DOI:
10.1039/C9SC05656D
(Edge Article)
Chem. Sci., 2020,
11, 1564-1572
Independent control over size, valence, and elemental composition in the synthesis of DNA–nanoparticle conjugates†
Received
7th November 2019
, Accepted 31st December 2019
First published on 2nd January 2020
Abstract
DNA–nanoparticle conjugates have found widespread use in sensing, imaging, and as components of devices. However, their synthesis remains relatively complicated and empirically based, often requiring specialized protocols for conjugates of different size, valence, and elemental composition. Here we report a novel, bottom-up approach for the synthesis of DNA–nanoparticle conjugates, based on ring-opening metathesis polymerization (ROMP), intramolecular crosslinking, and template synthesis. Using size, valence, and elemental composition as three independent synthetic parameters, various conjugates can be obtained using a facile and universal procedure. Examples are given to show the usefulness of these conjugates as sensing probes, building blocks for self-assembly, and as model particles for structure–property relationship studies.
Introduction
DNA–nanoparticle (DNA–NP) conjugates have attracted significant interest1 in large part because they combine two unique and complementary functionalities; namely linking the strong and selective pairing of DNA with remarkable optoelectronic properties and catalytic activity of NPs. This combination enables many applications, including the programmable construction of 3D structures,2 development of sensing probes,3 and demonstration of targeted therapeutic and diagnostic agents.4 To fully unleash the potential of DNA–NP conjugates, it is important to control the size and valence of the NPs, because both variables significantly affect the structure and properties of the resultant conjugates. At the same time, the elemental composition of the NPs, which can be organic (polymer), noble metal, metal oxides, or other inorganic materials, is obviously a critical determinant of the physical and catalytic properties of the material.
A possible but challenging synthetic approach is to build the DNA–NP conjugates up from the molecular level, allowing a finer level of control over all aspects of their preparation. However, this is a relatively difficult approach, because NP nucleation and growth are complicated processes that are governed by many parameters, including but not limited to concentration, temperature, pH, surfactant type, and time.5 Considering this complexity, a viable bottom-up strategy would involve template synthesis of NPs,6 as the formation of NPs is largely controlled by the corresponding templates, yet it remains a problem for adding functional groups on the afforded NPs.
We recently reported such a bottom-up strategy wherein a single-chain organic nanoparticle (ONP)7 templated the synthesis of a monovalent DNA–AuNP conjugate.8 Considering the highly tunable parameters in the polymer scaffold synthesis and post-functionalization, differently sized and functionalized templates can be easily obtained. Thus, we envisioned the possibility of expanding this strategy to more metal NPs with different sizes and functionalities by preparing different templates. Ideally, the size of NPs can be adjusted by using ONPs of different sizes; the functional valence of NPs can be tuned by adjusting the number of DNA strands on the DNA–ONP conjugates; and the elemental composition of the NPs can be changed by using different noble metal precursors. This will create a powerful strategy allowing independent control of multiple structural features, including particle size, valence, and elemental composition. Here we report our efforts to develop this new, versatile strategy and some proof-of-concept applications of the afforded NPs.
Experimental section
Materials and methods
Detailed information of the chemicals and instrumentation methods used in this work can be found in the ESI.†
Synthetic protocols
ONP synthesis.
Syntheses of the functional monomers (M1–4)9 used in ring-opening metathesis polymerizations (ROMP) and the chain-transfer agent (CTA)7 have been previously reported. The detailed synthesis of ONP templates can be found in the ESI,† including the polymerization, allyl functionalization, ring-closing metathesis (RCM), dihydroxylation and DNA conjugation procedures.
DNA activation and functionalization.
A detailed procedure is provided in the ESI.† MALDI-TOF analysis was used to characterize the product. A peak with a +337 Da shift compared to the original DNA-S-S-CH2CH2CH2OH was found.
Conjugation of DNA to ONP.
This step is analogous to our reported procedure,8 with the detailed protocol available in the ESI.†
Growth of metal nanoparticles on ONP templates.
Metal nanoparticles were prepared using different chlorometallates through sucrose or ascorbic acid reduction with a representative protocol following. For AuNP prepared by using sucrose as reductant, 10 μL polymer template (50 μM) was diluted using 940 μL citrate buffer (5 mM, pH = 3). Then, 25 μL HAuCl4 (10 mM) was added. The solution was mixed and incubated at room temperature for 10 min and 25 μL of a 1 M aqueous sucrose solution was added to reduce the chloroaurate. For control samples, citrate buffer was used instead of the polymer template solution. All of the samples for scanning electron microscopy (SEM) and transmission electron microscopy (TEM) were allowed to react overnight. For AuNP prepared using ascorbic acid as reductant, 5 μL polymer template (50 μM) was added in 445 μL citrate buffer (5 mM, pH = 3). Then, 25 μL HAuCl4 (10 mM) was added. The solution was mixed, incubated at room temperature for 10 min, and 25 μL of a 100 mM aqueous ascorbic acid solution was added to reduce chloroaurate. PtNP and PdNP were prepared analogously using PtCl62− and PdCl42− as metal sources. Small variations of the concentrations of the citrate, metal source, and reducing agent may be needed to optimize the experimental conditions for different batches of ONP samples.
Results and discussion
Synthesis of DNA–ONP conjugates of different sizes
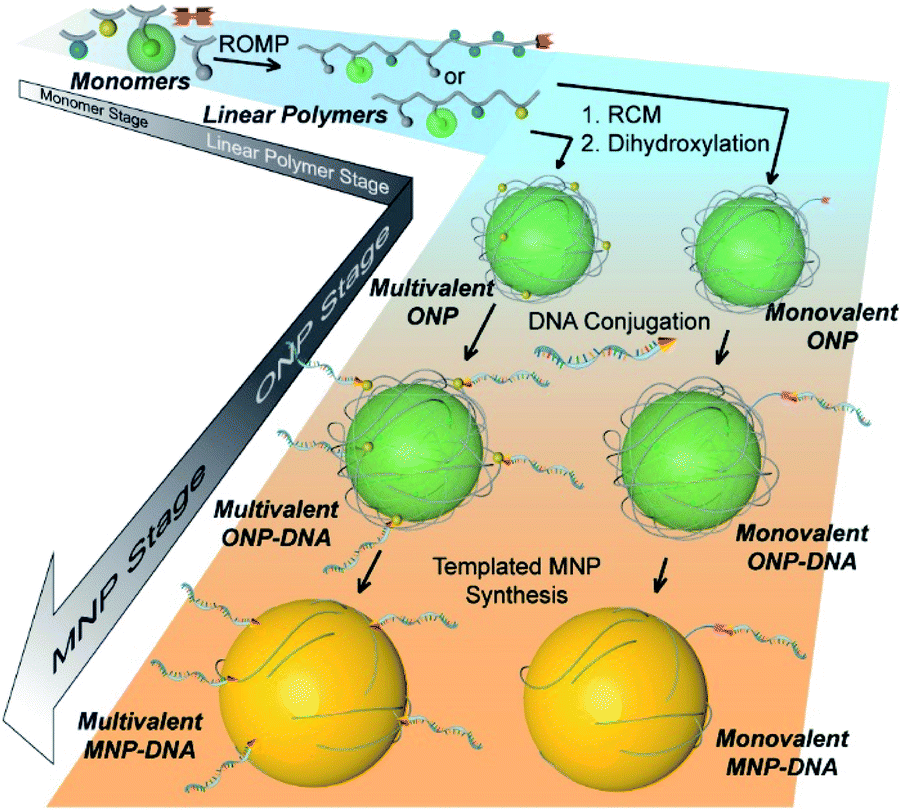
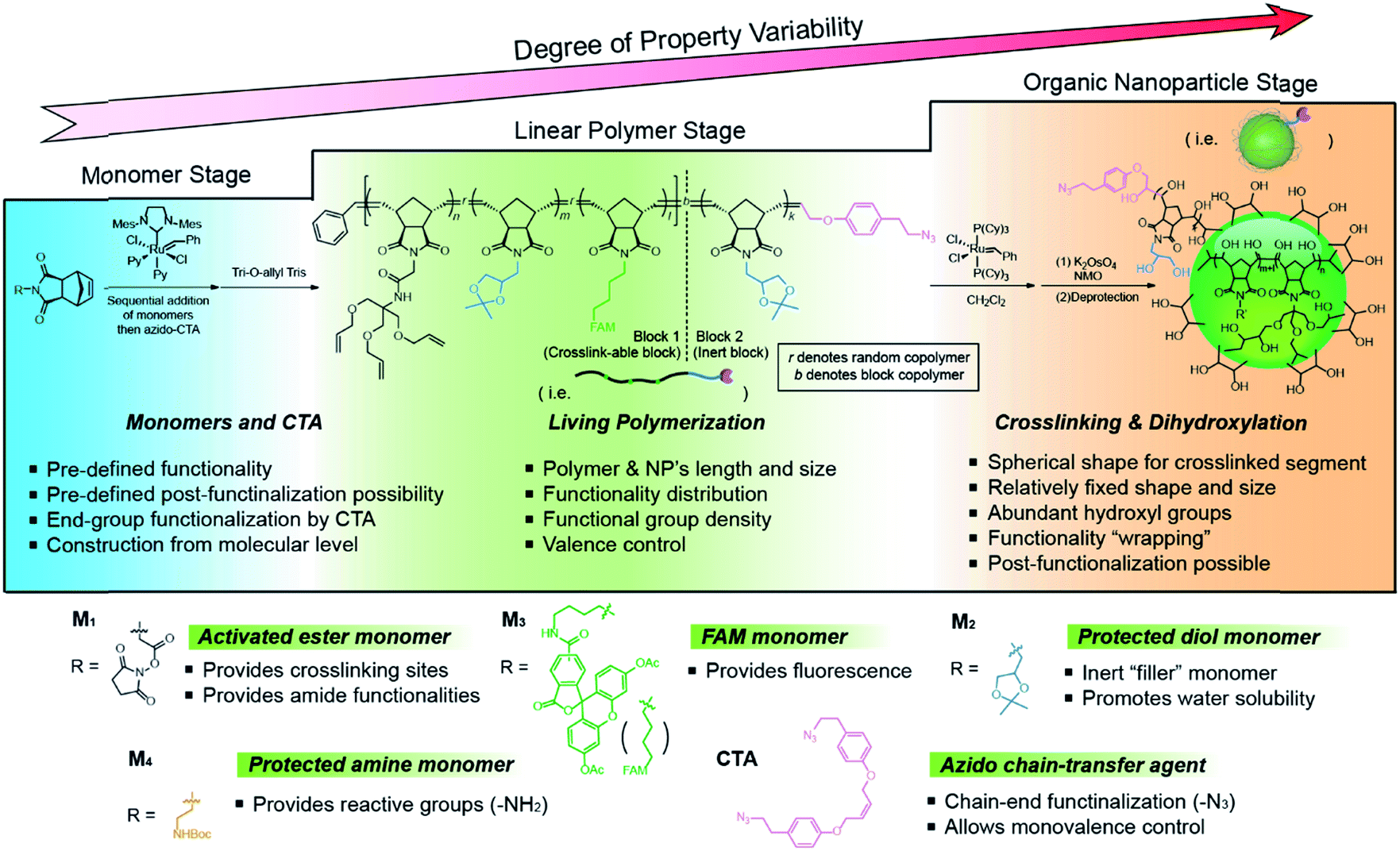
The bottom-up synthetic approach to DNA–NP conjugates relies on four distinct synthetic stages: monomer synthesis, ruthenium-mediated ring opening metathesis polymerization (ROMP), crosslinking and template-induced metallization (Fig. 1). The polymeric template synthesis largely follows our previously reported strategy for ONP synthesis using ROMP and RCM.7 Thus, we prepared four norbornene derivatives as functional monomers (M1–4)9 and an azido-functionalized cis-alkene as chain-transfer agent.7,10 Living ROMP was applied to the functional monomers and the polymer was end-capped by the CTA, generating a linear polymer with azide functional group on one end (Fig. 2). This two-staged copolymerization produced two distinct blocks, with one block containing activated ester for later crosslinking and the other block containing only inert monomer M2. By doing this, a non-crosslinkable, azide-containing tail can be generated for easy conjugation.11 Both molecular weight and crosslinking density can be used to tune the size of the final ONP. Considering that the crosslinking density adjustment is less straightforward and can affect the porosity of the nanoparticle, we chose to tune the ONP size solely by adjusting the monomer to initiator ratio (M/I ratio), which resulted in polymers with different molecular weights in a highly controllable manner.
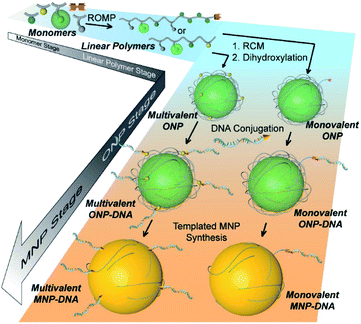 |
| | Fig. 1 Cartoon illustration of the step-wise, bottom-up strategy of functional DNA–nanoparticle synthesis. | |
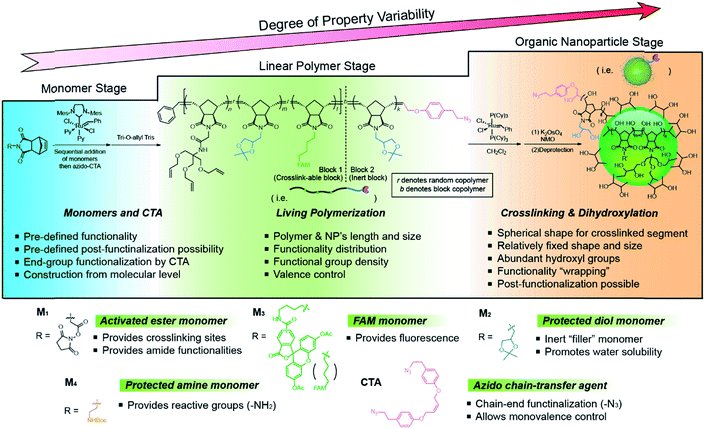 |
| | Fig. 2 Structures of monomers and CTA used in this work and a detailed synthetic scheme of the functional ONP. | |
Thus, by fixing the molar ratio of M1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :M2 at 1:2 in the crosslinkable block but adjusting the equivalence of catalyst used in the polymerization, which was followed by crosslinking and dihydroxylation, monovalent ONPs of different sizes with a single reactive tail were produced (Fig. 3a and Table 1). The parent linear polymers of ONPs could be characterized by GPC (Fig. S1, ESI†), and the curve overlay clearly indicated that the molecular weight (e.g. chain length) increased as the monomer to initiator ratio increased, giving peaks at shorter retention times. TEM and atomic force microscopy (AFM) were used to characterize the final ONPs. In the TEM images (Fig. 3b–g and S2†), all three ONPs were observed as roughly spherical nanoparticles with different diameters (ONPL > ONPM > ONPS). AFM in PeakForce Tapping mode at ambient conditions showed that the heights of ONPs were smaller than the diameter observed by TEM (Fig. 3h–j, S3 and S4†), which may result from the deformation of the ONP under the tapping mode.
:M2 at 1:2 in the crosslinkable block but adjusting the equivalence of catalyst used in the polymerization, which was followed by crosslinking and dihydroxylation, monovalent ONPs of different sizes with a single reactive tail were produced (Fig. 3a and Table 1). The parent linear polymers of ONPs could be characterized by GPC (Fig. S1, ESI†), and the curve overlay clearly indicated that the molecular weight (e.g. chain length) increased as the monomer to initiator ratio increased, giving peaks at shorter retention times. TEM and atomic force microscopy (AFM) were used to characterize the final ONPs. In the TEM images (Fig. 3b–g and S2†), all three ONPs were observed as roughly spherical nanoparticles with different diameters (ONPL > ONPM > ONPS). AFM in PeakForce Tapping mode at ambient conditions showed that the heights of ONPs were smaller than the diameter observed by TEM (Fig. 3h–j, S3 and S4†), which may result from the deformation of the ONP under the tapping mode.
 |
| | Fig. 3 (a) Illustration of the synthesis of different-sized monovalent ONP-N3. (b–d) Negatively stained TEM images of ONPS/M/L. (e–g) Brightfield TEM images of ONPS/M/L using ultrathin carbon layer grid. (h–j) AFM images of ONPS/M/L using PeakForce Tapping mode under ambient condition. | |
Table 1 Control of molecular weight and size of azido-capped monovalent ONPs by varying M/I ratios (initiator = 1)
| ONP |
Block 1 |
Block 2 |
M
n
by GPCa (kDa) |
Diameter by TEM (nm) |
|
M
1
|
M
2
|
M
3
|
M
4
|
M
3
|
|
M
n
of ONPs were calculated based on the GPC-measured Mn of their corresponding parent linear block copolymers.
|
| Mono-ONPS |
15 |
30 |
3 |
— |
5 |
19 |
6–10 |
| Mono-ONPM |
45 |
90 |
3 |
— |
10 |
48 |
15–20 |
| Mono-ONPL |
90 |
180 |
3 |
— |
20 |
87 |
30–40 |
| Multi-ONPM |
50 |
75 |
3 |
25 |
— |
47 |
15–20 |
Valence control of the DNA–NP conjugates
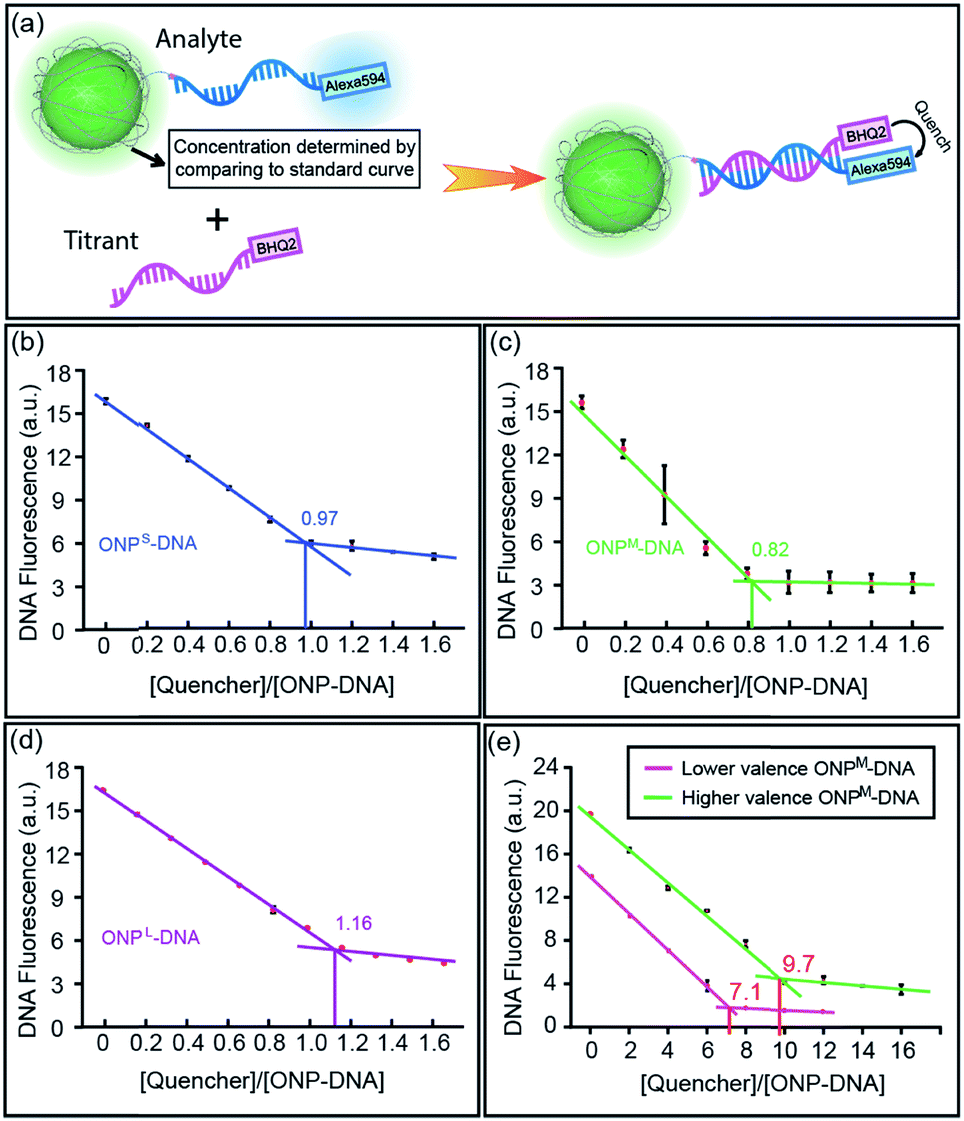
From the monovalent ONP-N3 synthesized above, it is straightforward to convert them into monovalent DNA–ONP conjugates by using copper-free click chemistry analogous to that reported previously.8,12 Thus, a bifunctional DNA strand 5′-Alexa594-M18bT20-SH-3′ (M18bT20 = TTG CTG AGT ATA ATT GTT-T20),12 was reacted with azadibenzocyclooctyne-maleimide (ADIBO-maleimide) through the highly efficient thiol-maleimide Michael addition, giving ADIBO on the DNA 3′-end, then conjugated to ONP-N3 through copper-free click chemistry (Fig. S5, ESI†). Unreacted ONPs were removed on a diethylaminoethyl cellulose (DEAE) column, followed by the isolation of the conjugates using ultracentrifugation.8 All three sized DNA–ONP conjugates, (DNA–ONPS/M/L), showed a clear narrow band in 1% agarose gel under FITC channel, with DNA–ONPS moving fastest and DNA–ONPL slowest (Fig. S7, ESI†). To validate the 1:1 ratio between DNA and ONP in the conjugates, a Black Hole Quencher 2 (BHQ2) modified complementary DNA strand (M18a-BHQ2, 5′-AAC AAT TAT ACT CAG CAA-BHQ2-3′) was titrated into the DNA–ONPS/M/L (Fig. 4a). The titration results are shown in Fig. 4b–d. With increasing concentrations of M18a-BHQ2, the fluorescence decreased for all three conjugates, with a critical transition point in the titration around a 1:1 ratio of quencher to ONP, indicating approximately one DNA strand per ONP.
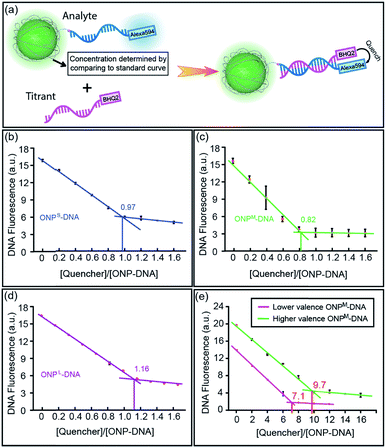 |
| | Fig. 4 (a) Cartoon illustration of the fluorescence titration experiment determining the ratio of DNA/ONP on different DNA–ONP conjugates. (b–d) Fluorescence titration results of DNA–ONPS/M/L. (e) Fluorescence titration results of the two polyvalent DNA–ONPM conjugates. | |
The strategy also allows control over NP valence in addition to size. Multivalent ONPs were prepared by introducing multiple reactive amino groups using M4 in the polymerization step (multi-ONPM in Table 1, Fig. S1 and S8 in ESI†). N-Succinimidyl 4-(maleimidomethyl)cyclohexanecarboxylate (SMCC) was used to conjugate thiolated DNA to multivalent ONP-(NH2)n (see Fig. S9 in ESI† for details). Briefly, the multivalent ONP-(NH2)n was first treated with an excessive amount of sulfo-SMCC to yield ONP-(maleimide)n, which was then reacted with thiolated DNA. The average number of DNA strands per ONP could be controlled either by tuning n of ONP-(NH2)n, which sets an upper limit on the valence of the final DNA–ONP conjugates, or by adjusting the equivalence of DNA in the conjugation. In this manner, ONPs with higher or lower valence numbers could be achieved. The titration curves of two polyvalent conjugates with different valence numbers also indicate the polyvalent nature of the conjugates (Fig. 4e). The two different multivalent DNA–ONPM's were both from ONPM-(NH2)25 but functionalized using different equivalents of thiolated DNAs (Table 1, multi-ONPM). In dynamic light scattering and ζ-potential studies, multivalent DNA–ONP (average valence = 9.7, same for gel and assembly study below) showed more negative ζ-potential, suggesting more negatively charged surface because of the conjugated DNA strands (Fig. S10, ESI†). In addition, multivalent DNA–ONP showed a faster-moving band on agarose gels (Fig. S11, ESI†). Lastly, in hybridization-mediated self-assembly studies, the polyvalent conjugate also exhibited a different assembly pattern than its monovalent analog, forming aggregates with AuNPs bearing complementary DNA strands (Fig. S12, ESI†).
Monovalent DNA–ONP conjugate as ratiometric probe
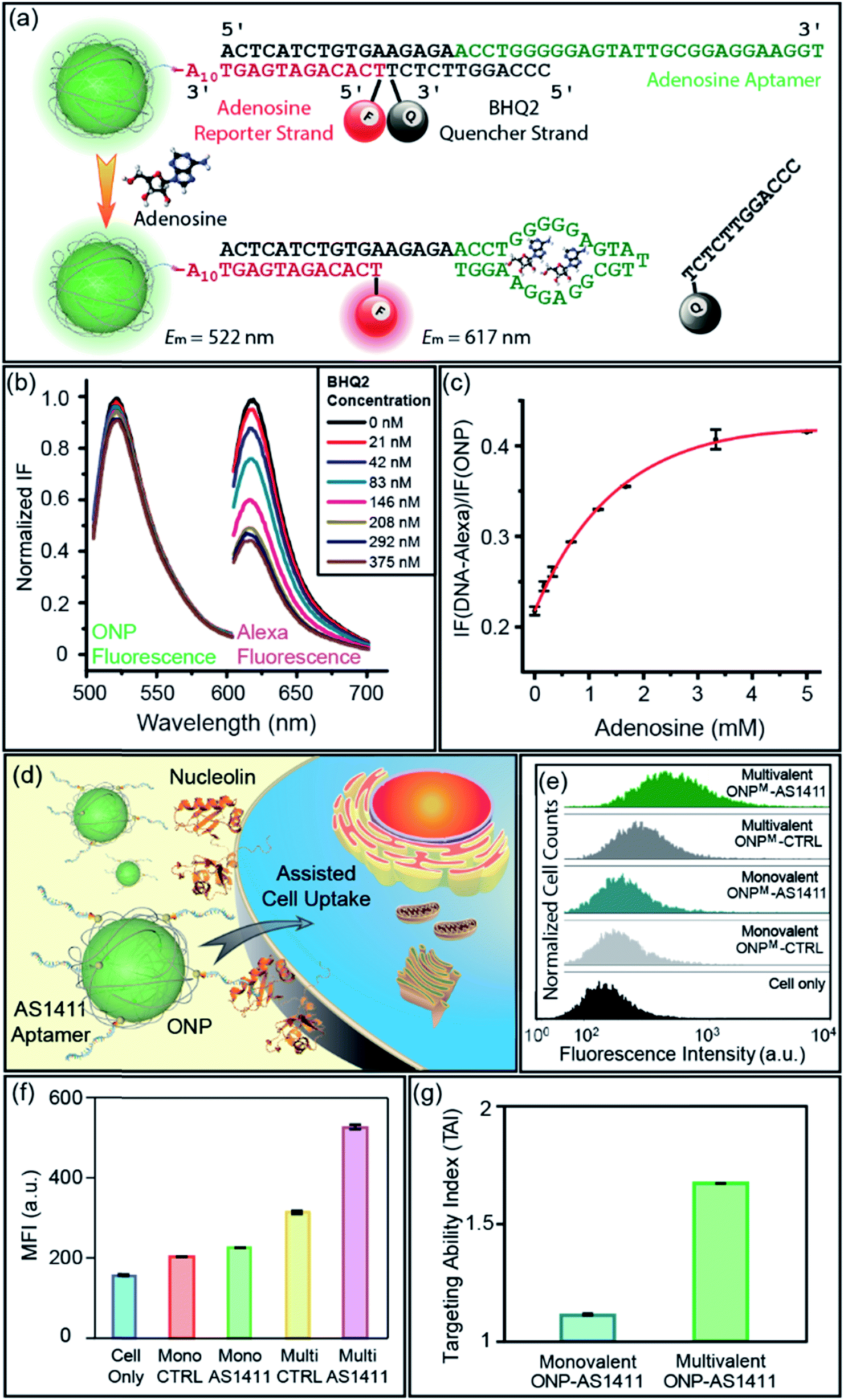
Having demonstrated the ability to control the size and valence of DNA–ONPs, we sought to explore their potential to serve as ratiometric sensors for quantitative detection in combination with DNA aptamers. Aptamers are widely used as highly specific recognition agents for the detection of small molecules and a broad range of larger bio-targets,13 but usually require a delivery platform for intracellular sensing.14 Utilizing the 1:1 stoichiometry of ONP and DNA, we designed a DNA–ONP sensing platform with the ONP as both the delivery vehicle and the internal standard.15 As a demonstration of the approach, an adenosine probe was constructed using an adenosine-binding aptamer (5′-ACT CAT CTG TGA AGA GAA CCT GGG GGA GTA TTG CGG AGG AAG GT-3′), a fluorescence reporter strand (5′-Alexa594-TCA CAG ATG AGT AAA AAA AAA A-SH-3′), and a quencher strand (5′-CCC AGG TTC TCT-BHQ2-3′) linked through conjugation and hybridization to monovalent ONPM (Fig. 5a).3b,16 The ONP:reporter ratio was determined to be ca. 1:1, validating the ratiometric design (Fig. 5b and S13†). The performance of the sensor was assessed by gradual addition of adenosine. Higher concentrations of adenosine resulted in increased fluorescence from Alexa594 with almost unchanged ONP fluorescence, as the aptamer–adenosine binding releases the BHQ2 quencher from the proximity of the fluorophore on the reporter strand and “lights-up” Alexa594. The response curve of the sensor showed a rapid increase at low adenosine concentration and a plateau around 3 mM (Fig. 5c). This adenosine sensor indicated the potential use of aptamer–ONP conjugates as a quantitative and ratiometric sensing platform for biomedical applications. In addition, this monovalent platform may provide higher sensitivity and specificity to track individual endogenous surface receptors, as multivalent particles have the ability to crosslink surface proteins and reduce receptor mobility.17
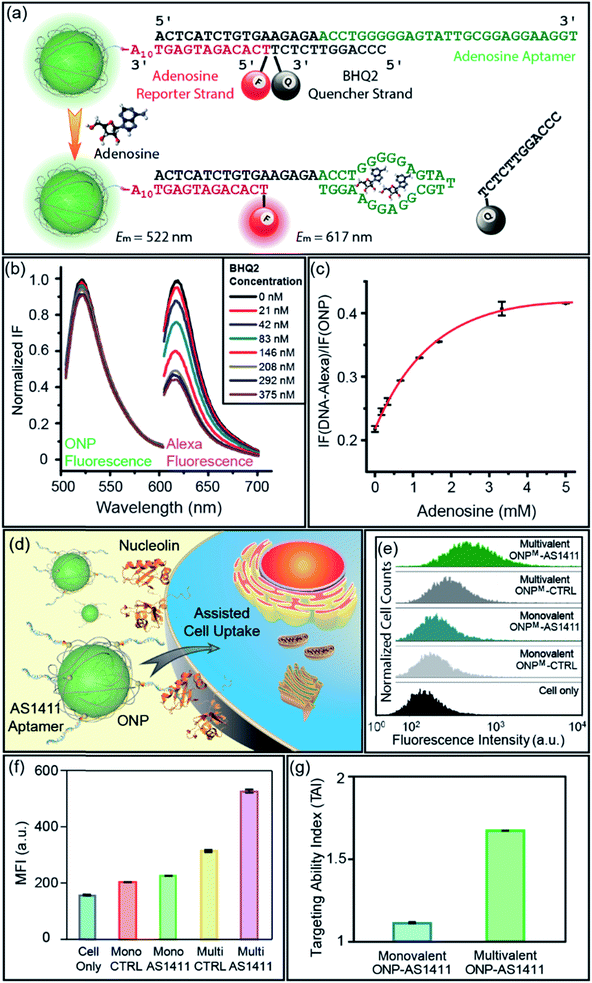 |
| | Fig. 5 (a) Illustration of the working mechanism of the ratiometric adenosine sensor. (b) BHQ2 quenching of Alexa dye during sensor assembly. (c) Response curve of the DNA–ONP ratiometric adenosine sensor against adenosine. (d) Illustration of the aptamer-assisted cellular uptake of ONP. (e) Flow cytometry curves overlay of the MCF-7 cells that had taken DNA–ONP. (f) Mean fluorescence intensity comparison of different DNA–ONP conjugates. (g) Targeting ability index comparison of multivalent and monovalent ONPs functionalized with AS1411. | |
Valence-adjustable DNA–ONP conjugates as cell uptake studying tool
In addition to the functionalities brought by DNA strands, the valence and chemical nature of ONP also play an important role in the properties of the corresponding DNA–NP. We recently reported that the size and lipophilicity of ONPs could be adjusted to tune their cell uptake rate in a regular way.9a DNA aptamers have been used as active targeting moieties for cell surface receptors, yet the effects of aptamer density on cell uptake is much less well explored.18 Given the ability to control the ONP valence using M4, it was of interest to examine the cellular uptake efficiency of aptamer–ONP conjugates as a function of valence. An aptamer named AS1411 (5′-GGT GGT GGT GGT TGT GGT GGT GGT GG-T10-SH-3′) was selected for conjugation to the ONPs as a targeting agent for cancer cells overexpressing nucleolin on their surface (Fig. 5d).13a,19 A scrambled strand of the same length (CTRL, 5′-GAG AAC CTG AGT CAG TAT TGC GGA GA-T10-SH-3′) was used as the non-targeting control. Both DNA strands were conjugated to monovalent and multivalent ONPs, generating two pairs of probes with different DNA density. The chemistry used to prepare these DNA–ONPM conjugates was the same as that described above (see also ESI†). The targeting ability of the conjugates was examined by flow cytometry after incubating the ONPs with MCF-7 cells for 2 h. The results were shown in Fig. 5e and f. To better quantify the targeting ability, the targeting ability index (TAI) is defined as:| | | TAI of ONP = MFI (AS1411-ONP)/MFI (CTRL-ONP) | (1) |
where MFI is the geometric mean fluorescence intensity of ONP-treated cells. A higher TAI indicates improved targeting and uptake with MCF-7 cells. The TAI of multivalent AS1411-ONPM samples was ca. 1.7, higher than the TAI of monovalent DNAM–AS1411 (ca. 1.1) (Fig. 5g), indicating that increasing the number of aptamers on the ONP (e.g., the valence of ONP) increases the targeted NP cell uptake. Interestingly, a higher level of uptake was observed for the multivalent CTRL-ONPM in comparison to the monovalent AS1411-ONPM suggesting that a higher loading of aptamer strands on the surface of the ONP should provide enhanced targeting efficiency.
Metal nanoparticle synthesis from ONP templates
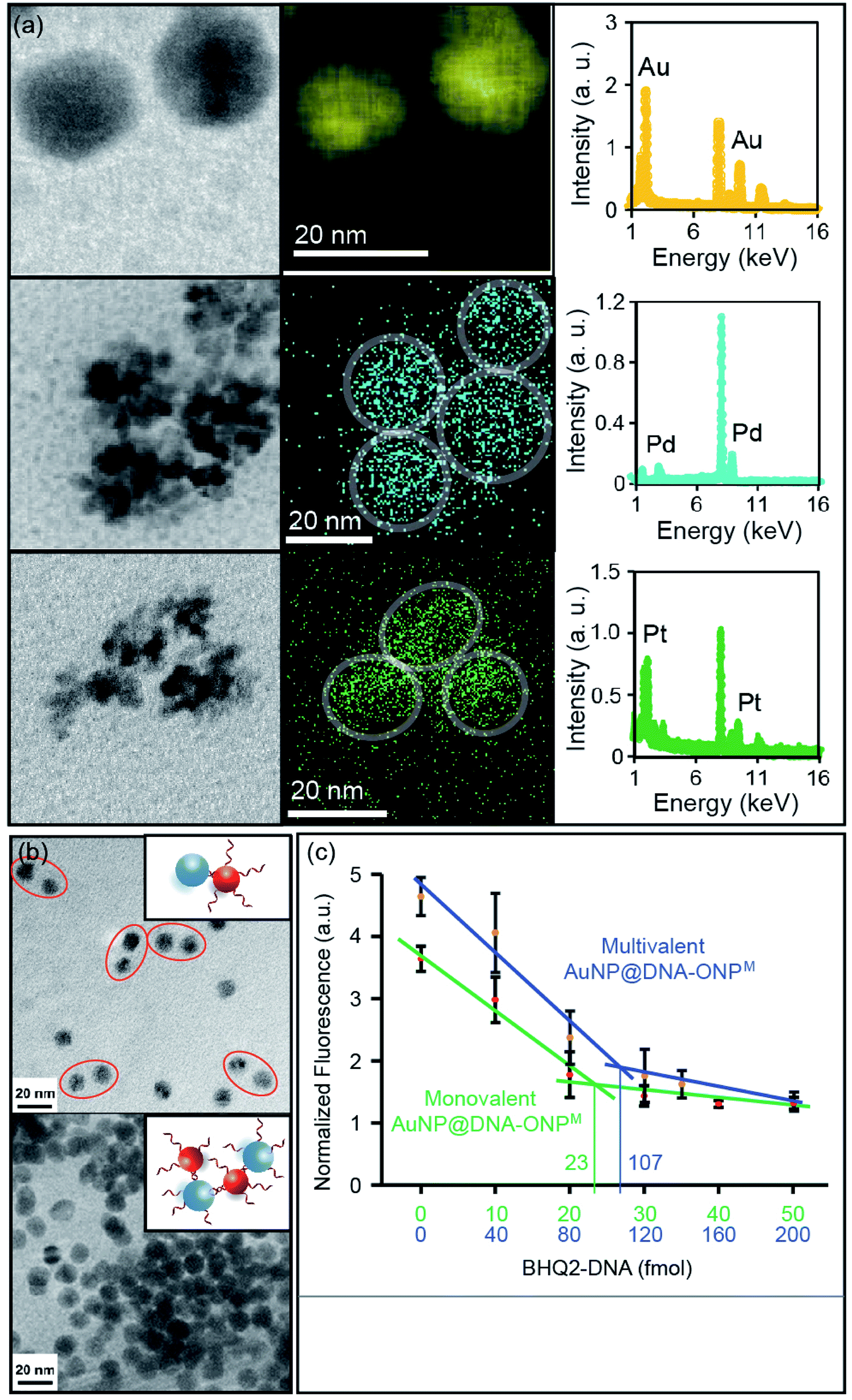
Metal nanoparticles (MNPs) are among the most widely used nanomaterials. Despite the many advances in preparing MNPs with different sizes and shapes, it remains challenging to achieve molecular level control over MNP surface functionality.20 We recently reported the conversion of monovalent DNA–ONP conjugates into DNA–MNP conjugates by a template-based approach,8 which suggested a possible way of converting organic-based DNA–NP conjugates into their metallic counterparts of different elemental compositions. A critical question is whether this process is applicable to other metals, sizes, and to multivalent DNA–ONP, and whether these parameters can be changed independently. To explore this possibility, other chlorometallates such as Na2PdCl4 and K2PtCl6 were tested for their potential to be deposited onto the ONP scaffolds with reduction. Indeed, PdNPs and PtNPs were successfully prepared using ONPM as a template (see ESI, including Fig. S14A and S15† for details). MNPs were characterized using scanning TEM (STEM) coupled with energy-dispersive X-ray spectroscopy (EDX). As shown in Fig. 6a, all three MNPs showed sizes of ca. 15–20 nm with their composition confirmed by EDX elemental mapping. PdNP and PtNP exhibited flower-shaped structures under STEM, possibly due to the high cohesive energy of Pd and Pt under the reduction conditions. The mechanism by which the nanoparticle forms has not been investigated, but likely involves an anion coordination-seeding-deposition process, analogous to that reported by Beer et al. (Fig. S16, ESI†).21 In this regard, the amide/imide groups may serve as coordinating groups.
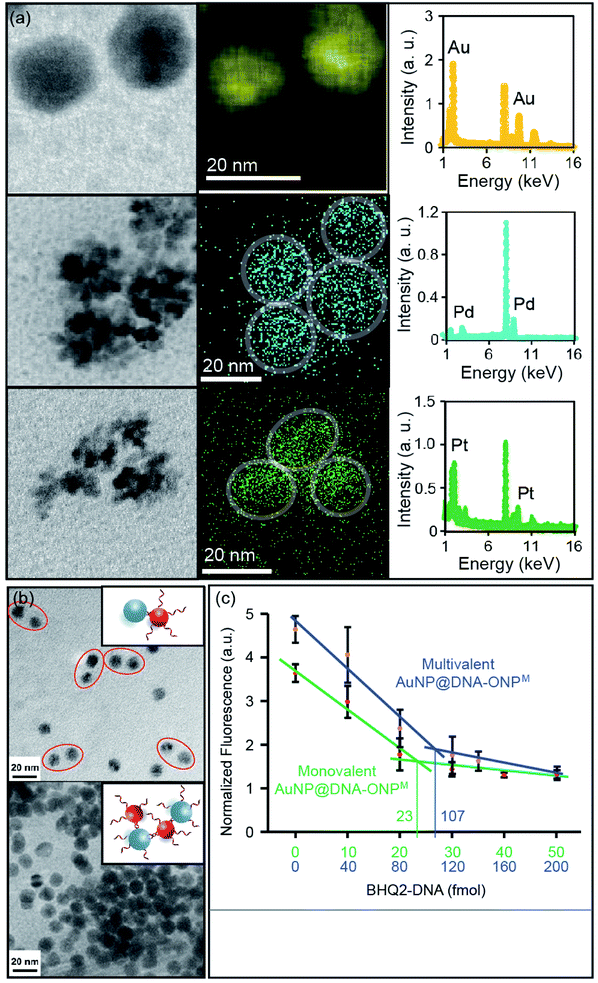 |
| | Fig. 6 (a) STEM and EDX element mapping of AuNP, PdNP and PtNP synthesized from ONPM. (b) Hybridization-mediated MNP dimerization using the AuNP@DNA–ONPM and commercial AuNP functionalized with complementary DNA strands. (c) Fluorescence titration plots of mono- and multivalent AuNP@DNA–ONPM. Note: the fluorescence intensity of the multivalent AuNP was normalized. Its actual fluorescence intensity was much stronger than the monovalent AuNP. | |
It is worth noting that an oligo-T20 sequence was inserted between the ONP and the functional part of the DNA, to provide a spacer for the MNP growth. The amount of metal ion used in the preparation was controlled to limit MNP growth within the ONP confines. Nonetheless, the T20 spacer ensured that the metal deposition would not interfere with the DNA strands, and allowed larger MNPs to be synthesized without disturbing the DNA functionality (Fig. S17, ESI†). To demonstrate that MNP growth occurred with preservation of the DNA biorecognition function, AuNPs were grown on ONPM–DNA conjugates (AuNP@DNA–ONPM, DNA = ERRed-M18bT20-SH). The observation of ERRed fluorescence served as evidence for the presence of DNA strands on the AuNP.
DNA strands on MNPs remain functional
Hybridization-mediated heterodimer or cross-linkage formation was achieved using monovalent and multivalent AuNP@DNA–ONPM, respectively, with commercial AuNPs containing complementary strands (Fig. 6b). Heterodimers were seen when monovalent AuNP@DNA–ONPM was used for assembly, whereas crosslinking and aggregation was observed when multivalent AuNP@DNA–ONPM was used. These assembly results clearly showed the preservation of DNA functionality after AuNP growth, and suggested the difference in valence between the monovalent and multivalent AuNPs from their corresponding templates. Fluorescence titrations were also conducted to quantify the number of DNA strands on the AuNP. Similar shaped plots were observed for both monovalent and multivalent AuNP@DNA–ONPM, but the fluorescence reached a plateau with ca. 5 times the concentration of quencher DNA for multivalent AuNP@DNA–ONPM compared to what was required for the monovalent analog (Fig. 6c). The amount of quencher DNA added matched the amount calculated from the estimated amount of AuNP. For PtNP and PdNP, it is difficult to estimate the amount of MNP formed because of their flower-shaped appearance, but similar fluorescence quenching plots could be clearly seen, indicating the proper functioning of the DNA on these MNPs (Fig. S18, ESI†).
Conclusions
In summary, a polymer-based, bottom-up strategy to synthesize various DNA–NP conjugates with tunable size, valence and elemental composition was established. Because of the precise chemical control over the polymeric scaffold, this strategy allows independent and simultaneous control over several essential parameters for DNA–NP conjugates, which are difficult to achieve by conventional methods. Using this strategy, nanoparticles of three different sizes (diameter = 6–10 nm, 15–20 nm and 30–40 nm, respectively), of three different valence numbers (monovalent and two different multivalent examples), and of four elemental compositions (organic, gold, platinum and palladium) were prepared. We demonstrated their potential application in assembly, ratiometric sensing, and targeted cellular uptake, and more potential applications of these DNA–NP conjugates can be envisioned. This new strategy should also be applicable to polymeric nanoparticles with other shapes and sizes and carrying other functionalities. Thus, more complicated nanoscale structures may become feasible using this template approach, enabling an even broader range of DNA–NP-based applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors thank the support from the National Science Foundation (CHE-1307404, CHE-1709718, and CTS-0120978), the US-Israel Binational Science Foundation (2014116), the National Institutes of Health (GM124316) and the National Natural Science Foundation of China (21877033, 21877032).
Notes and references
-
(a) C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607 CrossRef CAS PubMed;
(b) A. P. Alivisatos, K. P. Johnsson, X. G. Peng, T. E. Wilson, C. J. Loweth, M. P. Bruchez and P. G. Schultz, Nature, 1996, 382, 609 CrossRef CAS PubMed;
(c) F. A. Aldaye, A. L. Palmer and H. F. Sleiman, Science, 2008, 321, 1795 CrossRef CAS PubMed.
-
(a) A. V. Pinheiro, D. R. Han, W. M. Shih and H. Yan, Nat. Nanotechnol., 2011, 6, 763 CrossRef CAS PubMed;
(b) A. Kuzyk, R. Schreiber, Z. Y. Fan, G. Pardatscher, E. M. Roller, A. Hogele, F. C. Simmel, A. O. Govorov and T. Liedl, Nature, 2012, 483, 311 CrossRef CAS PubMed;
(c) S. J. Tan, M. J. Campolongo, D. Luo and W. L. Cheng, Nat. Nanotechnol., 2011, 6, 268 CrossRef CAS PubMed;
(d) D. Nykypanchuk, M. M. Maye, D. van der Lelie and O. Gang, Nature, 2008, 451, 549 CrossRef CAS PubMed;
(e) G. Tikhomirov, S. Hoogland, P. E. Lee, A. Fischer, E. H. Sargent and S. O. Kelley, Nat. Nanotechnol., 2011, 6, 485 CrossRef CAS PubMed;
(f) W. L. Cheng, M. J. Campolongo, J. J. Cha, S. J. Tan, C. C. Umbach, D. A. Muller and D. Luo, Nat. Mater., 2009, 8, 519 CrossRef CAS PubMed.
-
(a) J. Zhang, S. Song, L. Wang, D. Pan and C. Fan, Nat. Protoc., 2007, 2, 2888 CrossRef CAS PubMed;
(b) J. Liu and Y. Lu, Angew. Chem., Int. Ed., 2006, 45, 90 CrossRef CAS PubMed.
-
(a) Z. D. Wang and Y. Lu, J. Mater. Chem., 2009, 19, 1788 RSC;
(b) C. Teller and I. Willner, Curr. Opin. Biotechnol., 2010, 21, 376 CrossRef CAS PubMed;
(c) R. Chhabra, J. Sharma, Y. Liu, S. Rinker and H. Yan, Adv. Drug Delivery Rev., 2010, 62, 617 CrossRef CAS PubMed;
(d) Y. W. C. Cao, R. C. Jin and C. A. Mirkin, Science, 2002, 297, 1536 CrossRef CAS PubMed;
(e) X. J. Zhao, R. Tapec-Dytioco and W. H. Tan, J. Am. Chem. Soc., 2003, 125, 11474 CrossRef CAS PubMed.
- N. T. K. Thanh, N. Maclean and S. Mahiddine, Chem. Rev., 2014, 114, 7610 CrossRef CAS PubMed.
- For some selected reports on template synthesis of nanoparticles, see:
(a) K. Suzuki, S. Sato and M. Fujita, Nat. Chem., 2010, 2, 25 CrossRef CAS PubMed;
(b) Y. Wei, S. Han, D. A. Walker, P. E. Fuller and B. A. Grzybowski, Angew. Chem., Int. Ed., 2012, 51, 7435 CrossRef CAS PubMed;
(c) Y. Jin and X. Gao, Nat. Nanotechnol., 2009, 4, 571 CrossRef CAS PubMed;
(d) M. Mertig, L. Colombi Ciacchi, R. Seidel, W. Pompe and A. De Vita, Nano Lett., 2002, 2, 841 CrossRef CAS.
- Y. Bai, H. Xing, G. A. Vincil, J. Lee, E. J. Henderson, Y. Lu, N. G. Lemcoff and S. C. Zimmerman, Chem. Sci., 2014, 5, 2862 RSC.
- H. Xing, Y. Bai, Y. Bai, L. H. Tan, J. Tao, B. Pedretti, G. A. Vincil, Y. Lu and S. C. Zimmerman, J. Am. Chem. Soc., 2017, 139, 3623 CrossRef CAS PubMed.
-
(a) Y. Bai, H. Xing, P. Wu, X. Feng, K. Hwang, J. M. Lee, X. Y. Phang, Y. Lu and S. C. Zimmerman, ACS Nano, 2015, 9, 10227 CrossRef CAS PubMed;
(b) P. R. Patel, R. C. Kiser, Y. Y. Lu, E. Fong, W. C. Ho, D. A. Tirrell and R. H. Grubbs, Biomacromolecules, 2012, 13, 2546 CrossRef CAS PubMed.
- C. W. Bielawski and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1 CrossRef CAS.
- E. Harth, B. V. Horn, V. Y. Lee, D. S. Germack, C. P. Gonzales, R. D. Miller and C. J. Hawker, J. Am. Chem. Soc., 2002, 124, 8653 CrossRef CAS PubMed.
- L. H. Tan, H. Xing, H. Chen and Y. Lu, J. Am. Chem. Soc., 2013, 135, 17675 CrossRef CAS PubMed.
-
(a) J. Liu, Z. Cao and Y. Lu, Chem. Rev., 2009, 109, 1948 CrossRef CAS PubMed;
(b) W. Tan, M. J. Donovan and J. Jiang, Chem. Rev., 2013, 113, 2842 CrossRef CAS PubMed;
(c) O. I. Wilner and I. Willner, Chem. Rev., 2012, 112, 2528 CrossRef CAS PubMed.
-
(a) D. S. Seferos, D. A. Giljohann, H. D. Hill, A. E. Prigodich and C. A. Mirkin, J. Am. Chem. Soc., 2007, 129, 15477 CrossRef CAS PubMed;
(b) P. Wu, K. Hwang, T. Lan and Y. Lu, J. Am. Chem. Soc., 2013, 135, 5254 CrossRef CAS PubMed;
(c) Z. Yang, K. Y. Loh, Y.-T. Chu, R. Feng, N. S. R. Satyavolu, M. Xiong, S. M. N. Huynh, K. Hwang, L. Li, H. Xing, X. Zhang, Y. R. Chemla, M. Gruebele and Y. Lu, J. Am. Chem. Soc., 2018, 140, 17656 CrossRef CAS PubMed.
-
(a) X. J. Peng, J. J. Du, J. L. Fan, J. Y. Wang, Y. K. Wu, J. Z. Zhao, S. G. Sun and T. Xu, J. Am. Chem. Soc., 2007, 129, 1500 CrossRef CAS PubMed;
(b) Z. C. Xu, K. H. Baek, H. N. Kim, J. N. Cui, X. H. Qian, D. R. Spring, I. Shin and J. Yoon, J. Am. Chem. Soc., 2010, 132, 601 CrossRef CAS PubMed.
-
(a) J. Liu and Y. Lu, Anal. Chem., 2004, 76, 1627 CrossRef CAS PubMed;
(b) Y. Xiang, A. J. Tong and Y. Lu, J. Am. Chem. Soc., 2009, 131, 15352 CrossRef CAS PubMed.
-
(a) M. Howarth, W. Liu, S. Puthenveetil, Y. Zheng, L. F. Marshall, M. M. Schmidt, K. D. Wittrup, M. G. Bawendi and A. Y. Ting, Nat. Methods, 2008, 5, 397 CrossRef CAS PubMed;
(b) J. Farlow, D. Seo, K. E. Broaders, M. J. Taylor, Z. J. Gartner and Y.-W. Jun, Nat. Methods, 2013, 10, 1203 CrossRef CAS PubMed.
-
(a) H. Liang, X.-B. Zhang, Y. Lyu, L. Gong, R. Wang, X. Zhu, R. Yang and W. Tan, Acc. Chem. Res., 2014, 47, 1891 CrossRef CAS PubMed;
(b) D. S. Lee, H. Qian, C. Y. Tay and D. T. Leong, Chem. Soc. Rev., 2016, 45, 4199 RSC.
-
(a) H. Xing, L. Tang, X. Yang, K. Hwang, W. Wang, Q. Yin, N. Y. Wong, L. W. Dobrucki, N. Yasui, J. A. Katzenellenbogen, W. G. Helferich, J. Cheng and Y. Lu, J. Mater. Chem. B, 2013, 1, 5288 RSC;
(b) Z. Cao, R. Tong, A. Mishra, W. Xu, G. C. L. Wong, J. Cheng and Y. Lu, Angew. Chem., Int. Ed., 2009, 48, 6494 CrossRef CAS PubMed.
-
(a) C. Kruger, S. Agarwal and A. Greiner, J. Am. Chem. Soc., 2008, 130, 2710 CrossRef PubMed;
(b) J. Cheon and J. H. Lee, Acc. Chem. Res., 2008, 41, 1630 CrossRef CAS PubMed;
(c) H. Wang, D. W. Brandl, P. Nordlander and N. J. Halas, Acc. Chem. Res., 2007, 40, 53 CrossRef CAS PubMed;
(d) A. H. Latham and M. E. Williams, Acc. Chem. Res., 2008, 41, 411 CrossRef CAS PubMed;
(e) K. M. L. Taylor-Pashow, J. Della Rocca, Z. G. Xie, S. Tran and W. B. Lin, J. Am. Chem. Soc., 2009, 131, 14261 CrossRef CAS PubMed.
- C. J. Serpell, J. Cookson, D. Ozkaya and P. D. Beer, Nat. Chem., 2011, 3, 478 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional characterization data and details on the synthesis, methods and reagents. See DOI: 10.1039/c9sc05656d |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Hang
Xing‡
abc,
Yunhao
Bai
a,
Li Huey
Tan
a,
Kevin
Hwang
a,
Ji
Li
a,
Yi
Lu
ab,
Hang
Xing‡
abc,
Yunhao
Bai
a,
Li Huey
Tan
a,
Kevin
Hwang
a,
Ji
Li
a,
Yi
Lu