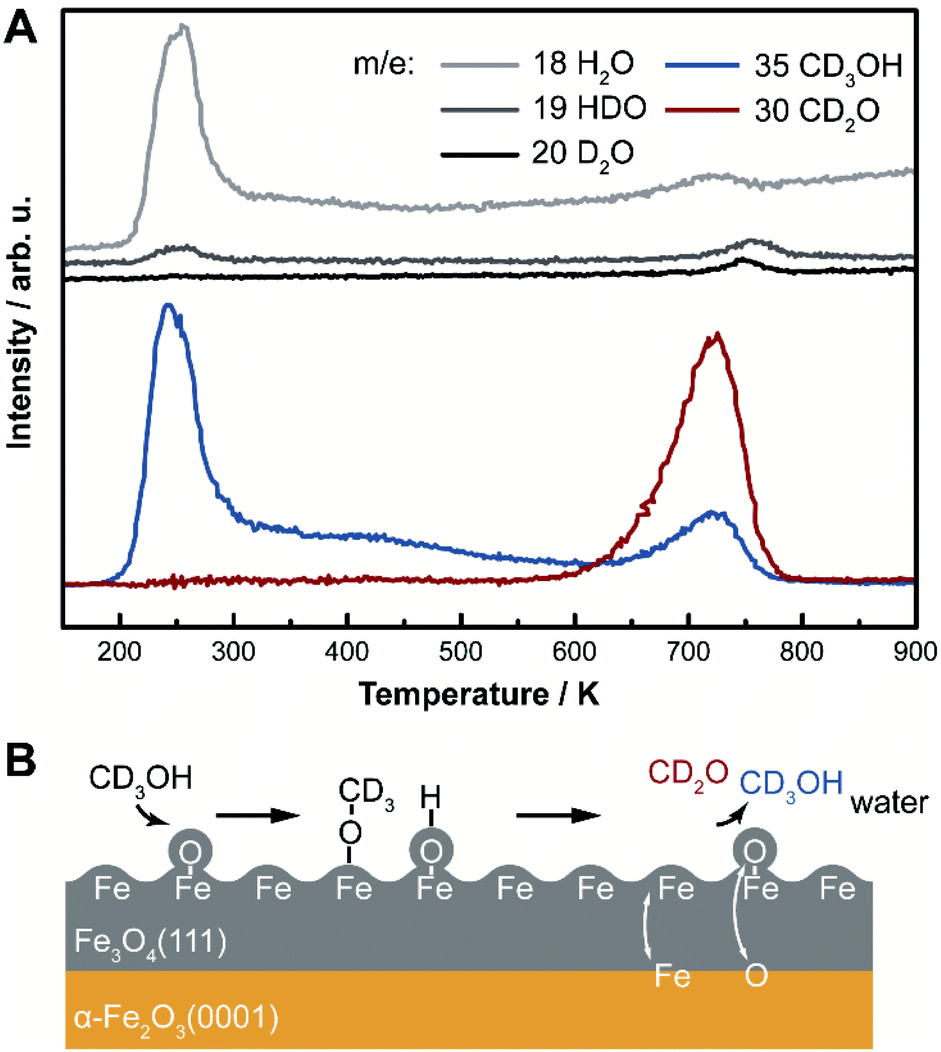 | ||
| Fig. 3 (A) Temperature programmed reaction data for methanol-d3 (CD3OH) show that methanol is dissociated and subsequently oxidized on as-prepared Fe3O4(111)/α-Fe2O3(0001). Traces of water, formaldehyde and methanol were tracked by mass-ion ratio indicated in the panel and corrected for fragmentation patterns. The heating rate is 1 K s−1. (B) The reaction mechanism is illustrated in a schematic. | ||
The formation of methoxy on Fe3O4(111), which is rich in Lewis acid–base pairs, was captured by in situ STM at room temperature (Fig. 4). The surface covered by Oad is shown in Fig. 4A. The atomic structure of the substrate (Fig. 4B) is not resolved during methanol exposure to the surface at room temperature due to the presence of weakly-bound methanol which is mobile on the surface, thus, producing a fuzzy image. After 21 min under 4 × 10−10 mbar methanol, the immobile bright features are attributed to methoxy formed from methanol dissociation. These features, as a single methoxy or a few in a group, have an apparent width from 1.0 to 3.0 nm and an apparent height of ∼0.3 nm (Fig. 4B). These methoxy species are stable on the surface after heating to 400 K (Fig. 4C), consistent with temperature programmed reaction results. The oxidation of methanol consumes oxygen from magnetite to form water through the Mars-van Krevelen mechanism. However, the oxygen adatoms reappear after heating the surface to 720 K (Fig. 4D), due to quick ion exchange with bulk and replenishment of Oad at the elevated temperature.
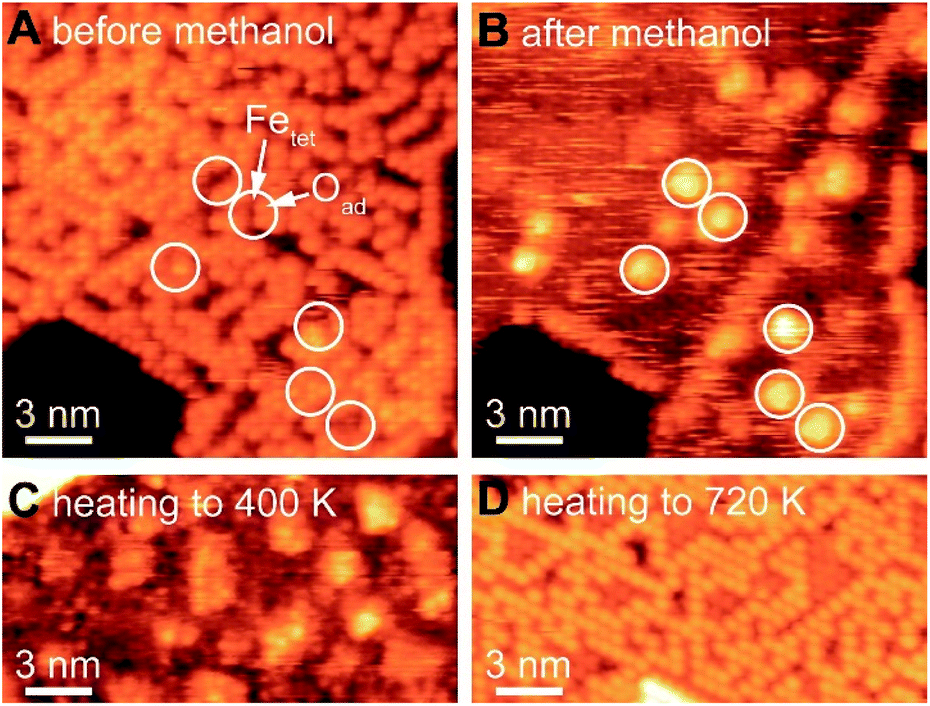 | ||
| Fig. 4 In situ STM of methanol dissociation on Fe3O4(111) films at room temperature tracks the location of active sites. The same surface was imaged (A) before and (B) after 21 min exposure of methanol. PMeOH = 4 × 10−10 mbar (after background subtraction); scanning conditions: −1.5 V, 0.015 nA; scanning from bottom to top. White circles highlight the same positions of methoxy species and interfaces of Fetet (dark in panel A) and Oad (bright in panel A). (C) and (D) are ex situ STM of the post methanol surface after being annealed to 400 K and 720 K, respectively. | ||
The as-prepared Fe3O4(111) surface contains Oad–Fetet interfaces, i.e. Lewis acid–base pairs, where methanol oxidation occurs readily. The active site for the first elementary step for methanol oxidation can be tracked by methoxy formation. These sites include both oxygen adatoms and nearby uncovered lattice iron measured by in situ STM (white circles in Fig. 4). In contrast, small domains with an alternative compact structure do not contain the Oad–Fetet interfaces is inert to O–H bond cleavage, as no methoxy was observed in these areas (Fig. S6†). The alternative compact structures are commonly assigned as O-terminated FeO islands,9,37 oxygen-terminated oxide surfaces,63 and Fe1−xO islands.19,40
Additionally, the Fe3O4(111) surface is active for other reactions, including water dissociation and formic acid decomposition.64 Fe3O4(111) films and bulk are known to dissociate water based on experimental and theoretical studies that dissociated OH− and H+ bound to surface Fe cations and O anions respectively.14,29,31,65 The dissociated OH− can further react with non-dissociated water to form complexes on a Fetet1 terminated films at low temperatures.57,66 The ring-like water–hydroxyl complexes, or (2 × 2) super structure, also forms but less stably67 on (√2 × √2)R45°-reconstructed surfaces of bulk Fe3O4(001) single crystals.68 At room temperature, the Fe3O4(001) surface is only covered by hydroxyls, which desorb as water at 520 K by abstraction of lattice oxygen.69 The reaction with water becomes more extensive on both (111) and (001) terminations with an increase of vapor pressure,21 and can lead to a lifting of the subsurface cation vacancy reconstruction on Fe3O4(001) surfaces.70
Conclusions
A well-ordered Fe3O4(111) film-terminated hematite is formed through reduction and heating. The surface termination is a mixture of O adatoms (Oad) and uncovered lattice Fetet, yielding Lewis base and acid sites, respectively. Reduced top layers are oxidized by annealing to 980 K in vacuo, an oxygen deficient environment. Rapid ion exchange occurs at these temperatures,52 especially cation diffusion, and oxygen-rich bulk leads to re-oxidation of surface layers. Further investigations on electronic structure by STM and DFT show a well separated density of states on Oad and Fetet, allowing identification of the two species.The mixture of Oad and Fetet sites impacts reactivity of chemical reactions that require both acid and base sites, illustrated by CD3OH oxidation. The surface is active for dissociation of the O–H bond near room temperature, to yield methoxy. In this process, the H can be transferred to the O adatoms to yield adsorbed OH and methoxy is bound to the exposed Fetet sites. The resulting methoxy is relatively immobile on the surface at room temperature, based on STM imaging. The Oad act as proton acceptors in the dissociative adsorption of methanol. The Lewis acidity of Fetet1 is theoretically predicted to accommodate methoxy species.71 Further oxidation with C–D cleavage occurs at 700 K, leading to the formation of formaldehyde and water.
The fundamental understanding of Fe3O4(111)/α-Fe2O3(0001) from in this work contributes to the mechanistic study of magnetite as a material for applications at surfaces and interfaces, especially for heterogeneous catalysis. Fe3O4(111), the most thermally stable facet, will dominate the termination of magnetite under equilibrium conditions. As surface termination can affect the present of Lewis acid–base pairs,72 work herein with a bulk of iron oxide is closely related to the magnetite catalysts or hematite catalysts under reducing conditions.
The dual Lewis site—meaning the structure contains an adjacent Lewis acid and base pair—is a promising feature in designing efficient, low-cost, and robust heterogeneous catalysts. The synergistic interactions between intermediates adsorbed on acid and base sites enhance catalytic efficiency in redox reactions, such as the industrially-important production of formaldehyde from methanol on iron oxide-based materials.73,74 In this regard, the Lewis base facilitates the formation of the oxidation intermediate at room temperature by O–H bond splitting, while the Lewis acid is the reaction of the final oxidation step. Designing catalyst materials with both Lewis sites in close proximity is a promising principle for multistep oxidation reactions of alcohols. Additionally, compared with catalysts that need a second metal component to form dual Lewis sites,75 the catalyst herein is free of sintering concerns at elevated temperatures.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was primarily supported by the U.S. Army Research Office under Grant No. W911NF1820143. The calculations were performed at the Oak Ridge Leadership Computing Facility (OLCF) and the National Energy Research Scientific Computing Center (NERSC), while XPS instrumentation support was provided by the Center for Functional Nanomaterials, Brookhaven National Laboratory, of the U.S. Department of Energy. Work by W. C. was supported by the Integrated Mesoscale Architectures for Sustainable Catalysis (IMASC), an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences under Award # DE-SC0012573. We would like to acknowledge Dr Robert J. Madix for his contributions in discussions.References
- Z. Wu, M. Li and S. H. Overbury, J. Catal., 2012, 285, 61–73 CrossRef CAS.
- M. Nolan, S. C. Parker and G. W. Watson, Surf. Sci., 2005, 595, 223–232 CrossRef CAS.
- W.-C. Huang, L.-M. Lyu, Y.-C. Yang and M. H. Huang, J. Am. Chem. Soc., 2012, 134, 1261–1267 CrossRef CAS PubMed.
- L. M. Rossi, A. D. Quach and Z. Rosenzweig, Anal. Bioanal. Chem., 2004, 380, 606–613 CrossRef CAS PubMed.
- W.-M. Zhang, X.-L. Wu, J.-S. Hu, Y.-G. Guo and L.-J. Wan, Adv. Funct. Mater., 2008, 18, 3941–3946 CrossRef CAS.
- M. Bibes and A. Barthelemy, IEEE Trans. Electron Devices, 2007, 54, 1003–1023 CAS.
- M. Hua, S. Zhang, B. Pan, W. Zhang, L. Lv and Q. Zhang, J. Hazard. Mater., 2012, 211–212, 317–331 CrossRef CAS PubMed.
- Y. S. Dedkov, M. Fonin, D. V. Vyalikh, J. O. Hauch, S. L. Molodtsov, U. Rudiger and G. Guntherodt, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 073405 CrossRef.
- Y. Tang, H. Qin, K. Wu, Q. Guo and J. Guo, Surf. Sci., 2013, 609, 67–72 CrossRef CAS.
- N. Mulakaluri, R. Pentcheva and M. Scheffler, J. Phys. Chem. C, 2010, 114, 11148–11156 CrossRef CAS.
- H. Sträter, H. Fedderwitz, B. Groß and N. Nilius, J. Phys. Chem. C, 2015, 119, 5975–5981 CrossRef.
- G. S. Parkinson, Surf. Sci. Rep., 2016, 71, 272–365 CrossRef CAS.
- N. G. Condon, F. M. Leibsle, A. R. Lennie, P. W. Murray, D. J. Vaughan and G. Thornton, Phys. Rev. Lett., 1995, 75, 1961–1964 CrossRef CAS PubMed.
- R. S. Cutting, C. A. Muryn, D. J. Vaughan and G. Thornton, Surf. Sci., 2008, 602, 1155–1165 CrossRef CAS.
- K. Adib, N. Camillone III, J. P. Fitts, K. T. Rim, G. W. Flynn, S. A. Joyce and R. M. Osgood Jr, Surf. Sci., 2002, 497, 127–138 CrossRef CAS.
- N. Camillone III, K. Adib, J. P. Fitts, K. T. Rim, G. W. Flynn, S. A. Joyce and R. M. Osgood, Surf. Sci., 2002, 511, 267–282 CrossRef.
- K. Adib, G. G. Totir, J. P. Fitts, K. T. Rim, T. Mueller, G. W. Flynn, S. A. Joyce and R. M. Osgood Jr, Surf. Sci., 2003, 537, 191–204 CrossRef CAS.
- H. Kuhlenbeck, S. Shaikhutdinov and H.-J. Freund, Chem. Rev., 2013, 113, 3986–4034 CrossRef CAS PubMed.
- M. Paul, M. Sing, R. Claessen, D. Schrupp and V. A. M. Brabers, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 075412 CrossRef.
- T. K. Shimizu, J. Jung, H. S. Kato, Y. Kim and M. Kawai, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 235429 CrossRef.
- T. Kendelewicz, P. Liu, C. S. Doyle, G. E. Brown Jr, E. J. Nelson and S. A. Chambers, Surf. Sci., 2000, 453, 32–46 CrossRef CAS.
- Z. Li, D. V. Potapenko, K. T. Rim, M. Flytzani-Stephanopoulos, G. W. Flynn, R. M. Osgood, X.-D. Wen and E. R. Batista, J. Phys. Chem. C, 2015, 119, 1113–1120 CrossRef CAS.
- M. Ritter and W. Weiss, Surf. Sci., 1999, 432, 81–94 CrossRef CAS.
- F. Genuzio, A. Sala, T. Schmidt, D. Menzel and H.-J. Freund, J. Phys. Chem. C, 2014, 118, 29068–29076 CrossRef CAS.
- H. Xiang, F. Shi, M. S. Rzchowski, P. M. Voyles and Y. A. Chang, Appl. Phys. Lett., 2010, 97, 092508 CrossRef.
- J. B. Moussy, S. Gota, A. Bataille, M. J. Guittet, M. Gautier-Soyer, F. Delille, B. Dieny, F. Ott, T. Doan, P. Warin, P. Bayle-Guillemaud, C. Gatel and E. Snoeck, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 174448 CrossRef.
- H. Hong, J. Kim, X. Fang, S. Hong and T.-C. Chiang, Appl. Phys. Lett., 2017, 110, 021601 CrossRef.
- X. Li, J. Paier, J. Sauer, F. Mirabella, E. Zaki, F. Ivars-Barceló, S. Shaikhutdinov and H. J. Freund, J. Phys. Chem. B, 2018, 122, 527–533 CrossRef CAS.
- Y. Joseph, W. Ranke and W. Weiss, J. Phys. Chem. B, 2000, 104, 3224–3236 CrossRef CAS.
- U. Leist, W. Ranke and K. Al-Shamery, Phys. Chem. Chem. Phys., 2003, 5, 2435–2441 RSC.
- K. T. Rim, D. Eom, S.-W. Chan, M. Flytzani-Stephanopoulos, G. W. Flynn, X.-D. Wen and E. R. Batista, J. Am. Chem. Soc., 2012, 134, 18979–18985 CrossRef CAS.
- A. R. Lennie, N. G. Condon, F. M. Leibsle, P. W. Murray, G. Thornton and D. J. Vaughan, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 10244–10253 CrossRef CAS PubMed.
- N. Berdunov, S. Murphy, G. Mariotto and I. V. Shvets, Phys. Rev. Lett., 2004, 93, 057201 CrossRef CAS PubMed.
- R. S. Cutting, C. A. Muryn, G. Thornton and D. J. Vaughan, Geochim. Cosmochim. Acta, 2006, 70, 3593–3612 CrossRef CAS.
- S. K. Shaikhutdinov, M. Ritter, X. G. Wang, H. Over and W. Weiss, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 11062–11069 CrossRef CAS.
- K. T. Rim, J. P. Fitts, T. Müller, K. Adib, N. Camillone III, R. M. Osgood, S. A. Joyce and G. W. Flynn, Surf. Sci., 2003, 541, 59–75 CrossRef CAS.
- K. T. Rim, T. Müller, J. P. Fitts, K. Adib, N. Camillone, R. M. Osgood, E. R. Batista, R. A. Friesner, S. A. Joyce and G. W. Flynn, J. Phys. Chem. B, 2004, 108, 16753–16760 CrossRef CAS.
- G. Ketteler, W. Weiss, W. Ranke and R. Schlögl, Phys. Chem. Chem. Phys., 2001, 3, 1114–1122 RSC.
- L. Schöttner, A. Nefedov, C. Yang, S. Heissler, Y. Wang and C. Wöll, Front. Chem., 2019, 7, 451 CrossRef.
- N. G. Condon, F. M. Leibsle, T. Parker, A. R. Lennie, D. J. Vaughan and G. Thornton, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 15885–15894 CrossRef CAS.
- F. Xu, I. Fampiou, C. R. O'Connor, S. Karakalos, F. Hiebel, E. Kaxiras, R. J. Madix and C. M. Friend, Phys. Chem. Chem. Phys., 2018, 20, 2196–2204 RSC.
- C. A. Walenta, A. S. Crampton, F. Xu, U. Heiz and C. M. Friend, J. Phys. Chem. C, 2018, 122, 25404–25410 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef PubMed.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- M. Methfessel and A. T. Paxton, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 3616–3621 CrossRef CAS PubMed.
- J. Noh, O. I. Osman, S. G. Aziz, P. Winget and J.-L. Brédas, Chem. Mater., 2015, 27, 5856–5867 CrossRef CAS.
- C. A. Walenta, A. S. Crampton, F. Xu, U. Heiz and C. M. Friend, J. Phys. Chem. C, 2018, 122, 25404–25410 CrossRef CAS.
- W. Weiss and M. Ritter, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 5201–5213 CrossRef CAS.
- S. Hallström, L. Höglund and J. Ågren, Acta Mater., 2011, 59, 53–60 CrossRef.
- S. Nie, E. Starodub, M. Monti, D. A. Siegel, L. Vergara, F. El Gabaly, N. C. Bartelt, J. de la Figuera and K. F. McCarty, J. Am. Chem. Soc., 2013, 135, 10091–10098 CrossRef CAS PubMed.
- Z. D. Sharp, Geology, 1991, 19, 653–656 CrossRef CAS.
- R. Dieckmann and H. Schmalzried, Berichte der Bunsengesellschaft für physikalische Chemie, 1977, 81, pp. 414–419 Search PubMed.
- C. Bamroongwongdee, M. Bowker, A. F. Carley, P. R. Davies, R. J. Davies and D. Edwards, Faraday Discuss., 2013, 162, 201–212 RSC.
- K. T. Rim, D. Eom, L. Liu, E. Stolyarova, J. M. Raitano, S.-W. Chan, M. Flytzani-Stephanopoulos and G. W. Flynn, J. Phys. Chem. C, 2009, 113, 10198–10205 CrossRef CAS.
- E. Zaki, F. Mirabella, F. Ivars-Barceló, J. Seifert, S. Carey, S. Shaikhutdinov, H.-J. Freund, X. Li, J. Paier and J. Sauer, Phys. Chem. Chem. Phys., 2018, 20, 15764–15774 RSC.
- P. Dementyev, K. H. Dostert, F. Ivars-Barcelo, C. P. O'Brien, F. Mirabella, S. Schauermann, X. Li, J. Paier, J. Sauer and H. J. Freund, Angew. Chem., Int. Ed. Engl., 2015, 54, 13942–13946 CrossRef CAS PubMed.
- L. Wang, T. Maxisch and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 195107 CrossRef.
- C. Lemire, S. Bertarione, A. Zecchina, D. Scarano, A. Chaka, S. Shaikhutdinov and H. J. Freund, Phys. Rev. Lett., 2005, 94, 166101 CrossRef CAS PubMed.
- P. A. Redhead, Vacuum, 1962, 12, 203–211 CrossRef CAS.
- R. J. Madix and S. G. Telford, Surf. Sci., 1995, 328, L576–L581 CrossRef CAS.
- A. Sala, H. Marchetto, Z. H. Qin, S. Shaikhutdinov, T. Schmidt and H. J. Freund, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 155430 CrossRef.
- J. Balajka, M. A. Hines, W. J. I. DeBenedetti, M. Komora, J. Pavelec, M. Schmid and U. Diebold, Science, 2018, 361, 786–789 CrossRef CAS PubMed.
- Y. Joseph, C. Kuhrs, W. Ranke, M. Ritter and W. Weiss, Chem. Phys. Lett., 1999, 314, 195–202 CrossRef CAS.
- F. Mirabella, E. Zaki, F. Ivars-Barceló, X. Li, J. Paier, J. Sauer, S. Shaikhutdinov and H.-J. Freund, Angew. Chem., Int. Ed., 2018, 57, 1409–1413 CrossRef CAS PubMed.
- E. Zaki, Z. Jakub, F. Mirabella, G. S. Parkinson, S. Shaikhutdinov and H.-J. Freund, J. Phys. Chem. Lett., 2019, 10, 2487–2492 CrossRef CAS PubMed.
- M. Meier, J. Hulva, Z. Jakub, J. Pavelec, M. Setvin, R. Bliem, M. Schmid, U. Diebold, C. Franchini and G. S. Parkinson, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E5642–E5650 CrossRef CAS PubMed.
- G. S. Parkinson, Z. Novotný, P. Jacobson, M. Schmid and U. Diebold, J. Am. Chem. Soc., 2011, 133, 12650–12655 CrossRef CAS.
- B. Arndt, M. Creutzburg, E. Grånäs, S. Volkov, K. Krausert, A. Vlad, H. Noei and A. Stierle, J. Phys. Chem. C, 2019, 123, 26662–26672 CrossRef CAS.
- X. Li and J. Paier, J. Phys. Chem. C, 2019, 123, 8429–8438 CrossRef CAS.
- S. Zhang, Z.-Q. Huang, Y. Ma, W. Gao, J. Li, F. Cao, L. Li, C.-R. Chang and Y. Qu, Nat. Commun., 2017, 8, 15266 CrossRef CAS.
- A. P. V. Soares, M. F. Portela and A. Kiennemann, Catal. Rev., 2005, 47, 125–174 CrossRef CAS.
- C. Brookes, P. P. Wells, G. Cibin, N. Dimitratos, W. Jones, D. J. Morgan and M. Bowker, ACS Catal., 2014, 4, 243–250 CrossRef CAS.
- V. K. Díez, C. R. Apesteguía and J. I. Di Cosimo, Catal. Today, 2000, 63, 53–62 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting results and movie M-1 are available for this paper. See DOI: 10.1039/c9sc06149e |
| This journal is © The Royal Society of Chemistry 2020 |